

Abstract
Cre-inducible mouse models are often utilized for the spatial and temporal expression of oncogenes. With the wide number of Cre recombinase lines available, inducible transgenesis represents a tractable approach to achieve discrete oncogene expression. Here, we describe a protocol for targeting Cre-inducible genes using a loxP-STOP-loxP approach to the ubiquitously expressed ROSA26 locus. Gene targeting provides several advantages over standard transgenic techniques, including a known site of integration and previously characterized pattern of expression. Historically, an inherent instability of ROSA26 targeting vectors has hampered the efficiency of developing ROSA26 knock-in lines. In this protocol, we provide individual steps for utilizing Gateway recombination for cloning, and detailed instructions for screening targeted ES cell clones. By following this protocol, one can achieve germline transmission of a ROSA26 knock-in line within several months.
Keywords: ROSA26, oncogene, Cre-inducible, Gateway, In-Fusion, Southern blotting, JM8A3 ES cells
INTRODUCTION
In the field of cancer biology and beyond, it is frequently necessary to ubiquitously or conditionally express genes in the mouse, whether it be to determine their function or to determine the consequences of their overexpression. Oftentimes, investigators employ pronuclear injection of transgenic cDNA driven by the desired promoter elements to achieve this result. While less labor intensive and faster, this technique is confounded by several factors, including the concatamerization of DNA and the random integration of the injected construct. Unless the site of integration is known, transgenes cannot be genotyped to distinguish homozygotes from heterozygotes without test crossing or performing costly copy number-based assays. Chromosomal position effects can dictate the level of transgene expression and founder-to-founder variance (Frese and Tuveson, 2007). Constructs that insert into actively transcribed loci can lead to disruption of endogenous transcripts, resulting in an independent phenotype. Gene conversion resulting from recombination within the concatamer can change copy number, and, therefore, expression of the transgene over subsequent generations of breeding. Each of these variables can be eliminated by targeting the transgenic DNA to a known locus through the use of homologous recombination (“knock-in”). In particular, many investigators have targeted transgenes to the reverse orientation splice acceptor 26 (ROSA26) locus for either ubiquitous or conditional transgene expression.
The conditional expression of ROSA26-targeted transgenes is often achieved through the placement of a loxP-poly(A)/STOP-loxP cassette upstream of the transgene of interest. Using this approach, the transgene is transcribed only following the Cre recombinase-mediated excision of the poly(A)/STOP sequence between the loxP sites. This system is particularly powerful due to the large number of inducible and tissue-specific Cre recombinase transgenic lines currently available (Nagy et al., 2009). However, our group and others (Hohenstein et al., 2008) have reported difficulty in generating conditional ROSA26 targeting vectors using traditional restriction digestion – ligation cloning with the pROSA26PA vector as previously described (Srinivas et al., 2001). This seems to be attributable to an instability resulting in recombination within the pROSA26PA vector. However, using the Gateway-compatible pRosa26-DEST vector, recombination-deficient E. coli, and lower incubation temperatures ameliorates this instability (Hohenstein et al., 2008). Here, we provide a protocol for the generation of a ROSA26 targeting vector for the Cre-inducible expression of transgenes and the preparation of DNA for electroporation into embryonic stem (ES) cells (Basic Protocol 1). We also provide protocols for the screening of ES cell clones for the successful targeting of the ROSA26 locus, using both PCR and Southern blotting (Basic Protocol 2). These screening steps are oftentimes either performed incompletely or skipped, and are the source of failed attempts to target the ROSA26 locus. By following these protocols, we have rapidly and successfully generated several targeting vectors and lines of Cre-inducible ROSA26 knock-in mice cost effectively in a short period of time.
NOTE: All protocols using live animals must first be reviewed and approved by an Institutional Animal Care and Use Committee (IACUC) or must conform to governmental regulations regarding the care and use of laboratory animals.
BASIC PROTOCOL 1: DESIGN AND CLONING OF ROSA26 TARGETING VECTORS
This protocol provides a description of how to clone a gene of interest into the Gateway-compatible, Cre-inducible (conditional) ROSA26 targeting vector pRosa26-DEST. It provides steps for first cloning the gene of interest and any desired epitope tags into a Gateway Entry vector, and steps for performing the reaction for shuttling the gene of interest into pRosa26-DEST. The steps of this protocol are outlined in Figure 1. Details for using a novel cloning approach (In-Fusion) are also provided. Finally, a protocol for the purification of DNA for electroporation into ES cells is described. Please see Critical Parameters for additional considerations in performing this protocol.
Figure 1.

Schematic of steps required for inserting a cDNA of interest into pRosa26-DEST. A cDNA is amplified using PCR to add In-Fusion (IF) adapter sequences. The In-Fusion reaction is then performed between the PCR product and linearized Entry vector to obtain a Gateway-ready Entry vector containing the cDNA. This vector is then combined with pRosa26-DEST in a LR reaction to obtain to targeting vector.
Note: several steps indicate that you should refer to the manufacturer’s instructions instead of detailing the required steps. Please follow the manufacturer’s instructions to perform this protocol successfully.
