

Abstract
The ability to accurately quantify proteins in formalin-fixed paraffin-embedded tissues using targeted mass spectrometry opens exciting perspectives for biomarker discovery. We have developed and evaluated a selectedreaction monitoring assay for the human receptor tyrosine-protein kinase erbB-2 (HER2) in formalin-fixed paraffin-embedded breast tumors. Peptide candidates were identified using an untargeted mass spectrometry approach in relevant cell lines. A multiplexed assay was developed for the six best candidate peptides and evaluated for linearity, precision and lower limit of quantification. Results showed a linear response over a calibration range of 0.012 to 100 fmol on column (R2: 0.99–1.00).The lower limit of quantification was 0.155 fmol on column for all peptides evaluated. The six HER2 peptides were quantified by selected reaction monitoring in a cohort of 40 archival formalin-fixed paraffin-embedded tumor tissues from women with invasive breast carcinomas, which showed different levels of HER2 gene amplification as assessed by standard methods used in clinical pathology. The amounts of the six HER2 peptides were highly and significantly correlated with each other, indicating that peptide levels can be used as surrogates of protein amounts in formalin-fixed paraffin-embedded tissues. After normalization for sample size, selected reaction monitoring peptide measurements were able to correctly predict 90% of cases based on HER2 amplification as defined by the American Society of Clinical Oncology and College of American Pathologists. In conclusion, the developed assay showed good analytical performance and a high agreement with immunohistochemistry and fluorescence in situ hybridization data. This study demonstrated that selected reaction monitoring allows to accurately quantify protein expression in formalin-fixed paraffin-embedded tissues and represents therefore a powerful approach for biomarker discovery studies. The untargeted mass spectrometry data is available via ProteomeXchange whereas the quantification data by selected reaction monitoring is available on the Panorama Public website.
MS based proteomics has traditionally been used to investigate complex biological systems, such as cell lines, plasma, or fresh-frozen tissues (1, 2). In the last decade however, MS proteomics has extended to the analysis of formalin-fixed paraffin-embedded (FFPE)1 tissues (3). Formalin fixation is the gold standard for sample storage in clinical pathology because it allows optimal preservation of the morphological features of the tissue and it is economically attractive (storage at room temperature over several years or decades) (4). Several studies have shown that although the individual peptides retrieved and identified from fresh-frozen and FFPE tissues may differ, the biological information obtained from both types of material in terms of number of proteins identified, cellular location and molecular function is very similar (5–10). A number of proteomics studies were reported, which used untargeted MS on FFPE tissues to compare diseased and healthy samples in the search for potential novel biomarkers (10). Nevertheless, these untargeted MS workflows do not allow performing accurate protein quantification on large numbers of samples. One option is to use targeted MS approaches, such as selected reaction monitoring (SRM), which are highly quantitative and reproducible over many samples (11, 12). Additionally, SRM assays allow a high level of multiplexing (several hundreds of peptides can be measured in parallel in a single analysis) (13). The lack of access to a sufficient number of high-quality samples annotated with comprehensive clinical data sets may be a limiting factor for preclinical exploratory phase biomarker studies (14). The possibility to use FFPE samples for MS-based proteomics, in particular for quantitative targeted approaches, would therefore open tremendous perspectives for performing large retrospective biomarker discovery and verification studies. Indeed, in addition to being widely available, most FFPE tissues are annotated with clinical data. Moreover, targeted MS workflows applied to FFPE samples are complementary to techniques requiring high-quality antibodies, such as immunohistochemistry (IHC) or reverse-phase protein arrays (RPPA). These techniques all rely on the measurement of the target protein, with SRM measuring one or ideally several peptides as surrogates of the protein (15, 16). In opposition to IHC and RPPA however, SRM does not rely on the presence of a specific antibody for analyte detection, thereby avoiding cross-reaction issues and making assay development relatively rapid and cost effective. Although SRM is less advanced for protein analysis than for small molecules quantification, the technique was demonstrated to be selective, reproducible, and highly quantitative over large dynamic ranges for proteins as well (17–19). However, although the equivalence of qualitative analyses performed on fresh-frozen and FFPE samples has been investigated and demonstrated, only a few studies evaluated quantitative targeted MS approaches in FFPE samples (20, 21). Targeted proteomics performed on FFPE tissues is still in its early days and known limitations of this technique include the loss of morphologic features of the tissue and an extensive sample preparation, causing a low sample throughput (20). Moreover, targeted proteomics quantifies peptides as surrogates of a protein, with the former not necessarily agreeing in absolute terms with the latter. This is true for bottom-up proteomics in general, but it is of particular importance for FFPE tissues.
In this study, we critically assessed the validity of targeted MS applied to peptide quantification in FFPE tissues. We developed and evaluated an SRM assay for the quantification of six peptides of the human receptor tyrosine-protein kinase erbB-2 (HER2) and compared the obtained results with those of standard methods used in clinical pathology, namely IHC and fluorescence in situ hybridization (FISH). HER2 was chosen as a candidate protein because its overexpression is routinely assessed in breast tumors in order to determine susceptibility to anti-HER2 treatment (22). Depending on the laboratory, HER2 overexpression can be assessed by IHC or the amplification of the corresponding gene (ERBB2) can be quantified by FISH (23). Several laboratories use both techniques for decision-making, as they are complementary.
In a first step, we developed an SRM assay for the quantification of HER2 peptides in archival clinical FFPE tumor tissues and assessed its analytical performance, including linearity, precision and lower limit of quantification (LLOQ). We then demonstrated the applicability of the method by quantifying HER2 peptides in a cohort of 40 FFPE tumor tissues expressing different levels of HER2 (selected based on ERBB2 gene amplification status). The samples originated from surgical resections performed on women with invasive mammary carcinomas. We thereby investigated several options to normalize the results in regard to sample size. Finally, in order to confirm the validity of SRM as a suitable method for protein quantification in FFPE tissues, we determined the agreement between data generated by SRM and data generated by IHC or FISH.
EXPERIMENTAL PROCEDURES
The untargeted mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (24) via the PRIDE partner repository with the data set identifier PXD002281 and under the project name “Identification of HER2 peptides for quantification in formalin-fixed paraffin-embedded breast cancer tissues”. The Skyline documents containing the targeted mass spectrometry data (including quantification of HER2 peptides in the cohort samples, linearity and LLOQ data) have been deposited on the Panorama server (25). Data can be accessed on the Panorama Public website using the following link: https://panoramaweb.org/labkey/Steiner.url.
Untargeted MS Analysis
Suitable HER2 peptides for targeted MS analysis were identified by untargeted MS analysis from untreated and formalin-fixed HER2-overexpressing cell lines (SKBR-3 and BT-474) (26). Nanoflow LC-MS/MS was performed on an LTQ Orbitrap Velos from Thermo Electron (Waltham, MA) equipped with a NanoAcquity HPLC system from Waters Corporation (Milford, MA). Peptides were trapped on a home-made 100 μm × 20 mm pre-column and separated on a gravity-pulled home-made emitter of dimension 75 μm × 150 mm (both columns packed with 5 μm 200 Å Magic C18 AQ chromatographic phase from Michrom (Bruker, Billerica, MA). The analytical separation was run for 65 min using a gradient of 0.1% formic acid in H2O (solvent A) and 0.1% formic acid in CH3CN (solvent B). The gradient was run as follows: 5% B for 1 min, 5–35% B in 54 min and 35–80% B in 10 min at a flow rate of 220 nL/min. Electrospray voltage was set to 1800 V. For MS survey scans, the orbitrap analyzer resolution was set to 60,000 and the ion population was set to 5 × 105 with an m/z window of 400 to 2000. A maximum of eight precursor ions were selected for collision-induced dissociation (CID) in the LTQ analyzer with a dynamic exclusion time set to 45 s. Ions were accumulated to a target value of 7 × 103 with an isolation width of 2 m/z and CID was performed at a normalized collision energy of 35%.
