

Abstract
The field of proteomics almost uniformly relies on peptide cation analysis, leading to an underrepresentation of acidic portions of proteomes, including relevant acidic posttranslational modifications. Despite the many benefits negative mode proteomics can offer, peptide anion analysis remains in its infancy due mainly to challenges with high-pH reversed-phase separations and a lack of robust fragmentation methods suitable for peptide anion characterization. Here, we report the first implementation of activated ion negative electron transfer dissociation (AI-NETD) on the chromatographic timescale, generating 7,601 unique peptide identifications from Saccharomyces cerevisiae in single-shot nLC-MS/MS analyses of tryptic peptides—a greater than 5-fold increase over previous results with NETD alone. These improvements translate to identification of 1,106 proteins, making this work the first negative mode study to identify more than 1,000 proteins in any system. We then compare the performance of AI-NETD for analysis of peptides generated by five proteases (trypsin, LysC, GluC, chymotrypsin, and AspN) for negative mode analyses, identifying as many as 5,356 peptides (1,045 proteins) with LysC and 4,213 peptides (857 proteins) with GluC in yeast—characterizing 1,359 proteins in total. Finally, we present the first deep-sequencing approach for negative mode proteomics, leveraging offline low-pH reversed-phase fractionation prior to online high-pH separations and peptide fragmentation with AI-NETD. With this platform, we identified 3,467 proteins in yeast with trypsin alone and characterized a total of 3,730 proteins using multiple proteases, or nearly 83% of the expressed yeast proteome. This work represents the most extensive negative mode proteomics study to date, establishing AI-NETD as a robust tool for large-scale peptide anion characterization and making the negative mode approach a more viable platform for future proteomic studies.
Global protein analysis continues to enjoy substantial technological leaps forward in its ability to characterize protein expression in a variety of organisms with both speed and sensitivity (1–4). Nevertheless, the impressive advances in protein sequence technology over past decades have rigidly adhered to positive electrospray ionization for MS1 analysis, limiting the scope of peptides and posttranslational modifications that can be analyzed. Widely utilized acidic mobile phases both permit stable and reproducible reversed-phase separations and also provide optimal conditions for ionization and detection of peptides and proteins that readily accept positive charge via protonation (i.e. basic species). Acidic peptides and proteins, however, favor deprotonation, making positive electrospray regimes ill suited for their characterization. Moreover, important classes of posttranslational modifications (PTMs), such as phosphorylation, sulfation, and glycosylation, can impart acidic properties to the peptides and proteins they modify, often producing entire classes of biomolecules that preferentially ionize as anions (5–10).
Electrospray ionization operated in the negative mode can generate multiply deprotonated species (11, 12); however, canonical collisional activation methods produce MS/MS spectra riddled with neutral losses and internal fragments that are difficult, if not impossible, to interpret (13–16). Alternatively, a number of emerging fragmentation techniques, including electron-based dissociation methods and photodissociation approaches, can generate sequence informative MS/MS spectra from peptide anions (17–23). Both negative electron transfer dissociation (NETD) and ultraviolet photodissociation (UVPD) have been employed in large-scale proteomic studies, enabling sequencing of thousands of unique peptides in a single experiment (24, 25). In NETD, the negative mode analog of electron transfer dissociation (ETD) (26), peptide anions are oxidized with reagent cations, causing the radical peptide anions to undergo electron rearrangement steps that often lead to cleavage of the C-Cα backbone bond, producing a●- and x-type product ions (18, 24, 27, 28). Sometimes, however, an electron is abstracted from the precursor anion and backbone cleavage is achieved, but the product ions are held together by intramolecular noncovalent interactions as long-lived charge-reduced species that do not separate. The probability of this phenomenon, called nondissociative negative electron transfer, is directly related to precursor anion charge density; as charge density decreases, i.e. the precursor mass-to-charge ratio (m/z) increases, so does the magnitude of nondissociative negative electron transfer, limiting the amount of sequence information derived from the NETD MS/MS event (29–31).
Many approaches have been explored to mitigate the effects of nondissociative electron transfer in ETD reactions of peptide cations, including collisional activation of all product ions (32, 33), activation of nondissociative electron transfer products (34, 35), elevated bath gas temperatures (36), and infrared photon bombardment concurrent to the ETD reaction (37). The last of these approaches, termed activated ion ETD (AI-ETD), has shown substantial promise for proteomics applications (38, 39). The concomitant IR photoirradiation disrupts the secondary gas-phase structure responsible for nondissociative electron transfer, increasing the efficiency of sequence-informative product ion generation; furthermore, the introduction of additional energy to the reaction during AI-ETD occurs only during the ion–ion reaction time, keeping the instrument cycle time as short as possible and reducing problematic hydrogen-atom rearrangements that can occur prior to fragment ion separation with other supplemental activation techniques (34, 37). Indeed, activated ion NETD (AI-NETD), which uses simultaneous IR irradiation during the NETD reaction, has been reported to improve peptide anion fragmentation for a handful of standard peptides (30), but a large-scale implementation of AI-NETD for negative mode shotgun proteomics has yet to be demonstrated.
We recently described the development of a multipurpose dissociation cell (MDC) specifically for improved ion–ion reactions on an ETD-enabled dual cell quadrupole ion trap-Orbitrap hybrid mass spectrometer, the same platform on which we reported the successful implementation of NETD for high-throughput peptide analyses (40). The MDC, equipped with a higher operating RF frequency and longer axial dimensions, allows faster ETD reaction times and larger precursor ion populations for improved fragment ion signal-to-noise. The placement of MDC is ideal for implementing AI-NETD on an NETD-capable mass spectrometer because a continuous wave CO2 laser can be easily introduced concentrically to the trapping volume of the MDC (39, 41, 42).
Here, we present the first description of NETD in the MDC reaction vessel, in addition to the first implementation of AI-NETD for large-scale peptide anion analysis. Analyzing complex mixtures of peptides from Saccharomyces cerevisiae whole cell lysates, we show that AI-NETD, in combination with judiciously chosen high-pH chromatographic conditions, enables the most robust analysis of peptide anions to date, identifying over 1,100 proteins in single-shot experiments. Furthermore, we extend our studies to investigate the performance of AI-NETD with five distinct proteases (trypsin, LysC, GluC, chymotrypsin, and AspN), providing the first large-scale investigation into the performance of multiple enzymes for peptide anion characterization. Using both single-shot analyses and deep sequencing via offline low-pH fractionation for each protease, we assert that entire proteomes can be investigated in the negative mode with AI-NETD, providing a new platform to thoroughly explore biologically relevant hypotheses, e.g. acidic PTM networks, which were previously inaccessible with canonical positive mode approaches.
EXPERIMENTAL PROCEDURES
Mass Spectrometry Instrumentation
The multipurpose dissociation cell (MDC) replaces the preexisting HCD cell in the ETD-enabled dual cell quadrupole linear ion trap-Orbitrap hybrid mass spectrometer system (Fig. 1C) (43, 44) (Thermo Fisher Scientific, San Jose, CA), retaining its basic geometry but requiring additional electronics to supply higher trapping rf voltages for faster reaction times, axial rf voltages for charge-sign independent trapping, and independently controlled DC (direct current) biases to its four sections. Offsets used for previous cation analysis with the MDC were inverted to accommodate injection and trapping of precursor anions rather than cations. Consistent with our earlier NETD work (24, 28), positive reagent ions were generated without hardware modification through optimized electron ionization/chemical ionization (EI/CI) source conditions, and reagent transmission through ion-transfer optics was tuned via automated optimization routines. Radical fluoranthene reagent cations were formed in a CI ion volume in the presence of nitrogen gas and were then accumulated in the back two sections of the MDC for NETD reactions. Following the NETD reaction via charge-sign independent trapping, the reaction was quenched by setting the center sections of the MDC to a positive DC offset (10 V), retaining anionic product ions and ejecting remaining cationic reagents. Product ions were then transferred to the C-trap for subsequent Orbitrap mass analysis using an extraction gradient analogous to that used for HCD scans. AI-NETD was performed by irradiating the trapping volume of the MDC during the entirety of the NETD reaction with a Firestar T-100 Synrad 100-W CO2 continuous wave laser (Mukilteo, WA). The laser was introduced into the cell via an excavated ion passage in the reagent ion transfer multipole and a ZnSe window that was installed concentric to the MDC. Using instrument firmware and modification to instrument code in conjunction with a gated laser controller, laser power output (in Watts) from the laser (10.6 μm) was modulated remotely through voltage inputs to the controller and was triggered to fire only during the NETD reaction as it was being conducted in the MDC. The nitrogen pressure in the MDC was lowered to a ΔN2 pressure of ∼0.1 × 10−10 Torr, as measured by the Penning gauge in the Orbitrap chamber, to prevent collisional cooling that negates the additional energy supplied by the infrared laser. Lowered nitrogen pressure also increased transmission of the fluoranthene reagent cation, so pressure in the MDC was held at similar levels for NETD analyses (ΔN2 of ∼0.3 × 10−10 Torr). For more detailed descriptions of the MDC and affixed laser, see previous work (39–41).
Fig. 1.
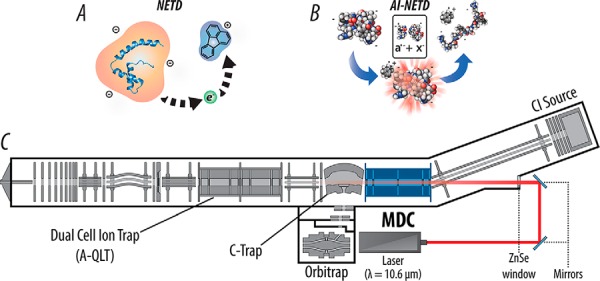
Modified linear ion trap-Orbitrap hybrid MS system for performing AI-NETD. (A) NETD, which involves oxidation of precursor peptide anions by a reagent cation, can now be performed in the mass-analyzing quadrupole linear ion trap (A-QLT) and multipurpose dissociation cell (MDC). (B) Modifying the MDC to perform NETD opens access to AI-NETD, in which anionic peptide precursors are concurrently irradiated with IR photons during the ion–ion reaction. This additional photoactivation disrupts peptide secondary gas-phase structure, increasing the efficiency of the NETD reaction. (C) Instrument schematic of the hybrid linear ion trap-Orbitrap mass spectrometer is modified with the MDC in place of the traditional HCD collision cell, in addition to an excavated beam path, ZnSe window, and affixed 10.6 μm CO2 laser for concentric irradiation of the MDC.