Materials
Gateway pENTR vector (Life Technologies)
One Shot ccdB Survival 2 T1R Competent Cells (Life Technologies, cat. no. A10460)
LB broth
Kanamycin
14 mL round-bottom polypropylene tubes
37°C shaking bacterial incubator
LB agar plates containing 50 μg/mL kanamycin
1.5 mL microcentrifuge tubes
Mini-prep plasmid DNA preparation kit
Vector containing cDNA for gene of interest
Oligonucleotide primers
AmpliTaq Gold 360 Master Mix (Life Technologies, cat. no. 4398881)
Thermal cycler
Restriction enzymes
0.5X TBE (from 10X TBE stock, see recipe)
1% TBE agarose gel with 0.5 μg/mL ethidium bromide
Gel electrophoresis apparatus and power source
UV light box
QIAquick Gel Extraction Kit (Qiagen, cat. no. 28704)
Microcentrifuge
Spectrophotometer
In-Fusion HD Cloning Plus Kit (Clontech, cat. no. 638909)
pRosa26-DEST vector (Addgene, plasmid 21189)
30°C shaking bacterial incubator
Gateway LR Clonase II Enzyme Mix (Life Technologies, cat. no. 11791-100)
TE buffer
SURE2 Supercompetent Cells (Agilent Technologies, cat. no. 200152)
Ampicillin
LB agar plates containing 100 μg/mL ampicillin
EndoFree Plasmid Maxi Kit (Qiagen, cat. no. 12362)
0.7% TBE agarose gel with 0.5 μg/mL ethidium bromide
0.5M EDTA, pH 8.0
Phenol:chloroform:isoamyl alcohol (25:24:1)
Benchtop centrifuge
Chloroform 3M NaOAc
100% and 70% ethanol
Generate Entry vector containing gene of interest
-
1
Identify, obtain, and prepare sufficient amounts (i.e. 5 μg) of an appropriate Gateway pENTR vector.
See Internet Resources for a selection guide. Note that most pENTR vectors contain the ccdB gene, and must be propagated in ccdB-resistant E.coli (i.e. One Shot ccdB Survival 2 T1R cells). We chose to use the pENTR1A Dual Selection Vector, which contains both the ccdB gene and Chloramphenicol resistance gene within the dual selection cassette for counterselection. -
2
Design primers to amplify your gene of interest from an existing cDNA clone or vector. The forward primer should contain a Kozak sequence [(G/A)NNATGG], and, if an N-terminal epitope tag (e.g. FLAG) is desired, the sequence for that modification. Similarly, the reverse primer should contain the sequence of any C-terminal epitope tags and a stop codon. Check to ensure that the gene sequence remains in-frame. Convert these primer sequences to In-Fusion primers using the online tool (http://bioinfo.clontech.com/infusion/convertPcrPrimersInit.do).
We have found that the In-Fusion system is a simpler and more effective approach than traditional restriction-ligation cloning. However, a traditional approach can be employed, and primers can be used to amplify the gene fragment as-is without conversion. We also used this step to add additional modifications to the fragment, an IRES-EGFP cassette for visual identification of cells expressing the gene of interest. This was achieved by first cloning the gene of interest into an IRES-EGFP-containing vector, and then designing the forward primer so that it contained the 5′ end of the gene of interest, and the reverse primer so that it contained the 3′ end of the IRES-EGFP cassette. Design 5′ and 3′ primers to cover ~15 bp of the desired amplicon. Avoid using GC-rich primers, and monitor their melting temperature. In-Fusion primers are typically between 30 – 40 bp, as they must contain both vector backbone homology arms and amplicon-homology sequences. Primers will be longer yet if they are used to add epitope tags. -
3
PCR amplify the gene of interest, containing desired modifications and In-Fusion adapters. PCR annealing temperatures should be optimized so that a single PCR product is obtained. We have amplified products of ~2 kb, but shorter or longer products should be equally compatible with this vector system, provided the PCR is optimized and the product sequence is verified.
We use AmpliTaq Gold 360 Master Mix per the manufacturer’s instructions, and typically find that the GC Enhancer is not necessary. Optimize the parameters for your specific gene of interest. -
4
Digest the pENTR vector using the restriction enzymes you specified during the InFusion primer design process. The enzymes should flank both sides of and remove the dual selection cassette from the pENTR vector.
We typically perform several digestion reactions to ensure that a sufficient amount of vector backbone is obtained for the subsequent steps. -
5
Gel purify both the PCR product and the digested vector. Run the products on a 1% agarose gel, and then excise the PCR product-containing band and vector backbone-containing band and place in separate 1.5 mL tubes. Perform the gel extraction using a kit of your choice, and elute in the smallest volume permissible. Determine the DNA concentration using a spectrophotometer.
We use the QIAquick Gel Extraction Kit and elute in 30 μL. -
6
Perform the In-Fusion reaction according to the manufacturer’s instructions. Use Stellar Competent cells (included in kit) for the highest efficiency. Sequence-verify clones for use in the next step.
Insert gene of interest into pRosa26-DEST
-
7
Propagate the pRosa26-DEST vector in SURE2 cells at 30°C in LB broth containing 100 μg/mL ampicillin. Make DNA from a mini- or maxi-prep using standard protocols.
We and others have noted that ROSA26-containing vectors are highly unstable. To ensure vector integrity, propagate in a recombination-deficient E. coli such as SURE2, and incubate at a lower temperature, as detailed below. Note that cultures will need to be incubated for longer periods of time. -
8
Perform the LR reaction (which will allow sequences between the attL sites of the Entry vector and attR sites of pRosa26-DEST to recombine in the presence of the LR enzyme) per the manufacturer’s instructions. Briefly, combine 150 ng of Entry clone, 150 ng of pRosa-DEST, TE to 8 μL, and 2 μL LR Clonase II mix, and incubate at room temperature for 4 hours. Add 1 μL of the supplied Proteinase K and incubate at 37°C for 10 minutes.