Peak lists were generated from raw data using the embedded software from the instrument vendor (extract_MSN.exe) and were subsequently submitted to EasyProt (v2.2), a platform that uses Phenyx (GeneBio, Geneva, Switzerland) for protein identification (27). Searches were conducted against the UniProtKB/SwissProt database (11.01.2011 and 13.06.2012 releases containing 20′252 and 20′237 reviewed entries respectively) specifying “Homo sapiens” taxonomy. The parent ion tolerance was set to 10 ppm. Cysteine carbamidomethylation was set as a fixed or variable modification whereas oxidized methionine was set as a variable modification. Trypsin was selected as the proteolytic enzyme, with one potential missed cleavage allowed. Peptide scores were set up to maintain the false positive peptide ratio below 5%. Alternatively, raw mass spectrometry data were processed using the SEQUEST search algorithm (SEQUEST version 27.0, revision 12, Thermo Electron) and searches were performed using the UniProtKB/SwissProt protein knowledgebase database (version 2011_07) filtered for “homo sapiens” (58,772 sequences concatenated with their decoy entries). Data were searched with a mass tolerance of ±5 ppm for parent ions and ± 1.0 Da for fragment ions. Methionines (reduced/oxidized; +15.9949 Da) were considered as differential modifications whereas cysteines were considered as fully carbamidomethylated (+57.0199 Da). False discovery rate was set to 1%. Only fully tryptic peptides with no more than one missed cleavage were considered for data analysis.
Targeted MS Method Development
The peptide sequences observed using the untargeted MS approach and suitable for developing an SRM assay (proteotypic and containing no methionine) were entered into Skyline v1.3 software (MacCoss Lab, Seattle, WA) (28) in order to build an SRM method for peptide screening. The Skyline method settings were set to allow doubly and triply charged precursor ions and singly charged “y” fragment ions, which are the most predominant ions generated by CID (15). The Skyline file was exported to the mass spectrometer to build an SRM method, which was used to analyze FFPE tissue extracts from three different HER2-overexpressing tumors spiked with light and heavy-labeled synthetic peptides. Individual peptides were assessed according to the following criteria: visual detection and co-elution of the light and heavy-isotope-labeled chromatographic peaks, reproducibility of retention time (Rt) (for a given peptide, the Rt for all replicates are within a window of 1 min) and reproducibility of peak areas (total peak area for all replicates within average peak area ± ∼30%). The six best performing peptides were selected to be used for quantification.
The reconstituted FFPE tissue extracts were analyzed by SRM on a Vantage triple stage quadrupole mass spectrometer from Thermo Electron equipped with a Dionex Ultimate 3 000 RSLCnano system (Thermo Electron). Peptides were separated on a 0.075 × 170 mm column (MS Wil, Wil, Switzerland) filled with 3 μm Reprosil-Pur C18-AQ (Dr Maisch, Ammerbuch-Entringen, Germany). The analytical separation was run for 24 min using a gradient of 0.5% acetic acid/2% acetonitrile (solvent A) and 0.5% acetic acid/80% acetonitrile (solvent B). The gradient was run as follows: 0–20% B in 11 min, 20–45% B in 13 min and 45–100% B in 0.1 min at a flow rate of 450 nL/min. The spray voltage was set to 1800 V, resolution was set to 0.7 Th for Q1 and Q3 (FWHM), whereas collision gas pressure was 1.5 mTorr. The final SRM method had a cycle time of 1 s for 88 transitions. Collision energy was individually optimized for each peptide and is displayed in supplemental Table S1.
The acquired SRM data were imported into Skyline v2.1 for peak integration and all further calculations for peptide quantification and analytical validation was performed using Microsoft Excel 2010. In addition, data was visualized using TIBCO Spotfire version 5.5 (TIBCO Software Inc., Boston, MA).
Linearity and LLOQ of the SRM assay were assessed using light and heavy labeled peptides in an artificial background matrix consisting of a bacterial protein digest as previously proposed by others (29) (see also supplemental Materials and Methods), whereas precision was calculated from replicate analyses of the 40 samples included in the study.
Verification Study in FFPE Breast Cancer Tissue Samples With HER2 Amplification
The study was approved by the ethical committee of the Canton of Geneva (protocol NAC 13–109). FFPE breast tumor resection samples were selected retrospectively from the archive of the Division of Clinical Pathology of the Geneva University Hospitals from 2001 to 2011. Samples included cases of tumorectomy or mastectomy from neo-adjuvant treatment-naïve women with invasive breast carcinomas, including 34 ductal, three lobular, one micropapillary, one mucinous, and one medullary. Exclusion criteria were male gender and administration of neo-adjuvant treatment.
Assessment of HER2 Amplification by FISH and HER2 Overexpression by IHC
For all cases, HER2 amplification was assessed by FISH as part of the routine procedure in the Division of Clinical Pathology. The cohort included 40 patients distributed in four groups of 10 patients each, according to their HER2 amplification status as measured by FISH: 10 patients with no HER2 amplification (FISH ratio <2, all ductal) and three groups of 10 patients, each with increasing HER2 gene amplification level, as determined by a FISH ratio of 2–4 (nine ductal and one medullary); 4–10 (nine ductal and one lobular); and >10 (six ductal, two lobular, one mucinous, one micropapillary), respectively. FISH was performed as follows: 4 μm slices were cut and mounted on superfrost glass slides. The HER2 FISH assay (02J01–36, Abbott Molecular, Abbott Park, IL) was performed as recommended by the manufacturer. In addition, HER2 overexpression was assessed by IHC for all cases. IHC was performed with antigen retrieval on the Benchmark XT automated stainer (Ventana, Mannheim, Germany). Prediluted antibody, ready-to-use for HER2 (790–2291, Clone 4B5, Ventana) were used as recommended by the manufacturer. HER2 FISH and IHC scoring were performed according to the American Society of Clinical Oncology/College of American Pathologists (ASCO/CAP) recommendations (30).
Two expert breast pathologists independently scored the IHC and FISH analyses under a microscope without digital quantification. For IHC: a score of 0 represents no staining observed or membrane staining that is incomplete and is faint/barely perceptible and within less than 10% of the invasive carcinoma; a score of 1+ represents an incomplete membrane staining that is faint/barely perceptible and within more than 10% of the invasive carcinoma; a score of 2+ represents circumferential membrane staining that is incomplete and/or weak/moderate and within more than 10% of the invasive tumor or complete and circumferential membrane staining that is intense and within less than 10% of the invasive tumor; a score of 3+ represents circumferential membrane staining that is complete, intense in more than 10% of the invasive tumor. For FISH, the number of HER2 and centromere (CEP17) probes per nucleus was counted in more than 40 invasive cancer cells. An absence of HER2 amplification corresponds to a HER2/CEP17 ratio < 2.0 with an average HER2 copy number < 4.0 signals/cell, whereas HER2 amplification corresponds to a HER2/CEP17 ratio ≥ 2.0 or a HER2/CEP17 ratio < 2.0 with an average HER2 copy number > 6.0 signals/cells.
Peptide Extraction of FFPE Samples
For peptide extraction, paraffin blocks containing the resected tumors were cut using a microtome according to the following scheme (Fig. 1): 4 μm thick slices were cut and mounted on superfrost glass slides, the first of which was stained using hematoxylin and eosin (H&E), the second being stored at −20 °C until measurement of HER2 expression by IHC. The three following 20 μm thick slices were cut and mounted on glass slides for dissection and protein extraction. One last 4 μm tissue slice was stained with H&E in order to confirm the presence of tumor tissue throughout all collected tissue slices. Only the infiltrative tumor area, omitting ductal or lobular in situ carcinoma was then delimited by a pathologist expert in breast pathology (J.-C.T.) on the first H&E slide. Scalpel macrodissection of the tumor was performed by superimposing each 20 μm slice with the H&E template. The dissected areas were deparaffinated and rehydrated using a series of UltraClear™ and graded alcohol baths, after which the rehydrated tissue was collected with a needle and stored at −80 °C until protein extraction.
Fig. 1.

Sample preparation and tumor excision from formalin-fixed paraffin-embedded tissue blocks.
A detailed protocol to generate and extract peptides from the FFPE tissue slices is provided in the supplemental Material and Methods. Briefly, the collected tissue recovered from a 20 μm thick slice was suspended in a Tris-Cl buffer, pH 8.5, containing RapiGest™ SF and 1,4-dithioerythritol (DTE) and the tubes were heated at 100 °C for 20 min. After sonication, samples were heated again at 80 °C for 2 h, after which they underwent a second round of sonication and were subjected to alkylation with iodoacetamide. Tryptic digestion was performed overnight at 37 °C. The samples were then acidified and desalted, after which the eluates were evaporated to dryness using a speed-vac concentrator and kept at −20 °C until use. The extracts were reconstituted in 50 μl of 2% (v/v) acetonitrile containing 0.5% (v/v) acetic acid and further diluted by a ratio 1:50. Heavy-labeled peptides were added to a final concentration of 0.2 fmol/μl. 5 μl of this solution (representing 0.2% of the total extract) were injected into the LC-MS system for analysis using the final SRM method described above.
