

Abstract
Monoclonal antibodies targeting GD2 ganglioside (GD2) have recently been approved for the treatment of high risk neuroblastoma and are extensively evaluated in clinics in other indications. This study illustrates how a therapeutic antibody distinguishes between different types of gangliosides present on normal and cancer cells and informs how synthetic peptides can imitate ganglioside in its binding to the antibody. Using high resolution crystal structures we demonstrate that the ganglioside recognition by a model antibody (14G2a) is based primarily on an extended network of direct and water molecule mediated hydrogen bonds. Comparison of the GD2-Fab structure with that of a ligand free antibody reveals an induced fit mechanism of ligand binding. These conclusions are validated by directed mutagenesis and allowed structure guided generation of antibody variant with improved affinity toward GD2. Contrary to the carbohydrate, both evaluated mimetic peptides utilize a “key and lock” interaction mechanism complementing the surface of the antibody binding groove exactly as found in the empty structure. The interaction of both peptides with the Fab relies considerably on hydrophobic contacts however, the detailed connections differ significantly between the peptides. As such, the evaluated peptide carbohydrate mimicry is defined primarily in a functional and not in structural manner.
Malignant transformation is universally accompanied by changes in cell surface glycosylation. A glycolipid, GD2 ganglioside (GD2)1, is one of the most prominent tumor-associated antigens, ranking in the 12th position of the NCI prioritized list of cancer vaccine targets (1). GD2 is embedded in the outer plasma membrane with its ceramide tail (fatty acid coupled sphingosine). The sugar moiety is exposed to the extracellular milieu and is composed of glucose (Glc; linked to ceramide), galactose (Gal) and N-acetylgalactosamine (GalNAc). Two additional sialic acid residues (N-acetylneuraminic acid, NeuAc) branch form Gal and provide GD2 with a negative charge (Fig. 1). Overexpression of GD2 is well documented in neuroblastoma, melanoma, certain osteosarcomas, small cell lung cancers, and soft tissue sarcomas (2–4).
Fig. 1.

Recognition of GD2 ganglioside by monoclonal antibody 14G2a at the cell surface. (top panel) Antigen combining region of 14G2a antibody recognizes the sugar moiety of GD2 ganglioside (yellow), which is exposed to the extracellular milieu. The lipid part of the ganglioside is buried inside the cell membrane. GD2 bound Fab structure determined in this study is shown in color. Fc fragment (PDB ID: 1igt) and membrane model derived from published data are shown in corresponding scale and colored gray. (bottom panel) Chemical structure of GD2 ganglioside and sugar ring nomenclature used throughout the study.
The concept of therapeutic targeting of GD2 is currently most advanced in neuroblastoma, the most common extracranial tumor of childhood. Neuroblastoma is a heterogenous and complex disease. Spontaneous remissions are sometimes observed, but more than a half of the patients are diagnosed with a high-risk neuroblastoma of poor prognosis. This highlights the demand for treatment modalities that would offer major clinical benefits for this group of patients (5). High and stable presence of GD2 on cancer cells in neuroblastoma and limited expression on relevant normal tissues (i.e. neurons, peripheral nerve fibers and skin melanocytes) allows diagnosis, detection of metastases, treatment monitoring and, most importantly, targeting of the tumor itself.
GD2-specific monoclonal antibodies have been extensively tested in clinics. This includes a mouse 14G2a antibody (IgG2a; derived from a mouse 14.18 antibody of IgG3 subclass), and improved modifications thereof including a chimeric antibody ch14.18, and recently a humanized antibody hu14.18K322A. Moreover, mouse 3F8 antibody (IgG3) and recently its humanized derivative hu3F8 were also evaluated. The antibodies were demonstrated to engage antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) against neuroblastoma (5). Additionally, direct cytotoxic effects were observed in neuroblastoma models (6). The results of a randomized clinical trial published in 2010, evaluating ch14.18, interleukin-2 and granulocyte and macrophage-colony stimulating factor combined with a standard maintenance agent 13-cis retinoic acid demonstrated significant improvement of outcome in high-risk neuroblastoma patients (7). Based on these and further findings, the Food and Drug Administration (FDA) has just recently approved Unituxin (dinutuximab; ch14.18) combination therapy for high risk neuroblastoma (8). Therefore, the standard care treatment protocols may now be extended with monoclonal antibodies targeting GD2 for a better expected outcome.
Antibodies against gangliosides other than GD2 are considered as potential therapeutic agents in different types of cancer. Ganglioside-specific antibodies are moreover involved in various types of autoimmune diseases (9). Nevertheless, the molecular mechanism of ganglioside recognition remains unknown because not a single crystal structure of antibody–ganglioside complex has been determined to date. In particular, it is not known how the specificity against GD2 is achieved in antibodies evaluated in clinics. Although crystal structures of empty ME36.1 antibody specific for GD2 and GD3 (10) and empty 3F8 antibody specific for GD2 (11) were determined, the conclusions concerning GD2 binding have to be treated with caution because of general limitations in reliable prediction of binding modes of complex, flexible ligands in dynamic pockets.
The success of GD2-specific antibodies in treatment of neuroblastoma fuels investigation on active immunization strategies. To overcome poor antigenicity of GD2, glycolipid surrogates including peptide mimetics are being developed. The idea of a peptide vaccine eliciting anticarbohydrate response has been precedented in the case of Group B Streptococcus polysaccharide (12). Multiple peptides mimicking GD2 in its binding to specific antibodies were selected using phage display (13, 14) and some have been demonstrated to elicit protective, GD2 directed response in preclinical studies. However, the structural basis of peptide-ganglioside mimicry and its relation to the potential of particular peptides to induce GD2 directed immune response remain unknown.
Here, we analyze the interactions guiding ganglioside recognition by an antibody and the structural basis of peptide-ganglioside mimicry. The crystal structure of Fab fragment of 14G2a antibody in a complex with the sugar moiety of GD2 ganglioside is provided and the binding mode is discussed in detail. Structure of an empty 14G2a antibody is reported for reference. The major conclusions are verified by directed mutagenesis and antibody variant with increased affinity toward GD2 is developed using structure guided approach. The binding modes of two largely divergent peptide mimics of GD2 (15) at the antigen-binding site of 14G2a antibody are reported and compared with that of the carbohydrate. Mouse 14G2a antibody was chosen for this study because it contains the same antigen binding region as the ch14.18 chimeric antibody recently approved by FDA (8).
EXPERIMENTAL PROCEDURES
Antibody Purification and Obtaining the Fab Fragment
The hybridoma cell line producing mouse 14G2a antibody (16) was kindly provided by R.A. Reisfeld from the Scripps Institute, La Jolla, CA. The antibody was purified from FBS-free medium using immobilized Protein G (GE Healthcare, Uppsala, Sweden). The preparation was dialyzed against 10 mm Tris HCl, pH 7.0 containing 50 mm NaCl. Fab fragments were obtained by digestion with immobilized papain (Pierce, Rockford, IL) for 4.5 h at 37 °C after supplementing the buffer with 14 mm cysteine and 1.4 mm EDTA. The Fab fragments were purified from the remaining uncleaved antibodies and Fc using immobilized protein A (Pierce).