-
9
Transform 1 μL of the LR reaction into 50 μL SURE2 cells according to the manufacturer’s instructions. Allow cells to recover for 1.5 hours at 30°C instead of 1 hour at 37°C. Plate on LB agar plates containing 100 μg/mL ampicillin and incubate overnight at 30°C. Colonies may not be visible until mid- to late-day.
-
10
Pick between 10 and 20 colonies to mini-prep and screen. Inoculate 5 mL of LB broth containing 100 μg/mL ampicillin for each clone and incubate overnight at 30°C to obtain cultures for mini-prep. Screen for correct insert orientation, length, and sequence using a combination of test restriction digestion and sequencing.
Save approximately 100 μL of each culture in a 1.5 mL tube and save at 4°C prior to performing the mini-prep. Correct clones can then be propagated from that culture without needing to re-transform the vector into E. coli. -
11
Prepare DNA from a correctly targeted clone using the EndoFree Maxi Plasmid Kit per the manufacturer’s instructions. Again, perform all bacterial incubations at 30°C. A large volume of culture (i.e. 400 mL) will likely be necessary to obtain sufficient plasmid amounts. Verify sequence integrity using restriction digestion and sequencing.
Checking the vector integrity prior to proceeding with the generation of mice is crucial. Use primers that read from the insert into the vector backbone, and primers around the vector backbone, to ensure that no deletions or inversions have occurred. Care taken at this step can prevent problems at later stages of the protocol.
Prepare DNA for electroporation
-
12
Digest 300 μg of pRosa26-DEST containing your gene of interest using an appropriate linearization enzyme. KpnI can be used if your insert did not contain a KpnI site. Otherwise, identify an alternative linearization enzyme. We have also used BbvCI, but this cuts within the diphtheria toxin A (DTA) cassette, negating the negative selection feature of the vector. Use 1–5 units of enzyme per μg of DNA. Incubate at the appropriate temperature for 6 hours.
-
13
Check for complete linearization by running a sample of both cut and uncut plasmid DNA on a 0.7% TBE agarose gel.
-
14
Stop the reaction by adding EDTA to 50mM.
-
15
Bring the total volume to 600 μL using TE.
-
16
Add 600 μL phenol:chloroform:isoamyl alcohol and mix until an emulsion forms. Centrifuge at 12,000xg for 5 minutes. Transfer the aqueous phase to a new tube. Repeat the extraction once using phenol:chloroform:isoamyl alcohol and once using chloroform only.
-
17
Add a 1/10 volume of 3M NaOAc and precipitate the DNA by adding a 2X volume of 100% ethanol. Pellet, rinse once with 70% ethanol, pellet, and then store in 100% ethanol at −20°C until ready to proceed.
We utilized a core facility for the electroporation of the linearized targeting vector into embryonic stem (ES) cells. However, if a core facility is not available, you can use various standard protocols (Tompers and Labosky, 2004; Bhatia et al., 2007). Note that the pRosa26-DEST vector contains a Neomycin resistance cassette, and ES cell clones should be selected for using G418. The ROSA26 locus targets with high efficiency, and we typically screen only 24–48 clones for successful targeting. We chose to electroporate into the JM8A3 ES cell line (Pettitt et al., 2009), a C57BL/6N cell line that is heterozygous for a corrected copy of the Agouti allele, which allows for the identification of ES cell-derived mice using coat color.
BASIC PROTOCOL 2: SCREENING FOR SUCCESSFUL TARGETING OF THE ROSA26 LOCUS
This protocol describes how to screen DNA from ROSA26-targeted ES cell clones using a PCR-based screen followed by Southern blotting (Figure 2). The steps between Basic Protocol 1 and Basic Protocol 2 involve the electroporation, selection, and expansion of ES cell clones, and are not described. While the PCR-based screen is not necessary, it can reduce the number of clones that must be analyzed by Southern blotting when a large number of clones are to be analyzed. Please see Commentary - Critical Parameters section for additional considerations in performing this protocol. The Commentary section also includes a discussion of ES cell line choice and chimera breeding strategies for germline transmission.
Figure 2.

Targeting the ROSA26 locus for inducible expression of a gene of interest. A) A cassette containing Neomycin as a selectable marker driven by the PGK promoter, followed by 4X poly A (pA) signals is flanked by loxP sequences (black triangles), and placed downstream of a splice acceptor (sa) site in the ROSA26 locus. A gene of interest may be followed by an intervening sequence and a reporter gene such as green fluorescent protein. B) All cells that will grow in neomycin are screened by PCR to determine if they contain the cassette. The Forward (F) primer in the 5′ homology arm also binds to WT ROSA sequence, but the Reverse (R) primer is downstream in unique sequence (does not bind to WT ROSA) (black arrows in A). C) Positive ES cell clones detected by PCR are expanded, and screened by Southern blot using a unique sequence probe from the 5′ end (black line), and from the 3′ end (black line). Restriction enzyme X and Y cut within the gene of interest to produce a smaller product than the wild type. The appropriate size predicts correctly targeted clones. These probes can be used to confirm correctly rearranged alleles after deletion of the floxed STOP cassette with a Cre.
