

Abstract
Accurate knowledge of retention time (RT) in liquid chromatography-based mass spectrometry data facilitates peptide identification, quantification, and multiplexing in targeted and discovery-based workflows. Retention time prediction is particularly important for peptide analysis in emerging data-independent acquisition (DIA) experiments such as SWATH-MS. The indexed RT approach, iRT, uses synthetic spiked-in peptide standards (SiRT) to set RT to a unit-less scale, allowing for normalization of peptide RT between different samples and chromatographic set-ups. The obligatory use of SiRTs can be costly and complicates comparisons and data integration if standards are not included in every sample. Reliance on SiRTs also prevents the inclusion of archived mass spectrometry data for generation of the peptide assay libraries central to targeted DIA-MS data analysis. We have identified a set of peptide sequences that are conserved across most eukaryotic species, termed Common internal Retention Time standards (CiRT). In a series of tests to support the appropriateness of the CiRT-based method, we show: (1) the CiRT peptides normalized RT in human, yeast, and mouse cell lysate derived peptide assay libraries and enabled merging of archived libraries for expanded DIA-MS quantitative applications; (2) CiRTs predicted RT in SWATH-MS data within a 2-min margin of error for the majority of peptides; and (3) normalization of RT using the CiRT peptides enabled the accurate SWATH-MS-based quantification of 340 synthetic isotopically labeled peptides that were spiked into either human or yeast cell lysate. To automate and facilitate the use of these CiRT peptide lists or other custom user-defined internal RT reference peptides in DIA workflows, an algorithm was designed to automatically select a high-quality subset of datapoints for robust linear alignment of RT for use. Implementations of this algorithm are available for the OpenSWATH and Skyline platforms. Thus, CiRT peptides can be used alone or as a complement to SiRTs for RT normalization across peptide spectral libraries and in quantitative DIA-MS studies.
The separation of peptides by reverse phase liquid chromatography (LC)1 prior to mass spectrometry detection has become an important technique for the in depth analysis of proteomes in data-dependent acquisition (DDA), data-independent acquisition (DIA), and targeted mass spectrometry workflows. In addition, the reproducibility of the retention and subsequent elution of a peptide from a reversed phase (e.g. C18-based) column at a specific time point along the acidic/organic gradient has led to the application of retention time (RT) as an orthogonal characteristic applicable to confident peptide identification (2, 3), targeted quantitative assay scheduling (4), and MS1-based quantitation (5). In targeted workflows, where an MS1 precursor and associated MS2 fragment masses (i.e. a transition group or peptide assay) are monitored and quantified with high sensitivity, accurate prior knowledge of peptide RT is a critical piece of information used for distinguishing between multiple extracted ion chromatogram (XIC) peaks that may occur along the chromatographic space. This is of particular concern in comparative experiments where it is crucial that signals arising from identical peptides are appropriately aligned and quantified across samples.
The importance of accurate RT knowledge is amplified in emerging DIA analytical strategies such as SWATH-MS (6, 7). With the SWATH-MS approach, large “peptide assay libraries” of peptide precursor and selected product ion transition groups, akin to those used in multiple reaction monitoring (MRM) experiments, are assembled from shotgun data sets. These peptide assay libraries are used to probe for and extract tens-to-hundreds of thousands of ion chromatograms from complex and convoluted chimeric MS2-fragmentation spectra derived from “all observable” peptides falling within predetermined mass windows (for instance, 25 m/z) (6). With this high-throughput in silico targeted ion extraction the currently available data analysis pipelines are challenged and orthogonal a-priori information (such as RT and fragment ion intensity information) is needed for correct identification of the target signal among multiple peak groups. Multiple peak group XIC signals may be detected because of amino acid modifications, closely related peptide isoforms, or simply the existence of multiple peptides with similar chemical properties and thus co-eluting transitions (8). Thus, the precise knowledge of RT is integral to the three major steps in the SWATH-MS analysis workflow: (1) alignment of peptide assay library and DIA-MS chromatograms; (2) prediction of assay library RT for accurate identification of target peptides in DIA-MS files; and (3) estimating confidence of peptide identifications for the accurate quantification of transition group XICs from DIA-MS files. A method proposed as a means to align chromatograms for DIA-MS analysis must be tested in each of these application steps.
Achieving maximum proteome coverage with SWATH-MS is dependent upon the assembly of comprehensive peptide assay libraries, and as such an active goal within the field is to build databases composed of every mass-spectrometry observable peptide within a given proteome (9–13). While requiring a large amount of resources up front to build these libraries, once assembled they become a permanent and exchangeable tool for targeted proteomic quantification. Furthermore, these peptide assay libraries are applicable for the quantification of both a single protein via SRM and for up to thousands of proteins and potentially their modified forms in a SWATH-MS experiment. Due to the importance of accurate RT knowledge for SWATH-MS, the portability of peptide assay libraries is critically dependent on the establishment of a uniform, normalized RT index to which the raw RTs in peptide assay libraries can be aligned.
Recently, Escher et al. introduced a set of synthetic peptides that are added to samples and used to normalize peptide RT across any reversed phase LC-gradient elution profile, but in their example with C18 as the stationary phase (iRT-C18). The normalization of RT by the synthetic iRT standards (SiRTs) allows for the meaningful transfer of peptide assay libraries between labs (1). The utility and accuracy of the RT alignment generated by the iRT-C18 approach has by now been demonstrated in multiple studies (14–16), and algorithms for transforming experimental peptide RTs to a normalized iRT-C18 space have been included in software tools used for both SRM-assay design (Skyline) and SWATH-MS data analysis (Skyline, OpenSWATH, Spectronaut).
Although the established iRT-C18 approach is effective and reproducible, a typical iRT-normalization relies upon the presence of spiked synthetic peptides within every sample analyzed, which presents a few drawbacks. First, the inclusion of the SiRT reference peptides in every single MS-injection becomes costly, especially for very large experiments and in high-throughput laboratories. Second, if the SiRT peptides are not included, or are of low signal quality, then automated data analysis of a measurement is currently impossible. Third, the SiRT approach precludes the inclusion of archived MS data, for example shotgun data collected prior to the development of the SiRT-based approach, in building assay libraries.
Here we present a simple solution to these limitations, whereby we have generated lists of common peptides highly conserved among most eukaryotes along with their normalized RT values (termed CiRT, for common iRT peptides). The peptides we have chosen are likely to be present in a range of eukaryotes from yeast to human across a number of experimental preparations and thus can be used to translate raw peptide RTs into a normalized iRT space. Furthermore, in our description of the assembly of these peptide lists we outline the workflow necessary for generating alternative RT calibration peptides for samples in which the particular endogenous CiRT peptides described here are not or are only partially present. In this article we have tested the performance of the CiRT method as it is applied to the three critical steps for DIA-MS targeted analysis: (1) peptide assay RT alignment; (2) peptide identification in DIA-MS data files; (3) quantitative accuracy based on CiRT alignment. Notably, for this work we have chosen to use the same normalized RT space established by Escher and colleagues (iRT-C18) such that currently existing SWATH-MS peptide assay libraries can be directly applied in ongoing projects. However, our approach is more general and applicable to any RT normalization scheme and the iRT-C18 scheme was chosen as a convenient reference framework for our comparisons.
EXPERIMENTAL PROCEDURES
Biological Sample Preparation
Data sets were generated from three separate biological sources.