Expression and Purification of Single-chain Variable Fragment of 14G2a (scFv-14G2a) and its Variants
Expression construct for scFv-14G2a was designed according to (17) with modifications. In brief, a synthetic, codon optimized gene encoding VH domain (Glu1-Ser113), a (Gly4Ser)3 linker, and VL domain (Asp1-Lys113) was obtained and cloned into pET22b(+) expression vector (Genscript, Piscataway, NJ). The pelB sequence was removed by overlap extension PCR and mutants were obtained by site directed mutagenesis. The proteins were expressed in Escherichia coli strain SHuffle T7 competent cells (NEB, Ipswich, MA). Bacteria were cultured in Luria-Bertani broth at 37 °C until OD600 reached 0.8. Protein expression was induced with isopropyl-β-d-1-thiogalactopyranoside (0.1 mm), the temperature was lowered to 25 °C and the culture was continued for 16 h. Bacterial cells were harvested by centrifugation, re-suspended in 50 mm Tris pH 7.9 containing 150 mm NaCl and lysed by sonication. Debris was removed by centrifugation and soluble scFv was recovered by metal affinity chromatography and dialyzed against PBS. The concentration of scFv variants was normalized at that of the wild type by SDS-PAGE densitometry and quantitative Western blotting (detected with HRP-labeled anti-His-tag mAbs, clone HIS-1, Sigma-Aldrich, Poznan, Poland).
Evaluation of GD2 Recognition by 14G2a Based scFv and its Variants by ELISA and Flow Cytometry
For ELISA, MaxiSorp strips (Nunc, Roskilde, Denmark) were coated with ethanol solubilized GD2 or GD3 (50 ng per well) by evaporating the solvent at room temperature and blocked with nonfat dry milk (NFDM). Tested samples (i.e. scFv variants, 14G2a antibody, and anti-GD3 mAb (clone ME.3.6, BD Biosciences, Warszawa, Poland)) were diluted in 0.01% NFDM and incubated overnight at 4 °C in the wells. Binding of scFv variants was detected by HRP labeled anti-His-tag mAb (clone HIS-1, Sigma-Aldrich). HRP labeled rabbit anti-mouse IgG (Sigma-Aldrich) was used to detect control antibodies. All washing steps were performed with 0.05% Tween-20 in PBS. Signal was developed using OptiELISA TMB substrate (BD Biosciences), the reaction was stopped with 0.18 m H2SO4 and the absorbance at 450 nm was recorded.
For flow cytometry, human neuroblastoma cell line CHP-134 (ECACC, London, UK) characterized by high expression of GD2 (6), mouse neuroblastoma NXS2 (kindly provided by L. Raffaghello, G. Gaslini Insititute, Genova, Italy) with heteregenous expression of GD2 (18), and GD2-negative human neuroblastoma SK-N-SH (ATCC; LCG Standards, Lomianki, Poland) (6) were used to analyze the binding of scFv variants. 14G2a was used as a control. The cells were collected by trypsinization, suspended in 2% FBS containing 0.1% NaN3 (used for all subsequent steps) and incubated with tested molecules for 30 min. on ice. Binding of scFv was detected with FITC-conjugated mouse anti-His mAb (Pierce), whereas that of 14G2a with FITC-conjugated, mouse IgG-specific goat F(ab′)2 (MP Biomedicals, Warszawa, Poland) by 20 min incubation on ice. The cells were fixed in 1% paraformaldehyde in PBS and signal was detected using flow cytometry (LSRFortessa, BD Biosciences). The data was analyzed using FACSDiva software (BD Biosciences).
Crystallization
Fab fragment was subject to size exclusion chromatography on Superdex 75 in 5 mm Tris HCl, pH 8.0 containing 50 mm NaCl and concentrated to 10 mg/ml. Peptide 1 (RCNPNMEPPRCWAAEGD) or peptide 2 (VCNPLTGALLCSAAEGD; both from Genscript) (15) were incubated overnight with the Fab fragment at 3:1 molar ratio at 4 °C and used for crystallization. GD2 sugar (GalNAcβ4(Neu5Acα8Neu5Acα3)Galβ4Glc; Elicityl, Crolles, France) was incubated with the Fab fragment in conditions similar to those used for both peptides. Antibody-GD2 complex was additionally purified by gel filtration using Superdex 75 equilibrated with 10 mm Tris HCl, 50 mm NaCl, pH 8.0, and concentrated to 10 mg/ml prior screening. Screening of crystallization conditions was performed using sitting drop vapor diffusion method and buffer sets available from Hampton Research (Aliso Viejo, CA) and prepared in house according to (19). In all cases crystals appeared in multiple different conditions after several days to few weeks and were further optimized for size and shape. Nevertheless, the majority of those crystals diffracted only to low resolution or the diffraction pattern demonstrated significant defects. This was especially true for the Fab ganglioside sugar complex where all evaluated crystals save one either diffracted poorly or contained only empty Fab.
Crystals obtained from the following conditions were used for structure determination: empty Fab (0.1 m Hepes pH 7.8 containing 20% PEG 4000 (w/v) and 10% isopropanol (v/v)), GD2 complex (0.1 m Bis-tris pH 6.5 containing 25% PEG 3350 (w/v) and 0.2 m NaCl), peptide 1 complex (0.1 m Bis-Tris pH 5.5 containing 18% PEG 5000 (w/v)), peptide 2 complex (P1 space group - 0.1 m Hepes pH 7.1 containing 20% PEG 4000 and 10% isopropanol; P21 space group - 0.1 m MES pH 6.5 containing 28% PEG 5000 and 0.2 m NH4SO4).
Structure Solution
The diffraction data were collected at 100 K at the EMBL beamline P13 at PETRA III (DESY, Hamburg, Germany) or using a rotating anode copper source (Rigaku, Sevenoaks, UK) for crystal containing peptide 1. Data was integrated using xds (20) or MOSFLM (21). Further computations were performed using programs contained in the CCP4 package (22). The data was scaled using SCALA (23). Initial phases were obtained by molecular replacement using Phaser (24) with Fab fragment (PDB ID: 1F8T) as a search model. The model was manually constructed in the resulting electron density maps using Coot (25). Restrained refinement was performed using Refmac 5.0 (26) and Phenix (27). Five percent of reflections were used for cross-validation analysis (28), and the behavior of Rfree was employed to monitor the refinement strategy. Water molecules were added using Coot and subsequently manually inspected. The final models were deposited to the Protein Data Bank under accession numbers: 4TRP, 4TUJ, 4TUK, 4TUL, and 4TUO. The data collection and refinement statistics are summarized in Table I.
Table I. Data collection and refinement statistics.
| Ligand | None | Peptide 1 | Peptide 2 | Peptide 2 | GD2 |
|---|---|---|---|---|---|
| (PDB ID) | (4TRP) | (4TUJ) | (4TUK) | (4TUL) | (4TUO) |
| Data collection | |||||
| Space group | P1 | P1 | P1 | P1 21 1 | P1 |
| Cell dimensions | |||||
| a, b, c (Å) | 42.93 | 43.38 | 41.58 | 42.68 | 58.09 |
| 46.93 | 69.82 | 42.54 | 64.17 | 61.09 | |
| 62.36 | 78 | 59.52 | 78.90 | 65.36 | |
| α, β, γ (°) | 110.81 | 111.45 | 86.01 | 90.00 | 107.79 |
| 92.8 | 101.14 | 73.92 | 99.46 | 113.43 | |
| 100.13 | 90.35 | 87.39 | 90.00 | 95.68 | |
| Resolution (Å)a | 57.86–1.25 (1.32–1.25) | 40.84–1.89 (1.99–1.89) | 57.09–1.6 (1.657–1.6) | 77.83–1.4 (1.45–1.4) | 56.28–1.55 (1.605–1.55) |
| Rmergea | 0.053 (0.457) | 0.069 (0.548) | 0.056 (0.260) | 0.0292 (0.176) | 0.068 (0.433) |
| I/σIa | 10.99 (2.71) | 9.66 (1.85) | 17.38 (3.76) | 14.52 (3.98) | 11.0 (2.7) |
| Completeness (%)a | 89.14 (86.70) | 90.92 (84.54) | 90.68 (90.66) | 98.96 (99.87) | 90.5 (89.7) |
| Redundancya | 3.8 (3.8) | 3.9 (3.9) | 3.8 (3.8) | 2.0 (2.0) | 3.8 (3.8) |
| Refinement | |||||
| Resolution (Å)a | 57.86–1.25 (1.32–1.25) | 40.84–1.89 (1.99–1.89) | 57.09–1.6 (1.657–1.6) | 77.83–1.4 (1.45–1.4) | 56.28–1.55 (1.605–1.55) |
| No. reflections | 419818 | 240380 | 179530 | 163073 | 381810 |
| Rwork/Rfree | 0.149/0.178 | 0.179/0.232 | 0.162/0.206 | 0.188/0.217 | 0.166/0.206 |
| No. atoms | 3797 | 6746 | 3706 | 3628 | 7332 |
| Protein | 3272 | 6313 | 3357 | 3311 | 6488 |
| Water | 525 | 433 | 348 | 300 | 688 |
| Average B-factor | 19.80 | 42.30 | 17.90 | 15.80 | 16.90 |
| Protein | 18.30 | 42.50 | 17.20 | 15.30 | 16.30 |
| Water | 28.60 | 39.60 | 25.30 | 21.30 | 23.00 |
| R.m.s. deviations | |||||
| Bond lengths (Å) | 0.005 | 0.012 | 0.021 | 0.023 | 0.021 |
| Bond angles (°) | 1.08 | 1.39 | 2.11 | 2.27 | 2.09 |
a Values in parentheses are for highest-resolution shell.