Materials
ROSA26-targeted ES cell clones, grown to confluency in 96-well plate
PBS
Humidified chamber
55°C incubator
ES cell lysis buffer (see recipe)
75mM NaCl in ice cold 100% ethanol, prepared fresh
70% ethanol
ddH2O
Spectrophotometer
1.5 mL microcentrifuge tubes
Oligonucleotide primers
Apex Hot Start 2X Master Mix, Blue (Genesee Scientific, cat. no. 42-148)
Thermal cycler
0.5X TBE (see recipe)
1% TBE agarose gel with 0.5 μg/mL ethidium bromide
Gel electrophoresis apparatus and power source
UV light box
Southern restriction enzyme cocktail (see recipe)
37°C incubator
6X DNA loading dye (see recipe)
TrackIt λ DNA/Hind III fragments (Life Technologies, cat. no. 10488-064)
0.8% TBE agarose gel with 0.5 μg/mL ethidium bromide
Fluorescent ruler
Crosslinking apparatus (e.g. Stratagene UV Stratalinker 2400)
Non-reactive glass dish (9″ x 13″ for a large gel)
Orbital shaker
Denaturing solution (see recipe)
Neutralizing solution (see recipe)
10X SSC (see recipe)
Nylon membrane, Hybond XL (GE Healthcare cat. no. RPN303S)
Upward capillary transfer apparatus
2X SSC, diluted from 10X SSC stock (see recipe)
Hybridization tubes
65°C hybridization oven
0.1X SSC, 1% SDS solution
Church-Gilbert hybridization solution (see recipe)
AmpliTaq Gold 360 Master Mix (Life Technologies, cat. no. 4398881)
Thermal cycler
QIAquick PCR Purification Kit (Qiagen, cat. no. 28104)
Purified probes (5 ng/μL)
Prime-It II Random Primer Labeling Kit (Agilent Technologies, cat. no. 300385)
[α–32P]dATP
Sonicated salmon sperm DNA (Life Technologies, cat. no. 15632-011)
Illustra ProbeQuant G50 Micro Columns (GE Healthcare, cat. no. 28-9034-08)
1X SSC, 0.1% SDS
0.2X SSC, 0.1% SDS
Geiger counter
Plastic sheet protectors
Film cassette with intensifying screens or phosphorimager
Autoradiography film
Aluminum foil
Film developer
Extract genomic DNA from ES cell clones
-
1
Start with ES cell clones grown to confluency in individual wells of a 96-well plate. Remove media and wash with PBS. Add 50 μL of ES cell lysis buffer per well and incubate at 55°C overnight in a humidified chamber (sealed plastic container containing moistened paper towels).
-
2
Add 100 μL of 75 mM NaCl in ice cold 100% ethanol (prepared fresh just before use) per well and incubate at room temperature until precipitated DNA is visible (approximately 30 minutes). Plates can be incubated at −80°C if DNA is not readily precipitating.
-
3
Place a stack of paper towels on top of the plate and slowly invert the plate to drain the solution. The DNA should remain attached to bottom of the plate.
If the plates are inverted too quickly, the DNA will not remain on the plate. Use caution and invert plate very slowly. -
4
Carefully add 100 μL of 70% ethanol to each well, being careful not to disturb the DNA. Invert as described above to remove ethanol. Repeat two times.
-
5
Allow the plate to air dry for ten minutes.
-
6
Dissolve the DNA in a sufficient volume of ddH2O and determine the concentration using a spectrophotometer. Transfer the DNA to 1.5 mL tubes and store at −20°C until ready for use.
Start with the lowest volume of water possible (i.e. 10 μL) and add more as necessary to dissolve the DNA. If the 260:280 or 260:230 ratios obtained from the spectrophotometer are poor, you may need to perform a phenol chloroform extraction to further purify the DNA. However, this must be done carefully as the DNA yield from a single well of a 96-well plate is low.
Perform PCR screen of ES cell clones
-
7
Dilute DNA to 50 ng/μL and use 2 μL per 20 μL PCR reaction. Perform the PCR using the ROSA26 PCR screen forward and reverse primers (Table 1) and Apex Hot Start 2X
Master Mix:
10 μL Apex Hot Start 2X Master Mix
2 μL ROSA26 PCR screen forward primer (10 μM stock)
2 μL ROSA26 PCR screen reverse primer (10 μM stock)
2 μL ES cell clone DNA (50 ng/μL)
4 μL sterile ddH2O
Amplify the PCR products using the following PCR parameters:
1 cycle: 5 minutes 95°C 35 cycles: 1 minute 95°C 1 minute 62°C 1.5 minutes 72°C 1 cycle: 10 minutes 72°C Hold at 4°C -
8
Run the PCR products on a 1% agarose gel. Bands of approximately 1.5 kb should appear for targeted clones. Identify PCR+ clones for subsequent Southern blot analysis.
These primers fall within the 5′ region of the ROSA26 genomic locus and within the targeting vector. This assay should not detect the endogenous ROSA26 locus, and will not identify some mistargeting events or problems with the 3′ portion of the locus. This PCR screen should only be used as a preliminary step to identify potentially positive clones.
Table 1.