(1) HEK293 (ATCC, Manassas, VA) cells (Human cell line) at ∼80% confluency were harvested on ice by pipetting in ice-cold PBS with 1 mm EDTA, pelleted by centrifugation for 3 min at 300 × g at 4 °C, and snap frozen. Cells were lysed on ice in HNN lysis buffer (0.5% Nonidet P-40, 50 mm HEPES, pH 7.5, 150 mm NaCl, 50 mm NaF, 200 μm NaVO3, 0.5 mm PMSF), and protease inhibitor mixture (Sigma, St. Louis, MO) and centrifuged at 16,100 × g for 15 min at 4 °C to remove insoluble material. Protein was precipitated from the lysate using 6 volumes of ice cold acetone for 1 h, pelleted by centrifugation at 16,100 × g for 15 min at 4 °C. The protein pellet was washed three times with 200 μl ice cold acetone with interspersed centrifugation, dried briefly by vacuum centrifugation at 45 °C, and resuspended in 8 m urea, 50 mm NH4HCO3 prior to digestion (described below).
(2) The yeast strain BY4741 MATa his3Δ leu2Δ met15Δ ura3Δ (Yeast cell line) was grown in S.D. medium until they reached an A600 of 0.8. The culture media was quenched by addition of trichloroacetic acid to a final concentration of 6.25% and the cells were harvested by centrifugation at 1500 × g for 5 min at 4 °C. The supernatants were discarded and the cell pellets were washed three times by centrifugation with cold (−20 °C) acetone to remove interfering compounds. The final cell pellets were resolubilized in lysis buffer containing 8 m urea, 0.1 m NH4HCO3 and 5 mm EDTA and cells were disrupted by glass bead beating (5 times 5 min at 4 °C) to lyse the cells prior to digestion (described below).
(3) Mouse immortalized vascular smooth muscle cells (VSMC, ATCC CRL-2797, Manassass, VA) were grown to 90% confluency and lysed by freeze/thaw cycling and cell scraping in ice-cold 8 m urea.
Preparation of Peptide Samples for LC MS
Proteins were reduced with either 10–12 mm DTT (Mouse, Yeast lysate) or 5 mm TCEP (HEK lysate) for 30 min at 37 °C and subsequently alkylated with iodoacetamide (10 mm, 40 mm and, 50 mm for HEK, yeast, and mouse lysates, respectively). Mouse lysates were diluted with 0.1 m NH4HCO3 to a 6 m Urea concentration and digested in 0.1 μg Lys-C (Roche, Indianapolis, IN) for 4 h at 37 °C prior to trypsin digestion. All samples were diluted to ∼1.5 M Urea by addition of 0.1 m NH4HCO3, after which sequencing grade modified trypsin (Promega, Madison, WI) was added at a 1:100 ratio of enzyme/substrate. Digestion of all samples was stopped by acidification with Formic acid. To test the alignment capabilities of the CiRT peptides on a library synthesized from a highly fractionated data set, peptides from the mouse lysate only were separated by basic reverse phase chromatography, collecting 80 fractions. The 80 fractions were reduced to 12 fractions by recombining fractions spaced far from each other in the organic elution gradient. Fractions were dried and re-suspended in 0.1% trifluoroacetic acid prior to desalting.
Acidified peptide mixtures derived from all three sources were desalted using reverse phase cartridges Sep-Pak C18 (Waters, Milford, MA) according to the following procedure; wet cartridge with 1 volume of 100% methanol, wash with 1 volume of 80% acetonitrile, equilibrate with 4 volumes of 0.1% formic acid, load acidified digest, wash 6 volumes of 0.1% formic acid, and elute with 1 volume of 50% acetonitrile in 0.1% formic acid. Peptides were dried using a vacuum centrifuge and resolubilized in 0.1% formic acid with synthetic iRT (SiRT) calibration peptides at a 1:20 v/v ratio (Biognosys, Schlieren, Switzerland).
Shotgun Data Dependent Mass Spectrometry and Assay Library Generation
Peptides were analyzed by LC MS over a 2 h gradient from 2–35% acetonitrile by an Eksigent NanoLC Ultra 2D Plus HPLC system coupled to an 5600 TripleTOF mass spectrometer (AB Sciex, Framingham, MA) operating in shotgun mode. The top 20 most intense MS1 precursors (collected between 360 and 1460 m/z for 250 ms) with charge states between 2 and 5 were selected for MS2 fragmentation, with a 15 s exclusion window. Fragment MS2 ions were collected for 100ms across a 50–2000 m/z range.
Raw profile mode wiff files were centroided and converted to mzML using the AB Sciex converter (v1.3) and subsequently converted to mzXML using msconvert (ProteoWizard, v3.04.238). Peptide sequences were assigned using parallel searches with the OMSSA and X!Tandem algorithms, searching against UniProtKB/TrEMBL databases of Human, Yeast, or Mouse proteins, appended with common contaminants and decoy sequences (Human 40,577 proteins and decoys, September 2013; Yeast 12,006 proteins and decoys, October 2013; Mouse 33,330 proteins and decoys, October 2013). Semitryptic peptides with up to two missed cleavages were allowed, with carbamidomethylation of cysteines set as a fixed modification and variable modifications of oxidized methionine and phosphorylation of tyrosine, serine, and threonine. Precursor and product ion mass errors were set to 30 ppm and 75 ppm, respectively. Search engine results were converted to pepXML format using omssa2pepXML (v2.1.9) or Tandem2XML (v4.6.0). Peptide spectral match probability scoring was modeled in PeptideProphet (v4.6.0), and the resulting interact.pepXML files of the two search engines were combined in iProphet (v4.6.0).
Peptide assay libraries were generated with SpectraST (v4.0) from the identified peptides with a Peptide Prophet probability > 0.95. The resulting spectrast .splib file was submitted as input to the custom spectrast2spectrast_irt.py converter script that was used to align RT to the normalized iRT_C18 space, initially using the SiRT peptides (1) and subsequently by substituting in the CiRT-reference peptides, generated as described below (note: this functionality has now been incorporated directly into the SpectraST (v5.0) workflow). A nonredundant, consolidated peptide assay library was created from each of the initial iRT normalized libraries with SpectraST. To test the feasibility of merging data sets derived from archived and diverse sources for library creation, two additional human-derived data sets, 11 OFFGEL fractions and 15 basic reverse phase fractions, generated from human cell lysates were accessed and downloaded from the PRIDE data repository (PXD000953, filenames beginning with PFARID and PXD000442, filenames containing MCF71-MCF715) (17, 18). All three data sets from the human-derived lysates were then merged into a single master library using the SpectraST library manipulation tool available in the trans proteomic pipeline. Spectral libraries were formatted for OpenSWATH using the custom script spectrast2tsv.py, assigning 5 y- or b-ion transitions to each peptide, and including peptides between m/z 350–2000, with charge states 1, 2, 3, or 4. The OpenSWATH tools ConvertTSVtoTraML and OpenSwathDecoyGenerator were then used to format the library for input to OpenSWATH and append shuffled decoys to the full assay libraries as described (7).
All custom python scripts are available at: https://github.com/msproteomicstools.
OpenSWATH tools can be found at: http://www.openswath.org.
