

Abstract
Massively parallel sequencing technologies have enabled characterization of genomic alterations across multiple tumor types. Efforts have focused on identifying driver mutations because they represent potential targets for therapy. However, because of the presence of driver and passenger mutations, it is often challenging to assign the clinical relevance of specific mutations observed in patients. Currently, there are multiple databases and tools that provide in silico assessment for potential drivers; however, there is no comprehensive resource for mutations with functional characterization. Therefore, we created an expert-curated database of potentially actionable driver mutations for molecular pathologists to facilitate annotation of cancer genomic testing. We reviewed scientific literature to identify variants that have been functionally characterized in vitro or in vivo as driver mutations. We obtained the chromosome location and all possible nucleotide positions for each amino acid change and uploaded them to the Cancer Driver Log (CanDL) database with associated literature reference indicating functional driver evidence. In addition to a simple interface, the database allows users to download all or selected genes as a comma-separated values file for incorporation into their own analysis pipeline. Furthermore, the database includes a mechanism for third-party contributions to support updates for novel driver mutations. Overall, this freely available database will facilitate rapid annotation of cancer genomic testing in molecular pathology laboratories for mutations.
Recent advances in DNA sequencing technology have enabled real-time cancer genomic testing of patients to facilitate matching genomic alterations to targeted therapies.1 Clinically relevant molecular subsets of cancer have been identified through initiatives such as The Cancer Genome Atlas and International Cancer Gene Consortium, which have helped characterize the landscape of cancer genomes and provided information on mutational patterns. Sequencing data have revealed that mutational rates vary within and across cancers, with lung and melanoma harboring a higher rate of somatic mutations due to tobacco and UV exposures, respectively.2,3 However, only a few represent driver mutations that provide a selective growth advantage, whereas most are passenger mutations. Therefore, it is often challenging to identify mutations that are clinically relevant drivers or potentially actionable through targeted therapies.
Although there are in silico approaches to predict potential drivers or alterations that are actionable, for accurate assessment, functional characterization of the mutation with alteration at the level of protein is essential. For industry or researchers, functional characterization is mandatory before pursuing drug development and testing a treatment hypothesis in clinical trials. To support the development of clinical trials for novel targeted therapies, molecular pathologists and genetics laboratory directors have implemented novel next-generation sequencing assays to test for hundreds of genes with appropriate analytic validity.4–9 However, for clinical interpretation, laboratory directors face hundreds of mutations and must rapidly annotate drivers versus passengers for their potential functional and clinical significance in their reports. There is currently no comprehensive pan-cancer database of driver mutations defined by genomic position linked with primary source literature. On the basis of our multidisciplinary experiences in cancer biology, medical oncology, and molecular diagnostics development, we have curated a database of mutations, the Cancer Driver Log (CanDL), that has mutations with proven functional characterization or that have been targeted clinically or pre-clinically by either existing therapies or investigational agents.
Materials and Methods
Mutation Categorization
We reviewed the list of genes from the Sanger Cancer Gene Census (http://cancer.sanger.ac.uk/cancergenome/projects/census; all URLs cited were last accessed on April 7, 2015) to categorize mutations that have been putatively implicated in cancer biology.10 From this selection, we curated mutations in oncogenes that have proven functional characterization and are associated with an increased or decreased response (resistance) to an approved therapy or investigational agent clinically or preclinically after manual review of the scientific literature.
We defined requirements to qualify as a driver mutation to include in vitro or in vivo experimentation (Table 1). For each gene, we obtained the following information: amino acid variant, cancer type, whether targetable clinically or preclinically, and the PubMed reference identification indicating functional evidence as listed in Table 1.
Table 1.