Sample Preparation
All protein lysates were derived from S. cerevisiae strain BY4741, as described previously (45). Briefly, cells were grown to an optimal density at 600 nm (OD600) of ∼0.6 and pelleted by centrifugation. A pellet was resuspended in lysis buffer (50 mm Tris, pH 8; 8 m urea; 75 mm sodium chloride; 10 mm sodium butyrate:protease and phosphatase inhibitor tablet (Roche Diagnostics, Indianapolis, IN)), and yeast cells were lysed by glass bead milling (Retsch GmbH, Haan, Germany). Two ml of acid-washed glass beads were combined with 2 ml of resuspended yeast cells in a 5 ml stainless steel container and shaken eight times at 30 Hz for 4 min with a 1 min rest in between. Lysate protein concentration was measured via BCA (Thermo Pierce, Rockford, IL), and yeast proteins were reduced through incubation in 5 mm dithiothreitol for 45 min at 58 °C. Free cysteines were alkylated in 15 mm iodoacetamide in the dark for 30 min. The alkylation was stopped with 5 mm DTT. For trypsin digestion, a 1 mg protein aliquot was digested overnight at room temperature in 1.5 m urea with trypsin (Promega, Madison, WI) added at a 1:50 (w/w) enzyme to protein ratio. A second trypsin addition was performed in the morning at a 1:100 (w/w) enzyme to protein ratio for 1 h. The digestion was quenched by the addition of TFA. For LysC digestion, a 1 mg protein aliquot was digested overnight at room temperature in 4 m urea with endo LysC (Wako Chemicals, Richmond, VA) at a 1:50 enzyme:protein ratio. Following overnight digestion, a second 1:50 aliquot of LysC was added and the digestion was allowed to proceed for 1 h. For GluC digestion, a 1 mg protein aliquot was digested overnight with 25 μg GluC (Roche) at room temperature in 0.5 m urea. Following overnight digestion, the sample was incubated with an additional 25 μg GluC for 1 h. For chymotrypsin digestion, a 1 mg protein aliquot was digested overnight with 12.5 μg of chymotrypsin (Promega) in 1 m urea. Following overnight digestion, an additional 12.5 μg of chymotrypsin was added to the sample, and the digestion was allowed to proceed for 1 h. For digestion with AspN, a 1 mg protein aliquot was incubated with 6 μg AspN (Roche) at room temperature overnight. Each digest was quenched by the addition of TFA and desalted over tC18 Sep-Pak cartridges (Waters, Milford, MA).
High-pH nLC-MS/MS
Five total reversed-phase solvent systems were tested for online high-pH separations. Ammonium hydroxide, ammonium formate, and piperidine were purchased from Sigma Aldrich (St. Louis, MO) and HPLC-grade water and acetonitrile were purchased from Fisher Scientific (Waltham, MA). The two ammonium formate solvent systems consisted of mobile phase A (5 mm ammonium formate in water) and mobile phase B (5 mm ammonium formate in 85% acetonitrile), basified to either pH 10 or pH 11.5 with ammonium hydroxide. Two different piperidine solvents systems used a mobile phase A of water and mobile phase B of 85% acetonitrile, 15% water, with either 5 mm or 10 mm piperidine in both A and B. Another 5 mm piperidine solvent system had the same mobile phase B, but mobile phase A consisted of 95% water, 5% DMSO with 5 mm piperidine. Reversed-phase columns were packed in-house using 75 μm inner diameter, 360 μm outer diameter bare fused silica capillary. A nanoelectrospray tip was laser pulled (Sutter Instrument Company, Novato, CA) and packed with 3.5 μm diameter, 130 Å pore size ethylene bridged hybrid C18 particles (Waters) to a length of 30–35 cm. The column was installed on a nanoACQUITY UltraPerformance LC (Waters) using a stainless steel ultra-high pressure union formatted for 360 μm outer diameter columns (IDEX). One microgram of sample was loaded onto the column in 95% A for 10 min at 400 nl min−1. Gradient elution was performed at 400 nl min−1, and gradients increased linearly from 5 to 30% B over 70 min, followed by an increase to 70% B at 76 min and a wash at 70% B for the 4 min. The column was then re-equilibrated at 5% B for 10 min.
Eluting peptide anions were converted to gas-phase ions by electrospray ionization at −1.5 kV with respect to ground, and the inlet capillary temperature was held at 300 °C. Survey scans of peptide precursors were collected over the 300–1,250 Th range with an automatic gain control (AGC) target value of 1,000,000, followed by data-dependent NETD MS/MS scans of the 10 most intense peaks (maximum injection times of 200 ms for both full and tandem MS scans). Precursors with charge states equal to one or unassigned were rejected. NETD reactions were performed in either the mass-analyzing quadrupole linear ion trap (A-QLT) or the MDC (50,000 or 100,000 AGC target values, respectively), followed by mass analysis in either the A-QLT or Orbitrap, as indicated in the text. The radical cation of fluoranthene was the reagent used for all NETD analyses. For NETD reactions conducted in the A-QLT, the reagent AGC target value was set to 1,000,000 and the default reaction time was set to 100 ms for z = −2 precursors, with reaction time scaling enabled as discussed in the text. For all MDC analyses, reagent accumulation times were set to 20 ms, and reaction times were as follows: 40 ms for z = −2, 30 ms for z = −3, 20 ms for z = −4, 15 ms for z = −5, and 10 ms for z = −6 and higher charge states. During AI-NETD reactions, the external CO2 continuous wave laser was triggered using instrument firmware and modification to instrument code in conjunction with a gated laser controller. The laser irradiated the trapping volume of the MDC during the entirety of the NETD reaction at 70% total output. Precursors were isolated using a ±0.9 Th isolation window, and an exclusion window of ±10 ppm was constructed around the monoisotopic peak of each selected precursor for 45 s. Resolving powers of 60,000 and 15,000 at 400 m/z were used for survey scans and MS/MS scans in the Orbitrap, respectively.
Low-pH nLC-MS/MS
For comparison to positive mode methods, single-shot HCD and ETD analyses were collected for tryptic yeast peptides. Reversed-phase columns were prepared as described above. Mobile phase A was 0.2% formic acid in water with 5% DMSO, and mobile phase B was 0.2% formic acid in acetonitrile. One microgram of sample was loaded onto the column in 95% A for 10 min at 400 nl min−1. Gradient elution was performed at 400 nl min−1, with the gradient increased linearly from 5 to 25% B over 70 min, followed by an increase to 70% B at 76 min and a wash at 70% B for the 4 min. Electrospray voltage was set to 2 kV with respect to ground, and the inlet capillary was held at 275 °C. Survey scans of peptide precursors were collected over the 300–1,250 Th range with an AGC target value of 1,000,000 and 60,000 resolution, followed by data-dependent HCD MS/MS scans of the 15 most-intense peaks or ETD MS/MS scans of the 10 most-intense peaks (maximum injection times of 75 ms and 200 ms for full and tandem MS scans, respectively). Precursors with charge states equal to 1 or unassigned were rejected. HCD and ETD MS/MS events were both performed in the MDC with an AGC target value of 100,000, followed by mass analysis in the Orbitrap at 15,000 resolution. Normalized collision energies of 30 were used for HCD events. The radical anion of fluoranthene was the reagent used for ETD reactions. Reagent accumulation times were set to 20 ms, and reaction times were analogous to NETD reaction times described above. Precursor isolation and dynamic exclusion were the same as above.
Low-pH Prefractionation
In addition to single-shot nLC-MS/MS runs, deep-sequencing analyses were performed on digests from each enzyme, leveraging low-pH RPLC offline fractionation for an orthogonal degree of separation prior to online high-pH chromatography. Peptides were fractionated on a Phenomenex (Torrance, CA) Gemini 5 μm, 110 Å pore size C18 column (250 × 4.6 mm) with 0.1% TFA in pure water and 80% acetonitrile (mobile phases A and B, respectively). The separation gradient had a flow rate of 0.8 ml min−1 starting at 5% B for 4 min. From 4 to 8 min, B was increased to 12% and then to 45% at 49 min. At 51 min, a 5 min wash of 100% B started, followed by 20 min of re-equilibration in 5% B. Fractions were collected every minute from 4 min to 54 min for a total of 50 fractions, which were then combined into 10 total fractions in concatenated fashion. Each set of fractions was run in triplicate.
Data Analysis
Tandem mass spectra were searched with the Open Mass Spectrometry Search Algorithm, which was previously modified to accommodate anionic peptide fragments and NETD spectra, which contain a●- and x-type product ions (24, 46). Prior to the Open Mass Spectrometry Search Algorithm search, spectra were “cleaned” such that charge-reduced product ions and neutral losses within the window 55 Da below and 5 Da above the charge-reduced peaks were removed in addition to a ±3 Da window around the unreacted precursor (47, 48). A multi-isotope search using three isotopes with a mass tolerance of ±125 ppm was used for precursors, and a monoisotopic mass tolerance of ±0.30 Da or ±0.02 Da was used for product ions (a●- and x-type) in the ion trap or Orbitrap, respectively. Oxidation of methionine was specified as a variable modification, while carbamidomethylation of cysteine was a set as a fixed modification. For all enzymes, three missed cleavages were allowed with the following specificity: trypsin, full with P-rule; LysC, full with P-rule; GluC, full DE; chymotrypsin, full with P-rule; and AspN, full. Data processing was performed using in-house software (COMPASS) designed for Open Mass Spectrometry Search Algorithm search outputs (49). Peptide spectral matches (PSMs) were made against the UniProt yeast database downloaded on September 29, 2014 (6,726 entries), which was concatenated with a reversed sequence version of the forward database. Peptides were filtered to a 1% false discovery rate using both e-value and precursor mass accuracy. When pooling spectra from multiple nLC-MS/MS analyses, the false discovery rate was calculated for the aggregate set of data rather than calculating a separate false discovery rate for each run prior to combining results. Information about identified peptides and proteins are available as Supplemental Material. Information pertinent to fragmentation evaluation for NETD and AI-NETD was extracted from MS/MS scans using a C# script developed in-house. Protein isoelectric points (pI) were calculated using ExPASy (http://www.expasy.org/). The same pipeline was used for positive mode comparisons, searching b- and y-type product ions for HCD and c- and z●-type product ions for ETD, with product ion search tolerances of ±0.02 Da. ETD spectra were cleaned (above). Large-scale positive mode data (50) were downloaded from Chorus (ID# 183) and analyzed with COMPASS. Raw files were searched as previously described.