In Silico Estimation of the Effect of Point Mutations Within 14G2a on its Affinity Toward GD2
Linear interaction energy (LIE) method (29) was used to estimate the impact of point mutations on the free energy (ΔG) of ligand binding. Energy data was derived from molecular dynamics (MD) simulations. Fragments missing in the 14G2a:GD2 crystal structure were added using Modeler (30). Mutations within 14G2a were introduced using VMD (31). The ligand topology was constructed using Automated Topology Builder (32) and adjusted to GROMOS96 (33) force field used in all simulations. Complexes were solvated and relaxed using steepest decent algorithm followed by MD with position restrains on protein backbone atoms. Each system was subjected to simulated annealing (SA) followed by a production run of unrestrained MD performed using GROMACS (34). See supplemental Information for details of computational procedures. The effect of each evaluated mutation on GD2 binding was estimated as ΔΔG = ΔGmutant_complex - ΔGWT_complex.
RESULTS
Crystal Structure of Fab Fragment of 14G2a Antibody
The crystal structure of Fab fragment of 14G2a antibody was refined to 1.25 Å resolution and contains a single molecule in the asymmetric unit (Table I). The protein adopts a characteristic immunoglobulin fold. The entire molecule is well defined by electron density safe for heavy-chain first-complementarity-determining region (CDR H1). The V-shaped antigen binding groove runs parallel to the interface of variable light and heavy chains. It is ca. 15 Å deep and filled at its bottom with a number of water molecules of clearly defined electron density. Three of these water molecules located at the very bottom of the cavity have temperature factors comparable to the surrounding residues suggesting their tight coordination. The antigen combining site is open to a solvent channel, which ensures that its structure is not significantly influenced by crystal packing interactions. This is important because the structure of empty 14G2a antibody serves as a reference to probe the binding mechanism of GD2 and mimicking peptides in further discussion.
Crystal Structure of 14G2a Fab - GD2 Sugar Complex
The crystal structure of 14G2a Fab fragment in complex with GD2 pentacarbohydrate moiety (Fig. 1) has been refined to 1.55 Å resolution (Table I). The structure accommodates two Fab molecules in the asymmetric unit, each containing a carbohydrate ligand. The entire CDR region and the ligand are well defined by electron density in each molecule. The carbohydrate binding mode in the two molecules contained in asymmetric unit is virtually identical and as such, both molecules are discussed collectively.
Recognition of GD2 at the antigen binding groove of 14G2a is facilitated by an extended network of direct and water mediated hydrogen bonds (Fig. 2). CDR loops H1, H2, L3 and L1 contribute the majority of the interactions with the ligand whereas loops H3 and L2 only approach NeuAc1. NeuAc2 is located at the bottom of the binding cavity and all capable atoms of the sialic acid are involved in hydrogen bonds with the antibody, including the sidechains of HAsn35, LSer96, LHis39, LLys55, and the main chain of HGly99 and HAsn33, as well as the surrounding water molecules. The sidechains of HTyr32 and LTyr37 contribute weak hydrophobic contacts with the sugar ring, but no CH-π stacking is involved. NeuAc1 is involved in hydrogen bond contacts with the sidechains of HAsn33 and HAsp52. Remarkably, the antigen binding groove does not tightly complement the GD2 sugar. A large cavity remains between CDR H2 and NeuAc1. This cavity is filled with water molecules, which are clearly defined by electron density and are characterized by relatively low temperature factors. The water molecules compensate the lack of direct complementarity of the antigen binding groove and the sugar by mediating hydrogen bonds between the sialic acid moiety of GD2 and CDR H2 of the antibody (Figs. 2B, 2C, and 6C). GalNAc contributes hydrogen bonds to the main chain carbonyl oxygens of LSer96, LThr97, and LVal99, and the sidechains of LHis31 and LArg32. Gal is located at the entrance of the binding cavity, but is still involved in hydrogen bond contacts with LArg32 and LAsn33 (in the second molecule contained in the asymmetric unit the disposition of LArg32 is influenced by crystal packing interactions such that it does not contact the ganglioside). Water mediated hydrogen bonds within the ganglioside sugar contribute to its rigid structure at the binding cavity. GD2 residues interconnected via a water molecule include NeuAc2 and NeuAc1, NeuAc1 and Gal, NeuAc1 and GalNAc, and Gal and Glc. The terminal Glc residue does not contribute to GD2-antibody interaction. It adopts distinct orientations in the two molecules contained in the asymmetric unit, each influenced in a different way by interactions with adjacent, symmetry related antibody molecules.
Fig. 2.

GD2 ganglioside binding mode at the antigen-combining site of 14G2a. (A) GD2 (yellow) recognition relies heavily on the hydrogen bond interactions with the main chain and the side chains of both the heavy (blue) and the light (green) chains of the antibody. Antibody residues contributing major interactions are shown as sticks. Hydrogen bonds are depicted as black dotted lines. Gray mesh represents Fo-Fc omit map for the ganglioside contoured at 1.5σ. (B) Schematic representation of the major interactions guiding the recognition and specificity of 14G2a antibody toward GD2 sugar. (C) Spatial view of interactions depicted schematically in panel B. (D) Schematic representation of the steric determinants that dictate the strict preference of 14G2a toward GD2, but not closely related gangliosides GD1b, GD3 and GM2.
Fig. 6.

Comparison of the binding modes of GD2 and peptide mimetics at the 14G2a antigen combining site. The general disposition of the main chain of peptide 1 (A) and peptide 2 (B) imitates the overall shape of the ganglioside sugar (yellow) because of general steric constraints imposed by the antigen binding cavity. However, the detailed interactions differ significantly as exemplified: (A–B) at the bottommost pocket of the binding cavity the carbohydrate is involved primarily in hydrogen bond interactions whereas both peptides expose hydrophobic side chains. (C) The water molecule mediated interactions of GD2 with CDR H2 are replaced by hydrophobic contacts in the case of peptide 1 (D) and peptide 2 (E). (all panels) Peptides are represented as a main-chain trace with the discussed residues shown as sticks.