Parameters Used to Normalize for Tumor Size
The percentage of tumor cells in the dissected area was evaluated by a pathologist and used as a normalization factor for all samples. In order to allow comparison between tumors of various sizes, four additional parameters expected to represent tumor size were acquired and evaluated for normalization. The first normalization parameter was the surface of the dissected tumor area and was calculated based on the H&E template slides, which were digitally scanned using the Pannoramic 250 Flash II scanner (3DHistech, Budapest, Hungary) and measured in μm using the Pannoramic Viewer software (3DHistech). A second normalization parameter was the total amount of peptides extracted in a 20 μm thick tissue slice quantified using the Pierce BCA Protein Assay Kit (Thermo Fischer Scientific). As a third parameter, the 40 samples were analyzed a second time by MS in fullscan mode (500–1000 m/z; scan time 1 s) with a similar but shorter gradient as for the SRM method (0–45% B in 5 min and 45–100% B in 0.1 min at a flow rate of 450 nL/min). The area under the curve (AUC) of the generated total ion current (TIC) was integrated between 19.0 and 24.0 min and used as the third parameter for normalization for tumor size. The fourth normalization parameter was cytokeratin 19 (KRT19), which is specifically expressed in breast epithelium (31). Three KRT19 peptides were monitored in parallel with the six HER2 peptides in the final SRM method. Calibration curves were prepared as for HER2 peptides, but in this case the heavy-labeled peptides were spiked at 2.0 fmol/μl in the reconstituted extract (10 fmol on column). The response was plotted in the same way as for HER2 and the amount of peptide in the total tumor extracts was calculated. The values obtained this way for the three KRT19 peptides were averaged in order to obtain a single KRT19 value for each sample (the values for the three KRT19 peptides significantly correlated with each other: Spearman correlation coefficients: 0.899–0.924; p values (two-tailed): 1.72E-17–3.32E-15; data not shown).
Statistical Analysis
Correlations and box plots representations were performed using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA).
Statistical analysis was performed as described previously (32) using statistical language R 2.10.1 (33). Principal Component Analysis, Spearman Correlation, and Mann-Whitney tests were performed using the package “stats” and graphical visualizations were performed using the package “lattice” (33). The employed Kappa coefficient (k) is a statistical measure for the agreement of two raters; in this case the raters are the IHC score or FISH category and the normalized HER2 peptide concentrations. The Kappa coefficient ranges from 0 (disagreement) to 1 (perfect agreement). Disagreements were weighted according to their squared distance from perfect agreement (34). Ordinal multinomial logistic regression: For each sample i, the IHC score (or FISH category) Yi and the normalized HER2 peptide concentration xi is determined. The IHC score (or FISH category) Y is categorical by definition. Each score follows a logical ordering by level of HER2 expression, from category j = 0 to 3+ (or 1 to 4 for FISH). Therefore the IHC HER2 score is an ordinal categorical response. The normalized HER2 peptide concentration xi is a continuous variable. The relationship between the IHC score and the normalized HER2 peptide concentration is adequately modeled by an ordinal multinomial logistic regression, which was performed as previously described (35, 36). This model aims to predict the probability of association of sample i to be assigned to category j based on xi, or pij = P(Yi = j). Because the probability for each of the samples is predicted for each of the four scores simultaneously, the probability distribution of the samples to be assigned to each of the four scores is called multinomial. Technically, this is resolved by fitting a binary logistic regression model for each score j in which scores 0 to j combines to form a single category and categories j+1 to 3+ form a second category. At any normalized HER2 peptide concentration xi, the probability of Y being at or below a particular score j is expressed as a cumulative probability, that reflects the HER2 score ordering: P(Y £ 0) £ P (Y £ 1+) £ P (Y £ 2+) £ P (Y £ 3+). Finally the probability P(Y = j) is estimated from P(Y = j) = P(Y £ j) − P(Y £ j − 1) (37).
RESULTS
Development of the Analytical Method
In order to identify the HER2 peptides generated from biological material containing the protein of interest, untargeted LC-MS analysis was performed on peptide extracts from untreated and formalin-fixed cell lines (SKBR-3 and BT-474). The peptides identified in this way are reported in Table I and supplemental Table S2.
Table I. Peptides identified by untargeted mass spectrometry in cell lines overexpressing HER2. N-terminal and C-terminal amino acid position are indicated in superscript. Peptides used for SRM development are in italic whereas peptides selected for the final SRM assay are in bold.

Light and heavy-labeled versions of the peptide sequences which are proteotypic to HER2 and contain no methionine were synthesized (marked in italic in Table I) and spiked into peptide extracts originating from three FFPE tumors overexpressing HER2 and screened using an SRM method generated as described in the Materials and Methods section. For each tumor, a total of 11 injections were performed: six replicates of a same tissue slice extract and one replicate of five additional extracts from adjacent tissue slices. Peptides were evaluated within the 11 analyses using the following criteria: presence of a clearly defined peak, co-elution of the light and heavy-labeled chromatographic peaks, reproducibility of Rt (for a given peptide, the Rt for all replicates needed to be within a window of 1 min) and reproducibility of peak areas (total peak area for all replicates needed to be within average peak area ± 30%). Peptides not meeting these criteria in more than one of the three FFPE tissue backgrounds were excluded. The six peptides selected for analytical evaluation are marked in bold in Table I.
Choice of Matrix for Calibration Curves and Assessment of Linearity
It is well known that components present in biological matrices can influence the ionization process in SRM assays. For the quantification of drugs and their metabolites, the traditional approach to account for matrix effects consists in building a calibration curve in a “blank” matrix of identical composition to that containing the unknown analyte. In the case of a drug, the surrogate matrix is usually blank plasma or urine originating from patients not taking the drug of interest. This is more complex in the case of protein quantification in tissues as the protein for quantification is present at endogenous levels. Although spiking over the endogenous levels and later subtracting these is possible, it introduces an additional step and increases error in the quantification process. In addition, although it is relatively easy to obtain large amounts of biological fluids for the purpose of building calibration curves, the repeated use of tissue samples beyond method development, for the sole purpose of building calibration curves in a representative matrix, does not appear practical and ethically justifiable. We therefore chose an alternative approach by using a commercially available protein extract from the thermophile archaea Pyroccoccus furiosus as a surrogate matrix. The suitability for HER2 quantification of this artificial matrix was assessed by preparing a calibration curve in the P. furiosus extract and by comparing the signal linearity and slope with a calibration curve constructed in a protein extract from three pooled ductal triple-negative breast carcinomas (TNBC), a breast cancer type known not to overexpress HER2 (38).
Linearity in both backgrounds was compared using a log10/log10 representation (supplemental Fig. S1). The average slopes and intercepts are reported in supplemental Table S3. When considering the entire calibration domain, the slope was systematically lower and the intercept higher in the TNBC matrix compared with the P. furiosus matrix. This was because of a flattening of the curve in the lower part of the calibration curve for the TNBC matrix (calibrators between 0.012 to 0.391 fmol on column), possibly because of physiological expression level of HER2 in TNBC tumors. However, when omitting the critical points and considering only the upper eight points of the calibration domain for both matrixes, a close agreement was observed between the two data sets with the slopes in the TNBC matrix ranging from 86% to 93% of the values observed in the P. furiosus extract. In addition, the response was linear for all six peptides across the entire calibration range in the P. furiosus extract using the log10/log10 representation (average slope: 0.960 - 1.001; average r2: 0.9875 - 0.9984; supplemental Table S3). Therefore, the P. furiosus background was subsequently used to perform method evaluation and sample quantification.
As a result, linearity was assessed in the P. furiosus background and plotted as a log10/log10 representation. For intra-day linearity, the r square (r2) values ranged from 0.990 to 1.000 and slopes ranged from 0.958 to 1.115 for all 6 peptides. For inter-day linearity, the r2 values ranged from 0.952 to 1.000 and slopes ranged from 0.958 to 1.212 for all six peptides. Linearity was observed over four orders of magnitude. Detailed information with respect to linearity, including dispersion of residuals, is shown in supplemental Table S4. Residual plots for the six peptides are shown in supplemental Fig. S2.
Lower Limit of Quantification and Precision
LLOQ was defined as the lowest peptide concentration that could be quantified with a coefficient of variation (CV) below 20% (39) based on three replicate injections. LLOQ was set to 31 amol/μl (0.155 fmol on column) for all peptides as their CVs were consistently below 20% for the full evaluation domain (0.155 fmol-5.0 fmol on column) that was selected for this work.