RESULTS
Negative Electron Transfer Dissociation in the Multipurpose Dissociation Cell
In our previous work using NETD for large-scale analyses of peptide anions (24), we conducted all NETD reactions in the high-pressure trap of the dual-cell mass-analyzing quadrupole linear ion trap (A-QLT) and subsequent mass analysis was performed in the low-pressure trap of the A-QLT (Fig. 1C). At the time, the sensitivity and speed of the A-QLT for analysis of tandem mass spectra offered more benefit than the higher resolution/accurate mass provided by Orbitrap mass analysis, especially considering the low precursor anion flux observed in those experiments; however, Orbitrap mass analysis did provide more confident spectral identification and was used in ensuing studies (28). Following these investigations, we described the multipurpose dissociation cell (MDC) that demonstrated improved ETD performance (faster reaction times and higher product ion signal-to-noise) for precursor cations (40). We hypothesized that the MDC could offer similar benefits for NETD analysis, granting access to the superior spectral quality of higher resolution/accurate mass product ion mass analysis in the Orbitrap without sacrificing the scan speed achieved with the A-QLT.
As described above, we modified the MDC to perform charge-sign-independent trapping of precursor anions and reagent cations, permitting NETD of multiply deprotonated peptides with radical fluoranthene cations (Fig. 1A) (51). Using a 5 mm piperidine solvent system (vide infra), we compared performance of the A-QLT and MDC for NETD analyses using 90-min shotgun nLC-MS/MS experiments on a complex mixture of S. cerevisiae peptides, performed in triplicate. Reflecting our previous studies for NETD reactions in the A-QLT, product ion mass analysis in the A-QLT outperformed product ion mass analysis in the Orbitrap (3,530 versus 3,134 peptides). Thus, all experiments using the A-QLT for NETD reactions also used the A-QLT for product ion mass analysis. All NETD reactions in the MDC, however, used the Orbitrap for product ion mass analysis, as dictated by instrument geometry. To compare directly to our previous work, we first used an NETD reaction time of 100 ms for all precursor charge states for A-QLT analyses but used dynamic reaction times for MDC reactions, as optimal reaction times scale with precursor charge (52). Because the MDC allows faster reaction times, only 40 ms were needed to achieve high-quality spectra for doubly deprotonated peptides compared with the 100 ms required for the reaction in the A-QLT. NETD experiments using the MDC as a reaction vessel afforded slightly more MS/MS scans on average than those using the A-QLT (12,494 versus 12,374, respectively) in addition to providing higher MS/MS success rates, i.e. percentage of tandem mass spectra that map back to sequence (29% versus 27%). With these advantages, NETD single-shot analyses in the MDC identified 3,805 unique peptides to the A-QLT's 3,411 unique peptides, both of which outmatched our previous results. Furthermore, we conducted a third set of experiments, this time enabling dynamic reaction times for NETD conducted in the A-QLT (scaled with precursor charge). With these conditions, NETD in the A-QLT averaged 12,582 MS/MS scans per run, illustrating the increase in scan speed afforded by scaled reaction times; however, the MS/MS success rates in these experiments also averaged 27%, matching that produced with a static reaction time in the A-QLT. Despite the increase in the number of MS/MS scans with dynamic reaction times enabled, the MDC still outperformed the A-QLT in the number of unique peptides identified (3,805 versus 3,530). These results illustrate the advantages the MDC provides for shotgun nLC-MS/MS peptide anion analyses, making high-quality, higher resolution/accurate mass NETD tandem mass spectra accessible for routine experiments.
Activated Ion NETD for Large-Scale Sequencing of Peptide Anions
Beyond the advantages the MDC provides for NETD alone, the ability to conduct NETD in this reaction vessel provides straightforward access to AI-NETD. The instrument geometry enables simple alignment of an external infrared laser that can be introduced concentric to the trapping volume of the MDC (39–42), facilitating concurrent photoactivation during NETD for improved fragmentation efficiency (Fig. 1B). Following laser alignment, we conducted another set of triplicate 90-min nLC-MS/MS experiments with yeast peptides, this time comparing NETD in the MDC to AI-NETD in the MDC. The improvement in peptide fragmentation was immediately apparent. Figure 2 provides an example of a peptide, NFNDPEVQGDMK, successfully identified at z = −2 and z = −3 in both NETD and AI-NETD analyses. The doubly deprotonated species of this 12-residue peptide has a moderate charge density (m/z 695.29), and fragmentation with NETD is somewhat limited (top left panel), providing only 45% peptide sequence coverage (as defined by number of bonds broken divided by total number of bonds); however, AI-NETD provides extensive fragmentation of the peptide, permitting straightforward annotation of fragment ions that provide 100% peptide sequence coverage (bottom left panel). NETD for the more charge-dense triply deprotonated species (m/z 463.19) provides more comprehensive fragmentation than with its doubly deprotonated counterpart, as expected (top right panel). AI-NETD maintains its high level of performance for the z = −3, too—again enabling 100% sequence coverage and providing more sequencing ions than NETD alone (bottom right panel).
Fig. 2.
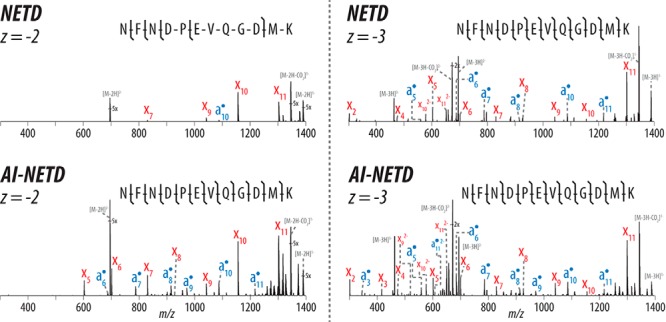
NETD- and AI-NETD-MS/MS spectra for the same peptide at z = −2 and −3. AI-NETD improves precursor-to-product ion conversion for enhanced peptide dissociation for precursors in lower charge states, increasing peptide sequence coverage in this case from 45% to 100%. NETD and AI-NETD both perform well on the same peptide at a higher charge (100% sequence coverage with both), although AI-NETD still provides a greater total number of sequencing ions. NETD and AI-NETD spectra are on the same scale for each precursor charge state.
The overall performance of AI-NETD for the large-scale analyses was just as compelling. The MS/MS success rate for AI-NETD experiments averaged nearly 53%, a drastic improvement over the 29% success rate of NETD alone. To demonstrate how AI-NETD could provide such a boost, we chose 409 unique peptides that were identified using both NETD and AI-NETD with z = −2 and compared the extent of fragmentation achieved with the two fragmentation techniques by extracting the product ions detected in each MS/MS spectrum. Figure 3 displays fragment maps for these spectra, providing a separate column for a●-type and x-type fragments. Each column is further divided into subcolumns, the number of which corresponds to the number of backbone bonds in the peptide. For example, a peptide that is eight residues long has seven columns representing the seven backbone bonds. The furthest most left subcolumn for the a●-type fragments represents fragment a●1, while the furthest most right subcolumn shows a●7. Conversely, the furthest most left subcolumn for x-type fragments shows the x7 fragment, whereas the right most subcolumn shows x1. The color scale for the two fragment ion types, shown at the bottom of the figure, indicates the intensity of the fragment. To permit comparisons among multiple spectra, the intensity of all fragments in a given spectrum was normalized to the intensity of the unreacted precursor detected in that spectrum; thus, the intensities of the fragment ions are reported as percentages of this intensity. Additionally, the peptides shown in this figure are first grouped by length, which is depicted by the number in parentheses on the far left side, and the peptides with the same length are organized by m/z values in ascending fashion. With this organization, the charge density of the peptide precursors decreases from top to bottom, making the charge density dependence trends of NETD apparent. As peptide length increases, the extent of fragmentation achieved with NETD, alone, decreases noticeably, corresponding to expected trends for decreasing charge density. By comparison, AI-NETD provides extensive fragmentation for nearly all peptides shown, even as charge densities of the precursors decrease, increasing the number and intensity of both a●-type and x-type fragments. By mitigating the detrimental charge density dependence of NETD, AI-NETD clearly offers advantages for peptide anion fragmentation, explaining the increase in MS/MS success rate between NETD and AI-NETD experiments.
Fig. 3.

A fragment map of peptides identified with both NETD and AI-NETD. Here, each row is a unique peptide so that the same row across all four columns represents the same peptide sequence. Each subcolumn corresponds a peptide backbone bond so that a peptide with eight residues has seven backbone bonds and thus seven subcolumns for both a●- and x-type product ions. The numbers in parenthesis to the left show peptide length in number of residues, and all peptides shown here are z = −2, meaning precursor charge density decreases from top to bottom. With NETD, a●- and x-type fragments decrease in number and intensity as precursor charge density decreases (i.e. as peptide length increases). AI-NETD maintains superior fragment ion generation even with decreasing precursor charge density, greatly increasing peptide dissociation and sequence coverage compared with NETD.
Importantly, these improvements afforded by AI-NETD translate not only to higher quality MS/MS spectra with more robust fragmentation but also to remarkable improvements in peptide and protein identifications. Compared with the 3,805 peptides reported above for NETD analysis using the MDC, AI-NETD identified 7,601 unique peptides, essentially doubling the number of peptide identifications achievable in the same amount of analysis time. Figure 4 summarizes the benefits of AI-NETD. The distribution of precursors across m/z space was similar for both NETD and AI-NETD runs (Fig. 4A), but the distribution of peptides successfully sequenced is noticeably more extensive with AI-NETD (Fig. 4B). Here, the peptide sequence coverage achieved for each doubly and triply deprotonated peptide identified with NETD or AI-NETD is plotted as a function of its m/z value. Successful identification of doubly deprotonated peptides with NETD dropped abruptly beyond 800 Th, and the sequence coverage achieved for these peptides decreased with higher m/z values. AI-NETD, on the other hand, successfully identified peptides across the entire m/z range and provided higher peptide sequence coverage, even maintaining 100% sequence coverage for doubly deprotonated peptides up to 1,000 Th. NETD did perform more favorably for triply deprotonated precursors than for doubly deprotonated ones, but AI-NETD remained distinctly superior for this population of peptides as well.