Rules for Mutational Entry into the Cancer Driver Log
| Rule No. | Details |
|---|---|
| 1 | Targeted clinically or in vitro with approved or investigational agent(s) |
| 2 | Associated with increased sensitivity to a drug in vitro or in vivo |
| 3 | Associated with resistance to a drug in vitro or in vivo |
| 4 | Ectopic expression induces transformation in vitro (eg, BAF3, NIH 3T3 cells) or in vivo (eg, animal models) |
| 5 | Inhibition of the mutant with a specific shRNA or an inhibitor decreases cell growth |
Identification of the Genomic Position of Genes and Domain Information
To obtain genomic positions, we used BackLocate (https://code.google.com/p/variationtoolkit/wiki/BackLocate). We entered genes and their associated amino acid changes and mapped them to all possible transcripts associated with that gene. Transcripts that failed to match the canonical amino acid at the specified site were discarded. To evaluate the performance of BackLocate, we manually inspected positions with the Integrative Genomics Viewer.11
We used ANNOVAR to identify the chromosome location, exons, nucleotide positions, and amino acid change.12 Positions not annotated by ANNOVAR were manually confirmed using the UCSC reference genes database. Variant alleles and external links were corroborated using custom Python scripts and the Biopython Entrez module (http://biopython.org/DIST/docs/api/Bio.Entrez-module.html). The final output was uploaded to the website (https://candl.osu.edu). To obtain domain information for proteins in the CanDL database, we used the BioMart tool of Ensembl release version 68 (http://useast.ensembl.org/biomart/martview) to obtain InterPro protein identification. The InterPro identification numbers were then used to extract the domain information using the InterPro BioMart-0.7 tool (http://www.ebi.ac.uk/interpro/biomart/martview), and custom Perl scripts were used to match protein domains in CanDL with InterPro.
In the future, we plan to update the database by using a semiautomated pipeline that combines data from genomic and protein databases using bioinformatics tools. Driver mutations identified from the literature are compiled into an input list for the pipeline. The pipeline uses the BackLocate tool from the Variation Toolkit to map out the genomic positions for protein alteration. These data would then be matched to the HUGO Gene Nomenclature Committee database (http://www.genenames.org), the Ensembl BioMart [Ensembl Genes 68 database, Homo sapiens (GRCh37.p8) data set], and the InterPro BioMart (InterPro Entry Annotation data set) to create a list of potential matches. Duplicate and mismatched results are automatically filtered and discarded. No fewer than two experts would then manually corroborate these entries before uploading into the CanDL database. Although partial automation helps to reduce the time for analyzing large data sets, no updates to the CanDL database will be performed without manual expert review to maintain data integrity and validity. We would aggregate new entries and plan to update the database on a quarterly basis.
Results
We have manually curated a database of driver mutations in cancers that are potentially actionable to support annotation in clinical genomic testing laboratories by molecular pathologists, laboratory directors, and bioinformaticians. Mutations in 56 genes with 330 distinct variants with 160 unique matching references across multiple cancers were identified for entry into CanDL. Entries can be searched by gene(s), searched by amino acid variants, or downloaded for custom analyses. Although the default output includes normal amino acid, peptide position, variant amino acid, cancer type, and the reference for a given gene, users have the option to customize output to include additional information (eg, exon, mutation coding DNA sequence, transcript). Importantly, mutations are defined by all potential genomic positions and nucleotide changes that may yield a given amino acid substitution that has been proven to be a functional driver. In addition, the references associated with matching alterations are categorized into four levels based on the strength of evidence of actionability (Table 2). Pathologists and laboratory directors seeking to annotate reports can either upload candidate mutations or download the entire log from the website.
Table 2.
Levels of Evidence for Mutations in the Cancer Driver Log
| Tier | Alteration details |
|---|---|
| 1 | Alteration has matching FDA-approved or NCCN-recommended therapy. |
| 2 | Alteration has matching therapy based on evidence from clinical trials, case reports, or exceptional responders. |
| 3 | Alteration predicts for response or resistance to therapy based on evidence from pr-clinical data (in vitro or in vivo models). |
| 4 | Alteration is a putative oncogenic driver based on functional activation of a pathway. |
FDA, Food and Drug Administration; NCCN, National Comprehensive Cancer Network.
We assessed the distribution and prevalence of mutations listed in CanDL across samples in cBioPortal, a database of large-scale cancer genomics data sets (Figure 1).13 Of the 56 genes in CanDL, 39 genes had matching alterations in cBioPortal. The heat map displays the number of samples in cBioPortal that had a corresponding mutation in CanDL distributed across a given gene (top 10 listed) and sample type. The most commonly observed mutations involved BRAF in thyroid and melanoma followed by KRAS in pancreas. However, because cBioPortal consists of large-scale data sets for the 50 most common cancers, the prevalence of curated mutations in CanDL is unknown in other less characterized cancer types.