Primer List
| Primer | Sequence (5′ – 3′) |
|---|---|
| ROSA26 PCR screen forward | GGCGGACTGGCGGGACTA |
| ROSA26 PCR screen reverse | GGGACAGGATAAGTATGACATCATCAAGG |
| ROSA26 5′ Southern probe F | GGCTCCTCAGAGAGCCTC |
| ROSA26 5′ Southern probe R | CCGGCTGTCTCACAGAAC |
| ROSA26 3′ Southern probe F | ACTTCCCACAGATTTTCGGTT |
| ROSA26 3′ Southern probe R | TCTCAAGCAGGAGAGTATAAAACTC |
Perform Southern blot analysis on PCR+ clones
-
9
Once a subset of potentially positive clones is identified using PCR, proceed with Southern blot analysis to confirm appropriate ROSA26 targeting. If sufficient prepared DNA remains from the previous step, it can be used. However, it is often easier to start with a duplicate 96-well plate.
-
10
Repeat steps 1–5 of the “Extract genomic DNA from ES cell clones” protocol on a duplicate ES cell clone-containing 96-well plate.
-
11
Instead of resuspending DNA, directly add 40 μL of Southern restriction enzyme cocktail to each of the desired (PCR+) wells of the plate. Prepare enough of the enzyme cocktail fresh to aliquot into the desired number of wells. Incubate at 37°C overnight in a humidified chamber.
See Critical Parameters, “Selection of Southern blot probes and restriction enzymes” for a discussion of probe location and restriction enzyme choice. We used EcoRV, which cuts within the vector, so when used in conjunction with the 5′ probe, gives bands of 11.5 kb for the wild-type locus and 4 kb for the targeted locus. EcoRV can also be used in conjunction with the 3′ probe, which gives bands of 11.5 kb for the wild-type locus, and 9.6 kb for the targeted locus. -
12
Add 8 μL of 6X DNA loading dye per well and load onto a 0.8% agarose gel. Include a lane of TrackIt λ DNA/Hind III fragments (prepared by first heating to 65°C for 10 minutes) as a ladder. Run gel at low voltage (i.e. 60–80V) until an adequate separation of DNA has been achieved. Take a picture of the gel with a fluorescent ruler to mark the positions of the DNA ladder bands relative to the bottom edge of the wells.
Use a gel comb with large wells when preparing the 0.8% agarose gel to ensure that the whole volume (48 μL) will fit into the well. Monitor separation by visualizing the position of the DNA ladder bands and/or by tracking the position of the bromophenol blue dye front. Appropriately digested DNA should form a long smear on the gel. A band of high molecular weight DNA indicates incomplete digestion. -
13
UV crosslink the gel using a crosslinking apparatus.
You may need to optimize crosslinking settings using your device. We use the Auto setting of the Stratagene UV Stratalinker 2400. -
14
Cut the gel to remove the wells and excess agarose, and notch the lower right corner for later orienting of the gel.
-
15
Place the gel in an appropriately sized non-reactive glass dish, add sufficient denaturing solution to cover, and soak for 45 minutes on an orbital shaker. Rinse with water and repeat with neutralizing solution.
-
16
Measure gel and cut the nylon membrane to size. Wet membrane with water and then equilibrate in 10X SSC.
-
17
Prepare an upward capillary transfer apparatus as previously described (Brown, 2001). Transfer overnight using 10X SSC.
We place a piece of plexiglass across the center of a large glass dish (or between two dishes), and fill the dish(es) with 10X SSC. A large piece of 3mm Whatman paper should be placed across the plexiglass, and should be long enough to wick up the solution from the dish below. The gel should be inverted onto this surface, followed by the prepared membrane, and additional pieces of 3mm Whatman paper cut to the same size as the gel, a layer of paper towels, topped by a sheet of plexiglass and a weight. Parafilm or plastic wrap should be used to ensure the paper towels do not touch the wick. -
18
Remove the membrane and mark the positions of the DNA ladder bands by using the ruler and referencing the gel image. Rinse the membrane in 2X SSC and UV crosslink using a crosslinking apparatus.
-
19
Put membrane into a hybridization tube and soak in 0.1X SSC, 1% SDS for at least 1 hour in a 65°C hybridization oven to remove ethidium bromide.
-
20
Prehybridize with Church-Gilbert buffer for at least 1 hour in a 65°C hybridization oven.
-
21
Prepare the probe by amplifying from mouse genomic DNA using AmpliTaq Gold 360 Master Mix and purify using the QIAquick PCR Purification Kit, both according to the manufacturer’s instructions. Dilute the probe to 5 ng/μL.
See Critical Parameters, “Selection of Southern blot probes and restriction enzymes” for a discussion of probe location. Probes can either be designed by the investigator, generated by PCR using the primer sequences listed in Table 1, or by digesting pROSA26-5′ as previously described (Soriano, 1999; Srinivas et al., 2001). -
22
Label the probe using the Prime-It II Random Primer Labeling Kit according to the manufacturer’s instructions.
-
23
Use Illustra ProbeQuant G50 Micro columns to remove unincorporated nucleotides according to the manufacturer’s instructions.
-
24
Add 100 μL of sonicated salmon sperm DNA, boil for 10 minutes, and then add to hybridization tube containing the membrane, along with 5 mL of fresh Church-Gilbert solution. Hybridize overnight at 65°C with constant rotation.
-
25
Wash with 1X SSC, 0.1% SDS (low stringency wash) for 30 minutes at 65°C with constant rotation. Repeat.
-
26
Wash with 0.2X SSC, 0.1% SDS (high stringency wash) for 30 minutes at 65°C with constant rotation. Continue washing (normally a total of 3 times) until membrane only gives localized readings on a Geiger counter.