Fig. 4.

AI-NETD outperforms NETD on a global scale. (A) The distribution of precursors selected for NETD or AI-NETD in a given run show roughly equivalent populations of peptides to fragment. (B) Using the PSMs generated from the runs shown in (A), peptide sequence coverage is plotted as a function of precursor m/z. AI-NETD increases the number of peptides identified, extends the m/z range that can generate successful PSMs, and provides overall higher peptide sequence coverage at given m/z values. (C) The combination of these improvements with AI-NETD at the peptide level translates to benefits at the protein level as well. AI-NETD characterized 1,106 proteins to NETD's 674. Beyond this, for proteins detected with both methods, AI-NETD overwhelmingly enhanced protein sequence coverage, by more than 50% in some cases.
Beyond the substantial improvements in fragmentation at the peptide level, AI-NETD also performed advantageously at the protein level. In triplicate single-shot 90 min analyses, AI-NETD identified 1,106 proteins in yeast, making it the first technique to achieve identification of more than 1,000 proteins using the negative mode approach. Comparatively, NETD identified 674 yeast proteins. Panel C of Fig. 4 illustrates the gain in protein sequence coverage afforded by AI-NETD for proteins identified using both methods. Here, protein sequence coverage represents number of total amino acids explained from peptide identifications divided by the total number of amino acids. The gain in sequence coverage is defined as sequence coverage with NETD subtracted from sequence coverage with AI-NETD for a given protein; thus, a negative value means that higher sequence coverage was seen with NETD. AI-NETD provided a gain in sequence coverage for the overwhelming majority of proteins, many of which were acidic in nature (as indicated by the x axis). In all, the enhancement in fragmentation that AI-NETD afforded for peptide anions unequivocally translates to more robust protein characterization, making it a premier tool for negative mode proteomics.
Selection of Robust High-pH Solvent System
Concurrent to our investigations into NETD and AI-NETD, we also explored how to increase precursor anion flux with different high-pH solvent systems. We previously employed ammonium formate solvents for high-pH separations, but charge state distributions favoring lowly charged precursors, degradation of silica-based packing materials, and low precursor flux were significant challenges with this system (24, 28). Although others have used similar systems with success, we also took note of several studies utilizing piperidine buffers for high-pH chromatography (20, 25, 53, 54). With this knowledge, we evaluated five different solvent systems using either piperidine or ammonium formate buffers over triplicate 90-min nLC-MS/MS analysis of yeast peptides with AI-NETD fragmentation. A 10 mm piperidine system and 5 mm ammonium formate, pH 10, were first compared. We observed increased peptide anion signal with piperidine solvents, so we prepared ammonium formate buffers that were basified to pH 11.5 with ammonium hydroxide to match the pH of the 10 mm piperidine system. In the same vein, we also prepared 5 mm piperidine solvents, reducing the pH of buffer A slightly to ∼11.3. Additionally, leveraging the recent descriptions of boosts provided by addition of DMSO to buffers for positive mode analyses, we assessed a 5 mm piperidine solvent system that contained 5% DMSO in buffer A.
Panel A of Fig. 5 displays base peak chromatograms from an experiment with each of the five solvent systems, highlighting the increases in signal, i.e. anion flux, observed with the piperidine buffers. The 5- to 10-fold improvements in base peak signal with piperidine solvents are reflected in the boosts observed in the number of tandem mass spectra, peptide spectral matches, and unique peptide identifications summarized in Table I. Panel B of Fig. 5 shows that, beyond increasing the number of MS/MS scans acquired, piperidine solvents shift the charge state distributions precursors to more highly charged (more negative) species, a phenomenon also reported for peptide standards and simple peptide mixtures (20, 53). Although increasing the pH of ammonium formate solvents to 11.5 did show a small expansion in precursor charge state distributions, this difference was minimal compared with the pH 10 ammonium formate solvents and failed to match that observed with piperidine. Interestingly, the 5 mm piperidine buffers with 5% DMSO provided neither the highest base peak signal nor a gain in peptide identifications, which are both advantages reported in positive mode analyses (45, 55, 56). As shown in Table I and Fig. 5B, DMSO in the 5 mm piperidine solvent did permit the acquisition of the most MS/MS scans and displayed the widest distribution of precursor charge states; however, peptide identifications were lower with the DMSO additive than both the other two piperidine solvents. Regardless, high-pH solvent systems that utilize piperidine performed consistently better than ammonium formate solvent systems, maximizing the number of unique peptides identified with AI-NETD.
Fig. 5.

A comparison of high-pH solvent systems used. (A) Base peak chromatograms show that choice of buffer additives, rather than pH alone, generate increased ionization with negative ESI. Piperidine solvents generate nearly an order of magnitude higher precursor ion signal than ammonium formate solvents. (B) Beyond increasing the number of precursors selected for MS/MS, piperidine solvents shift the charge state distributions of precursor anions to be more highly charged (more negative) than ammonium formate solvents. Notably, DMSO in the 5 mm piperidine solvent system expanded the distribution of charge states to more highly charged precursors, rather than collapsing it to lower charges as reported in positive electrospray.
Table I. Summary of AI-NETD experiments with five different high-pH solvent systems.
| Solvent system | 5 mm ammonium formate pH 10 | 5 mm ammonium formate pH 11.5 | 10 mm piperidine pH ∼ 11.5 | 5 mm piperidine +5% DMSO, pH ∼ 11.3 | 5 mm piperidine pH ∼ 11.3 |
|---|---|---|---|---|---|
| Total MS/MS scans | 30,818 | 32,752 | 38,856 | 40,865 | 39,318 |
| Total peptide spectral matches (PSMs) | 10,123 | 10,371 | 15,492 | 16,606 | 20,099 |
| Total unique peptides | 3,659 | 3,924 | 5,920 | 5,208 | 7,601 |
Multiple Proteases for Single-Shot Negative Mode Proteomics
Even with the robust analyses afforded by the combination of AI-NETD with prudently chosen piperidine high-pH solvents, use of just one protease limits the portion of the proteome that is accessible in a given experiment. The value of multiple proteases for canonical positive mode shotgun proteomics has been shown by us and others (50, 57–59); however, negative mode studies to date have largely failed to capitalize on the advantages offered by use of multiple enzymes for protein digestion. Instead, most studies rely mainly on trypsin for enzymatic digestion, although GluC has also been used. In this study, we extended our success with AI-NETD for tryptic peptides to single-shot analyses of peptides derived from four other proteases (LysC, GluC, chymotrypsin, and AspN) with the goals of exploring the amenability of different proteases to negative mode experiments and increasing the proteomic depth that can be achieved with negative mode analyses.
Table II summarizes the results from triplicate 90-min AI-NETD nLC-MS/MS experiments that analyzed five different complex yeast peptide mixtures, each from one of the five proteases investigated. Immediately evident is the superior performance of trypsin and LysC, both of which enabled the identification of more than 1,000 proteins. Similar performance of these two enzymes is unsurprising considering their related proteolytic specificity C-terminal to lysine (trypsin and LysC) and arginine (trypsin) residues. GluC, which cleaves C-terminal to glutamic acid (and at slower rates, aspartic acid (60)), enabled the identification of more than 4,200 unique peptides, a greater than 7-fold increase over our previous NETD results with the enzyme (24). These peptides mapped back to 857 proteins, which also outmatches the best results achieved to date for any protease in negative mode approaches (25). Chymotrypsin and AspN performed considerably well, too, illustrating the flexibility AI-NETD can offer for peptide anion characterization. Toward our goal of increasing proteomic depth, we batched the results from these five proteases together, providing nearly 21,000 unique peptide identifications. This combination of results provided valuable depth at the protein level, bolstering the number of proteins identified by ∼23% and providing a jump in average protein sequence coverage from 23.6% to 34.4% over analysis with trypsin alone.
Table II. AI-NETD performance for peptides generated from five different proteases in single-shot experiments. Proteolytic specificity for each enzyme is shown.

Beyond the beneficial information derived from the multiple protease approach for proteome characterization, we were also curious how AI-NETD performed for fragmenting families of peptides that were chemically distinct due to their proteolytic origins. We constructed fragment maps (vide supra) for AI-NETD fragmentation of peptides from all five enzymes and used results from trypsin as a point of reference. The trends in fragmentation between peptides from trypsin, LysC, chymotrypsin, and AspN were similar (data not shown); the fragmentation for GluC peptides, however, seemed to be more extensive than that seen with trypsin (Fig. 6A). Here, we looked at peptides of relatively moderate and long length (12 and 24 residues, respectively), considering two precursor charge states for each. All peptides of the given length and charge are represented for peptides from both enzymes. This includes 443, 98, 94, and 65 tryptic peptides and 323, 146, 43, and 34 GluC peptides for length 12, z = −2; length 12, z = −3; length 24, z = −3; and length 24, z = −4, respectively. AI-NETD appears to produce fragment ions in greater number and intensity for peptides derived from GluC compared with trypsin, which is especially notable for z = −3 peptides that are 12 amino acids (AA) long. Juxtaposed to tryptic peptides with basic C termini, GluC peptides have a C-terminal acidic residue (D/E), meaning at least one negative charge is fixed at the C terminus. This could explain the improved electron-driven fragmentation seen for GluC peptides, especially for shorter peptides where proximity of backbone bonds to this C-terminal negative charge is greater. Broadening this idea beyond the subset of peptides investigated in panel A, we calculated peptide sequence coverage values for all identified tryptic and GluC peptides, z = −2 through −4. Density plots in Fig. 6B show the frequency of peptides characterized with a given sequence coverage, serving as a surrogate for extent of fragmentation. AI-NETD fragmentation shows a similar trend for z = −2 peptides from trypsin and GluC, favoring higher sequencing coverage; however, AI-NETD with GluC peptides maintains high peptide sequence coverage for z = −3 and −4 peptides while the sequence coverage for tryptic peptides is much more evenly distributed, mirroring the more-extensive fragmentation seen with the subset of GluC peptides in panel A.
Fig. 6.