Figure 1.

Prevalence of the Cancer Drive Log (CanDL) mutations in cBioPortal. The color shade in each box corresponds to the number of samples in cBioPortal that had at least one CanDL mutation for a given gene and tumor type. The gradient scale is on the right. Genes were sorted in descending order, and the top 10 are represented on the y axis. The x axis represents tumor type.
Next, we characterized the distribution of protein domains involved in this curated data set of driver mutations. Using custom Perl scripts, we matched the proteins in the CanDL database with their domain names and coordinates using Ensembl and InterPro. We obtained domain information for 50 proteins in the CanDL database and plotted the frequency of mutations across different domains as defined by InterPro (Figure 2). Nearly 50% percent of the mutations were observed in protein kinase domains. Notably, the rest were made up of alterations distributed across other domains underscoring the contribution of nonkinase mutations as drivers.
Figure 2.
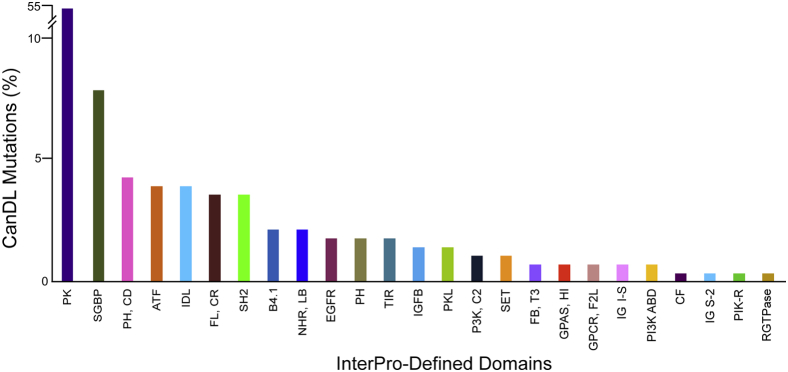
Distribution of the Cancer Driver Log (CanDL) mutations across protein domains. The distribution of domains for mutations that are listed in CanDL, as defined by InterPro database, are shown. The y axis (// denotes break) represents the percentage of genes. Protein kinase (PK) domain mutations were the most common followed by small GTP-binding (SGBP) domain mutations. ATF, Armadillo-type fold; B4.1, band 4.1 domain; CF, coagulation factor 5/8 C-terminal type domain; EGFR, epidermal growth factor receptor, L domain; FB, T3, fibronectin type III; FL, CR, Furin-like cysteine-rich domain; GPAS, HI, G-protein α-subunit, helical insertion; GPCR, G protein coupled receptor; F2L, family 2-like; IDL, isopropylmalate dehydrogenase–like domain; IG I-S, immunoglobulin I set; IG S-2, immunoglobulin subtype 2; IGFB, insulin-like growth factor binding protein, N-terminal; NHR, LB, nuclear hormone receptor, ligand-binding; P3K, C2, phosphatidylinositol 3-kinase, C2 domain; PH, pleckstrin homology domain; PH, CD, phosphatidylinositol 3-/4-kinase, catalytic domain; PI3K ABD, phosphatidylinositol 3-kinase adaptor-binding domain; PIK-R, PIK-related kinase; PK, protein kinase domain; PKL, protein kinase–like domain; RGTPase, Rho GTPase activation protein; SET, SET domain; SGBP, small GTP-binding protein domain; SH2, SH2 domain; TIR, Toll/interleukin-1 receptor homology domain.
Discussion
Advances in next-generation sequencing technologies have enabled widespread adoption of analytically validated cancer genomic testing in molecular pathology.4–9 Although the ability to detect genomic alterations across hundreds of genes has improved throughput and decreased cost, in clinical practice it is a challenge to distinguish and annotate driver from passenger mutations based on functional characterization. As molecular testing evolves to pan-cancer testing across multiple tumor histologic types, it will become increasingly difficult to have expertise across different diseases and pathways in which mutations may occur. Currently, laboratory directors must manually research individual mutations to determine the status as a driver or passenger and facilitate clinical interpretation. Therefore, the role of the CanDL database is to provide a laboratory director, molecular pathologist, or bioinformatician with a simple approach for rapid annotation based on genomic positions for driver mutations with direct literature evidence of functional characterization.