The analytical method precision was evaluated by calculating the CV of three repeated injections for each of the 40 tumor tissue extracts included in the study. Overall, CVs for all samples, including those with values below LLOQ, ranged from 0.6% to 75.6% for all peptides (Table IIA and supplemental Fig. S3). Considering only values above LLOQ, all measurements had CVs below 20%, except for peptides FVVIQNEDLGPASPLDSTFYR and SGGGDLTLGLEPSEEEAPR. For peptide FVVIQNEDLGPASPLDSTFYR, 47.2% of the measurements at or above LLOQ had CVs below 20% whereas 81.5% of the measurements for the peptide SGGGDLTLGLEPSEEEAPR had CVs below 20%.
Table II. Analytical method precision and whole procedure precision. (A) Analytical method precision calculated for the six HER2 peptides quantified in 40 tumors. (B) Whole procedure precision calculated for the six HER2 peptides from nine of the 40 tumors.
| Peptide sequence | Range peptide amount in tissue extract [pmol] | Reproducibility: Range coefficient of variation [%] |
|---|---|---|
|
A. Analytical method precision (n = 40) | ||
| ELVSEFSR | 0.014–2.73 | 1.1–51.5 |
| FVVIQNEDLGPASPLDSTFYR | 0.026–4.70 | 3.0–58.1 |
| GIWIPDGENVK | 0.009–0.507 | 3.7–52.6 |
| GLQSLPTHDPSPLQR | 0.011–2.25 | 1.3–40.5 |
| SGGGDLTLGLEPSEEEAPR | 0.022–3.11 | 2.4–75.6 |
| SLTEILK | 0.007–2.65 | 0.6–29.9 |
|
B. Whole procedure precision, including sample preparation (n = 9) | ||
| ELVSEFSR | 0.015–0.589 | 16.1–43.6 |
| FVVIQNEDLGPASPLDSTFYR | 0.053–1.22 | 14.7–59.6 |
| GIWIPDGENVK | 0.011–0.141 | 10.2–64.0 |
| GLQSLPTHDPSPLQR | 0.019–0.316 | 12.3–57.1 |
| SGGGDLTLGLEPSEEEAPR | 0.025–0.706 | 18.8–87.0 |
| SLTEILK | 0.010–0.549 | 6.8–31.9 |
The reproducibility of the entire procedure, including sample preparation, was evaluated by extracting three adjacent tissue slices for a subset (n = 9) of the 40 samples and by calculating the CVs from a single injection from each of the three tissue slices (Table 2B). Because the replicates were constituted of three adjacent but slightly different tissue slices, the threshold to evaluate the procedure reproducibility was set to 35% (39) to reflect this fact. Overall, CVs ranged from 6.8% to 87.0% for all values, including those below LLOQ. Two peptides, GIWIPDGENVK and SLTEILK, showed CVs below 35% for all measurementsat or above LLOQ. For two peptides, ELVSEFSR and SGGGDLTLGLEPSEEEAPR, 66.7% of the measurements at or above LLOQ showed CVs below 35% whereas the peptides FVVIQNEDLGPASPLDSTFYR and GLQSLPTHDPSPLQR performed less well with 14.3 and 0% of the measurements, respectively, below 35% CV. Individual CVs for all samples are reported in supplemental Table S5.
Quantification of the Six HER2 Peptides
The average peptide amounts measured in the 40 tumor extracts are reported in supplemental Table S6. The retrieved amounts for a given peptide spanned approximately two orders of magnitude. In addition, in any given sample, the amounts measured among all peptides were highly variable, with the amount of the most abundant peptide on average 6 times higher than the amount of the least abundant peptide (data not shown). However, Spearman rank correlation analysis showed a significant and positive correlation for all six peptides across all samples (Spearman correlation coefficients: 0.832 - 0.977; p values (two-tailed): 4.40E-27 - 2.92E-11), indicating that the quantitative data obtained for each peptide followed similar trends (Table III).
Table III. Spearman correlation matrix for the six HER2 peptides. Correlation coefficients (ρ) are indicated in bold whereas p values (two-tailed) are indicated in italic characters.
| Amount FVVIQNEDLGPASPLDSTFYR in total tumor extract [fmol] | 0.959 | ||||
| 1.67E-22 | |||||
| Amount GIWIPDGENVK in total tumor extract [fmol] | 0.915 | 0.935 | |||
| 1.52E-16 | 9.90E-19 | ||||
| Amount GLQSLPTHDPSPLQR in total tumor extract [fmol] | 0.904 | 0.938 | 0.903 | ||
| 1.36E-15 | 5.15E-19 | 1.56E-15 | |||
| Amount SGGGDLTLGLEPSEEEAPR in total tumor extract [fmol] | 0.902 | 0.895 | 0.851 | 0.832 | |
| 1.80E-15 | 6.65E-15 | 3.67E-12 | 2.92E-11 | ||
| Amount SLTEILK in total tumor extract [fmol] | 0.977 | 0.968 | 0.939 | 0.901 | 0.896 |
| 4.40E-27 | 2.13E-24 | 3.29E-19 | 2.37E-15 | 5.84E-15 | |
| Amount ELVSEFSR in total tumor extract [fmol] | Amount FVVIQNEDLGPASPLDSTFYR in total tumor extract [fmol] | amount GIWIPDGENVK in total tumor extract [fmol] | amount GLQSLPTHDPSPLQR in total tumor extract [fmol] | amount SGGGDLTLGLEPSEEEAPR in total tu mor extract [fmol] |
Agreement Between SRM Data and IHC or FISH Data
The distribution of IHC scores within FISH categories is represented in Fig. 2. While the HER2 FISH assay is designed to quantitatively determine HER2 gene amplification and the HER2 IHC assay provides a semi-quantitative score based on the extent and intensity of cell membrane staining, the HER2 SRM assay reports absolute peptide amounts extracted from the macrodissected tumor area. The reported amounts by SRM need to be corrected for the presence of nontumoral cell types in the macrodissected area, such as fibroblasts, macrophages, and lymphocytes. We therefore adjusted the HER2 peptide amounts to the tumor content in order to obtain peptide amounts as if the sampled area was containing only tumor cells. Additionally for comparison with IHC and FISH, it is necessary to normalize for the size of the dissected area as a large tumor expressing basal levels of HER2 might actually contain greater absolute HER2 peptide amounts than a small tumor with high overexpression of HER2. We therefore considered four different parameters representative of tumor size and used either one of these four parameters to normalize the SRM data. These include tumor surface (mm2), total peptide amount (μg) measured by a colorimetric assay, TIC generated by full-scan mass spectrometry (arbitrary unit) and KRT19 amount (pmol) in the tumor extract based on the average measurement by SRM of three KRT19 peptides. This led to four distinct normalized SRM data sets, which in turn were matched to IHC and FISH data. The SRM data for all samples before and after normalization as well as the measures of the normalization parameters can be found in supplemental Table S6, whereas IHC scores and FISH ratios can be found in supplemental Table S7. A Spearman rank correlation analysis showed that all four normalization parameters were significantly and positively correlated with each other (Spearman correlation coefficients: 0.557–0.897; p values (two-tailed): 4.96E-15–1.87E-04) (Fig. 3). It is of note however that the KRT19 peptide amounts correlated less well with all other three parameters (Spearman correlation coefficients: 0.557–0.572; p values (two-tailed): 1.15E-04–1.87E-04).
Fig. 2.
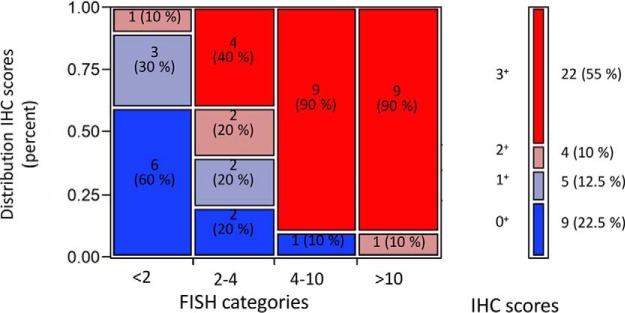
Distribution of IHC scores within FISH categories.
Fig. 3.

Correlation between normalization factors. The Spearman correlation coefficient and p value for each pair is reported in the insert.