Comparison of single-shot AI-NETD for peptides produced by either trypsin or GluC. (A) AI-NETD fragment map for peptides (12 and 24 amino acids in length) derived from both trypsin and GluC digestions. The numbers in parenthesis to the left indicate peptide charge. (B) Density plots for peptide sequence coverage for PSMs from trypsin and GluC. (C) The larger histogram shows the distribution of peptide lengths from an in silico digest for trypsin and GluC. The inset displays the distribution of lengths of peptides identified in the trypsin and GluC experiments. The numbers in parenthesis in the respective legends show the average peptide length for each protease. (D) The MS/MS success rate (blue) for GluC peptides is significantly lower than tryptic peptides (p < .01, indicated by *), while the ratio of unique peptides to total PSMs detected (red) is not statistically different (p < .05).
Although it appears AI-NETD fragments GluC peptides more extensively, results with GluC do not rival those achieved with trypsin. Intrigued by this, we performed an in silico digest of the yeast proteome for both proteases, allowing up to two missed cleavages with a minimum peptide length of six amino acids and a maximum length of 75 amino acids. The larger histogram in Fig. 6C shows that the distribution of possible tryptic peptides favors shorter peptides (average length of 20.6 residues) while GluC produces fewer total peptides and does not favor short peptides as drastically (average length of 27.8 residues). The inset in Fig. 6C displays a histogram of peptide lengths for peptides actually identified in AI-NETD analyses, providing the complementary experimental measurement to the theoretical data derived from the in silico digest. Interestingly, despite the notable difference in distributions of peptide length from the in silico digestion, the experimental distributions of peptide length are very similar, with average peptide lengths of 16 and 15 residues for trypsin and GluC, respectively. We surmise that lower peptide identifications with GluC compared with trypsin is a function of the population of peptides generated by GluC rather than the fragmentation achieved for these peptides. Although GluC peptides may fragment better, a smaller portion of peptides derived from GluC are in the ideal range of peptide length for AI-NETD, which appears to be 10–25 residues. This translates to a higher percentage of fragmentation events (i.e. MS/MS scans) occurring on larger, more difficult-to-sequence peptides for GluC. In fact, Fig. 6D supports this, showing that the ratio of unique peptides to total PSMs is the same for the two enzymes, but the MS/MS success rate is significantly lower (p < .01) for GluC. Additionally, our entire workflow was originally optimized for tryptic peptides, including sample preparation and chromatographic conditions, which could also contribute to this discrepancy. These observations may explain why, even with superior fragmentation and higher peptide sequence coverage for the peptides we do sequence, fewer peptides are ultimately identified using GluC as a protease rather than trypsin. Even so, this does not make AI-NETD analyses of GluC peptides any less valuable; rather, these results suggest that combinations of proteases can be used not only to enhance proteomic depth but also to access the advantages AI-NETD can provide for peptides with distinct chemical properties.
Deep Sequencing in the Negative Mode with Offline Low-pH Fractionation and Multiple Proteases
Encouraged by these results, we sought to improve upon the proteomic depth we could achieve with purely negative mode techniques. All previous large-scale peptide anion analyses have used online one-dimensional high-pH chromatography for single-shot experiments, similar to the approaches we have described thus far. Common practice in traditional proteomic experiments is to increase the achievable proteomic depth by decreasing sample complexity via online and/or offline fractionation (61, 62); we used the same logic to fractionate peptide mixtures from trypsin, LysC, GluC, chymotrypsin, and AspN digestions prior to nLC-MS/MS analysis with AI-NETD. In positive mode approaches, offline high-pH reversed-phase fractionation provides an orthogonal mode of separation to the online acidic reversed-phase chromatographic conditions used for nLC-MS/MS analysis (63). It holds that the two should remain orthogonal even if the order in which they are performed is inverted. Thus, we employed a simple low-pH reversed-phase fractionation system to separate a complex mixture of peptides into 50 fractions, which were then concatenated into 10 total fractions for subsequent negative mode nLC-MS/MS analysis. This fractionation was done for a digestion from each protease.
The offline fractionation approach extraordinarily improved peptide and protein identification for all five proteases with AI-NETD (Fig. 7). Where single-shot AI-NETD experiments with tryptic peptides produced 7,601 unique peptide identifications and 1,106 proteins, analysis of the same mixture of tryptic peptides split equally into 10 fractions enabled the identification of 36,713 unique peptides and 3,467 proteins. Figure 7A shows the results achieved from analysis of 10 fractions for each of the five proteases. Here, the area of the circle represents the total number of proteins identified. The circles along the diagonal present the results for the proteases individually. This figure also displays the average percentage of protein sequence coverage observed with each enzyme with a color gradient. Further, panel A presents pairwise comparisons that show the combination of results from two proteases batched together. This analysis allows an evaluation of how combinations of different proteases affect the number and coverage of proteins detected, illustrating the degree of orthogonality of each protease with the others. Intriguingly, the combination of trypsin and GluC proteases for this large-scale, deep-sequencing approach provided the largest number of proteins identified in these pairwise comparisons with an average protein sequence coverage of 35.14%. The combination of trypsin and chymotrypsin, however, provided the greatest average sequence coverage (36.5%) even though it did not appreciably increase the number of proteins identified with trypsin alone. This analysis provides insight into what combinations of proteases, whether they cleave at basic, acidic, or hydrophobic residues, may be the most beneficial as negative mode approaches continue to advance. Panel B shows the number of total PSMs collected for each enzyme, providing some concept of the complexity of peptide mixtures produced from each. When integrating the identifications from all five enzymes into a batched analysis of proteins identified, AI-NETD facilitated the characterization of 3,730 proteins with an average sequence coverage of 43.9%. This represents nearly 83% of the expressed yeast proteome (estimated to be ∼4,500 proteins (64)), demonstrating that comprehensive proteome analysis, which has been previously confined to positive mode analyses, can be achieved in the negative mode via peptide fragmentation with AI-NETD. Figure 7C shows the proportions of these 3,730 proteins with a given protein sequence coverage; while approximately half of the proteins had sequence coverage under 40%, an appreciable fraction of these protein identifications had excellent sequence coverage (80–100%).
Fig. 7.
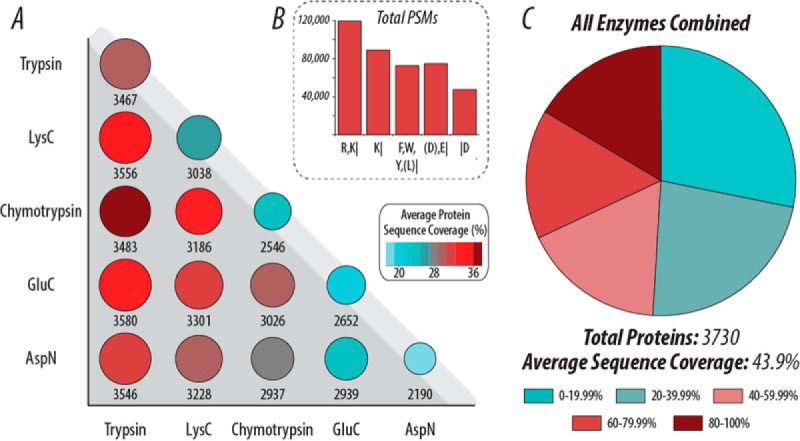
Proteome coverage in the negative mode with various proteases using AI-NETD and low-pH fractionation. (A) Pairwise comparisons of the number of proteins (area of circle) and average protein sequence coverage (color) when using different proteases, illustrating the degree of orthogonality of each protease with the others. The circles along the top edge (light gray background) show each enzyme by itself. (B) The total number of PSMs for each protease. (C) When combining all PSMs from the five enzymes, AI-NETD characterizes over 80% of the yeast proteome (, proteins). The pie chart here shows what proportions of these proteins had sequence coverage in the given range.
Comparing Positive and Negative Mode Data
Although the data presented above stand alone as a demonstration of what can be considered realistic and achievable in negative mode proteomics, a natural and valuable extension lies in the comparison of these large-scale peptide anion analyses to large-scale positive mode experiments. First, we performed triplicate single-shot analyses of yeast tryptic peptides using HCD and ETD fragmentation for comparison of positive mode data to AI-NETD. Both HCD and ETD were performed in the MDC to provide the most direct comparison possible, thus keeping both reaction cell and mass analyzer (i.e. the Orbitrap) consistent with the negative mode experiments. Figure 8A displays the number of peptides identified with each method and shows the overlap in peptides between the three fragmentation types. Surprisingly, AI-NETD performed as well as, if not better, than ETD in the number of peptides identified (7,601 versus 7,414). Only one-third of the total peptides sequenced by ETD and NETD were identified in both methods, highlighting the complementarity of the positive and negative mode approaches for electron-driven dissociation techniques. Furthermore, although HCD produced more peptide identifications than both ETD and AI-NETD, the overlap in peptides was higher for the two positive mode techniques—83% of ETD peptides were also identified with HCD while 68.5% of AI-NETD peptides were also seen in the HCD data. This provides clear evidence that negative mode analysis with AI-NETD is fully capable of affording greater orthogonality to positive mode collision-based peptide identification than offered by positive mode ETD, maximizing the number of peptides than can be identified in a sample (although we note that more vigorous and extensive comparisons are need to fully explore this issue).
Fig. 8.

Comparison of AI-NETD to positive mode analyses. (A) Overlap in yeast tryptic peptides identified in HCD, ETD, and AI-NETD single-shot experiments. The number of unique peptides identified with each fragmentation type are indicated in italics below the appropriate label. (B) Distribution of protein sequence coverages achieved with combinations of trypsin with different proteases in positive mode analyses with CID and ETD fragmentation compared with positive mode data combined with negative mode data with AI-NETD (gray background) using only trypsin. The dotted line shows the highest average sequence coverage. (C) Overlap in yeast proteins identified using positive mode (CID and ETD) and negative mode (AI-NETD) analyses. For both sets of proteins, peptides from five different proteases were batched together. (D) Proteins detected in the positive mode data from (C) are rank ordered by percentage sequence coverage and plotted in blue, with an average sequence coverage of 41.1%. The protein sequence coverage achieved for a given protein when combining the positive and negative mode data is plotted in red, highlighting the gain in sequence coverage afforded by the addition of negative mode analyses. To the far right, sequence coverages are shown for the 272 proteins that were not detected in positive mode analyses but were characterized with AI-NETD.