-
27
Place membrane into a plastic sheet protector, and then expose to autoradiography film in a cassette with intensifying screens. Wrap in foil and store in −80°C freezer overnight. Remove from the freezer and allow to come to room temperature before developing. Depending on presence or absence of bands on film, decide if exposure needs to be longer or shorter.
Alternatively, a phosphorimager can be used to image blots prepared using this technique. Other non-radioactive methods, such as utilizing digoxigenin or biotin-labeled probes followed by chemiluminiscent, colorimetric, or infrared imaging can be used, but may have varying degrees of sensitivity and should by optimized by your laboratory.
REAGENTS AND SOLUTIONS
2X Phosphate Buffer Stock Solution
134g Na2HPO4-7H2O
4mL 85% H3PO4
Bring to 1L with dH2O
Adjust pH to 7.2
Store at room temperature
6X DNA Loading Dye
30% glycerol (v/v)
0.25% bromophenol blue (w/v)
0.25% xylene cyanol (w/v)
Store at room temperature
10X TBE
1M Tris
1M Boric acid
0.02M EDTA (disodium salt)
pH adjusted to 8.3
Store at room temperature
20X SSC
0.3 M Sodium citrate
3 M NaCl
pH adjusted to 7
Store at room temperature
Church-Gilbert Hybridization Solution
50 mL 2X Phosphate Buffer
35 mL 20% SDS
0.2 mL 0.5M EDTA, pH 8.0
1g BSA, Fraction V
14.8 mL H2O
Aliquot into 50 mL tubes and store at −20°C
Thaw at 68°C just prior to use
Denaturing Solution
0.5 M NaOH
1.5 M NaCl
Store at room temperature
ES Cell Lysis Buffer
10 mM Tris, pH 7.5
10 mM EDTA, pH 8.0
10 mM NaCl
0.5% Sarcosyl
2 mg/ml Proteinase K (added fresh each time)
Store buffer (without Proteinase K) at room temperature
Neutralizing Solution
1 M Tris-Cl
3 M NaCl
pH adjusted to 7.2
Store at room temperature
Southern Restriction Enzyme Cocktail
1X appropriate buffer for restriction enzyme
1 mM spermidine
100 μg/mL bovine serum albumin (BSA)
10–15 units of restriction enzyme
Bring up to 40 μL with sterile ddH2O
Make fresh just before use
COMMENTARY
Background Information
The ROSA26 locus was first identified through a murine promoter trap screen, where the promoterless splice acceptor (SA) β-galactosidase/neomycin phosphotransferase (β-geo) reporter gene can be transcribed only when inserted into an active transcription unit. Subsequent X-gal staining determined the spatial and temporal expression pattern of the trapped promoter. The ROSA26 line was identified as one of many with a ubiquitous pattern of expression, complete penetrance, and no variation in expression (Friedrich and Soriano, 1991).
The ROSA26 genomic region produces three transcripts. Transcript 1 produces a 1,170 nt cDNA, and transcript 2 produces a 412 nt cDNA. However, neither transcript has an identifiable open reading frame (ORF). A third transcript, transcribed from the complementary strand (termed transcript AS) produces a 2 kb cDNA and contains a 1605 nt ORF which is highly conserved from C. elegans to humans. The two noncoding transcripts are disrupted by the gene trap insertion at ROSA26, which is localized to the shared intron 1 of transcripts 1 and 2, but the coding transcript AS is unaffected; its reverse orientation does not permit it to be trapped by the SA of ROSAβgeo (Zambrowicz et al., 1997). No function has been identified for the two noncoding ROSA26 transcripts, and there is no apparent phenotype in heterozygous or homozygous gene trapped mice. Because of this feature, transgene targeting to the ROSA26 locus has been widely used over the last two decades due to the advantages over classical transgenic mice. A problem with classical transgenes that insert randomly into the genome is “position effects”, the influence of surrounding regulatory elements on transgene expression. Even further, classical transgenes tend to insert as a concatemer that can change copy number and thus, expression, during subsequent generations of breeding. Thus, multiple founder lines had to be analyzed to ensure that any effect was due to the transgene, and not the site of insertion, and to analyze variations in gene expression. Knocking into the ROSA26 locus provides a defined genomic site without variable influence on gene regulation, and ensures expression from a single copy of the gene, thus eliminating the need for multiple founder lines.
At the time of this publication, over 700 annotated gene trapped ROSA26 alleles were listed in the Mouse Genome Database (MGD) (Blake et al., 2014), which is reflective of the wide utility and adaptability of targeting this locus. The ROSA26 locus has traditionally been used for ubiquitous or conditional expression of genes, but has also been exploited for alternative techniques like Cre reporting and cell lineage tracing (Soriano, 1999; Srinivas et al., 2001; Mao et al., 1999, 2001; Safran et al., 2003; Luche et al., 2007; Muzumdar et al., 2007; Yamamoto et al., 2009; Shioi et al., 2011; Imayoshi et al., 2012; Hasegawa et al., 2013; Abe et al., 2011), tetracycline and tamoxifen-inducible Cre expression (Jullien et al., 2008; Bäckman et al., 2009), RNAi (Hohenstein et al., 2008; Casola, 2010; Kleinhammer et al., 2013), quantification of homologous recombination (Sukup-Jackson et al., 2014), and reporting of DNA methylation activity (Ueda et al., 2014).