Binding of GD2 induces large structural rearrangement within the antibody. The entry of the binding groove in the empty structure is ca. 25 Å wide whereas in the ligand bound structure it is only ca. 21 Å wide (Fig. 3). Interestingly, the disposition of the majority of the sidechains exposed into the binding groove relative to their direct neighbors remains almost identical in both structures. Instead, it is the entire CDR loops that all cooperatively close around the ligand upon binding. Interaction with the ligand also stabilizes CDR H1. A part of this loop is not defined by electron density in the ligand free structure, but the entire region is ordered in the carbohydrate containing structure. NeuAc2 displaces a number of water molecules, which constitute a well-defined network in the ligand free structure. At the same time ligand binding organizes a defined water structure in a pocket formed by CDR H2, L3 and the ligand itself, which the cavity is not present in a ligand free structure. Overall, GD2 binding by 14G2a is best described by an induced fit model involving cooperative closing of all CDR loops around the ligand and reorganization of a water structure within the antigen binding cavity.
Fig. 3.
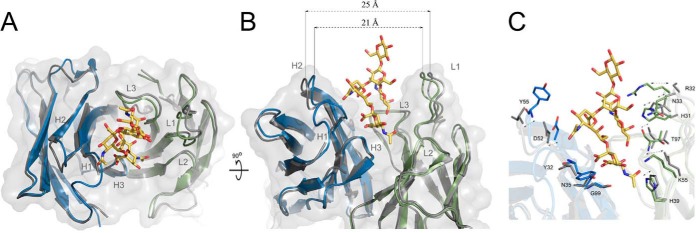
Ganglioside binding to 14G2a follows an induced fit mechanism. (A) Overlay of ligand free (gray) and GD2 (yellow) containing (heavy and light chains in blue and green, respectively) structures of 14G2a antibody. Ganglioside binding induces narrowing of the V shaped binding groove associated with cooperative closing of antibody CDR loops around the ligand. (B) The entrance to the groove is 25 Å wide in the ligand free structure and only 21 Å wide in the ligand containing structure. The largest movement is associated with CDR loops H2 and L1, but all other loops also close around the ligand. (C) Same view as in panel B, but the sidechains that undergo most pronounced translations and rearrangements upon ligand binding are shown in ligand free (gray) and ligand bound (color) antibody structures.
Directed Mutagenesis of scFv-14G2a Supports the Conclusions Drawn from Cocrystal Structure and Allows to Manipulate the scFv Affinity Toward GD2
To validate the above described binding mode of GD2, we have evaluated single residue mutants designed to abolish the most prominent intermolecular interactions observed in the cocrystal structure. Because of the complicated nature of genetic manipulations within hybridoma cell lines, to allow straightforward generation and expression of mutants we have devised a recombinant expression system in bacteria for the antigen binding region of 14G2a in a form of a single chain variable fragment (scFv). Analogically to 14G2a, scFv-14G2a (wild type) selectively recognized GD2 over GD3 as determined by ELISA and flow cytometry (data not shown). Expectedly, all the mutants designed to abolish prominent interactions observed within the crystal structure (i.e. HAsn33Ala, HAsn35Ala and LSer96Ala) have completely lost their ability to interact with GD2 both immobilized on plastic surface (as evaluated by ELISA) and present within the cell membrane of the CHP-134 cells (as evaluated by flow cytometry) (Fig. 4 and data not shown).
Fig. 4.

Relative affinity and specificity of scFv variants toward GD2. (A) Binding of scFv variants to GD2 as evaluated by ELISA. Plastic surface was coated with GD2 and incubated with particular scFv, which binding was detected using HRP-conjugated anti-His tag antibody; WT - wild type scFv, Control - signal from wells incubated with anti-His tag antibody alone. The statistical significance in ELISA signals between the wild type scFv-14G2a and its mutants was evaluated by t test (p values are indicated as follows p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***). (B, C) Binding of scFv variants to GD2 on the cell surface. CHP-134 (high GD2 expression), NXS2 (heteregenous expression of GD2) and SK-NS-H (GD2-negative) neuroblastoma cell lines were first incubated with evaluated scFv variants and then with FITC-labeled anti-His tag antibody. Staining was measured by flow cytometry. (B) Representative histograms showing binding of WT and HGlu101Lys variants of scFv (5 μg/ml) to tested neuroblastoma cells (dark gray) overlaid with control staining of the respective cells with FITC-conjugated anti-His tag antibody alone (light gray). Based on signals from cells stained with the latter antibody alone, positive cell pools were determined (gate P7) for which median fluorescence intensity (MFI) and/or percentage of staining (%) values were measured (as indicated on the plots). (C) MFI signals for single cell populations stained with particular scFv and FITC-conjugated anti-His tag antibody, or the latter antibody alone (control).
Having obtained a confirmation that conclusions predictive of in vitro activity can be drawn from the co-crystal structure, we attempted to modify the affinity of 14G2a toward GD2.To this end, in silico preselection of a set of single residue variants within the binding region was performed by comparing the calculated binding energies to that calculated for the wild type complex (see supplemental Fig. S1, S2). The most promising variants (i.e. LHis31Asn, HAla50Lys, LHis54Arg, HSer59Lys, HGlu101Lys) were expressed as scFvs and their relative affinity toward GD2 was evaluated. When tested by ELISA, LHis31Asn variant has completely lost its ability to bind GD2 (data not shown) and the binding of HAla50Lys was significantly reduced compared with the wild type (Fig. 4A). The affinity of LHis54Arg and HSer59Lys was comparable to the wild type, slightly decreased in numerical values, but this decrease has not reached statistical significance. Most importantly, however, the binding of HGlu101Lys was significantly improved compared with the wild type (Fig. 4A).
Comparable effects were observed when the ability of scFv variants to detect GD2 on the surface of CHP-134 and NXS2 cells was tested by flow cytometry (Fig. 4C). The above data collectively demonstrates that the provided here crystal structure fully explains the binding mode of GD2, which allows rational modification of antibody's affinity.
Crystal Structures of 14G2a Fab in Complex with GD2-Mimicking Peptides
Two peptide mimics of GD2 largely different in their primary structure were crystallized in complex with 14G2a antibody. The structure containing peptide 1 accommodates two antibody molecules in the asymmetric unit. The peptide ligand is well-defined by electron density in both molecules except for its C-terminal part, which extends beyond the binding site. The binding mode is virtually identical in both molecules contained in the asymmetric unit, which are therefore discussed collectively.
The major interactions with the antibody are contributed within the disulfide cyclized moiety of the peptide (Cys2-Cys11) whereas the extended fragment toward the C terminus is not defined by the electron density beyond Ala14 (Fig. 5A). The N-terminal part of the ligand contacts the antibody via a network of direct and water molecule mediated hydrogen bonds whereas hydrophobic contacts dominate beyond Glu7. The N-terminal amine of peptide 1 donates hydrogen bond to the sidechain of HAsn33 whereas the carbonyl of Arg1 accepts a hydrogen bond form the mainchain amine of HAsn33. In the second molecule contained in the asymmetric unit the interactions at the N terminus additionally involve HAsp52 and HGly31. The sidechain of Arg1 is not defined by electron density indicating that it does not significantly contribute to the interaction with the antibody. The sidechain of Asn3 is involved in hydrogen bonds with HGly99, the sidechain of HGlu101 and peptide residue n+2. A water molecule mediates hydrogen bond contacts between the carbonyl group of Pro4 and sidechain atoms of LTyr37, and LAsn35 or LLys55, respectively in the two molecules contained in the asymmetric unit. The main chain carbonyl of Asn5 accepts a hydrogen bond from the sidechain of LHis54 whereas the carbonyl of Met6 accepts hydrogen bonds from the sidechains of LHis39 and LSer96. The sidechain of Met6 anchors deep in the binding groove into a pocket formed by the sidechains of LHis39, LHis54 and the main chain between HGly99 and HGlu101. Peptide 1 tightly fills the antigen binding cleft on the side of CDR H1, H3, L1, and L2 but poorly explores the cavity on the side of CDRs L3 and H2. A spacious, almost 15 Å deep pocket is present between the ligand and CDR loops L3 and H2. The sidechain of Glu7 points into the cavity that is filled with water molecules of well-defined electron density, which mediate additional interactions between the ligand (Glu7) and the antibody. The peptide-antibody interaction surface changes into primary hydrophobic beyond Glu7. Pro8 is involved in prominent ring stacking contact with LTyr37. Residues 9 and 10 do not contribute significantly to the interaction with the antibody, are solvent exposed and characterized by relatively large temperature factors. Cys2 and Cys 11 do not contribute significantly to the interaction surface either, however provide a cyclic scaffold of the peptide by maintaining a disulfide crosslink. The indole ring of Trp12 provides a significant apolar interface by stacking against the β-sheet surface at the bottom of CDR H2. This closes the entrance to the above described water filled cavity on the side of CDR H2, however the cavity remains open to the solvent on the side of CDR L3. Ala13 fills a small opening within the ligand itself. Further residues are not involved in ligand–antibody interaction and are undefined by electron density beyond Ala14 (Ala13 in the second molecule contained in the asymmetric unit).