To further our comparison to positive mode methods, we examined the overlap in peptides and proteins characterized in our deep-sequencing experiments to those identified in an extensive published proteomic analysis of yeast peptides that used offline fractionation and multiple proteases, two key components of our deep proteome sequencing with AI-NETD (50). Additionally this study utilized decision tree logic to tailor fragmentation (either CID or ETD) to each peptide, maximizing the chances of an MS/MS scan being successfully mapped to sequence (65). All of these components make this data set one of the most robust available characterizations of the yeast proteome, with the added benefit of the ability to compare multiple proteases in positive and negative modes. Figure 8B shows the distribution of percentage protein sequence coverages achieved for n proteins in the positive mode when using trypsin and various combinations of peptides from other proteases [trypsin(+) and proteaseX(+), where proteaseX is LysC, GluC, AspN, or ArgC]. Also included in that plot is the combination of tryptic peptides from the positive mode data and the AI-NETD data set [trypsin(+) and trypsin(-)] (gray background). Compellingly, using one protease with both the positive and negative modes outperforms all other positive mode combinations of trypsin with a different protease. Not only does the trypsin(+) and trypsin(-) combination identify more proteins than any trypsin (+) and proteaseX(+) combination, but it also provides the greatest median (29.17%) and average (34.02%) percentage sequence coverage—indicating that negative mode analyses for tryptic peptides can provide more orthogonality to positive mode experiments than the use of different proteases. For the other three proteases in common between the two studies (LysC, GluC, and AspN), the addition of negative mode analyses with AI-NETD increases “positive mode only” protein sequence coverage with LysC from 23.72% to 30.97%, with GluC from 19.57% to 24.10%, and with AspN from 20.98% to 23.34%.
Finally, we examined the overlap of all proteins from all five proteases characterized in the positive mode data to all proteins from all five proteases in the AI-NETD data set, which is depicted in Fig. 8C. The degree of overlap in proteins identified is noticeably large (∼85% of all proteins identified were seen in both data sets), which is not wholly unsurprising as >90% of the expressed yeast proteome is represented. What this overlap fails to display, however, is the difference in the populations of peptides sequenced. Of the 106,861 unique peptides identified between the two experiments, only 19,697 of them (∼18%) were detected in both data sets. To visualize how this impacts protein sequence coverage, Fig. 8D displays all proteins identified in the positive mode data, rank ordered by percentage protein sequence coverage (blue circles). For each protein there is a corresponding red circle that shows the sequence coverage achieved when including the negative mode data with the positive mode data set; thus, the magnitude of the difference along the y axis between a red and blue circle for any given protein shows the gain in percentage protein sequence coverage provided by the inclusion of the AI-NETD data. The average percentage sequence coverage for the positive mode data is 41.1%, which is boosted by nearly 12 percentage points to 52.7% by the addition of the negative mode analyses. This combination of positive and negative mode data puts the average sequence coverage above 50%, a mark that neither data set reached on their own. The highest gain in percentage sequence coverage was 61.7%, starting at 18.29% sequence coverage with positive mode methods alone and going to 80% sequence coverage with combined positive and negative mode analyses. This 174 residue protein, calcineurin subunit B—which is a calcium-dependent, calmodulin stimulated protein phosphatase that confers calcium sensitivity—is a notably acidic protein (pI ∼ 4.36) with 36 negatively charged residues (Asp and Glu) and only 21 basic residues (Lys and Arg). Many proteins that showed comparable gains in sequence coverage shared similar characteristics in isoelectric point and residue frequencies, illustrating the ability of AI-NETD to add coverage to the acidic portions of the proteome that may be missed by positive mode methods. The far right of the graph in Fig. 8D shows sequence coverage for 272 proteins that were only characterized by the inclusion of the AI-NETD data. Interestingly, many of these proteins are membrane proteins, including mitochondrial membrane, Golgi apparatus membrane, and other transport proteins; this characterization of membrane proteins via negative mode methods may point to an interesting avenue to explore in future investigations. In all, these comparisons demonstrate that large-scale negative mode analyses with AI-NETD are a valuable complement to positive mode methods, perhaps even offering more orthogonality than ETD or the use of multiple proteases.
DISCUSSION
Negative mode approaches for the characterization of peptide anions offer a valuable dimension to proteomic analyses (21, 24, 25, 66), especially as biologically relevant posttranslational modifications and other analytes that pose challenges to canonical positive mode techniques continue to emerge (7, 53, 67–72). Platforms for shotgun analysis of complex mixtures of peptide anions have been introduced, most notably using UVPD and NETD; however, these approaches have yet to provide considerable proteomic depth (fewer than ∼800 total proteins identified in a given experiment), restricting the degree to which peptide anion characterization can benefit the proteomic community.
With a new implementation of AI-NETD, we have introduced a robust negative mode platform for the characterization of over 1,100 proteins in a eukaryotic system (yeast) using 90-min single-shot analyses. Moreover, we demonstrate that AI-NETD is compatible with a diverse array of commonly used proteases, increasing proteomic depth (> 1,350 proteins) and degree of protein characterization, i.e. sequence coverage, achievable in single-shot experiments. The ability to utilize several proteases makes AI-NETD an especially viable technique for analysis of PTMs, where access to chemically distinct peptides or a combinatorial pattern of sequences may greatly increase confidence in identification and localization. This approach may also prove beneficial for more extensive characterization of proteins that are challenging to current techniques, e.g. proteins with dominantly acidic sequences or those with both highly hydrophobic and hydrophilic regions.
Integral to the improvements observed with our AI-NETD work was the selection of a high-pH solvent system that increased anion flux and provided optimal distribution of precursor charge states. Previous studies have shown the benefits of piperidine buffers for anion analysis, but this work is the first to demonstrate the consequential effects it can have on large-scale shotgun proteomic experiments. Based on the benefits we and others observed with DMSO (45, 55, 56), we expected the addition of DMSO to our piperidine solvents would provide an additional boon to our negative mode work. This was not the case, however, as the piperidine solvents with DMSO included provided the fewest numbers of unique peptide identifications among the three piperidine systems tested. It has been suggested that the benefits of DMSO in positive mode comes from the charge state coalescence for peptide cations, making signal more concentrated for a fewer number of charge states and reducing redundant sampling of the same peptide with different m/z values. In the negative mode, DMSO appears to have the opposite effect, expanding the charge state distribution of peptide precursors sampled for MS/MS events (Fig. 5B). The base peak intensity of the chromatogram from 5 mm piperidine solvent with 5% DMSO was slightly lower than 5 mm piperidine alone (Fig. 5A), also juxtaposing the trends observed in positive mode. Noticeable, though, is that the greatest number of MS/MS scans was taken with the piperidine/DMSO buffers than with any other system. These results suggest that DMSO is further spreading signal among many charge states for peptide precursor anions, rather than collapsing it as in positive mode. Thus, it is possible that DMSO does not merely condense a signal into lower charge states, as was suggested based on positive mode studies but that it makes the precursor charge “more negative” or “less positive” than before. Surely validation of this hypothesis requires further exploration beyond the scope of this work, but this observation highlights an unexpected outcome that emphasizes the value negative mode proteomics can have as a complementary tool to positive mode techniques.
Additionally, challenges with column longevity and hindered performance of LC pumps have been reported for high-pH reversed-phase separations for negative mode proteomics (7, 24, 73). We previously struggled with both precolumn and analytical column degradation due to the instability of silica in basic conditions. In this work, we eliminated the precolumns that used silica frits and employed a polymer-silica hybrid reversed-phase packing material that is stable at both acidic and basic conditions (74). With these modifications, our columns often lasted a week or more of constant runs, showing impressive longevity compared with our previous column setup that lasted roughly a day. LC maintenance was required periodically, including changing of silica capillaries lines prior to the analytical column, but this still permitted straightforward and consistent data collection for our single-shot and deep-sequencing experiments.
In summary, we demonstrated that AI-NETD, in conjunction with robust high-pH separations, multiple proteases, and offline low-pH prefractionation, can be leveraged to characterize the large majority (>80%) of the yeast proteome, matching the average sequence coverage observed in similar positive mode experiments using multiple fragmentation types (∼45%) (50). Moreover, AI-NETD analyses provided a substantial improvement in protein sequence coverage (an average of ∼12% but as much as ∼62%) over what could be achieved with positive mode methods alone, even those utilizing a multiple protease approach. That being said, we do not see negative mode proteomics replacing any of the wide array of positive mode approaches, much less competing with the acquisition rate and proteomic depth they can achieve (45, 75, 76); instead, we foresee negative mode proteomics continuing to advance in sensitivity and speed—especially as robust fragmentation techniques like AI-NETD are implemented on the newest generations of instruments—serving as a powerful complement to traditional positive mode methods. Most importantly, this work demonstrates that large-scale analysis of peptide anions can be used to characterize nearly an entire proteome, enabling a much more thorough investigation of previously intractable portions of the proteome and critical PTMs that will greatly benefit from analysis in the negative mode.
Supplemental Information
All raw files and annotated for spectra single peptide protein identifications from these experiments are available on Chorus (Project ID 879). Supplemental information includes details for peptide and protein identifications for all experiments.
Supplementary Material
Acknowledgments
We thank Jae Schwartz, John E.P. Syka, Chad Weisbrod, and Chris Mullen of Thermo in addition to all members of the Coon group for helpful discussions, especially Tim Rhoads and Catie Minogue.
Footnotes
Author contributions: N.M.R., M.J.R., C.M.R., M.S.W., and J.J.C. designed the research; N.M.R., M.J.R., and A.L.R. performed the research; N.M.R., M.J.R., C.M.R., D.J.B., and M.S.W. contributed new reagents or analytic tools; N.M.R., M.J.R., N.W.K., and A.S.H. analyzed data; and N.M.R. and J.J.C. wrote the paper.
* We gratefully acknowledge support from Thermo Fisher Scientific and NIH grant R01 GM080148. N.M.R. was funded through an NSF Graduate Research Fellowship (DGE-1256259). C.M.R was funded by an NSF Graduate Research Fellowship and NIH Traineeship (T32GM008505). A.L.R. was supported by an NIH-funded Genomic Sciences Training Program (5T32HG002760).
1 The abbreviations used are:
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- PTM
- posttranslational modification
- ETD
- electron transfer dissociation
- AI-ETD
- activated ion electron transfer dissociation
- NETD
- negative electron transfer dissociation
- AI-NETD
- activated ion negative electron transfer dissociation
- UVPD
- ultraviolet photodissociation
- HCD
- higher-energy collisional dissociation
- MDC
- multipurpose dissociation cell
- A-QLT
- mass-analyzing quadrupole linear ion trap
- ESI
- electrospray ionization
- nLC-MS/MS
- nanoflow liquid chromatography-tandem mass spectrometry
- DC
- direct current
- AGC
- automatic gain control
- pI
- isoelectric point.