Identification of CiRT Reference Peptides
Two separate peptide assay libraries comprising 6728 yeast and 9565 human peptides that were derived from cell lysates (confidence of identification set to <1% FDR) were generated as described above. The raw RT of each peptide was normalized to the iRT-C18 space using the traditional biognosys SiRT reference peptides (Biognosys, Schlieren, Switzerland). In a separate analysis, we ran a query of a theoretical tryptic digest of the entire Swiss-Prot protein database, selecting all annotated species from bacteria up the phylogenetic tree to homo sapiens to generate a table organized with a peptide amino acid sequence in one column and the number of times that peptide sequence occurs across all species in the database in a second column, rank ordered by the most commonly occurring peptide sequences at the top of the list. Although the surrounding amino acids of a given conserved peptide sequence may differ across species, by specifically querying the peptide sequences produced by theoretical tryptic digest we ensured that the peptide sequence reported is consistent with the most common method for peptide sample preparation used in mass spectrometry experiments. To identify the CiRT list, the peptides common between the yeast and human libraries were cross-referenced against the 500 most commonly occurring tryptic peptide sequences from the UniProtKB/Swiss-Prot database query. The result of this three way comparison was a list of mass-spectrometry accessible peptides that are present in both yeast and human cell lysates, that were also among the most commonly occurring (i.e. highly conserved) peptides across the entire Swiss-Prot protein database (CiRT; supplemental Table S1). We also included a handful of trypsin autolysis peptides, as these are very commonly detected as a consequence of typical MS sample preparation, for a final CiRT list of 113 peptides. The reference iRT value for each CiRT peptide was calculated as an average of the iRT value calculated from the identification of that peptide in the yeast and human lysates.
SWATH Mass Spectrometry and OpenSWATH Data Analysis
Peptides analyzed by SWATH-MS were acquired as described previously (6, 15). Briefly, the TripleTOF 5600 mass spectrometer was tuned to allow a quadrupole resolution of 25 m/z mass selection. Precursor MS1 ions were grouped into 26 m/z windows across a 400–1200 m/z range, creating a set of one precursor scan and 32 MS/MS fragmentation scans with 0.5 m/z overlap at either end of a given window. SWATH MS2 spectra were collected between 100–2000 m/z within each window, with collision energy determined as that appropriate for a 2+ charged ion centered within the window with a spread of 15. Total duty cycle was ∼3.4 s, with 100 ms accumulation time set for each of the 32 SWATH scans in high sensitivity mode and one high resolution survey scan at the beginning of each cycle.
Data were analyzed using the recently published OpenSWATH workflow, described in detail elsewhere (7, 15). Briefly, raw wiff files from SWATH-MS acquisitions were converted in profile mode to mzXML using msconvert. This conversion results in wrongly annotated precursor isolation windows, and was fixed with the custom script fix_swath_windows.py. The repaired mzXML file was then split into 33 files (one for each SWATH window, plus the precursor scans) using the custom script split_mzXML_intoSWATH.py, and each mzXML file was subsequently converted to mzML with msconvert. The resulting mzMLs were input into the OpenSWATH workflow, along with the SiRT or a custom internal iRT alignment TraML file, and the appropriate transition library assay TraML file (yeast, human, mouse, or SGS). The candidate peptide signals from the OpenSWATH output were then classified using the pyprophet implementation of the mProphet algorithm (v0.10.0, https://pypi.python.org/pypi/pyprophet/0.10.0) (17). Only peak group identifications with an assay q-value < 1% were included for all downstream analyses.
Assessment of Quantitative Accuracy using the SWATH-MS Gold Standard Data Set
The effect of iRT alignment on quantitative accuracy by SWATH-MS was assessed on the previously described SWATH-MS Gold Standard (SGS) data set, details for which can be found in Röst et al. (7). Briefly, 340 isotopically labeled “heavy” peptides along with the iRT-kit (Biognosys) were spiked into human or yeast cell lysate at increasing twofold concentration increments from 0.116 fmol up to 60 fmol. Lysates spiked with the peptides at each concentration were analyzed in triplicate SWATH-MS acquisitions. An assay library containing each of the spiked-in peptides, aligned to normalized iRT-C18 space, was used for targeted extraction on each SGS data file. XICs and peak groups were generated for each peptide, when observable, using the OpenSWATH workflow with the RT normalization performed in duplicate extractions, once with the iRT-kit peptides and once with the internal custom iRT peptides. Peak groups that did not reach the q-value threshold below 1% q-value were given a zero value. Peptide intensity at each concentration in the twofold dilution series was normalized to the intensity value from the highest concentration (30 fmol/ul). The Log (base 2) values of normalized peptide intensities (nLog2) were averaged across technical replicates. Box and whisker plots were generated to test whether the observed intensity of peptides fit the expected linear pattern of a twofold increase in nLog2 intensity between any two dilution steps, which with perfect quantification should be equal to 1. To compare the accuracy of quantification between CiRT and SiRT normalized data sets, the error of quantification was calculated as described in equation 1:
The average error of all peptides observed between any two dilution steps was calculated for the CiRT and SiRT normalized data sets, and one-way ANOVA was used to examine any differences in quantitative error between the CiRT and SiRT normalized data sets.
Algorithmic Refinements to Improve the Robustness of the Automated RT Normalization
First, through manual inspection of OpenSWATH subscores (described in Rost et al., 7) for high and low quality peptide transition groups, we optimized a filter function that was implemented as an extension to the preliminary scoring of the existing OpenSwathRTNormalizer component tool in the OpenSWATH workflow. This filter uses a simplified linear model with pretrained fixed weights (instead of the semi-supervised learning linear discriminant analysis) and is optimized to filter and remove low-scoring candidate signals without requiring RT information. Second, to automatically remove outlier signals introduced by false positive peptide peak group identifications that might introduce noise in the linear regression, we added two additional outlier removal methods. The originally implemented jackknife method optimizing the coefficient of determination of the linear regression was supplemented with an algorithm selecting the largest residual and an implementation and adaptation of the Random Sample Consensus (RANSAC) algorithm (20). Third, to ensure that the selected endogenous normalization peptides and smoothed linear regression cover the whole retention time range, the algorithms require that at least one sampled peptide is present in eight out of ten bins that collectively span the entire retention time range. This option can be enabled in the OpenSwathRTNormalizer component of the OpenSWATH tool using the parameter “-best_peptides” and an assay library of candidate alignment peptides and their assigned iRT values.
We then created a benchmark data set to test the accuracy and efficiency of the different algorithms for RT peptide selection and RT normalization. We selected one data set where we manually annotated the position of 113 endogenous CiRT peptides (if they could be detected at all in the samples analyzed) to create a “ground truth” value for this file. The original CiRT assay library was complemented with a varying number of additional known false assays (up to a 10-fold excess) created with one of two methods (“assay decoy” or “iRT decoy”). The assay decoys were generated using the OpenSwathDecoyGenerator (7) and appended in ratios from 1:1 to 1:10. These decoys were used to assess the optimized filter function for undetectable peptides. The “iRT decoys” were generated and appended in ratios from 1:1 to 1:10 by duplication of the target assays and randomized shuffling of the iRT coordinates within the list. The goal was here to generate assays that will result in detected signals but with wrong iRT coordinates, challenging the outlier removal algorithms even more.
The linear regressions and transformations from iRT to RT space as computed by the different algorithms were assessed by using the target CiRT assay library as input for the algorithms without any decoys as ground truth. The performance of the algorithms was judged by computing accuracy and recall (using manually validated datapoints and treating the problem as a classical classification problem). In addition, we report the standard deviation of the difference between the predicted retention times using the computed model and the true, known retention time as an objective goodness-of-fit measure.
Deposition of Data
All unpublished data have been deposited for public access: http://www.peptideatlas.org/PASS/PASS00724.
RESULTS
Our goal was to develop a set of commonly detected peptides for internal RT calibration in DIA workflows. The integration of internal RT standards into the DIA workflow involves the following steps (summarized in Fig. 1): (1) normalizing RT within ion libraries using CiRT; (2) predicting and scoring assay peak group RT in a SWATH-MS data file; (3) ensuring CiRT-based RT prediction does not adversely effect quantitative accuracy by SWATH-MS.
Fig. 1.