Fig. 5.

Binding of ganglioside peptide mimetics at the antigen combining site of 14G2a. Recognition of peptide 1 (A, pink) and peptide 2 (B, orange) relies on hydrogen bond and hydrophobic interactions. The peptides are represented as main chain trace and the antibody is shown in ribbon representation. For clarity, only the most important residues mediating the antibody-peptide interaction are shown in stick representation. The ligand residue located at the bottommost pocket of the binding groove is indicated by an arrow. Antibody model orientation and color coding same as in Fig. 2.
Structures of 14G2a Fab fragment in complex with peptide 2 have been solved using crystals that belonged to two different space groups. Both structures are largely identical indicating that crystal packing interactions do not significantly influence the observed binding mode. Both structures are discussed collectively unless indicated otherwise.
The ligand is well defined by electron density aside from its C-terminal part in the P21 structure. The disposition of peptide 2 within the antigen binding groove is roughly comparable to that observed for peptide 1 (Fig. 5). Both peptides contain a characteristic apolar anchor at the very bottom of the binding groove provided by Leu5 in peptide 2 and corresponding Met6 in peptide 1, which are both further stabilized by hydrogen bond between the main chain carbonyl and the sidechain of LHis39. Analogically to peptide 1, loops H1 and H3 contact peptide 2 primarily via hydrogen bonds whereas the interactions with loop L1, L3 and H2 are primarily hydrophobic. Despite the overall similarity, the particular details of peptide–antibody interaction differ significantly between peptides 1 and 2. The N-terminal amine of peptide 2 donates hydrogen bonds to the sidechains of HAsn33 and HAsp52 and an intermolecular hydrogen bond to the main chain carbonyl of Leu10. Val1 occupies a small pocket between CDR H1 and H2 and its main chain carbonyl contacts the sidechain of HAsn33. Hydrogen bonds connect the sidechain of Asn3 with the main chain atoms of HGly31 and HAsn33 whereas the main chain carbonyl of Asn3 accepts intramolecular hydrogen bond from backbone amide of residue 6 and a weaker bond from the backbone amide of residue 7. Pro4 is not involved in any significant contacts. The above interactions do not directly resemble those found in the corresponding region of peptide 1 containing structure. Peptide 2 contacts the light chain side of the binding groove utilizing hydrogen bonds between the carbonyl of Thr6 and the side chain of LSer96 and a water mediated bond connecting the amide hydrogen of Ala8 with the main chain of loop L3. Peptide 1 contributes only hydrophobic interactions in this region. The sidechain of Thr6 is directed into the binding cavity and is involved in hydrogen bond contacts with buried water molecules comparably to the corresponding Glu7 in peptide 1. However, the water filled cavity in peptide 2 containing structure is only residual compared with that found in peptide 1 containing structure. This is because in peptide 1, beyond Glu7, the main chain flips on top of its N-terminal part creating a large cavity between the peptide and CDR loops H2 and L3 whereas in peptide 2 the main chain is oriented differently and better explores the CDR L3 side of the binding cavity leaving no free space in this region. In peptide 1 containing structure the cavity is partially filled with the sidechain of Trp12 on the side of CDR H2 whereas in peptide 2 the side chains of Leu9 and Leu10 occupy the same location, but being more bulky, they replace most water molecules found in this region in peptide 1 containing structure. As such, the large, water filled cavity observed in peptide 1 containing structure is reduced to two water molecules only in the structure containing peptide 2. One of those waters mediates the interaction of Thr6 with the sidechain of HAsn33 and the second mediates intrapeptide interactions. Further than Cys11, peptide 2 is defined by electron density only in the P1 structure. The fragment creates a lid partly covering the entrance of the ligand binding cavity and stabilized by hydrogen bond interactions with the sidechains of LHis31 and LAsn33 at the very top of CDR L1. Because the lid is further influenced by interactions with symmetry related molecules and is not defined by electron density in the P21 structure, we believe that it is an artifact of crystal packing unlikely to be preserved in solution.
The Nature of Peptide–Carbohydrate Mimicry
A generic similarity in the binding modes of GD2 and mimetic peptides is imposed by the spatial restraints of the binding pocket (Fig. 6A, 6B). However, analysis of specific interactions reveals neither structural correlation nor direct functional equivalence at the atomic level. The N-terminal amine groups of both peptides are located in a similar position as the amine group of NeuAc1 and all donate hydrogen bonds to the sidechain of HAsn33. However, in both peptides the primary amine is involved in additional hydrogen bonds whereas the secondary amine of the carbohydrate donates only a single hydrogen bond. In peptide 2 the sidechain of Asn3 mimics one of the hydrogen bonds contributed by the carboxyl group of NeuAc2 to the backbone amine of HAsn33. However, the difference in the chemical character of involved moieties results in additional interactions of Asn3 not observed in the carbohydrate structure. The sidechain of Asn3 in peptide 1 is found in a different orientation and does not contribute interactions comparable to that of a carbohydrate. The bottommost pocket of the antibody binding groove is filled with a hydrophobic sidechain of the ligand in the case of both peptides (Fig. 5), whereas in the carbohydrate containing structure this cavity is occupied by a polar etheneamide moiety of NeuAc2 (Fig. 2A). The etheneamide is stabilized by two hydrogen bonds, which are not directly mimicked by the tested peptides. A week hydrophobic interaction of the etheneamide methyl group is explored more efficiently by the hydrophobic side chains in both peptides (Fig. 6A, B). Compared with the empty structure, carbohydrate binding induces conformational rearrangement in LLys55 sidechain such that it donates a hydrogen bond to NeuAc2. In turn, LLys55 does not directly interact with the ligand in the peptide 1 containing structure whereas its interaction with peptide 2 does not resemble that with the carbohydrate. Neither of the two peptides mimic the prominent hydrogen bond interactions of a glycerin moiety of NeuAc2 with HAsn35. Furthermore, although GalNac interacts with the light chain primarily via hydrogen bonds, peptide 1 is involved only in hydrophobic contacts in this region. Although peptide 2 contributes a single hydrogen bond to the light chain in the discussed region, this interaction does not directly mimic any of the contacts contributed by GalNac. Of further significant differences, the ordered water structure mediating the interaction of the GD2 and loop H2 is replaced by direct hydrophobic interactions in case of both peptides (Fig. 6C-6E). In turn, whereas the carbohydrate is involved in direct interaction with CDR L3, in the peptide 1 containing structure the interaction with CDR L3 is mediated through a number of ordered water molecules. The above comparison concerned the bottom part of the binding pocket where some interactions between the mimetic peptides and the antibody resemble to a certain extent those of the carbohydrate. In turn, the dispositions and interactions of the studied ligands at the outer part of the pocket are entirely dissimilar.