REFERENCES
- 1. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 2. Kim M. S., Pinto S. M., Getnet D., Nirujogi R. S., Manda S. S., Chaerkady R., Madugundu A. K., Kelkar D. S., Isserlin R., Jain S., Thomas J. K., Muthusamy B., Leal-Rojas P., Kumar P., Sahasrabuddhe N. A., Balakrishnan L., Advani J., George B., Renuse S., Selvan L. D., Patil A. H., Nanjappa V., Radhakrishnan A., Prasad S., Subbannayya T., Raju R., Kumar M., Sreenivasamurthy S. K., Marimuthu A., Sathe G. J., Chavan S., Datta K. K., Subbannayya Y., Sahu A., Yelamanchi S. D., Jayaram S., Rajagopalan P., Sharma J., Murthy K. R., Syed N., Goel R., Khan A. A., Ahmad S., Dey G., Mudgal K., Chatterjee A., Huang T. C., Zhong J., Wu X., Shaw P. G., Freed D., Zahari M. S., Mukherjee K. K., Shankar S., Mahadevan A., Lam H., Mitchell C. J., Shankar S. K., Satishchandra P., Schroeder J. T., Sirdeshmukh R., Maitra A., Leach S. D., Drake C. G., Halushka M. K., Prasad T. S., Hruban R. H., Kerr C. L., Bader G. D., Iacobuzio-Donahue C. A., Gowda H., Pandey A. (2014) A draft map of the human proteome. Nature 509, 575–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wilhelm M., Schlegl J., Hahne H., Moghaddas Gholami A., Lieberenz M., Savitski M. M., Ziegler E., Butzmann L., Gessulat S., Marx H., Mathieson T., Lemeer S., Schnatbaum K., Reimer U., Wenschuh H., Mollenhauer M., Slotta-Huspenina J., Boese J. H., Bantscheff M., Gerstmair A., Faerber F., Kuster B. (2014) Mass-spectrometry-based draft of the human proteome. Nature 509, 582–587 [DOI] [PubMed] [Google Scholar]
- 4. Richards A. L., Merrill A. E., Coon J. J. (2015) Proteome sequencing goes deep. Curr. Opin. Chem. Biol. 24C, 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Engholm-Keller K., Larsen M. R. (2013) Technologies and challenges in large-scale phosphoproteomics. Proteomics 13, 910–931 [DOI] [PubMed] [Google Scholar]
- 6. Dalpathado D. S., Desaire H. (2008) Glycopeptide analysis by mass spectrometry. Analyst 133, 731–738 [DOI] [PubMed] [Google Scholar]
- 7. Robinson M. R., Moore K. L., Brodbelt J. S. (2014) Direct identification of tyrosine sulfation by using ultraviolet photodissociation mass spectrometry. J. Am. Soc. Mass Spectrom. 25, 1461–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ewing N. P., Cassady C. J. (2001) Dissociation of multiply charged negative ions for hirudin (54–65), fibrinopeptide B, and insulin A (oxidized). J. Am. Soc. Mass Spectrom. 12, 105–116 [DOI] [PubMed] [Google Scholar]
- 9. Kiraga J., Mackiewicz P., Mackiewicz D., Kowalczuk M., Biecek P., Polak N., Smolarczyk K., Dudek M. R., Cebrat S. (2007) The relationships between the isoelectric point and: Length of proteins, taxonomy and ecology of organisms. BMC Genomics 8, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leymarie N., Zaia J. (2012) Effective use of mass spectrometry for glycan and glycopeptide structural analysis. Anal. Chem. 84, 3040–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamashita M., Fenn J. (1984) Negative ion production with the electrospray ion source. J. Phys. Chem. 750, 4611–4615 [Google Scholar]
- 12. Straub R. F., Voyksner R. D., Voyksner D. (1993) Negative ion formation in electrospray mass spectrometry. J. Am. Soc. Mass Spectrom. 4, 578–587 [DOI] [PubMed] [Google Scholar]
- 13. Bowie J. H., Brinkworth C. S., Dua S.. Collision-induced fragmentations of the (M-H)- parent anions of underivatized peptides: An aid to structure determination and some unusual negative ion cleavages. Mass Spectrom. Rev. 21, 87–107 [DOI] [PubMed] [Google Scholar]
- 14. Brinkworth C. S., Dua S., McAnoy A. M., Bowie J. H. (2001) Negative ion fragmentations of deprotonated peptides: Backbone cleavages directed through both Asp and Glu. Rapid Commun. Mass Spectrom. 15, 1965–1973 [DOI] [PubMed] [Google Scholar]
- 15. Steinborner S. T., Bowie J. H. (1996) A comparison of the positive- and negative-ion mass spectra of bio-active peptides from the dorsal secretion of the Australian red tree frog, Litoria rubella. Rapid Commun. Mass Spectrom. 10, 1243–1247 [DOI] [PubMed] [Google Scholar]
- 16. Steinborner S. T., Bowie J. H. (1997) The negative ion mass spectra of MH-ions derived from caeridin and dynastin peptides. Internal backbone cleavages directed through Asp and Asn residues. Rapid Commun. Mass Spectrom. 11(3), 253–258 [Google Scholar]
- 17. Budnik B. A., Haselmann K. F., Zubarev R. A. (2001) Electron detachment dissociation of peptide di-anions: An electron–hole recombination phenomenon. Chem. Phys. Lett. 342, 299–302 [Google Scholar]
- 18. Coon J. J., Shabanowitz J., Hunt D. F., Syka J. E. P. (2005) Electron transfer dissociation of peptide anions. J. Am. Soc. Mass Spectrom. 16, 880–882 [DOI] [PubMed] [Google Scholar]
- 19. Kalli A., Grigorean G., Håkansson K. (2011) Electron induced dissociation of singly deprotonated peptides. J. Am. Soc. Mass Spectrom. 22, 2209–2221 [DOI] [PubMed] [Google Scholar]
- 20. Madsen J. A., Kaoud T. S., Dalby K. N., Brodbelt J. S. (2011) 193-nm photodissociation of singly and multiply charged peptide anions for acidic proteome characterization. Proteomics 11, 1329–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoo H. J., Wang N., Zhuang S., Song H., Håkansson K. (2011) Negative-ion electron capture dissociation: Radical-driven fragmentation of charge-increased gaseous peptide anions. J. Am. Chem. Soc. 133, 16790–16793 [DOI] [PubMed] [Google Scholar]
- 22. Antoine R., Joly L., Tabarin T., Broyer M., Dugourd P., Lemoine J. (2007) Photo-induced formation of radical anion peptides. Electron photodetachment dissociation experiments. Rapid Commun. Mass Spectrom. 21, 265–268 [DOI] [PubMed] [Google Scholar]
- 23. Kjeldsen F., Silivra O. A., Ivonin I. A., Haselmann K. F., Gorshkov M., Zubarev R. A. (2005) C alpha-C backbone fragmentation dominates in electron detachment dissociation of gas-phase polypeptide polyanions. Chemistry. 11, 1803–1812 [DOI] [PubMed] [Google Scholar]
- 24. McAlister G. C., Russell J. D., Rumachik N. G., Hebert A. S., Syka J. E., Geer L. Y., Westphall M. S., Pagliarini D. J., Coon J. J. (2012) Analysis of the acidic proteome with negative electron-transfer dissociation mass spectrometry. Anal. Chem. 84, 2875–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Madsen J. A., Xu H., Robinson M. R., Horton A. P., Shaw J. B., Giles D. K., Kaoud T. S., Dalby K. N., Trent M. S., Brodbelt J. S. (2013) High-throughput database search and large-scale negative polarity liquid chromatography-tandem mass spectrometry with ultraviolet photodissociation for complex proteomic samples. Mol. Cell. Proteomics 12, 2604–2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Syka J. E., Coon J. J., Schroeder M. J., Shabanowitz J., Hunt D. F. (2004) Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 101, 9528–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huzarska M., Ugalde I., Kaplan D. A., Hartmer R., Easterling M. L., Polfer N. C. (2010) Negative electron transfer dissociation of deprotonated phosphopeptide anions: Choice of radical cation reagent and competition between electron and proton transfer. Anal. Chem. 82, 2873–2878 [DOI] [PubMed] [Google Scholar]
- 28. Rumachik N. G., McAlister G. C., Russell J. D., Bailey D. J., Wenger C. D., Coon J. J. (2012) Characterizing peptide neutral losses induced by negative electron-transfer dissociation (NETD). J. Am. Soc. Mass Spectrom. 23, 718–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Good D. M., Wirtala M., McAlister G. C., Coon J. J. (2007) Performance characteristics of electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics 6, 1942–1951 [DOI] [PubMed] [Google Scholar]
- 30. Shaw J. B., Kaplan D. A., Brodbelt J. S. (2013) Activated ion negative electron transfer dissociation of multiply charged peptide anions. Anal. Chem. 85, 4721–4728 [DOI] [PubMed] [Google Scholar]
- 31. Shaw J. B., Madsen J. A, Xu H., Brodbelt J. S. (2012) Systematic comparison of ultraviolet photodissociation and electron transfer dissociation for peptide anion characterization. J. Am. Soc. Mass Spectrom. 23, 1707–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xia Y., Han H., McLuckey S. A. (2008) Activation of intact electron-transfer products of polypeptides and proteins in cation transmission mode ion/ion reactions. Anal. Chem. 80, 1111–1117 [DOI] [PubMed] [Google Scholar]
- 33. Frese C. K., Altelaar A. F. M., van den Toorn H., Nolting D., Griep-Raming J., Heck A. J., Mohammed S. (2012) Toward full peptide sequence coverage by dual fragmentation combining electron-transfer and higher-energy collision dissociation tandem mass spectrometry. Anal. Chem. 84, 9668–9673 [DOI] [PubMed] [Google Scholar]