A schema depicting the process of identifying internal iRT normalization peptides. Top Panel: Samples of lysate are digested with trypsin, spiked with synthetic iRT calibration peptides (SiRT), and analyzed by data-dependent mass spectrometry. The most abundant peptides, or those identified across multiple species, are selected as candidates and iRT values are assigned using the linear regression created by referencing the external iRT calibrant peptides. Middle panel: Retention time is normalized across spectral libraries by replacing the SiRT peptides with the selected CiRT peptides in the SpectraST iRT normalization step. For libraries generated from multiple fractions of peptides, larger CiRT lists are required. Bottom panel: Prediction of retention time in SWATH-MS data files using CiRTs requires very high intensity, low signal-to-noise calibration candidates. A filtered list of CiRT normalization peptides, created either manually or via the newly written algorithm for CiRT refinement described here, are extracted from the SWATH-MS data and a linear regression is computed to transform iRT to observed RT for that file. The subsequent linear equation is used to predict retention time of a given peptide in the assay library within a user-specified confidence window, typically ± 5 min. Candidate peak groups selected within this window are scored using the OpenSWATH scoring algorithm, where the difference between the experimental retention time relative to the predicted retention time of a given peptide is scored and contributes to the composite score used to assess confidence in peak group identification.
Normalization of Assay Library RTs Using Internal CiRT Peptides
One utility of internal RT standards is to be able to convert the raw RT of a given shotgun sample to a normalized RT for easy transfer of peptide transition libraries across laboratories and experimental set ups. We tested the capabilities of the CiRT-reference peptides to normalize RT in a total of five different data sets: Two unfractionated whole cell lysate samples generated from human and yeast cell lines; one data set comprised of 12 separate shotgun files generated from a basic reverse phase fractionation of mouse VSMC digest; and two additional data sets comprised of 11 OFFGEL fractions and 15 basic reverse phase fractions generated from human cell lysates that were accessed and downloaded from the PRIDE data repository. To perform the RT normalization step, CiRT-reference peptides were formatted for insertion into the spectrast2spectrast_irt.py script (supplemental Table S4) as a comma separated list containing only the exact peptide sequence followed by a colon and then the assigned iRT value (e.g. PEPTIDE1:iRT1, PEPTIDE2:iRT2, etc).
First, we compared the CiRT normalization peptides to exogenous SiRT standards for library RT alignment. Ninety-eight (86%) of the 113 possible CiRT peptides were identified in and used to fit the yeast library iRT normalization and 84 (74%) could be detected and used to fit the human library normalization. The errors between the predicted RT (calculated from the normalized iRT-value) and the actual observed RT (ΔRT) were comparable between CiRT-peptide and SiRT-peptide alignments (Fig. 2A). For all normalization strategies, the interquartile range for overall error in RT-prediction from normalized iRT was within ± 30 s.
Fig. 2.
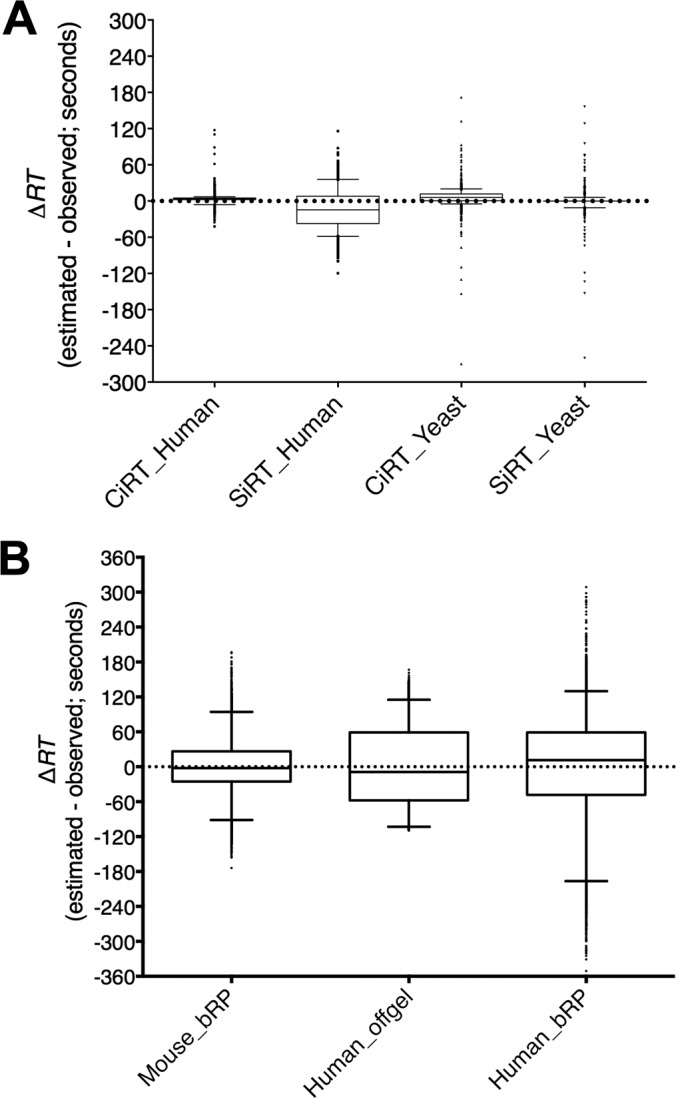
Assay library retention time normalization between synthetic and internal calibration peptides. The difference between observed and predicted retention time (ΔRT) was calculated for each peptide included in the assay libraries generated from the same raw files of human and yeast lysate (A), and from three different sets of fractionated peptides from mouse (generated in house) and human cell lysate (data downloaded from the PRIDE MS data repository), separated by either basic reverse phase (bRP) or OFFGEL fractionation methods (B). Retention times were predicted separately using linear regression equations created from a large list of 113 common iRT (CIRT) peptides or the 11 synthetic iRT peptides (SiRT). Data are presented as box and whisker plots, with the middle quartiles surrounding the median for the entire assay library represented by the box, whiskers showing the 95% data range and the upper and lower 2.5% of all values plotted as individual data points. Note: for some data sets the distribution of ΔRT was within such a small range that the box and whisker plots appear as a single horizontal line. Y-axis ranges were set at values that demonstrate the full range of error.
Next, we tested whether the endogenous CiRT peptides could be used to normalize RT within a series of peptide sets fractionated from a starting sample of trypsin digested complex lysate. This presents a unique challenge for the use of endogenous internal reference peptides, because these normalization reference peptides are present prior to peptide fractionation, and the starting list must be large enough to ensure distribution of reference peptides with adequate coverage of the chromatogram in each fraction. We tested the capability of CiRT peptides to align 12 shotgun runs from separate peptide fractions of mouse vascular smooth muscle cell lysate that had been separated by basic reverse phase (bRP) fractionation, as well as 11 samples of OFFGEL fractionated peptides and 15 samples of bRP fractionated peptides both derived from human cell lysate that were accessed via the PRIDE data repository (PXD000953 and PXD000442, respectively). The CiRT peptide alignments were successful for all three sets of files, with each separate alignment exhibiting R2 values > 0.92. Tables of the peptides included for alignment of each fraction for the mouse and human OFFGEL data set are shown in supplemental Tables S2 and S3. Overall, the CiRT alignment predicted RT within ± 60 s for the majority of assay library peptides generated from the fractionated data sets (Fig. 2B).