The lack of significant mimicry between the peptide ligands and GD2 at the level of atomic interactions with the antibody is accompanied by pronounced differences in the overall binding mechanism. Recognition of GD2 carbohydrate follows an induced fit mechanism eliciting significant structural rearrangement within the CDR region. Contrary, the binding of either of the tested peptides is not associated with any significant rearrangement within CDR. Instead, even though the interaction details differ significantly between the peptides, both perfectly complement the antibody binding groove as found in the empty structure. Thus, peptide recognition by the antibody is best described by the “key and lock” mechanism. Further of difference, carbohydrate binding relies in its major part on direct and water mediated hydrogen bonds and no significant hydrophobic interactions are involved. Contrary, the evaluated peptides rely equally on hydrogen bonds and hydrophobic interactions in their binding to 14G2a antibody.
DISCUSSION
Gangliosides proved valuable targets in cancer immunotherapy. Changes in GD2 expression characteristic for the tumor cells were recently successfully explored to specifically target neuroblastoma in clinics. Immunotherapies directed at GD2, GM2, GM3, and other gangliosides are being evaluated in different types of cancer. However, the structural understanding of antibody–ganglioside recognition is limited. Crystal structures of GD2, GD3, and GM3 specific antibodies have been determined in a ligand free form (10, 11, 35). The presumed antigen binding modes were modeled, but were lacking empirical validation because no experimentally determined antibody structure containing the target ganglioside was available to date. Several structures of carbohydrate specific, protective antibodies targeting the surface antigens of human pathogens have been determined in complex with their target ligands (36, 37), but these structures did not provide suitable homology models of ganglioside recognition by an antibody. This is because the steric features of short, branched ganglioside sugars, directionally restrained by the proximity of the cell membrane, were not reflected in any of the experimentally determined structures. This study provides a detailed insight into the structural basis of ganglioside recognition by mouse 14G2a antibody, which contains an identical antigen binding region as chimeric ch14.18 antibody recently approved by FDA for the treatment of neuroblastoma (8). The major conclusion is that GD2 binding follows an induced fit mechanism requiring a conformational rearrangement involving all CDR loops and reorganization of water structure within the cavity. The majority of the interaction surface is contributed by NeuAc2, GalNAc and NeuAc1, which explains the exclusive specificity of 14G2a toward GD2 compared with other gangliosides. Of the most structurally related gangliosides, GD1b contains an additional Gal residue, which is sterically incompatible with the determined mode of carbohydrate binding by 14G2a whereas both GD3 and GM2 each lack either of the two residues that significantly contribute to the interaction surface, GalNAc and NeuAc2, respectively (Fig. 2D). The particular interactions within 14G2a:GD2 interface are highly cooperative because interference within particular hydrogen bonds mediating the interaction (by mutating respective residues to alanine) in all cases resulted in complete loss of affinity of the mutant toward GD2 despite the fact that multiple other hydrogen bonds presumably should not have been affected by each particular mutation.
Despite the highly hydrophilic character of most sugars, the structural studies on carbohydrate recognition by various binding proteins and enzymes highlight the importance of hydrophobic interactions in complementing the networks of hydrogen bond contacts. A common feature of the binding surface and a notable contribution to its energy is provided by CH-π stacking of sugar rings and aromatic protein sidechains (38). Examples most relevant to this study include clostridial neurotoxin in complex with GD1 sugar (39) and sialic acid receptor of immune cells (siglec-7) in complex with GT1b analog (40). In both structures the ganglioside is recognized in a relatively shallow hollow compared with the pronounced groove found in 14G2a. The interaction surface includes several direct and water mediated hydrogen bonds and a significant CH-π stacking of tryptophan indole and galactose or glucose, respectively in neurotoxin and siglec-7. Here we demonstrated that GD2 recognition by 14G2a is based primarily on direct and water mediated hydrogen bonds without significant involvement of hydrophobic interactions and no stacking interactions whatsoever. Large part of the sugar ligand is buried in a deep pocket within the antibody. However, the carbohydrate antigen does not explore the entire binding pocket of 14G2a. Instead, a network of tightly coordinated water molecules mediates the interaction of NeuAc1 and loop H2. Water mediated antigen recognition was previously observed in a structure of a polysialic-acid complex with a specific antibody (41). However, apart from that general parallel, the modes of binding of the short, branched GD2 sugar in a deep pocket of 14G2a and a long, linear polysialic-acid in a shallow groove of mAb735 are entirely disparate. Molecular modeling predicted that water mediated contacts between loop H2 of 14G2a and NeuAc1 of GD2 can be favorably replaced by direct intermolecular interaction by exchanging short sidechains of HAla50 or HSer59 by a long sidechain of lysine. However, the in silico prediction was only partially verified in vitro. The affinity of HSer59Lys differed only insignificantly form that of the wild type. Therefore, presumably HSer59Lys mutation truly induced a direct contact between loop H2 and GD2 sugar, however, this has not translated into increased affinity. In turn, HAla50Lys almost completely lost affinity toward GD2 demonstrating that in this instance the in silico prediction proved inadequate.
In further attempt to improve 14G2a affinity toward GD2, LHis31Asn substitution was designed to exchange favorable hydrogen bond interaction of the imidazole ring within the sidechain of LHis31 and GalNAc, with comparable, but expectedly more favorable interaction of the carboxamide group within the sidechain of asparagine. This approach has failed, however, because LHis31Asn have lost its affinity toward GD2. Another variant, LHis54Arg, was designed to substitute a water network mediated interaction of LHis54 by an expectedly more favorable direct interaction. In this case no significant change in affinity was noted compared with the wild type. This suggests that most probably a direct interaction has been induced but, this has not resulted in significant increase in activity. Most interestingly, however, replacing glutamic acid at position 101 in the heavy chain with lysine residue resulted in significant increase in affinity of HGlu101Lys toward GD2 compared with the wild type. Based on in silico modeling we hypothesize that such a change replaces a water mediated contact between the sidechain of HGlu101 and NeuAc2 with a more energetically favorable direct interaction (supplemental Fig. S3).
Overall, the in vitro activity of three out of five in silico designed and in vitro tested mutants was qualitatively consistent with in silico prediction, but only a single mutant showed quantitative correspondence. Because meaningful modeling of extended protein–carbohydrate interactions and especially adequately assessing the contribution of particular residues to the overall binding energy within complex systems is relatively complicated we consider this result a reasonable success. Improved affinity of HGlu101Lys may likely lead to improved tumor selectivity and as such scFv-14G2a(HGlu101Lys) may find utility as a carrier molecule for cytotoxic agents (42).
The fact that peptides can mimic carbohydrates in their binding to target proteins has been first demonstrated by Oldenburg and colleagues (43). Given the diverse potential applications of such peptides, a significant number of mimetics have been described since. Nevertheless, the nature of peptide-carbohydrate mimicry remains poorly understood at molecular level. Two principle mechanisms were proposed by Johnson and Pinto (14): structural mimicry, whereby the peptide presents a similar arrangement of functional groups as the carbohydrate, and functional mimicry, whereby the peptide utilizes an alternative binding mode to that of the carbohydrate. Comparative analysis of a limited number of available crystal structures of antibody Fab fragments in complex with mimicking peptides and relevant carbohydrates (44, 45) indicates that the actual mechanism is best described as partial structural mimicry. However, a more definitive conclusion awaits characterization of a wider variety of examples (14). In particular, only linear carbohydrates spanning in an extended fashion the relatively shallow antibody binding grooves were characterized to date. More difficult targets of peptide mimicry, the chemically complex, branched carbohydrates exploring deep binding cavities within the antibody were not evaluated in this context prior to this study. Moreover, it has not been previously demonstrated how two peptides largely different in their primary structure may interchangeably explore the combining site of a particular antibody.