In order to determine whether the data generated by SRM matched with the data obtained by IHC or FISH, peptide amounts calculated and normalized as described above were plotted against the IHC scores or the FISH categories (supplemental Fig. S4A and S4B respectively). The obtained pattern when plotted against IHC scores or FISH ratio categories was found to be reproducible between the six peptides. As expected from the correlation analyses, using either one of the four normalization factors also led to similar distribution patterns. The agreement of SRM HER2 peptide levels and IHC scores or FISH ratios was tested using ordinal logistic regression. The predicted IHC or FISH for each sample was compared with the respective experimental IHC scores and FISH ratios using kappa coefficients (Table IV). The overall percentage of agreement between predicted and observed scores was also determined. When comparing SRM data with IHC, the highest kappa coefficient was obtained for peptide ELVSEFSR using normalization parameter 3 (AUC of TIC): κ = 0.874 (95% CI (percentile, 10,000 bootstraps) 0.673–0.887) with an agreement of 77.5% (95% CI (percentile, 10,000 bootstraps) 62.5–82.5%). When using SRM to predict FISH ratio categories, the highest kappa coefficient was obtained for peptide GLQSLPTHDPSPLQR using normalization parameter 2 (BCA assay): κ = 0.687 (95% CI (percentile, 10,000 bootstraps) 0.265–0.705) with an agreement of 47.5% (95% CI (percentile, 10,000 bootstraps) 32.5–63.3%). Although the different groups seemed to follow a clearer increasing trend when using SRM to predict FISH scores, the prediction accuracy was actually lower than when using SRM to predict IHC (Table IV). This is probably because of the larger overlap between the different FISH ratio categories than between the different IHC scores. In addition for the two best performing peptides (ELVSEFSR for IHC and GLQSLPTHDPSPLQR for FISH), the agreement was also calculated separately for each IHC score or FISH ratio (supplemental Table S7). Peptide ELVSEFSR best predicted IHC scores 0+ and 3+ with correct classification rates of 88.9% and 95.5%, respectively, whereas peptide GLQSLPTHDPSPLQR best classified tumors within the first category (nonamplified) with 100% correct classification. However, this peptide was less suited to predict the other 3 FISH categories (correct classification of 10–50%).
Table IV. Agreement between SRM peptide amounts and IHC scores (A) or FISH ratio categories (B). k factors, p values (for κ factor) and percentage agreement are reported for all six peptides and for all four normalization factors tested. The highest κ factor for prediction of IHC score and FISH category respectively are highlighted in bold characters.
| Peptide sequence |
Normalization parameter 1 (tumor surface) |
Normalization parameter 2 (BCA assay) |
Normalization parameter 3 (AUC total ion current) |
Normalization parameter 4 (average KRT19 peptides) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| κ factor | p value κ | agreement [%] | κ factor | p value κ | agreement [%] | κ factor | p value κ | agreement [%] | κ factor | p value κ | agreement [%] | |
|
A. Agreement of SRM peptide amount with IHC score | ||||||||||||
| ELVSEFSR | 0.728 | 3.3E-06 | 70.0 | 0.817 | 2.1E-07 | 72.5 | 0.874 | 2.9E-08 | 77.5 | 0.741 | 1.7E-06 | 72.5 |
| FVVIQNEDLGPASPLDSTFYR | 0.715 | 3.5E-06 | 67.5 | 0.830 | 1.5E-07 | 77.5 | 0.712 | 2.6E-06 | 70.0 | 0.687 | 7.5E-06 | 70.0 |
| GIWIPDGENVK | 0.741 | 1.7E-06 | 72.5 | 0.712 | 2.6E-06 | 70.0 | 0.674 | 8.8E-06 | 67.5 | 0.687 | 7.5E-06 | 70.0 |
| GLQSLPTHDPSPLQR | 0.707 | 5.3E-06 | 67.5 | 0.809 | 3.0E-07 | 80.0 | 0.805 | 2.7E-07 | 77.5 | 0.622 | 6.0E-05 | 67.5 |
| SGGGDLTLGLEPSEEEAPR | 0.712 | 2.6E-06 | 70.0 | 0.712 | 2.6E-06 | 70.0 | 0.726 | 2.2E-06 | 70.0 | 0.635 | 2.8E-05 | 67.5 |
| SLTEILK | 0.728 | 3.3E-06 | 70.0 | 0.815 | 2.4E-07 | 72.5 | 0.779 | 7.6E-07 | 72.5 | 0.789 | 1.9E-07 | 75.0 |
|
B. Agreement of SRM peptide amount with FISH category | ||||||||||||
| ELVSEFSR | 0.602 | 7.0E-05 | 40.0 | 0.619 | 2.8E-05 | 45.0 | 0.592 | 7.6E-05 | 42.5 | 0.589 | 5.8E-05 | 50.0 |
| FVVIQNEDLGPASPLDSTFYR | 0.642 | 2.6E-05 | 42.5 | 0.603 | 3.9E-05 | 42.5 | 0.568 | 1.5E-04 | 45.0 | 0.600 | 5.5E-05 | 47.5 |
| GIWIPDGENVK | 0.647 | 2.4E-05 | 47.5 | 0.610 | 4.1E-05 | 42.5 | 0.585 | 9.3E-05 | 50.0 | 0.605 | 4.2E-05 | 45.0 |
| GLQSLPTHDPSPLQR | 0.667 | 1.2E-05 | 47.5 | 0.688 | 3.3E-06 | 47.5 | 0.634 | 2.9E-05 | 42.5 | 0.593 | 6.8E-05 | 47.5 |
| SGGGDLTLGLEPSEEEAPR | 0.617 | 4.1E-05 | 42.5 | 0.664 | 9.3E-06 | 45.0 | 0.592 | 7.6E-05 | 42.5 | 0.614 | 3.7E-05 | 47.5 |
| SLTEILK | 0.664 | 1.2E-05 | 45.0 | 0.625 | 2.3E-05 | 45.0 | 0.607 | 4.4E-05 | 45.0 | 0.623 | 2.5E-05 | 42.5 |
For the best performing peptides according to the comparison with IHC (ELVSEFSR) and FISH (GLQSLPTHDPSPLQR), we additionally assessed the ability of SRM to predict tumor classification according to the ASCO/CAP criteria used to define HER2 amplification for eligibility for anti-HER2 treatment (30) (Fig. 4 and supplemental Table S8). Classification for HER2 amplification is defined as follows: IHC score 0+ or 1+ or 2+ and nonamplified by FISH is considered as HER2 nonamplified; IHC score 3+ or 2+ and amplified by FISH is considered as HER2 amplified. The SRM data for both peptides sensitively predicted the tumors' HER2 amplification status (ASCO/CAP) with a threshold of 24.2 fmol and 1.002 fmol of normalized ELVSEFSR and GLQSLPTHDPSPLQR respectively, as observed in supplemental Fig. S5A and S5B. Using this classification, there were only three false positives for peptide ELVSEFSR and four false negatives for peptide GLQSLPTHDPSPLQR (Fig. 4 and supplemental Table S7). Interestingly, four or five tumors (depending on the peptide used) that were classified as amplified by FISH but with an IHC score ≤ 1 were predicted as negative by SRM (marked in yellow in supplemental Table S7) whereas the four tumors with the equivocal HER 2+ IHC score were classified as positive or negative depending on the peptide considered.
Fig. 4.

Boxplots depicting the agreement of the peptides (A) ELVSEFSR and (b) GLQSLPTHDPSPLQR levels with IHC scores, FISH categories, or HER2 amplification defined according to the ASCO criteria: negative: IHC scores 0+, 1+ and 2+ FISH non amplified; positive: IHC score 3+, 2+, and FISH amplified. The amount of each peptide was adjusted using the appropriate normalization parameter.
DISCUSSION
Protein quantification in FFPE tissues using targeted MS is a relatively new field with only few studies reported comparing SRM with other technologies (20, 21). In this paper, we described the development and evaluation of an SRM assay for the quantification of HER2 peptides in FFPE tissues and assessed the agreement between SRM and the two standard methods used in pathology, namely IHC and FISH. Compared with techniques such as IHC, SRM has the advantages of allowing a high level of multiplexing and does not rely on the availability of an antibody for detection of the analyte. It is also more quantitative compared with IHC, which only returns a semiquantitative score between 0 and 3. Although a previous study has measured HER2 in FFPE tissues (40), the present study is complementary in several ways. The main differences consist in the number of peptides monitored per protein, normalization aspects as well as statistical analysis of the data.