Normalization and Prediction of RT in SWATH-MS Files Using CiRT
In the SWATH-MS targeted data analysis workflow, accurate alignment of the experimental RT to the peptide assay iRT is crucial to enable sensitive detection of peak group candidates. To perform the RT-normalization of a SWATH-MS file in the OpenSWATH workflow, ion chromatograms are first extracted for the RT-reference peptides. In order to confidently and accurately assign peak group identity for a normalization peptide, it is imperative that (1) the peptide is present in high abundance within the sample so that the peak group has high intensity and low signal-to-noise and (2) there is no ambiguity in peak group assignment. If a peptide from the normalization set is not present in the SWATH-MS sample or is present in a moderate to low abundance, this can lead to misidentification of a peak group, erroneous RT assignment for this peptide and failure of the RT normalization step.
To address these requirements, we first generated a refined list of 14 CiRT peptides for SWATH-MS data set normalization (CiRT-SW) that represents the top most abundant and unambiguous peptides from the 113 CiRT peptide list. We selected these manually, comparing the extracted transition group chromatograms of only the CiRT peptides across the full chromatographic timespan from SWATH-MS runs generated from human, yeast, and mouse cell lysates using Skyline software. It is important to note that this was not a full OpenSWATH analysis, as we did not attempt to score and quantify the peptides using the full algorithm scoring method, we simply manually selected the peptides for high intensity, matching fragment ion co-elution and equal distribution across the chromatogram. To clarify this point, the total extracted ion chromatograms of the 14 peptides selected for the CiRT_SW list, as well as examples of peptides not selected, are provided in supplemental Fig. S1. This selection resulted in a set of high intensity, high signal-to-noise, and unambiguous CiRT peptides for use in RT normalization of most SWATH-MS files by any experimenter (Table I).
Table I. CiRT_SW peptides.
| Peptide sequence | Protein | iRT |
|---|---|---|
| DSYVGDEAQSK | Actin | −14.8 |
| AGFAGDDAPR | Actin | −8.7 |
| ATAGDTHLGGEDFDSR | Heat shock protein SSA3 | 5.2 |
| VATVSLPR | Trypsin | 13.4 |
| ELISNASDALDK | ATP-dependent molecular chaperone HSP82 | 25.1 |
| IGPLGLSPK | 60S ribosomal protein L12 | 29.4 |
| TTPSYVAFTDTER | Heat shock protein SSA3 | 34.8 |
| VC[160]ENIPIVLC[160]GNKVDVK | GTP-binding nuclear protein GSP1/CNR1 | 55.0 |
| DLTDYLMK | Actin | 59.8 |
| LGEHNIDVLEGNEQFINAAK | Trypsin | 60.0 |
| SYELPDGQVITIGNER | Actin | 66.9 |
| YFPTQALNFAFK | ADP/ATP translocase | 93.5 |
| SNYNFEKPFLWLAR | GTP-binding nuclear protein GSP1/CNR1 | 94.0 |
| DSTLIMQLLR | 14-3-3 protein | 101.8 |
In our initial test of the CiRT method, the refined CiRT SW list was used to predict RT and extract target peptides in a yeast, human, and mouse cell SWATH-MS data file, respectively. Independent t-tests comparing the ΔRT values for peptides identified in SWATH analyses performed using the two different alignment methods (CiRT versus SiRT) indicated that the accuracy of the RT prediction, was significantly different between sets (Fig. 3A; t = 9.36, p < 0.0001 comparing ΔRT values for CiRT and SiRT aligned peptides in human lysates, t = 43.35, p < 0.0001 comparing ΔRT values for CiRT and SiRT in yeast lysates, and t = 6.77, p < 0.0001). Neither the SiRT nor the CiRT peptide sets indicated bias toward higher accuracy of alignment, whereas the SiRT normalization set exhibited higher absolute error in the human sample (mean ΔRT of 14.5 ± 15 standard deviation versus mean ΔRT of 23.9 ± 94 standard deviation for CiRT and SiRT aligned peptides, respectively). Conversely the CiRT set exhibited higher absolute error in the yeast sample (mean ΔRT of 21.9 ± 43.2 standard deviation versus mean ΔRT of 16.2 ± 50.1 standard deviation for CiRT and SiRT, respectively) and the mouse sample (mean ΔRT of 35.6 ± 109.6 standard deviation versus mean ΔRT of 23.2 ± 106.7 standard deviation for CiRT and SiRT, respectively).
Fig. 3.

Accuracy between retention time prediction methods for peak groups extraction from SWATH-MS data files. A, The difference between observed and predicted retention time (ΔRT) of each of the confidently identified (FDR < 1%) peak groups defined by an RT-normalized assay library and extracted from a human, yeast, or mouse SWATH-MS run are compared between conditions where the assay library and SWATH-MS normalization were performed with synthetic iRT (SiRT) or common internal iRT (CiRT) normalization peptides. Data are presented as box and whisker plots, with the middle quartiles surrounding the median for the entire assay library represented by the box, whiskers showing the 95% data range and the upper and lower 2.5% of all values plotted as individual data points. B, Correlation between the intensity of a given peptide as determined by the SiRT or CiRT normalization procedure. Each dot represents the summed intensity of all transitions extracted for a given peptide peak group from the same raw file, with the only difference being the method of RT normalization. C, Distribution of matching and mis-matching peptide peak group intensities for the human (left) and yeast (middle) and mouse (right) derived samples. Pie charts depict overall distribution peptides with matching or mismatching intensity values between CiRT and SiRT aligned data sets. Horizontal bars show distribution of peptides among different categories explaining mismatched intensity values.
As an additional and important proof of principle, we tested whether the CiRT method could be used to normalize RT across multiple files from diverse sources (e.g. different labs, different fractionation methods, etc), ultimately yielding a merged library capable of improved and accurate SWATH-MS quantification. We analyzed the same human derived SWATH file described above against a larger, expanded library of 18,521 proteotypic peptides representing 4063 proteins (compared with 9107 peptides from 2195 proteins in the smaller Human library derived from a single, unfractionated cell lysate DDA-MS file). Using the expanded library, we identified an additional 3,899 unique peptides and 774 proteins from the same human SWATH-MS sample originally queried against the smaller library of human peptides (Fig. 4).
Fig. 4.
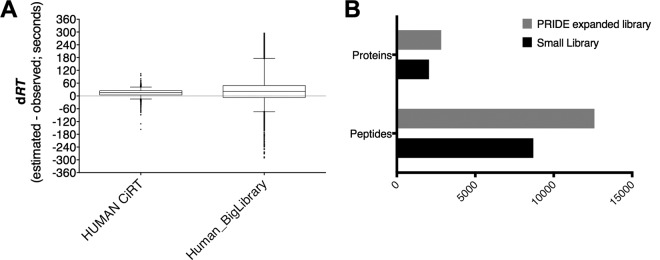
Expansion of peptide assay library using archived data sets improves SWATH-MS quantitative depth. A, The precision of RT prediction within the same SWATH-MS data file was slightly lower but still comparably accurate between a larger peptide assay library comprised of human peptide data from three diverse sources and the original, small human library built from the same lysate digest as the SWATH-MS data being analyzed. B, Use of the expanded library increased the number of nonredundant peptide sequences and corresponding proteins quantified from the same DIA-MS data file relative to the smaller library.