Critical Parameters
Basic Protocol 1
Choice of E. coli for propagating vectors
While standard bacterial strains like DH5α are sufficient for standard subcloning protocols, the use of appropriate competent E. coli strains is critical for the success of this protocol. Nearly all Gateway vectors carry the ccdB gene, whose product leads to cell death by inhibiting DNA gyrase (Bernard and Couturier, 1992; Bernard et al., 1993). Therefore, any vectors containing the ccdB gene must be propagated in ccdB-resistant E. coli for vector stock generation, and ccdB-sensitive E. coli after cloning removes the selection cassette. There are a large number of suitable strains available, including the one listed in this protocol. Any strain, whether chemically or electro-competent, is appropriate as long as it is ccdB-resistant. Furthermore, for steps following the Gateway recombination reaction (LR reaction), we suggest using recombination deficient E. coli such as SURE2 or Stbl3. These strains are engineered for the propagation of unstable clones, and lack recombination genes that would otherwise catalyze deletions between repeating sequences found in the vector backbone. Incubating at a lower temperature (30°C) may also help prevent unwanted recombination events. Note that cultures will take longer to become turbid, and colonies will take longer to appear on plates.
Primer design
Primer design choices will vary widely depending on the template source of your cDNA, desired epitope tags, and subcloning technique. While short epitope tags can easily be added with PCR primers, longer modifications may require subcloning through an intermediate vector prior to inserting into the Entry vector. For instance, we added IRES-EGFP downstream of the gene of interest (GOI) by first subcloning into the multiple cloning site of vector pBTG (Addgene plasmid 15037), and then amplifying GOI-IRES-EGFP by designing the forward primer against the 5′ end of GOI and the reverse primer against the 3′ end of EGFP. Using the In-Fusion system, these primers can easily be converted using an online tool, and the PCR product can easily be inserted into an Entry vector linearized with the chosen enzymes. If a traditional restriction digestion/ligation approach is desired, the PCR primers should be designed to contain compatible restriction enzyme recognition sequences. All clones should be verified using a combination of test restriction digestion and sequencing. Verify that all sequences remain in-frame following cloning manipulations.
Choice of ES cell line for electroporation
The choice of ES cell line for targeting is dependent on the individual experiment, expertise of the laboratory, or resources available at core laboratories or collaborating institutions or companies. To avoid lengthy backcrossing after germline transmission, ES cells of the desired murine genetic background should be employed if possible. We developed our line on a C57BL/6N genetic background, and chose JM8A3 ES cells for electroporation (Pettitt et al., 2009).
Basic Protocol 2
Selection of Southern blot probes and restriction enzymes
When choosing the location of Southern blot probes, they should be homologous to the genomic DNA outside of the homology arms of the targeting vector. Probes homologous to sequences within the homology arms can be used with caution (in conjunction with appropriate restriction enzymes), but will detect both the endogenous genomic locus and any mistargeted vector sequence. Two separate Southern blots, one with a probe falling on the 5′ side of the targeted genomic locus, and one falling on the 3′ side of the targeted genomic locus, should be used to ensure the integrity of gene targeting. Ideal Southern blot probes should be approximately 1 kb in length and should be unique with no homology to other genomic loci. Repetitive sequences at the ROSA26 genomic locus makes the design of long probes difficult; here, we describe relatively small probes, and membranes must be monitored carefully during washing steps to ensure thorough but not over-washing. PCR amplification of probes from wild-type genomic DNA is sufficient to generate probes, though the PCR reaction must be optimized so that only one product is obtained, and products should be carefully purified using a PCR purification kit or gel extraction kit to remove excess nucleotides and primers. Alternatively, probes can be cloned into a vector (using simple techniques such as TOPO or TA cloning) and released by digesting with restriction enzymes to generate probes. Other groups have described the digestion of vector pROSA26-5′ for the generation of probes, and those protocols can be followed if desired (Soriano, 1999; Srinivas et al., 2001).
The sequences of both the wild-type and targeted genomic locus should also be analyzed to determine the restriction enzymes which will yield different and easily discernable bands on the Southern blot. This can be achieved by identifying a restriction enzyme site that is introduced by the targeting vector, and will change the banding pattern of digested successfully targeted DNA. For example, EcoRV cuts on both the 5′ and 3′ sides of the ROSA26 locus, but a novel EcoRV site is introduced by the targeting vector. Therefore, wild-type DNA will yield a larger (11.5 kb) band, but successfully targeted DNA will yield a much smaller (4 kb) band. These band sizes can be easily distinguished on a Southern blot. If a smaller difference in band size is anticipated, the gel should be run for the appropriate length of time and the location of the DNA size marker should be carefully marked on the membrane during the early stages of the Southern blotting process.