In a first step, we performed discovery proteomics analysis of relevant and increasingly complex samples in order to determine which HER2 peptides could be extracted and detected in untreated and in formalin-fixed material. Although this approach is more time-consuming, it is cost effective because it reduces the number of standard peptides to be synthesized, while maintaining a high chance of selecting peptides performing well in the mass spectrometer. From the 18 synthetic HER2 peptides evaluated, six were chosen as candidates for the development of an SRM assay based on their analytical performance. For an optimal specificity of the SRM assay, three peptides per protein and three transitions per peptide should typically be monitored (15, 16). This was not limiting in the case of a large protein such as HER2, but it might represent an obstacle in the case of a small protein with only few observable proteotypic peptides. In this study, we deliberately chose to focus on the non-modified form of the peptides although HER2 is annotated in the Uniprot database as containing several phosphorylation and glycosylation sites. Indeed, it is the overexpression of HER2 which is measured in clinical pathology for diagnostic purposes, rather than the extent of phosphorylation. Therefore, although two of the peptides included in the SRM assay contain a phosphorylation site (SGGGDLTLGLEPSEEEAPR and GLQSLPTHDPSPLQR), we did not attempt to evaluate to which extent these peptides were phosphorylated. It would have been possible and relatively straightforward to include phosphorylated peptides in the assay and it is technically possible to retrieve phosphopeptides from FFPE tissues (41). However, the question remains whether this would be a reliable approach for FFPE tissues. As previously discussed, pre-analytical factors which are relevant to formalin fixation (such as ischemic time, fixation duration, or temperature), are not standardized and are very likely to influence the levels of phosphorylation (42–44).
In a second step, we evaluated the performance of the analytical method by assessing linearity, LLOQ and reproducibility of the assay. We validated the use of a bacterial protein extract as a reliable matrix for the preparation of the standard curve. Calibration curves were consistently lower in the TNBC extract than in the P. furiosus matrix. This effect might be explained by a stronger signal suppression occurring in the more complex human tissue in contrast to the bacterial P. furiosus extract. Nevertheless, signal linearity was reproducible in the P. furiosus background over three injection replicates of a single calibration curve as well as for three injections of calibration curves prepared separately on different days. Reproducibility of the calibration curve is of particular importance when comparing samples that have been analyzed on different days or in different batches. Therefore, the P. furiosus extract was considered to be suitable for quantification purposes.
LLOQ was determined for all six peptides as the lowest determined concentration with a precision below 20% as recommended for the development of MS-based assays for research use (Tier 2) (39). Indeed, CVs of standards spiked in a P. furiosus background were systematically below 20% down to the low amol range. The measured LLOQ standards however were not always accurate in regard to the nominal concentrations of the standards (data not shown), which would be a limiting factor if the assay were to be used in a clinical diagnostic or bioanalysis laboratory (Tier 1). In the absence of a suitable internal protein standard in the FFPE tissue of interest to evaluate digestion efficiency and recovery, however, true accuracy is likely to remain elusive and may not be achievable in this type of assay (39).
Regarding the precision of the SRM assay, four of the six HER2 peptides showed CVs below 20% for all replicate measurements on tumor extracts at or above LLOQ. The impact of the sample preparation process on the SRM assay precision was also evaluated (inter-batch precision) (39). In this experiment, sample replicates were obtained from the same tumor but were collected from adjacent and therefore slightly different tissue slices. This inevitably introduces a higher variability compared with biofluids, where sample replicates could be easily derived from the same tube and hence would show an identical protein composition. We therefore chose a threshold of 35%, instead of 20%, for data interpretation in this case. As expected, the CVs obtained when considering the entire sample preparation procedure were larger than those observed for the analytical precision of the SRM assay alone. However, for two of the six peptides, the CVs were all below 35%. The average CV for sample preparation reproducibility across all six peptides was 32% whereas the median was 30%.
Using the developed SRM assay, the six HER2 peptides were quantified in 40 breast carcinoma tissue extracts with varying HER2 gene amplification levels. The results generated by SRM were compared with those of standard clinical pathology methods. A first observation from the data acquired in these samples was that different peptides returned different absolute amounts within a given sample. This discrepancy might reflect a differential accessibility to trypsin during the digestion step and/or a lower recovery in the sample processing step for given peptides, thereby leading to lower absolute amounts extracted from the tissue. Nevertheless, although an absolute tissue protein concentration cannot be derived with this method (because of the impossibility of spiking an internal standard in the FFPE tissue before extraction to compensate for incomplete digestion), the observation that relative changes in peptide amounts were highly correlated indicated that peptide concentrations are suitable surrogate markers of protein concentrations. Work is in progress to develop statistical methods allowing reliable extrapolation of relative protein tissue concentrations from data sets of peptide quantification. In certain cases, provided that verification studies demonstrate the validity of this approach, one might consider using the peptide itself as a biomarker rather than the whole protein.
A large subset of the 40 tumor samples analyzed had peptide concentrations below LLOQ. This was not totally unexpected and sensitivity was known to be a limiting factor of the SRM technology. The dynamic range that can be achieved by the method is indeed restricted by the complexity of the protein mixture and sample load on column. However, although only samples with high levels of HER2 (typically of IHC score 3+) were quantified with optimal precision, values below LLOQ should be taken with caution rather than systematically disregarded because they may still provide useful information, in particular because CVs below 20% were observed for some of those data points.
In the absence of a standard procedure to account for sample size, four different parameters were considered for normalization. All four parameters significantly correlated with each other, although KRT19 showed poorer correlation coefficients when compared with the three other normalization parameters (Fig. 3). It was to be expected that general parameters representing the surface of extracted tissue, the total amount of peptides, or the TIC of extracted peptides all highly correlated with each other. The poorer correlation of KRT19 as well as the lower agreement with IHC data is probably explained by the fact that KRT19 is expressed in the cancerous cells but not in the other cells present in the sample. The macrodissected tumor region contains a majority of tumor cells expressing KRT19 but also other cells (lymphocytes, fibroblasts, etc…) not expressing KRT19. Therefore, KRT19 is not accurately representing the total amount of cells in the sample. Depending on the sample, the proportion of KRT19 positive cells will change, explaining the lower correlation of KRT19 with the other parameters. Interestingly, normalization using tumor surface performed as well as the other procedures. We expected tumor surface to correlate less well as it is the only parameter acquired prior to sample preparation, and it therefore does not reflect the variability introduced by the sample preparation procedure. However, as the normalization parameters correlated highly with each other except with KRT19, size normalization using tumor surface would be a convenient method because it can be easily measured and would not be affected by any potential bias introduced by sample preparation.
In our study, we investigated the agreement of SRM peptide levels with IHC scores or FISH ratios. All six peptides showed similar trends and agreed with IHC and FISH data. Nevertheless, comparison of SRM data with IHC scores showed that SRM could not successfully distinguish the lower IHC scores (0+, 1+, and 2+) from each other. These results may reflect the semi-quantitative (nonlinear) nature of IHC scoring, which has been calibrated and standardized against reference cell lines that express a known number of HER2 molecules to facilitate interpretation of immunostains (45). While an IHC score 0+ (negative staining) corresponds to cells containing less than 20,000 receptors (the basal level of HER2 expression in normal breast epithelial cells), IHC scores of 1+ and 2+ (cells containing ∼100,000 and 500,000 receptors, respectively) reflect relatively subtle differences in HER2 protein expression that might not be clearly distinguished by SRM. An IHC score of 3+ (∼2,300,000 receptors per cell that would show an intense, complete membranous staining in >10% of the tumor) comprises a much broader spectrum of HER2 protein expression levels (23). Interestingly, comparison of FISH categories with SRM data only provided accurate prediction for the non-amplified HER2 tumors. There was a clear increase of the observed levels of HER2 peptides as the HER2 gene was increasingly amplified but differences between FISH categories were not significant. Conversely, there was an excellent agreement between SRM data and a classification based on HER2 amplification as defined using the ASCO/CAP criteria (amplified: IHC scores 3+, 2+ with FISH ratio>2; non-amplified: IHC scores 0+, 1+, 2+ with FISH ratio<2). The two assays' best performing peptides were independently able to differentiate the two classes of tumors from each other with an AUC of the receiver operating characteristic (ROC) curve above 0.96 (supplemental Fig. S5A and S5B). Overall, SRM data tended to agree more with IHC scores than with FISH categories, possibly reflecting the fact that an overexpressed HER2 gene does not necessarily result in a level of HER2 protein overexpression that is clinically sufficient to trigger an anti-HER2 treatment. Accordingly and depending on the peptide used, four or all five tumors with FISH amplified but with an IHC score ≤ 1 were predicted as negative by SRM. All four tumors with IHC score 2+ (which, according to ASCO/CAP guidelines, need to be assessed by FISH to determine the HER2 amplification status) were classified by SRM as positive or negative depending on the peptide considered. However, this result might have been explained by the very few IHC 2+ samples available in this study. Overall, this may indicate that the current recommendation that the HER2 amplification status be assessed by IHC or FISH, with the latter considered as the gold standard, might show exceptions. Indeed, the nonconcordance between protein expression and gene amplification is about 17% for IHC scores of 2+ (46).