Comparison of the Quantitative Accuracy of CiRT Versus iRT Normalization
The SWATH-MS technique is a method of label free quantification. The RT alignment between assay library and SWATH-MS data file is a critical component of peptide identification and subsequent quantification, and therefore inaccurate alignments may negatively influence quantitative accuracy. Given the observed differences in ΔRT between SiRT and CiRT peptide sets, we examined whether these differences were sufficiently large to influence the identification and subsequent quantification of peptides by SWATH-MS. To do this, we compared the peak group intensities of each peptide identified with < 1% FDR between the different RT normalization sets (Fig. 3B). For the human lysate, 530 peptides (6.9%) had different intensities between the two RT normalization sets. Among these, 328 were detected with > 99% confidence of identification exclusively in the CiRT normalized data file, and 20 found only in the SiRT data file. An additional 208 peptides with different peak group XIC intensity values were detected in both CiRT and iRT normalized data files, but the actual observed RT between these peptides were different, suggesting erroneous or ambiguous peak group identification. Examination of the ΔRTs for these peptides indicated a roughly equivalent likelihood that the SiRT or CiRT peptide set more accurately predicted RT. Finally, the remaining 74 peptides (0.8%) had equal values for observed RT, and the difference in intensity appears to be because of the peak location at the left or right border of the RT window used for peak extraction, which can result in slight differences in the extracted intensity for some but not all transition group peaks. The quantitative discrepancies were similarly distributed for the yeast sample, with 2.6% of all peptides found to have different intensity between the CiRT and SiRT alignments. These discrepancies were attributed to a subset of peptides in both standards: 63 peptides identified only in the CiRT aligned data and 8 peptides only identified in SiRT aligned data, with 51 (0.8%) of peptides identified in both alignments but with different observed retention times and 46 (0.7%) detected in both alignments but with the same retention time. For the mouse sample, a higher overall percentage of discrepancies was found (8.1%), with the majority of error attributed to the observation of 365 peptides only detected in the SiRT-aligned and 165 peptides detected only in the CiRT_SW aligned data.
Taken together, these data indicate that slight differences in the accuracy of RT prediction by a given RT alignment set does not appear to substantially bias the overall distribution of quantitative errors in the SWATH-MS data set. This observation can be explained in part by the use of multiple orthogonal peak group scoring parameters in the OpenSWATH analysis software, of which RT is only one component. Finally, it is important to note that most discrepancies occur in the lower intensity range (e.g. for less confident peptide identifications), and that the percentage of quantitative differences between peptides in the SiRT or CiRT-aligned data were 10% or fewer of all peptides quantified in each sample.
To further ensure that the CiRT-SW normalization achieves comparable quantitative sensitivity and accuracy relative to SiRT normalization, we compared quantitative performance of the two RT alignment methods on a “gold standard” data set composed of 10 twofold dilution steps of 340 chemically synthesized stable isotope standard peptides, beginning at a concentration of 30 fmol/μl and ending at 0.058 fmol/μl (described in detail in Methods). The median and distribution of normalized log2 (nLog2) intensities for the peptides observed at each dilution step are shown in Fig. 5A. A comparison of the slope m in the linear relationship describing observed and expected mean nLog2-intensity (Fig. 5B) showed no significant differences in quantitative accuracy between the RT alignment by SiRT or CiRT, as indicated by overlap in the S.D. of the slopes (m = 0.889 ± 0.018 for SiRT and m = 0.865 ± .019 for CiRT in Yeast lysate, m = 1.031 ± .025 for SiRT and m = 1.037 ± .030 for CiRT in human cell lysate). The mean error of quantification (Fig. 5C) indicated that although there were noticeable matrix effects as well as an expected decrease in accuracy and increase in variability at the lowest concentrations, the error in CiRT and SiRT normalized data at each dilution step and in either matrix were not significantly different. These data indicate that the chosen RT normalization approach (CiRT versus SiRT) used did not significantly contribute to any errors in quantification.
Fig. 5.

Use of internal retention time prediction peptides does not alter the accuracy of peptide quantitation by SWATH-MS. A, Synthetic, heavy peptides were spiked into cell lysate from either human-derived (upper panel) or yeast (lower panel) cells at progressively twofold decreasing concentrations from 30 femtomoles to .058 femtomoles on column. Assay libraries, normalized to iRT, for the 340 synthetic peptides were used to extract peak groups from SWATH files, and the intensity of a given peak group was normalized to its observed intensity at 30 fmol and set to log scale with a base of 2. Perfectly accurate quantification would therefore represent a single unit increase between dilution steps. Quantification subsequent to synthetic iRT prediction is shown on the left and the CiRT shown on the right. B, Comparison of the linear estimate comparing the mean observed normalized Log2 Intensities plotted against that which would be expected based on the actual concentration of heavy peptide spiked into a given sample. Each data point represents the mean ± standard deviation (S.D.) of nLog2 intensity across all peptides observed at a given concentration, C, A plot of the mean absolute error calculated for each dilution step of the 10 × 2-fold dilution series (described in methods). No peptides were detected in the lowest concentration, and as such there are 8 dilution steps used to calculate quantification error see methods for the equation used for error estimation). Values are presented as mean ± S.D. at each dilution step, for each RT normalization method (CiRT versus SiRT) in Yeast and Human samples.
Performance and Robustness of the Automated RT Normalization Algorithms
Although the CiRT lists compiled and tested as described above were chosen to be generalizable to multiple species and experimental designs so that many experimenters can immediately implement them for internal RT calibration of DIA-MS files, these particular peptides will not be found in every possible experimental sample. For experimental preparations in which the CiRT_SW peptides are not present, researchers can use other candidate internal reference peptides from their library to predict RT for peptides assayed in SWATH-MS files. This process is not trivial, as we have mentioned above, endogenous peptides can exhibit more variation in signal quality among a given set of reference RT standards relative to synthetic peptides, and in our case we needed to hand curate the CiRT_SW peptides to identify the best candidates for SWATH-MS normalization. We have therefore amended the workflow in order to automate the selection of endogenous peptide standards for SWATH-MS normalization from a large list of candidate standards. This modified approach has now been implemented as a new filter function in the OpenSwathRTNormalizer tool (see Methods).
Using a benchmark data set (described in Methods), we evaluated the performance of the new filter function designed to remove low-scoring signals and select the best reference peptides for RT prediction using a combination of three outlier removal algorithms: jackknife, largest residual removal and Random Sample Consensus (RANSAC)(20). We found that the filter function confidently removes low-scoring signals (originating from decoys) at least up to a tenfold excess of false signals. The best-performing algorithm (iterative Jackknife with removal of low-quality peaks) was able to produce robust regression models even in the presence of a very large number of false signals. Specifically, the measured error of the linear model increased only minimally from 3.76 to either 3.81 or 3.86 for the two different methods of adding false signal (Fig. 5A, red and green curves) whereas a substantially larger increase in the model error was observed when not removing low-quality peaks (Fig. 6A, cyan and magenta curves). These results suggest that the new method, implemented in the publicly available development version of the OpenSWATH software, is robust enough to deal with nonoptimal lists of potential CiRT candidates provided by an experimentor and filter a substantial amount of noise in the data to select the best reference peptides for RT normalization.
Fig. 6.
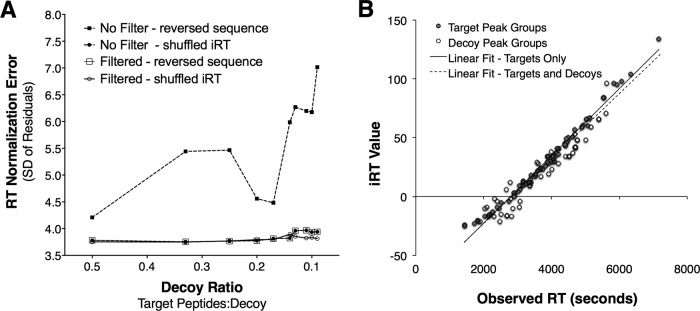
Robustness of the computed linear alignment in the presence of noise signal when using a jackknife approach for outlier removal. A, Increasing the number of noise signals from 2× to 10× impacts the error of the linear model (measured as standard deviation of the residuals of the correct signals) only substantially if peak quality is not taken into account. If a peak quality threshold is used and low-quality peaks are discarded (open symbols), the error of the linear model is almost constant even if a large number of false signals are present. B, Example robust regression in the presence of a 10-fold excess of noise peaks using the jackknife approach to remove outlier signal. In red are all known correct datapoints and in black are noise datapoints that by chance correlate with the correct ones and thus pull through the filtering. The dashed black regression line was obtained from all shown datapoints (R2 = 0.95), the solid black regression line was the regression model obtained from only the known correct datapoints (R2 = 0.98). The measured error of the linear model is 3.86 (whereas the error when only using the known correct datapoints is 3.76) and while achieving an accuracy of 94.9%.