Our results demonstrate that although certain aspects of structural mimicry are present in binding of both evaluated peptides at the 14G2a combining site, the observed mimicry is best described by the “functional” model (supplemental Table S1). First, binding of GD2 follows an induced fit mechanism whereas the interaction of both peptides with the antibody is best described by the “key and lock” model. Second, the binding of both peptides relies significantly on hydrophobic interactions that are marginal in the carbohydrate–antibody interaction surface. Third, the carbohydrate and both peptides utilize significantly different patterns of hydrogen bonds in their interactions with the antibody. Finally, although water mediated interactions are utilized by both GD2 and peptide 1, their location within the binding grove is significantly different. Peptide 2 binding relies considerably less on water mediated interactions. The functional rather than structural nature of the mimicry is further corroborated by the fact that the tested peptide mimetics interact only with 14G2a, but not other tested antibodies specific for GD2 (15). Therefore the observed mimicry is receptor rather than antigen dependent, as required by the “functional” model.
Active immunization against GD2 has been proposed as an alternative strategy to target neuroblastoma. This assumption is based on observations that elevated levels of GD2-specific antibodies correlate with better prognosis (46) and the clinical success of passive immunization (7, 8). Given the notoriously low immunogenicity of carbohydrates, the concept of peptide mimetic vaccination has been evaluated. The relevance of such approach was demonstrated by Pincus and collaborators (12) who demonstrated that peptides mimicking group B Streptococcus polysaccharide may provide active, protective immunization against the bacteria. However, as demonstrated here and by others (44, 45) the interaction of mimetic peptides and the carbohydrate with the target antibody may differ significantly. Moreover, as demonstrated in this study, mimetic peptides characterized by different primary structure differ significantly in their interaction modes. As such, it is not surprising that the validity of particular peptides for active immunization also varies. For example Fest and colleagues demonstrated that immunization with a GD2 ganglioside mimetic peptide (designated MD) resulted in protective antibodies recognizing GD2 (18). Using a different peptide (47-LDA), Wierzbicki et al. have shown that the immunization resulted in protective CD8+ T-cell response directed against CD166, in addition to GD2 cross-reactive antibodies (47). These results stress the importance of understanding the structural basis of peptide–carbohydrate mimicry. Our study provides a detailed insight into how the tested peptides imitate GD2 in its binding to specific antibody. The provided here structural data perfectly explains the results of previous mutagenesis studies within the predecessor of peptide 1 (for mutagenesis data see (15); for a detailed discussion see supplemental Information and Table S2). Collectively, this information shall facilitate rational modification of peptides to improve their utility in active immunization. First, however, the correlation between the extent of structural mimicry and utility for active immunization remains to be established.
Supplementary Material
Acknowledgments
We thank R.A. Reisfeld (Scripps Institute, CA) for the 14G2a hybridoma cell line, L. Raffaghello (G. Gaslini Institute, Genova, Italy) for the NXS2 cell line, G. Pompidor (EMBL, Hamburg, Germany) for his assistance at the beamline, M. Bzowska (Jagiellonian University, Krakow, Poland) for help with flow cytometry analysis, and K. Falkowski (Jagiellonian University, Krakow, Poland) for the English language correction.
Footnotes
Author contributions: I.H., M.Z. and G.D. designed and initiated the research; I.H., H.R. and G.D. provided key reagents and materials; I.H. and P.Go. purified and crystallized the proteins; G.D. and P.Gr. collected X-ray data; P.Gr., P.Go. and G.D. processed X ray data, refined and analyzed the structures; M.K. performed in silico modeling; G.D. designed scFv constructs; P.Go. and P.Gr. cloned, expressed and purified scFv variants; I.H. performed ELISA and flow cytometry analyses; I.H. and P.Gr. wrote the first draft and G.D. wrote the final manuscript; all authors commented on, or edited the manuscript.
* This work was supported in part by grants UMO-2011/01/D/NZ1/01169 (to G.D.) from the National Science Center, N302 034 31/3063 (to I.H.) from the Polish Ministry of Science and Higher Education and institutional grants WBBB 8 (to H.R.) and BW37/137 (to I.H.). P.Gr. was supported by the European Union within the SET project. The research was carried out with the equipment purchased thanks to the financial support of the European Union structural funds (grants POIG.02.01.00-12-064/08 and POIG.02.01.00-12-167/08). Faculty of Biochemistry, Biophysics and Biotechnology of the Jagiellonian University is a partner of the Leading National Research Center supported by the Ministry of Science and Higher Education of the Republic of Poland.
 This article contains supplemental Figs. S1 to S3, Tables S1 and S2, and Information.
This article contains supplemental Figs. S1 to S3, Tables S1 and S2, and Information.
The authors declare no competing financial interests.
1 The abbreviations used are:
- GD2
- GD2 ganglioside
- Glc
- glucose
- Gal
- galactose
- GalNAc
- N-acetylgalactosamine
- NeuAc
- N-acetylneuraminic acid
- ADCC
- antibody-dependent cell-mediated cytotoxicity
- CDC
- complement-dependent cytotoxicity
- CDR
- complementarity-determining region.