There are several complementary databases or tools that facilitate annotation of germline variants, somatic mutation prevalence, gene-drug associations, linkage of selected hot spot mutations with drugs, and in silico prediction tools for potential driver function (Table 3). ClinVar identifies associations between germline alterations and disease phenotypes.14 cBioPortal and COSMIC provide a means for visualization and determination of frequency and spectrum of somatic mutations across publically available cancer genomics data.13,15 My Cancer Genome (mycancergenome.org) and MD Anderson Cancer Center personalized cancer therapy (https://pct.mdanderson.org) web resources deliver detailed, comprehensive summaries on selected driver mutations with therapeutic implications, including a list of potential clinical trials. The Drug-Gene Interaction Database16 and Therapeutic Targeted Database17 mine existing resources to generate known and potential gene target-drug associations. In addition, there are a few databases that incorporate in silico prediction tools to identify novel driver mutations in cancer. Cancer Driver Annotation predicts missense driver mutations in cancers based on a set of 96 structural, evolutionary, and gene features using functional prediction algorithms, such as SIFT (sorting intolerant from tolerant) and CHASM (cancer-specific high-throughput annotation of somatic mutations).18 DriverDB incorporates annotation databases and algorithms with sequencing data to help identify and visualize driver genes related to particular tumor type or mutation profile across a cancer type.19 The MutSigCV algorithm ranks mutations that occur more often than based on chance and background mutation processes.3 Together, all these resources can be used to investigate and provide a clinical interpretation for a given somatic mutation.
Table 3.
Complementary Tools for Evaluating Cancer Mutations and Therapy Associations
| Type | Database | URL | Function |
|---|---|---|---|
| Hot spot mutations with therapeutics | My Cancer Genome | http://www.mycancergenome.org | Provides list of selected cancer driver mutations with therapeutic implications, including available clinical trials |
| MD Anderson Cancer Center Personalized Cancer Therapy | https://pct.mdanderson.org | Tool for physicians and patients to assess potential therapy options based on specific tumor biomarkers | |
| Mutation prevalence databases | cBioPortal13 | http://www.cbioportal.org | Provides visualization, analysis, and download of cancer genome data sets |
| COSMIC15 | http://cancer.sanger.ac.uk/cancergenome/projects/cosmic | Store and display somatic mutation information relating to human cancers | |
| In silico prediction | DriverDB19 | http://driverdb.ym.edu.tw/DriverDB | Incorporates large-scale databases and algorithms to identify driver genes |
| Cancer Driver Annotation18 | http://bioinformatics.mdanderson.org/main/CanDrA | Predicts cancer driver missense mutations based on 96 structural, evolutionary, and gene features | |
| MutSigCV3 | http://www.broadinstitute.org/cancer/cga/mutsig | Identifies genes that were mutated more often than expected by chance based on background mutation data | |
| Drug-gene associations | Therapeutic Targeted Database17 | http://bidd.nus.edu.sg/group/cjttd | Provides information on known and explored targets across diseases with potential drugs |
| Drug-Gene Interaction Database16 | http://dgidb.genome.wustl.edu | Mines existing resources to generate known and potential drug-gene associations | |
| Germline | ClinVar14 | http://www.ncbi.nlm.nih.gov/clinvar | Aggregates information about sequence variation and its association with to human health |
Considering the large number of somatic mutations in cancers, annotation is a challenging task if performed ad hoc. CanDL allows users to download all the genes in the database or selected genes as a comma-separated values file for incorporation into their own pipeline for annotation. Users can also submit mutations to the website for ad hoc assessment. We acknowledge that there are some limitations in this first release of CanDL. We have focused on single-nucleotide substitutions in oncogenes and not on somatic fusions and copy number alterations that may also represent driver alterations. In addition, because mutations in tumor suppressors are typically deleterious, including frame shifts and truncations, the degree of functional characterization for each unique mutation is limited but can often instead be inferred. For now, tumor suppressors, such as BRCA1, NF1, and PTEN, will require individual assessment.