- 34. Swaney D. L., McAlister G. C., Wirtala M., Schwartz J. C., Syka J. E., Coon J. J. (2007) Supplemental activation method for high-efficiency electron-transfer dissociation of doubly protonated peptide precursors. Anal. Chem. 79, 477–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Coon J. J. (2009) Collisions or electrons? Protein sequence analysis in the 21st century. Anal. Chem. 81, 3208–3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pitteri S. J., Chrisman P. A., McLuckey S. A. (2005) Electron-transfer ion/ion reactions of doubly protonated peptides: effect of elevated bath gas temperature. Anal. Chem. 77, 5662–5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ledvina A. R., McAlister G. C., Gardner M. W., Smith S. I., Madsen J. A., Schwartz J. C., Stafford G. C. Jr., Syka J. E., Brodbelt J. S., Coon J. J. (2009) Infrared photoactivation reduces peptide folding and hydrogen-atom migration following ETD tandem mass spectrometry. Angew Chem. Int. Ed. Engl. 48, 8526–8528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ledvina A. R., Beauchene N. A., McAlister G. C., Syka J. E., Schwartz J. C., Griep-Raming J., Westphall M. S., Coon J. J. (2010) Activated-ion electron transfer dissociation improves the ability of electron transfer dissociation to identify peptides in a complex mixture. Anal. Chem. 82, 10068–10074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ledvina A. R., Rose C. M., McAlister G. C., Syka J. E., Westphall M. S., Griep-Raming J., Schwartz J. C., Coon J. J. (2013) Activated ion ETD performed in a modified collision cell on a hybrid QLT-Oribtrap mass spectrometer. J. Am. Soc. Mass Spectrom. 24, 1623–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rose C. M., Russell J. D., Ledvina A. R., McAlister G. C., Westphall M. S., Griep-Raming J., Schwartz J. C., Coon J. J., Syka J. E. P. (2013) Multipurpose dissociation cell for enhanced ETD of intact protein species. J. Am. Soc. Mass Spectrom. 24, 816–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Riley N. M., Westphall M. S., Coon J. J. (2015) Activated Ion Electron Transfer Dissociation for Improved Fragmentation of Intact Proteins. Anal. Chem. 87(14), 7109–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao Y., Riley N. M., Sun L., Hebert A. S., Yan X., Westphall M. S., Rush M. J. P., Zhu G., Champion M. M., Medie F. M., Champion P. A., Coon J. J., Dovichi N. J. (2015) Coupling capillary zone electrophoresis with electron transfer dissociation and activated ion electron transfer dissociation for top-down proteomics. Anal. Chem. 87, 5422–5429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Michalski A., Damoc E., Lange O., Denisov E., Nolting D., Müller M., Viner R., Schwartz J., Remes P., Belford M., Dunyach J.-J., Cox J., Horning S., Mann M., Makarov A. (2012) Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol. Cell. Proteomics 11, O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McAlister G. C., Berggren W. T., Griep-Raming J., Horning S., Makarov A., Phanstiel D., Stafford G., Swaney D. L., Syka J. E., Zabrouskov V., Coon J. J. (2008) A proteomics grade electron transfer dissociation-enabled hybrid linear ion trap-Orbitrap mass spectrometer. J. Proteome Res. 7, 3127–3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hebert A. S., Richards A. L., Bailey D. J., Ulbrich A., Coughlin E. E., Westphall M. S., Coon J. J. (2014) The one hour yeast proteome. Mol. Cell. Proteomics 13, 339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 47. Good D. M., Wenger C. D., McAlister G. C., Bai D. L., Hunt D. F., Coon J. J. (2009) Post-acquisition ETD spectral processing for increased peptide identifications. J. Am. Soc. Mass Spectrom. 20, 1435–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Good D. M., Wenger C. D., Coon J. J. (2010) The effect of interfering ions on search algorithm performance for electron-transfer dissociation data. Proteomics 10, 164–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wenger C. D., Phanstiel D. H., Lee M. V., Bailey D. J., Coon J. J. (2011) COMPASS: A suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 11, 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Swaney D. L., Wenger C. D., Coon J. J. (2010) Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J. Proteome Res. 9, 1323–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Coon J. J., Syka J. E. P., Schwartz J. C., Shabanowitz J., Hunt D. F. (2004) Anion dependence in the partitioning between proton and electron transfer in ion/ion reactions. Int. J. Mass Spectrom. 236, 33–42 [Google Scholar]
- 52. Rose C. M., Rush M. J. P., Riley N. M., Merrill A. E., Kwiecien N. W., Holden D. D., Mullen C., Westphall M. S., Coon J. J. (2015) A calibration routine for efficient ETD in large-scale proteomics. J. Am. Soc. Mass Spectrom., doi: 10.1007/s13361-015-1183-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Flora J. W., Muddiman D. C. (2001) Selective, sensitive, and rapid phosphopeptide identification in enzymatic digests using ESI-FTICR-MS with infrared multiphoton dissociation. Anal. Chem. 73, 3305–3311 [DOI] [PubMed] [Google Scholar]
- 54. Ganisl B., Taucher M., Riml C., Breuker K. (2011) Charge as you like! Efficient manipulation of negative ion net charge in electrospray ionization of proteins and nucleic acids. Eur. J. Mass. Spectrom. (Chichester, Eng.) 17, 333–343 [DOI] [PubMed] [Google Scholar]
- 55. Hahne H., Pachl F., Ruprecht B., Maier S. K., Klaeger S., Helm D., Médard G., Wilm M., Lemeer S., Kuster B. (2013) DMSO enhances electrospray response, boosting sensitivity of proteomic experiments. Nat. Methods 10, 989–991 [DOI] [PubMed] [Google Scholar]
- 56. Meyer J. G., A Komives E. (2012) Charge state coalescence during electrospray ionization improves peptide identification by tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 23, 1390–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Choudhary G., Wu S. L., Shieh P., Hancock W. S. (2003) Multiple enzymatic digestion for enhanced sequence coverage of proteins in complex proteomic mixtures using capillary LC with ion trap MS/MS. J. Proteome Res. 2, 59–67 [DOI] [PubMed] [Google Scholar]
- 58. Biringer R. G., Amato H., Harrington M. G., Fonteh A. N., Riggins J. N., Huhmer A. F. (2006) Enhanced sequence coverage of proteins in human cerebrospinal fluid using multiple enzymatic digestion and linear ion trap LC-MS/MS. Br. Funct. Genomic Proteomic 5, 144–153 [DOI] [PubMed] [Google Scholar]
- 59. Leitner A., Reischl R., Walzthoeni T., Herzog F., Bohn S., Förster F., Aebersold R. (2012) Expanding the chemical cross-linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol. Cell. Proteomics 11, M111.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jakoby T., van den Berg B. H., Tholey A. (2012) Quantitative protease cleavage site profiling using tandem-mass-tag labeling and LC-MALDI-TOF/TOF MS/MS analysis. J. Proteome Res. 11, 1812–1820 [DOI] [PubMed] [Google Scholar]
- 61. Washburn M. P., Wolters D., Yates J. R. 3rd (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 62. Zhang Y., Fonslow B. R., Shan B., Baek M. C., Yates J. R. 3rd (2013) Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 113, 2343–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang F., Shen Y., Camp D. G., Smith R. D. (2012) High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev. Proteomics 9, 129–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ghaemmaghami S., Huh W.-K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 65. Swaney D. L., McAlister G. C., Coon J. J. (2008) Decision tree–driven tandem mass spectrometry for shotgun proteomics. Nat. Methods 5, 959–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kjeldsen F., Hørning O. B., Jensen S. S., Giessing A. M., Jensen O. N. (2008) Towards liquid chromatography time-scale peptide sequencing and characterization of post-translational modifications in the negative-ion mode using electron detachment dissociation tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 19, 1156–1162 [DOI] [PubMed] [Google Scholar]
- 67. Hersberger K. E., Håkansson K. (2012) Characterization of O-sulfopeptides by negative ion mode tandem mass spectrometry: Superior performance of negative ion electron capture dissociation. Anal. Chem. 84, 6370–6377 [DOI] [PubMed] [Google Scholar]
- 68. Huang Y., Yu X., Mao Y., Costello C. E., Zaia J., Lin C. (2013) De novo sequencing of heparan sulfate oligosaccharides by electron-activated dissociation. Anal. Chem. 85, 11979–11986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Leach F. E. 3rd, Wolff J. J., Xiao Z., Ly M., Laremore T. N., Arungundram S., Al-Mafraji K., Venot A., Boons G. J., Linhardt R. J., Amster I. J. (2011) Negative electron transfer dissociation Fourier transform mass spectrometry of glycosaminoglycan carbohydrates. Eur. J. Mass Spectrom. (Chichester, Eng.) 17, 167–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Madsen J. A., Ko B. J., Xu H., Iwashkiw J. A., Robotham S. A., Shaw J. B., Feldman M. F., Brodbelt J. S. (2013) Concurrent automated sequencing of the glycan and peptide portions of O-linked glycopeptide anions by ultraviolet photodissociation mass spectrometry. Anal. Chem. 85, 9253–9261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nwosu C. C., Strum J. S., An H. J., Lebrilla C. B. (2010) Enhanced detection and identification of glycopeptides in negative ion mode mass spectrometry. Anal. Chem. 82, 9654–9662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wolff J. J., Leach F. E. 3rd, Laremore T. N., Kaplan D. A., Easterling M. L., Linhardt R. J., Amster I. J. (2010) Negative electron transfer dissociation of glycosaminoglycans. Anal. Chem. 82, 3460–3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Greer S. M., Cannon J. R., Brodbelt J. S. (2014) Improvement of shotgun proteomics in the negative mode by carbamylation of peptides and ultraviolet photodissociation mass spectrometry. Anal. Chem. 86(24), 12285–12290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wyndham K. D., O'Gara J. E., Walter T. H., Glose K. H., Lawrence N. L., Alden B. A., Izzo G. S., Hudalla C. J., Iraneta P. C. (2003) Characterization and evaluation of C18 HPLC stationary phases based on ethyl-bridged hybrid organic/inorganic particles. Anal. Chem. 75, 6781–6788 [DOI] [PubMed] [Google Scholar]
- 75. Kelstrup C. D., Jersie-Christensen R. R., Batth T. S., Arrey T. N., Kuehn A., Kellmann M., Olsen J. V (2014) Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra-high-field Orbitrap mass spectrometer. J. Proteome Res. 13, 6187–6195 [DOI] [PubMed] [Google Scholar]
- 76. Scheltema R. A., Hauschild J.-P., Lange O., Hornburg D., Denisov E., Damoc E., Kuehn A., Makarov A., Mann M. (2014) The Q Exactive HF, a benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol. Cell. Proteomics 13, 3698–3708 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.