In conclusion, we were able to develop and evaluate an SRM assay for peptides in FFPE tissues and demonstrate that the results generated by SRM agree with those generated in clinical practice by IHC, which also reports a measure of protein expression. This study confirms the validity of using SRM to quantify proteins in FFPE tissues. As it was stated for proteins in biological fluids, the possibility to use SRM for protein quantification in FFPE tissues should open great perspectives for protein biomarker discovery studies (11, 12). However, some questions remain to be addressed. Although the detection of clearly overexpressing HER2 tumors was successfully achieved by SRM, distinction between low- and mildly overexpressers might represent more of a challenge. Increasing measurement precision at lower LLOQ might be achieved, for example, by using the following generation of mass spectrometer instruments. In parallel, increasing the sample number in the study would increase statistical significance and should thus enable a better separation between the different IHC and FISH categories. Moreover, the use of a laser-capture microdissection (LCM) step prior to SRM analysis could provide increased spatial resolution and therefore lead to a more homogeneous sample, enriched in HER2. The use of LCM however raises its own issues, including sensitivity (because fewer cells are available for analysis) and especially an even longer sample preparation procedure. As a final note, SRM applied to FFPE tissues opens many new perspectives, not only for biomarker discovery and validation. Indeed, a better understanding of the molecular mechanisms involved in complex pathologies will lead to improved diagnosis, prognosis and patient stratification and might ultimately also lead to the discovery of novel therapeutic targets. The technology is currently being applied to a larger cohort in order to investigate patient stratification in breast cancer.
Supplementary Material
Acknowledgments
We thank Tom Dunkley and Arno Friedlein for constructive discussions during the course of the study.
Footnotes
Author contributions: C.S., J.T., P.C., P.L., and A.D. designed research; C.S. performed research; C.S. and A.D. analyzed data; C.S. wrote the paper; J.T. and T.A.M. identified samples, performed pathology analysis; J.L. performed statistical data analysis; S.K. helped generate the data; M.V., L.R., D.H., and P.C. advised on and supported the study; P.L. and A.D. reviewed study, assisted drafting the manuscript.
* CS was supported by a Roche Post-doctoral Fellowship.
 This article contains supplemental Material and Methods, Figs. S1 to S5, and Tables S1 to S9.
This article contains supplemental Material and Methods, Figs. S1 to S5, and Tables S1 to S9.
1 The abbreviations used are:
- FFPE
- formalin-fixed paraffin-embedded
- HER2
- human receptor tyrosine-protein kinase erbB-2
- SRM
- selected reaction monitoring
- IHC
- immunohistochemistry
- RPPA
- reverse-phase protein array
- FISH
- fluorescence in situ hybridization
- H&E
- hematoxylin and eosin
- LLOQ
- lower limit of quantification
- Rt
- retention time
- ASCO/CAP
- American Society of Clinical Oncology/College of American Pathologists
- DTE
- 1,4-dithioerythritol
- KRT19
- cytokeratin 19
- TNBC
- triple-negative breast carcinoma
- CV
- coefficient of variation
- AUC
- area under the curve
- LCM
- laser-capture microdissection
- ROC
- Receiver operating characteristic
- TIC
- total ion current.
REFERENCES
- 1. Shao S., Guo T., Aebersold R. (2014) Mass spectrometry-based proteomic quest for diabetes biomarkers. Biochim. Biophys. Acta 1854, 519–527 [DOI] [PubMed] [Google Scholar]
- 2. Drabovich A. P., Martinez-Morillo E., Diamandis E. P. (2014) Toward an integrated pipeline for protein biomarker development. Biochim. Biophys. Acta 1854, 677–686 [DOI] [PubMed] [Google Scholar]
- 3. Gustafsson O. J., Arentz G., Hoffmann P. (2014) Proteomic developments in the analysis of formalin-fixed tissue. Biochim. Biophys. Acta 1854, 559–580 [DOI] [PubMed] [Google Scholar]
- 4. Fox C. H., Johnson F. B., Whiting J., Roller P. P. (1985) Formaldehyde fixation. J. Histochem. Cytochem. 33, 845–853 [DOI] [PubMed] [Google Scholar]
- 5. Tanca A., Pagnozzi D., Addis M. F. (2012) Setting proteins free: progresses and achievements in proteomics of formalin-fixed, paraffin-embedded tissues. Proteomics Clin. Appl. 6, 7–21 [DOI] [PubMed] [Google Scholar]
- 6. Crockett D. K., Lin Z., Vaughn C. P., Lim M. S., Elenitoba-Johnson K. S. (2005) Identification of proteins from formalin-fixed paraffin-embedded cells by LC-MS/MS. Lab. Invest. 85, 1405–1415 [DOI] [PubMed] [Google Scholar]
- 7. Guo T., Wang W., Rudnick P. A., Song T., Li J., Zhuang Z., Weil R. J., DeVoe D. L., Lee C. S., Balgley B. M. (2007) Proteome analysis of microdissected formalin-fixed and paraffin-embedded tissue specimens. J. Histochem. Cytochem. 55, 763–772 [DOI] [PubMed] [Google Scholar]
- 8. Sprung R. W. Jr., Brock J. W., Tanksley J. P., Li M., Washington M. K., Slebos R. J., Liebler D. C. (2009) Equivalence of protein inventories obtained from formalin-fixed paraffin-embedded and frozen tissue in multidimensional liquid chromatography-tandem mass spectrometry shotgun proteomic analysis. Mol. Cell. Proteomics 8, 1988–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanca A., Pagnozzi D., Burrai G. P., Polinas M., Uzzau S., Antuofermo E., Addis M. F. (2012) Comparability of differential proteomics data generated from paired archival fresh-frozen and formalin-fixed samples by GeLC-MS/MS and spectral counting. J. Proteomics 77, 561–576 [DOI] [PubMed] [Google Scholar]
- 10. Giusti L., Lucacchini A. (2013) Proteomic studies of formalin-fixed paraffin-embedded tissues. Expert. Rev. Proteomics 10, 165–177 [DOI] [PubMed] [Google Scholar]
- 11. Carr S. A., Anderson L. (2008) Protein quantitation through targeted mass spectrometry: the way out of biomarker purgatory? Clin Chem 54, 1749–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Borchers C. H., Parker C. E. (2010) Improving the biomarker pipeline. Clin. Chem. 56, 1786–1788 [DOI] [PubMed] [Google Scholar]
- 13. Domon B., Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 [DOI] [PubMed] [Google Scholar]
- 14. Hinestrosa M. C., Dickersin K., Klein P., Mayer M., Noss K., Slamon D., Sledge G., Visco F. M. (2007) Shaping the future of biomarker research in breast cancer to ensure clinical relevance. Nat. Rev. Cancer 7, 309–315 [DOI] [PubMed] [Google Scholar]
- 15. Lange V., Picotti P., Domon B., Aebersold R. (2008) Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gallien S., Duriez E., Domon B. (2011) Selected reaction monitoring applied to proteomics. J. Mass Spectrom. 46, 298–312 [DOI] [PubMed] [Google Scholar]
- 17. Kuzyk M. A., Smith D., Yang J., Cross T. J., Jackson A. M., Hardie D. B., Anderson N. L., Borchers C. H. (2009) Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteomics 8, 1860–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumar V., Barnidge D. R., Chen L. S., Twentyman J. M., Cradic K. W., Grebe S. K., Singh R. J. (2010) Quantification of serum 1–84 parathyroid hormone in patients with hyperparathyroidism by immunocapture in situ digestion liquid chromatography-tandem mass spectrometry. Clin. Chem. 56, 306–313 [DOI] [PubMed] [Google Scholar]