Upon investigation of individual regression models, even in the presence of a large amount of false signals (tenfold excess of decoys relative to targets), the new algorithm was able to retrieve almost all of the correct signals and produce a linear model of high quality (R2 > 0.95) (see Fig. 6B). The automated selection of target signals from noise was made with an accuracy of 94.9%, determined from the observation of 62 (5%) decoy peak groups that were incorrectly retained in the linear regression, out of a total of 1364 peak groups initially entered (black dots in Fig. 6B), with 1221 decoy peak groups correctly discarded by the filter function. Of the 81 “known true” target signals from the input data, 74 peak groups were correctly retained and used for the regression, representing a 91.3% recall rate for this data set (red dots in Fig. 6B). As noted above, the regression models obtained from the noisy data were extremely similar to the ones obtained using only the 81 “known true” target data points without the decoys added (black and dotted red lines in Fig. 6B). Thus, we show that the enhanced filter function in the OpenSwathRTNormalizer tool can automatically select correct candidate peptides for accurate RT alignment from a large input list containing up to 90% false signals. To test the practical utility of the novel algorithm, we repeated the OpenSWATH analysis of each yeast, human, and mouse file originally aligned by the CiRT_SW peptides, and instead entered the entire set of 113 CiRT peptides (CiRT_ALL), allowing the -best_peptides function to automatically select ideal candidates for alignment yielded slightly less precise alignment of RT, demonstrated by a slightly higher dRT value distribution relative to that of the CiRT_SW, however the quantitative outcome was essentially comparable to alignments performed using the manually selected peptides (supplemental Fig. S2).
DISCUSSION
The CiRT peptides identified in this manuscript provide a method to normalize RT for peptide identification and quantification in SWATH data analysis across experiments performed on most eukaryotic species. The endogenous CiRT peptides described here provide an alternative to the use of the spiked-in SiRT standard peptides in every single sample of a SWATH experiment. We show here that the CiRT peptides are detected in human, mouse, and yeast protein samples, are therefore represent tryptic peptide sequences that are highly conserved across eukaryotic species and generalizable to many common experimental preparations. We have tested the CiRT method and shown that these peptides effectively align the raw RTs of a peptide transition library to the normalized iRT space, and further that RT prediction and normalization by the CiRT peptides results in peptide identification and quantification that is comparably accurate to that achieved with SiRT peptides.
We based the CiRT approach on the original concept introduced by Escher et al. (1). In the design of their 11 synthetic peptides for RT alignment, they applied specific criteria to optimize the performance of their peptides: (1) intensity of the ionized peptide; (2) absence of amino acids prone to modification; (3) nonnaturally occurring peptide sequences; and (4) distribution of peptide RT across the chromatogram. The selection of internal CiRT peptides differed slightly from that of Escher et al. in that the peptides were selected first-and-foremost based on their frequently occurring and evolutionarily conserved sequence of amino acids across many eukaryotic samples, with the additional caveat that the peptides are readily observed by MS analysis of our own human and yeast samples. We could have potentially expanded the list of CiRT candidates by cross referencing the SWISS-PROT conserved peptide list with other MS data repositories such as NIST or PRIDE, however we were constrained at the time of the study by the requirement for both (1) a TripleTOF generated DDA spectrum for each peptide in order to compose a transition group library and (2) an iRT value for each peptide, preferably iRTs from multiple observations in different sample matrices (e.g. human, mouse, yeast lysates). As the volume of appropriate and publically accessible data sets increase we believe that a list of CiRT reference peptides for iRT alignment can be expanded by our own group as well as the greater MS community.
The CiRT peptides are endogenous to the samples, and as such, in assigning iRT values to the CiRT candidates we allowed for two common modifications that occur very typically MS samples preparations: oxidization of methionine and carbamidomethylation of cysteines. Allowing these commonly occurring modification states among the CiRT peptides makes them more consistent with the likely state of the peptide in an actual sample. This criteria differs from that necessary for the SiRTs, because the assigned iRT for SiRT peptides is based on their unmodified state, and they are exogenously added to sample, so any modification to them would result in altered RT properties and render them useless or even detrimental for iRT alignment. With CiRTs, the iRT value corresponding to the exact modified (or unmodified, as was commonly the case) sequence was determined from experimental data based on the most commonly observed state of the peptide in actual biological samples. For both the manual selection of CiRT_SW peptides and in the refined algorithm we have applied similar criteria as Escher et al., specifically that the CiRT peptides are well distributed across the chromatographic range, and that their extracted peak groups exhibit high intensities so they can be easily distinguished from noise and false positives.
The CiRT endogenous peptide sets can be used synergistically with exogenous SiRT standards or as an alternative RT standard in many eukaryotic samples. The iRT assigned to the CiRT peptides in the current manuscript was calculated as an average of iRT assigned from the observation of the peptide in our experimental preparations of a human, yeast, and mouse sample. Although it is unexpected that iRTs for a given peptide will vary substantially, it may be prudent to acquire one or more test samples spiked with the SiRT-peptides and determine experiment-specific iRT values for the endogenous CiRT peptides. Once calibrated in this way, within a given experimental design internal CiRTs can be used rather than spiking SiRT peptides into every single sample as a cost-efficient strategy for RT normalization.
In addition, this approach will allow access to archived LC MS data sets that were generated without standards or with different internal standards, as we have demonstrated with the inclusion of two data sets downloaded from the PRIDE data repository. These can be normalized to iRT space and used for building of peptide assay libraries. This approach is applicable even when the candidate peptides are spread out between multiple fractions, granted the list of candidates is sufficiently large to result in even spread of RT normalization peptides across all fractions. We demonstrated this concept with the alignment of RT across two separate fractionated data sets acquired from different laboratories and combined with our own data file from human cell lysate to generate a merged and expanded human library, which notably enhanced peptide detection and quantification by SWATH-MS analysis.
Finally, we have ensured the generalizability of the method for workflows in which these specific CiRT peptides are not detected (e.g. nontryptic digestions, PTM enriched samples, affinity purifications) by refining the “OpenSwathRTNormalizer” algorithm for automated selection of internal RT alignment reference peptides. In our example, the refined algorithm was able to normalize RT using a list where as few as 10% of internal RT reference peptides candidates were actually present in the sample. Therefore, when the CiRT list described here does not apply, alternative lists of internal RT alignment candidates can be generated by the user to be appropriate for their experimental preparation (e.g. immunoprecipitations, PTM enrichments) or organism. The amended “OpenSwathRTNormalizer” algorithm with the “best_peptides” feature will select a refined subset of the user-defined RT alignment candidates from their input list for RT normalization in subsequent SWATH experiments.
The CiRT peptide lists are provided in the appropriate formats for insertion into the OpenSWATH conversion and SWATH-MS iRT-normalization programs, and downloads of these formatted lists are provided in the supplemental material, as are brief instructions for their insertion into the workflow (supplemental Table S4). The algorithms for internal iRT selection from large candidate lists for SWATH-MS iRT alignment are also now implemented in new version of OpenSWATH (www.openswath.org), which will allow users to generate custom RT normalization sets ideally applicable to virtually any experiment and workflow.