REFERENCES
- 1. Cheever M. A., Allison J. P., Ferris A. S., Finn O. J., Hastings B. M., Hecht T. T., Mellman I., Prindiville S. A., Viner J. L., Weiner L. M., Matrisian L. M. (2009) The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 15, 5323–5337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roth M., Linkowski M., Tarim J., Piperdi S., Sowers R., Geller D., Gill J., Gorlick R. (2014) Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer 120, 548–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yoshida S., Fukumoto S., Kawaguchi H., Sato S., Ueda R., Furukawa K. (2001) Ganglioside G(D2) in small cell lung cancer cell lines: enhancement of cell proliferation and mediation of apoptosis. Cancer Res. 61, 4244–4252 [PubMed] [Google Scholar]
- 4. Wu Z. L., Schwartz E., Seeger R., Ladisch S. (1986) Expression of GD2 ganglioside by untreated primary human neuroblastomas. Cancer Res. 46, 440–443 [PubMed] [Google Scholar]
- 5. Cheung N. K., Dyer M. A. (2013) Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 13, 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Horwacik I., Durbas M., Boratyn E., Wegrzyn P., Rokita H. (2013) Targeting GD2 ganglioside and aurora A kinase as a dual strategy leading to cell death in cultures of human neuroblastoma cells. Cancer Lett. 341, 248–264 [DOI] [PubMed] [Google Scholar]
- 7. Yu A. L., Gilman A. L., Ozkaynak M. F., London W. B., Kreissman S. G., Chen H. X., Smith M., Anderson B., Villablanca J. G., Matthay K. K., Shimada H., Grupp S. A., Seeger R., Reynolds C. P., Buxton A., Reisfeld R. A., Gillies S. D., Cohn S. L., Maris J. M., Sondel P. M. (2010) Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 363, 1324–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dhillon S. (2015) Dinutuximab: First global approval. Drugs 75, 923–927 [DOI] [PubMed] [Google Scholar]
- 9. Willison H. J., Yuki N. (2002) Peripheral neuropathies and anti-glycolipid antibodies. Brain 125, 2591–2625 [DOI] [PubMed] [Google Scholar]
- 10. Pichla S. L., Murali R., Burnett R. M. (1997) The crystal structure of a Fab fragment to the melanoma-associated GD2 ganglioside. J. Struct. Biol. 119, 6–16 [DOI] [PubMed] [Google Scholar]
- 11. Ahmed M., Goldgur Y., Hu J., Guo H. F., Cheung N. K. (2013) In silico driven redesign of a clinically relevant antibody for the treatment of GD2 positive tumors. PLoS One 8, e63359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pincus S. H., Smith M. J., Jennings H. J., Burritt J. B., Glee P. M. (1998) Peptides that mimic the group B streptococcal type III capsular polysaccharide antigen. J. Immunol. 160, 293–298 [PubMed] [Google Scholar]
- 13. Horwacik I., Rokita H. (2012) Application of molecular mimicry to target GD2 ganglioside, in Neuroblastoma - Present and Future, Hiroyuki S., Editor. InTech; 251–270 [Google Scholar]
- 14. Johnson M. A., Pinto B. M. (2008) Structural and functional studies of peptide-carbohydrate mimicry. Top. Curr. Chem. 273, 55–116 [DOI] [PubMed] [Google Scholar]
- 15. Horwacik I., Kurcinski M., Bzowska M., Kowalczyk A. K., Czaplicki D., Kolinski A., Rokita H. (2011) Analysis and optimization of interactions between peptides mimicking the GD2 ganglioside and the monoclonal antibody 14G2a. Int. J. Mol. Med. 28, 47–57 [DOI] [PubMed] [Google Scholar]
- 16. Mujoo K., Kipps T. J., Yang H. M., Cheresh D. A., Wargalla U., Sander D. J., Reisfeld R. A. (1989) Functional properties and effect on growth suppression of human neuroblastoma tumors by isotype switch variants of monoclonal antiganglioside GD2 antibody 14.18. Cancer Res. 49, 2857–2861 [PubMed] [Google Scholar]
- 17. Fields C., O'Connell D., Xiao S., Lee G. U., Billiald P., Muzard J. (2013) Creation of recombinant antigen-binding molecules derived from hybridomas secreting specific antibodies. Nat. Protoc. 8, 1125–1148 [DOI] [PubMed] [Google Scholar]
- 18. Fest S., Huebener N., Weixler S., Bleeke M., Zeng Y., Strandsby A., Volkmer-Engert R., Landgraf C., Gaedicke G., Riemer A. B., Michalsky E., Jaeger I. S., Preissner R., Forster-Wald E., Jensen-Jarolim E., Lode H. N. (2006) Characterization of GD2 peptide mimotope DNA vaccines effective against spontaneous neuroblastoma metastases. Cancer Res. 66, 10567–10575 [DOI] [PubMed] [Google Scholar]
- 19. Jancarik J., Kim S. H. (1991) Sparse matrix sampling: a screening metod for crystallization of proteins. J. Appl. Cryst. 24, 409–411 [Google Scholar]
- 20. Kabsch W. (2010) Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Battye T.G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. (2011) iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 24. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brünger A. T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 [DOI] [PubMed] [Google Scholar]
- 29. Aqvist J., Medina C., Samuelsson J. E. (1994) A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 7, 385–391 [DOI] [PubMed] [Google Scholar]
- 30. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2006) Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics Chapter 5: Unit 5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 32. Koziara K.B., Stroet M., Malde A.K., Mark A.E. (2014) Testing and validation of the Automated Topology Builder (ATB) version 2.0: Prediction of hydration free enthalpies. J. Comput. Aided Mol. Des. 28, 221–233 [DOI] [PubMed] [Google Scholar]
- 33. Lin Z., van Gunsteren W. F. (2013) Refinement of the application of the GROMOS 54A7 force field to beta-peptides. J. Comput. Chem. 34, 2796–2805 [DOI] [PubMed] [Google Scholar]
- 34. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., Lindahl E. (2013) GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Talavera A., Eriksson A., Okvist M., López -Requena A., Fernández -Marrero Y., Pérez R., Moreno E., Krengel U. (2009) Crystal structure of an anti-ganglioside antibody, and modelling of the functional mimicry of its NeuGc-GM3 antigen by an anti-idiotypic antibody. Mol. Immunol. 46, 3466–3475 [DOI] [PubMed] [Google Scholar]
- 36. Cygler M., Rose D. R., Bundle D. R. (1991) Recognition of a cell-surface oligosaccharide of pathogenic Salmonella by an antibody Fab fragment. Science 253, 442–445 [DOI] [PubMed] [Google Scholar]
- 37. Villeneuve S., Souchon H., Riottot M. M., Mazie J. C., Lei P., Glaudemans C. P., Kovac P., Fournier J. M., Alzari P. M. (2000) Crystal structure of an anti-carbohydrate antibody directed against Vibrio cholerae O1 in complex with antigen: molecular basis for serotype specificity. Proc. Natl. Acad. Sci. U. S. A. 97, 8433–8438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Asensio J. L., Arda A., Canada F. J., Jimenez-Barbero J. (2013) Carbohydrate-aromatic interactions. Acc. Chem. Res. 46, 946–954 [DOI] [PubMed] [Google Scholar]
- 39. Benson M. A., Fu Z., Kim J. J., Baldwin M. R.. Unique ganglioside recognition strategies for clostridial neurotoxins. J. Biol. Chem. 286, 34015–34022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Attrill H., Imamura A., Sharma R. S., Kiso M., Crocker P. R., van Aalten D. M. (2006) Siglec-7 undergoes a major conformational change when complexed with the alpha(2,8)-disialylganglioside GT1b. J. Biol. Chem. 281, 32774–32783 [DOI] [PubMed] [Google Scholar]
- 41. Nagae M., Ikeda A., Hane M., Hanashima S., Kitajima K., Sato C., Yamaguchi Y. (2013) Crystal structure of anti-polysialic acid antibody single chain Fv fragment complexed with octasialic acid: insight into the binding preference for polysialic acid. J. Biol. Chem. 288, 33784–33796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adams G. P., Schier R., Marshall K., Wolf E. J., McCall A. M., Marks J. D., Weiner L. M. (1998) Increased affinity leads to improved selective tumor delivery of single-chain Fv antibodies. Cancer Res. 58, 485–490 [PubMed] [Google Scholar]
- 43. Oldenburg K. R., Loganathan D., Goldstein I. J., Schultz P. G., Gallop M. A. (1992) Peptide ligands for a sugar-binding protein isolated from a random peptide library. Proc. Natl. Acad. Sci. U. S. A. 89, 5393–5397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vyas N. K., Vyas M. N., Chervenak M. C., Bundle D. R., Pinto B. M., Quiocho F. A. (2003) Structural basis of peptide-carbohydrate mimicry in an antibody-combining site. Proc. Natl. Acad. Sci. U. S. A. 100, 15023–15028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tapryal S., Gaur V., Kaur K. J., Salunke D. M. (2013) Structural evaluation of a mimicry-recognizing paratope: plasticity in antigen-antibody interactions manifests in molecular mimicry. J. Immunol. 191, 456–463 [DOI] [PubMed] [Google Scholar]
- 46. Cheung N. K., Guo H. F., Cheung I. Y. (2000) Correlation of anti-idiotype network with survival following anti-G(D2) monoclonal antibody 3F8 therapy of stage 4 neuroblastoma. Med. Pediatr. Oncol. 35, 635–637 [DOI] [PubMed] [Google Scholar]
- 47. Wierzbicki A., Gil M., Ciesielski M., Fenstermaker R. A., Kaneko Y., Rokita H., Lau J. T., Kozbor D. (2008) Immunization with a mimotope of GD2 ganglioside induces CD8+ T cells that recognize cell adhesion molecules on tumor cells. J. Immunol. 181, 6644–6653 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.