Compiling the CanDL database has revealed the bias associated with characterization of driver mutations in oncogenes. Because most cancer mutations are located in kinase domains (Figure 2), most curated genomic databases have focused on functional characterization of high-yield kinase mutations. In contrast, pan-cancer analyses of genomic databases have revealed insight into gain-of-function mutations in domains and regions that are not typically studied. For example, Grabiner et al20 used publically available cancer genomic databases to identify missense mutations in the MTOR gene with clusters of recurrent mutations across all cancer subtypes and found functional activation of MTOR through mutations in nonkinase domains. In addition, Carpten et al21 reported novel activating mutations in the pleckstrin homology domain of AKT1, encoding a serine-threonine protein kinase. Kin-Driver, a curated database of activating and inactivating mutations in protein kinases, exemplifies the kinase bias associated with known oncogenic drivers. Although fairly comprehensive and presented in an uncomplicated interface, it is limited to kinase domain mutations, and therefore driver mutations in nonkinase domains are missed (eg, pleckstrin homology domain mutation in AKT1).22
We anticipate an expanding collection of driver mutations based on study of exceptional responders to targeted therapies, application of functional genomic screens, novel in silico approaches, and pan-cancer data sets that will facilitate identification and functional characterization of additional noncanonical drivers. For this reason, we have enabled CanDL to accept user submissions of novel mutations and feedback on existing alterations to keep the database up to date and accurate. Users can submit novel mutations to the CanDL website (http://candl.osu.edu/contact) with information on the gene, amino acid variant, cancer type, and associated reference(s). User submissions would be independently corroborated by our team and uploaded to the database.
Conclusion
We have created a curated database of cancer drivers. CanDL is not intended to replace the other available resources. On the other hand, it should be viewed as complementary in our ongoing efforts to support annotation and clinical interpretation and thus identify those variants that are most relevant to patient care. A concerted effort among pathologists, oncologists, and cancer researchers is essential to further identify and translate novel targetable alterations and thus realize the promise of personalized cancer medicine.
Acknowledgments
We thank the Comprehensive Cancer Center, Arthur G. James Cancer Hospital, and Richard A. Solove Research Institute at The Ohio State University Wexner Medical Center and The Ohio Supercomputer Center for supporting this study. Special thanks to Brianna Hunstad and Darshna Bhatt for administrative support for the team.
S.D., J.W.R., and S.R. conceived the research study. S.D., E.Z., and J.D. curated and reviewed mutation data for the CanDL database. E.K., S.D., and J.M. developed the CanDL website. J.M., E.Z., and E.S. obtained and annotated genomic positions and domains. J.M. and E.Z. generated figures. S.D., E.S., J.D., J.W.R., and S.R. reviewed the information used to generate figures and tables. S.D. and S.R. wrote and edited the manuscript. S.R. supervised the research.
Footnotes
Supported by the American Society of Clinical Oncology Young Investigator Award (S.D.), a Pelotonia postdoctoral fellowship (E.S.), American Cancer Society grant MRSG-12-194-01-TBG (S.R.), Prostate Cancer Foundation grant, NHGRI UM1HG006508-01A1 (S.R.), Fore Cancer Research, and Pelotonia.
Disclosures: S.R. receives funding from Novartis and Ariad Pharmaceuticals for conduct of clinical trials. S.R. has immediate family members who own stock in Johnson and Johnson.