Supplementary Material
Acknowledgments
We thank L. Gillet for providing and preparing the yeast samples used in the current manuscript, and L. Gillet, P. Navarro who worked along with the co-authors of the current manuscript H. Rost & G. Rosenberger to prepare and acquire the SWATH Gold Standard data set, as described in (7).
Footnotes
Author contributions: S.J.P., H.R., G.R., B.C.C., K.R., J.E.V., and R.A. designed research; S.J.P., H.R., G.R., and L.M. performed research; S.J.P., H.R., G.R., L.M., D.A., and K.R. contributed new reagents or analytic tools; S.J.P., H.R., G.R., D.A., V.V., and R.A. analyzed data; S.J.P., H.R., G.R., B.C.C., J.E.V., and R.A. wrote the paper.
* This work was supported in part by an European Molecular Biology Organization short term fellowship ASTF 456-2013 awarded to S.J.P. H.L.R. was funded by ETH Zurich (ETH-30 11-2). G.R. was funded by the Swiss Federal Commission for Technology and Innovation CTI (13539.1 PFFLI-LS). K.R. acknowledges support from Canadian Institute of Health Research, FRN: MFE123700.The project was also supported in part by the SNSF (Grant# 3100A0-688 107679), the European Research Council (Grant# ERC-2008-AdG 233226) to R.A, the US NHLBI Proteomics Contract HHSN268201000032C and R01 (R01 AR41135–18) to JVE and salary support by the National Marfan Foundation Victor E McKusik Post-Doctoral Fellowship to S.J.P.
 This article contains supplemental Figs. S1 and S2 and Tables S1 to S4.
This article contains supplemental Figs. S1 and S2 and Tables S1 to S4.
1 The abbreviations used are:
- LC
- Liquid Chromatography
- RT
- Retention Time
- SiRT
- Synthetic indexed Retention Time standard peptides
- CiRT
- Common internal Retention Time standard peptides
- LC
- Liquid Chromatography
- DDA
- Data-Dependent Acquisition
- DIA
- Data-Independent Acquisition
- iRT-C18
- Defined, unitless space described in Escher et al (1)
- TPP
- Trans Proteomic Pipeline
- dRT
- Difference between observed and predicted retention time.
REFERENCES
- 1. Escher C., Reiter L., MacLean B., Ossola R., Herzog F., Chilton J., MacCoss M. J., Rinner O. (2012) Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 12, 1111–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Klammer A. A., Yi X., MacCoss M. J., Noble W. S. (2007) Improving tandem mass spectrum identification using peptide retention time prediction across diverse chromatography conditions. Anal. Chem. 79, 6111–6118 [DOI] [PubMed] [Google Scholar]
- 3. Pfeifer N., Leinenbach A., Huber C. G., Kohlbacher O. (2009) Improving peptide identification in proteome analysis by a two-dimensional retention time filtering approach. J. Proteome Res. 8, 4109–4115 [DOI] [PubMed] [Google Scholar]
- 4. Gallien S., Peterman S., Kiyonami R., Souady J., Duriez E., Schoen A., Domon B. (2012) Highly multiplexed targeted proteomics using precise control of peptide retention time. Proteomics 12, 1122–1133 [DOI] [PubMed] [Google Scholar]
- 5. Bateman N. W., Goulding S. P., Shulman N. J., Gadok A. K., Szumlinski K. K., MacCoss M. J., Wu C. C. (2014) Maximizing peptide identification events in proteomic workflows using data-dependent acquisition (DDA). Mol. Cell. Proteomics 13, 329–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gillet L. C., Navarro P., Tate S., Rost H., Selevsek N., Reiter L., Bonner R., Aebersold R. (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111 016717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rost H. L., Rosenberger G., Navarro P., Gillet L., Miladinovic S. M., Schubert O. T., Wolski W., Collins B. C., Malmstrom J., Malmstrom L., Aebersold R. (2014) OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 32, 219–223 [DOI] [PubMed] [Google Scholar]
- 8. Rost H., Malmstrom L., Aebersold R. (2012) A computational tool to detect and avoid redundancy in selected reaction monitoring. Mol. Cell. Proteomics 11, 540–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karlsson C., Malmstrom L., Aebersold R., Malmstrom J. (2012) Proteome-wide selected reaction monitoring assays for the human pathogen Streptococcus pyogenes. Nat. Commun. 3, 1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lam H., Aebersold R. (2011) Building and searching tandem mass (MS/MS) spectral libraries for peptide identification in proteomics. Methods 54, 424–431 [DOI] [PubMed] [Google Scholar]
- 11. Picotti P., Clement-Ziza M., Lam H., Campbell D. S., Schmidt A., Deutsch E. W., Rost H., Sun Z., Rinner O., Reiter L., Shen Q., Michaelson J. J., Frei A., Alberti S., Kusebauch U., Wollscheid B., Moritz R. L., Beyer A., Aebersold R. (2013) A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature 494, 266–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schubert O. T., Mouritsen J., Ludwig C., Rost H. L., Rosenberger G., Arthur P. K., Claassen M., Campbell D. S., Sun Z., Farrah T., Gengenbacher M., Maiolica A., Kaufmann S. H., Moritz R. L., Aebersold R. (2013) The Mtb proteome library: a resource of assays to quantify the complete proteome of Mycobacterium tuberculosis. Cell Host Microbe 13, 602–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Picotti P., Aebersold R. (2012) Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Meth. 9, 555–566 [DOI] [PubMed] [Google Scholar]
- 14. Huttenhain R., Soste M., Selevsek N., Rost H., Sethi A., Carapito C., Farrah T., Deutsch E. W., Kusebauch U., Moritz R. L., Nimeus-Malmstrom E., Rinner O., Aebersold R. (2012) Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci. Transl. Med. 4, 142ra194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collins B. C., Gillet L. C., Rosenberger G., Rost H. L., Vichalkovski A., Gstaiger M., Aebersold R. (2013) Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14-3-3 system. Nat. Meth. 10, 1246–1253 [DOI] [PubMed] [Google Scholar]
- 16. Liu Y., Huttenhain R., Surinova S., Gillet L. C., Mouritsen J., Brunner R., Navarro P., Aebersold R. (2013) Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics 13, 1247–1256 [DOI] [PubMed] [Google Scholar]
- 17. Rosenberger G., Koh C. C., Guo T., Röst H. L., Kouvonen P., Collins B. C., Heusel M., Liu Y., Caron E., Vichalkovski A., Faini M., Schubert O. T., Faridi P., Ebhardt H. A., Matondo M., Lam H., Bader S. L., Campbell D. S., Deutsch E. W., Moritz R. L., Tate S., Aebersold R. (2014) A repository of assays to quantify 10,000 human proteins by SWATH-MS. Scientific Data. 1, Article no. 140031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Segura V., Medina-Aunon J. A., Mora M. I., et al. (2014) Surfing transcriptomic landscapes. A step beyond the annotation of chromosome 16 proteome. J. Proteome Res. 13, 158–172 [DOI] [PubMed] [Google Scholar]
- 19. Teleman J., Roest H. l., Rosenberger G., Schmitt U., Malstroem L., Malstroem J., Levander U. (2015) DIANA - algorithmic improvements for analysis of complex peptide sample data-independent acquisition MS data. Bioinformatics 31, 555–562 [DOI] [PubMed] [Google Scholar]
- 20. Fischler M. A., Bolles R. C. (1981) Random sample consensus: a paradigm for model fitting with applications to image analysis and automated cartography. Commun. ACM 24, 381–395 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.