- 19. Kushnir M. M., Rockwood A. L., Roberts W. L., Abraham D., Hoofnagle A. N., Meikle A. W. (2013) Measurement of thyroglobulin by liquid chromatography-tandem mass spectrometry in serum and plasma in the presence of antithyroglobulin autoantibodies. Clin. Chem. 59, 982–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Steiner C., Ducret A., Tille J. C., Thomas M., McKee T. A., Rubbia-Brandt L., Scherl A., Lescuyer P., Cutler P. (2013) Applications of mass spectrometry for quantitative protein analysis in formalin-fixed paraffin-embedded tissues. Proteomics 14, 441–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Catenacci D. V., Liao W. L., Thyparambil S., Henderson L., Xu P., Zhao L., Rambo B., Hart J., Xiao S. Y., Bengali K., Uzzell J., Darfler M., Krizman D. B., Cecchi F., Bottaro D. P., Karrison T., Veenstra T. D., Hembrough T., Burrows J. (2014) Absolute quantitation of Met using mass spectrometry for clinical application: assay precision, stability, and correlation with MET gene amplification in FFPE tumor tissue. PLoS ONE 9, e100586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gradishar W. J., Anderson B. O., Blair S. L., Burstein H. J., Cyr A., Elias A. D., Farrar W. B., Forero A., Giordano S. H., Goldstein L. J., Hayes D. F., Hudis C. A., Isakoff S. J., Ljung B. M., Marcom P. K., Mayer I. A., McCormick B., Miller R. S., Pegram M., Pierce L. J., Reed E. C., Salerno K. E., Schwartzberg L. S., Smith M. L., Soliman H., Somlo G., Ward J. H., Wolff A. C., Zellars R., Shead D. A., Kumar R. (2014) Breast cancer version 3.2014. J Natl Compr Canc Netw 12, 542–590 [DOI] [PubMed] [Google Scholar]
- 23. Hicks D. G., Kulkarni S. (2008) HER2+ breast cancer: review of biologic relevance and optimal use of diagnostic tools. Am. J. Clin. Pathol. 129, 263–273 [DOI] [PubMed] [Google Scholar]
- 24. Vizcaino J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Rios D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolomé S., Apweiler R., Omenn G. S., Martens L., Jones A. R., Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sharma V., Eckels J., Taylor G. K., Shulman N. J., Stergachis A. B., Joyner S. A., Yan P., Whiteaker J. R., Halusa G. N., Schilling B., Gibson B. W., Colangelo C. M., Paulovich A. G., Carr S. A., Jaffe J. D., MacCoss M. J., MacLean B. (2014) Panorama: a targeted proteomics knowledge base. J. Proteome Res. 13, 4205–4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Subik K., Lee J. F., Baxter L., Strzepek T., Costello D., Crowley P., Xing L., Hung M. C., Bonfiglio T., Hicks D. G., Tang P. (2010) The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical analysis in breast cancer cell lines. Breast Cancer 4, 35–41 [PMC free article] [PubMed] [Google Scholar]
- 27. Gluck F., Hoogland C., Antinori P., Robin X., Nikitin F., Zufferey A., Pasquarello C., Fétaud V., Dayon L., Muller M., Lisacek F., Geiser L., Hochstrasser D., Sanchez J. C., Scherl A. (2013) EasyProt–an easy-to-use graphical platform for proteomics data analysis. J. Proteomics 79, 146–160 [DOI] [PubMed] [Google Scholar]
- 28. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hembrough T., Thyparambil S., Liao W. L., Darfler M. M., Abdo J., Bengali K. M., Taylor P., Tong J., Lara-Guerra H., Waddell T. K., Moran M. F., Tsao M. S., Krizman D. B., Burrows J. (2012) Selected reaction monitoring (SRM) analysis of epidermal growth factor receptor (EGFR) in formalin fixed tumor tissue. Clin. Proteomics 9, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wolff A. C., Hammond M. E., Hicks D. G., Dowsett M., McShane L. M., Allison K. H., Allred D. C., Bartlett J. M., Bilous M., Fitzgibbons P., Hanna W., Jenkins R. B., Mangu P. B., Paik S., Perez E. A., Press M. F., Spears P. A., Vance G. H., Viale G., Hayes D. F. (2013) Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J. Clin. Oncol. 31, 3997–4013 [DOI] [PubMed] [Google Scholar]
- 31. Chu P. G., Weiss L. M. (2002) Keratin expression in human tissues and neoplasms. Histopathology 40, 403–439 [DOI] [PubMed] [Google Scholar]
- 32. Assadi M., Lamerz J., Jarutat T., Farfsing A., Paul H., Gierke B., Breitinger E., Templin M. F., Essioux L., Arbogast S., Venturi M., Pawlak M., Langen H., Schindler T. (2013) Multiple protein analysis of formalin-fixed and paraffin-embedded tissue samples with reverse phase protein arrays. Mol. Cell. Proteomics 12, 2615–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. R Development Core Team (2008) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 34. Gamer M., Lemon J., Fellows I. (2009) irr: Various Coefficients of Interrater Reliability and Agreement, R package version 0.82 R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 35. Venables W. N., Ripley B. D. (2002) Modern Applied Statistics with S, Springer, New York, NY [Google Scholar]
- 36. Faraway J. J. (2006) Extending the linear model with R: generalized linear, mixed effects and nonparametric regression models, Chapman & Hall/CRC, Boca Raton, FL [Google Scholar]
- 37. Agresti A. (2007) An Introduction to Categorical Data Analysis (Wiley Series in Probability and Statistics), Wiley-Interscience, Hoboken, NJ [Google Scholar]
- 38. Foulkes W. D., Smith I. E., Reis-Filho J. S. (2010) Triple-negative breast cancer. N. Engl. J. Med. 363, 1938–1948 [DOI] [PubMed] [Google Scholar]
- 39. Carr S. A., Abbatiello S. E., Ackermann B. L., Borchers C., Domon B., Deutsch E. W., Grant R. P., Hoofnagle A. N., Hüttenhain R., Koomen J. M., Liebler D. C., Liu T., MacLean B., Mani D. R., Mansfield E., Neubert H., Paulovich A. G., Reiter L., Vitek O., Aebersold R., Anderson L., Bethem R., Blonder J., Boja E., Botelho J., Boyne M., Bradshaw R. A., Burlingame A. L., Chan D., Keshishian H., Kuhn E., Kinsinger C., Lee J. S., Lee S. W., Moritz R., Oses-Prieto J., Rifai N., Ritchie J., Rodriguez H., Srinivas P. R., Townsend R. R., Van Eyk J., Whiteley G., Wiita A., Weintraub S. (2014) Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell. Proteomics 13, 907–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hembrough T., Thyparambil S., Liao W. L., Darfler M. M., Abdo J., Bengali K. M., Hewitt S. M., Bender R. A., Krizman D. B., Burrows J. (2013) Application of selected reaction monitoring for multiplex quantification of clinically validated biomarkers in formalin-fixed, paraffin-embedded tumor tissue. J. Mol. Diagn. 15, 454–465 [DOI] [PubMed] [Google Scholar]
- 41. Gámez -Pozo A., Sanchez-Navarro I., Calvo E., Diaz E., Miguel-Martin M., López R., Agullo T., Camafeita E., Espinosa E., López J. A., Nistal M., Vara J. A. (2011) Protein phosphorylation analysis in archival clinical cancer samples by shotgun and targeted proteomics approaches. Mol. Biosyst. 7, 2368–2374 [DOI] [PubMed] [Google Scholar]
- 42. Thompson S. M., Craven R. A., Nirmalan N. J., Harnden P., Selby P. J., Banks R. E. (2013) Impact of pre-analytical factors on the proteomic analysis of formalin-fixed paraffin-embedded tissue. Proteomics Clin. Appl. 7, 241–251 [DOI] [PubMed] [Google Scholar]
- 43. Fowler C. B., O'Leary T. J., Mason J. T. (2013) Toward improving the proteomic analysis of formalin-fixed, paraffin-embedded tissue. Expert Rev Proteomics 10, 389–400 [DOI] [PubMed] [Google Scholar]
- 44. Gündisch S., Hauck S., Sarioglu H., Schott C., Viertler C., Kap M., Schuster T., Reischauer B., Rosenberg R., Verhoef C., Mischinger H. J., Riegman P., Zatloukal K., Becker K. F. (2012) Variability of protein and phosphoprotein levels in clinical tissue specimens during the preanalytical phase. J. Proteome Res. 11, 5748–5762 [DOI] [PubMed] [Google Scholar]
- 45. Ross J. S., Slodkowska E. A., Symmans W. F., Pusztai L., Ravdin P. M., Hortobagyi G. N. (2009) The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist 14, 320–368 [DOI] [PubMed] [Google Scholar]
- 46. Cuadros M., Villegas R. (2009) Systematic review of HER2 breast cancer testing. Appl. Immunohistochem. Mol. Morphol. 17, 1–7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.