References
- 1.Garraway L.A. Genomics-driven oncology: framework for an emerging paradigm. J Clin Oncol. 2013;31:1806–1814. doi: 10.1200/JCO.2012.46.8934. [DOI] [PubMed] [Google Scholar]
- 2.Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frampton G.M., Fichtenholtz A., Otto G.A., Wang K., Downing S.R., He J. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luthra R., Patel K.P., Reddy N.G., Haghshenas V., Routbort M.J., Harmon M.A., Barkoh B.A., Kanagal-Shamanna R., Ravandi F., Cortes J.E., Kantarjian H.M., Medeiros L.J., Singh R.R. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica. 2014;99:465–473. doi: 10.3324/haematol.2013.093765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh R.R., Patel K.P., Routbort M.J., Reddy N.G., Barkoh B.A., Handal B., Kanagal-Shamanna R., Greaves W.O., Medeiros L.J., Aldape K.D., Luthra R. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn. 2013;15:607–622. doi: 10.1016/j.jmoldx.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Beadling C., Neff T.L., Heinrich M.C., Rhodes K., Thornton M., Leamon J., Andersen M., Corless C.L. Combining highly multiplexed PCR with semiconductor-based sequencing for rapid cancer genotyping. J Mol Diagn. 2013;15:171–176. doi: 10.1016/j.jmoldx.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 8.Pritchard C.C., Salipante S.J., Koehler K., Smith C., Scroggins S., Wood B., Wu D., Lee M.K., Dintzis S., Adey A., Liu Y., Eaton K.D., Martins R., Stricker K., Margolin K.A., Hoffman N., Churpek J.E., Tait J.F., King M.C., Walsh T. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16:56–67. doi: 10.1016/j.jmoldx.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cottrell C.E., Al-Kateb H., Bredemeyer A.J., Duncavage E.J., Spencer D.H., Abel H.J., Lockwood C.M., Hagemann I.S., O'Guin S.M., Burcea L.C., Sawyer C.S., Oschwald D.M., Stratman J.L., Sher D.A., Johnson M.R., Brown J.T., Cliften P.F., George B., McIntosh L.D., Shrivastava S., Nguyen T.T., Payton J.E., Watson M.A., Crosby S.D., Head R.D., Mitra R.D., Nagarajan R., Kulkarni S., Seibert K., Virgin HWt, Milbrandt J., Pfeifer J.D. Validation of a next-generation sequencing assay for clinical molecular oncology. J Mol Diagn. 2014;16:89–105. doi: 10.1016/j.jmoldx.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forbes S.A., Beare D., Gunasekaran P., Leung K., Bindal N., Boutselakis H., Ding M., Bamford S., Cole C., Ward S., Kok C.Y., Jia M., De T., Teague J.W., Stratton M.R., McDermott U., Campbell P.J. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2014;43 doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffith M., Griffith O.L., Coffman A.C., Weible J.V., McMichael J.F., Spies N.C., Koval J., Das I., Callaway M.B., Eldred J.M., Miller C.A., Subramanian J., Govindan R., Kumar R.D., Bose R., Ding L., Walker J.R., Larson D.E., Dooling D.J., Smith S.M., Ley T.J., Mardis E.R., Wilson R.K. DGIdb: mining the druggable genome. Nat Methods. 2013;10:1209–1210. doi: 10.1038/nmeth.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu F., Han B., Kumar P., Liu X., Ma X., Wei X., Huang L., Guo Y., Han L., Zheng C., Chen Y. Update of TTD: Therapeutic Target Database. Nucleic Acids Res. 2010;38:D787–D791. doi: 10.1093/nar/gkp1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mao Y., Chen H., Liang H., Meric-Bernstam F., Mills G.B., Chen K. CanDrA: cancer-specific driver missense mutation annotation with optimized features. PLoS One. 2013;8:e77945. doi: 10.1371/journal.pone.0077945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng W.C., Chung I.F., Chen C.Y., Sun H.J., Fen J.J., Tang W.C., Chang T.Y., Wong T.T., Wang H.W. DriverDB: an exome sequencing database for cancer driver gene identification. Nucleic Acids Res. 2014;42:D1048–D1054. doi: 10.1093/nar/gkt1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grabiner B.C., Nardi V., Birsoy K., Possemato R., Shen K., Sinha S., Jordan A., Beck A.H., Sabatini D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014;4:554–563. doi: 10.1158/2159-8290.CD-13-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carpten J.D., Faber A.L., Horn C., Donoho G.P., Briggs S.L., Robbins C.M., Hostetter G., Boguslawski S., Moses T.Y., Savage S., Uhlik M., Lin A., Du J., Qian Y.W., Zeckner D.J., Tucker-Kellogg G., Touchman J., Patel K., Mousses S., Bittner M., Schevitz R., Lai M.H., Blanchard K.L., Thomas J.E. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 22.Simonetti F.L., Tornador C., Nabau-Moreto N., Molina-Vila M.A., Marino-Buslje C. Kin-Driver: a database of driver mutations in protein kinases. Database (Oxford) 2014;2014 doi: 10.1093/database/bau104. [DOI] [PMC free article] [PubMed] [Google Scholar]