Microinjection of positive clones and germline transmission testing
Breeding schemes will vary depending on the ES cell line used. Here, we provide a brief discussion of the JM8A3 ES cell line (Pettitt et al., 2009). This line simplifies breeding schemes due to the targeted repair of one C57BL/6 nonagouti allele. The dominant repaired Agouti gene can then be used to identify offspring derived from targeted ES cells. When successfully targeted JM8A3 cells (A/a; Tyr+/+) are microinjected into C57BL/6-Tyrc-Brd (a/a; Tyrc/c) albino blastocysts for chimera generation, the degree of agouti coat color contribution in the resultant chimeras is typically indicative of the likelihood of subsequent germline transmission (Figure 3A–B). Germline transmission testing can be performed by breeding individual chimeric males with two C57BL/6-Tyrc-Brd (a/a; Tyrc/c) albino females in a 1–2 week rotation mating scheme. ES cell-derived pups can be identified at birth by the presence of dark eyes (Figure 3C), and will later develop black (a/a; Tyr+/c) or agouti (A/a; Tyr+/c) coats, but still must be genotyped for the targeted ROSA26 allele, which will have segregated separately. These animals will not be pure C57BL/6N, but rather a mix of C57BL/6N and C57BL/6-Tyrc-Brd. However, if pure C57BL/6N animals are desired, high coat color contribution chimeric males or proven germline transmission chimeras can be bred using the same scheme to C57BL/6N (a/a; Tyr+/+) females. All resulting pups will have black or agouti coats, but all agouti pups are definitively derived from targeted ES cells and should be genotyped. These animals will be on a pure C57BL/6N background. We often continue to backcross to C57BL/6N for several generations to establish a line free of potential second-site mutations introduced by the manipulation of ES cells.
Figure 3.
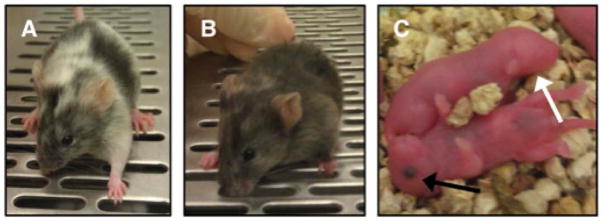
Representative images of ROSA26 knock-in chimeras and progeny generated using JM8A3 ES cells. A) An ~50% coat color contribution chimera. B) An ~90% coat color contribution chimera. C) A representative white-eyed (white arrow), C57BL/6-Tyrc-Brd albino blastocyst-derived pup, and a representative black-eyed (black arrow), C57BL/6N JM8A3-derived pup.
Troubleshooting
Please see Table 2 for troubleshooting.
Table 2.
Troubleshooting
| Problem | Possible Cause | Solution |
|---|---|---|
| pENTR or pRosa26-DEST vectors do not grow | Inappropriate E. coli used Vector stock mutated |
Make sure to propagate vectors in ccdB-resistant E. coli. Procure new vector stock. |
| No PCR product obtained when cloning into pENTR | Inappropriate annealing temperature | Perform a gradient PCR to identify the appropriate annealing temperature. Primers may be long and may require higher temperatures. |
| No colonies observed when cloning into pENTR | Incomplete digestion of pENTR vector PCR product sequence is incorrect |
Digest with fresh restriction enzyme or for a longer period of time. Verify complete linearization using gel electrophoresis. Sequence the PCR product to ensure that In-Fusion adapters or restriction enzyme sites are successfully added. |
| No colonies observed after the Gateway LR reaction | pRosa26-DEST or pENTR-GOI vectors not pure LR enzyme is old LB-Amp plates not incubated for long enough pENTR-GOI is mutated |
Check the quality of the DNA using a spectrophotometer and re-purify if necessary. Repeat reaction with fresh enzyme. Keep plates in the incubator until the end of the day. Sequence both the insert and portions of the pENTR backbone to ensure vector integrity. |
| Insufficient DNA obtained from maxi prep | Insufficient culture volume | Between 400 – 500 mL of culture may be necessary. Use extra resuspension, lysis, and neutralization solution and filtration columns if necessary. |
| Insufficient DNA obtained from 96-well plate during ES cell screening | DNA lost during inversion of plate | Invert the plate very slowly onto paper towels. If DNA is still lost, solution can be aspirated from the wells individually. |
| No bands obtained during PCR screening of ES cell clones | Inappropriate Taq polymerase or thermal cycling conditions | Use a hot start Taq polymerase and optimize thermal cycling conditions in your laboratory. |
| Poor signal on Southern blot | Probe specific activity is low | Use fresh [α 32P]dATP and check the specific activity of the probe via scintillation counting. |
| Bands not easily discernable on Southern blot | Shearing of DNA during preparation Incomplete digestion of DNA Gel not run long enough Membranes not washed sufficiently Intensifying screens not using when exposing membrane to film |
Handle DNA carefully and avoid pipetting up and down to prevent random shearing of DNA. Incubate DNA with enzyme for longer to ensure complete digestion. Run gel at a low voltage for a long period of time. The bands of the DNA ladder should be well separated. Wash membranes until activity is localized on the membrane. Prewarm the wash buffers. Use intensifying screens in cassettes when exposing membranes to film, and store at −80°C during exposure. |
Anticipated Results
Using traditional restriction digestion and ligation and the vectors described by Srinivas et al., 2001, we were unable to successfully generate a ROSA26 targeting vector due to instability of the vector and apparent self-recombination within the vector backbone. Using the protocol described here, however, we were able to achieve an 80–90% correct clone rate following the LR reaction between pENTR-GOI and pRosa26-DEST. The resultant vector readily targeted the ROSA26 locus, and typically 30% or more of the neomycin-resistant ES cell clones were identified as correctly targeted using Southern blotting. In our hands, we have found that the ROSA26 locus targets better than other genomic loci (perhaps due to its ubiquitous expression and an open, permissive chromatin state), and we have screened as few as 24 – 48 clones to obtain multiple positive clones. More clones may need to be screened depending on the ES cell line and expertise of the individual performing the ES cell culture, electroporation, clone selection, and expansion. We have also achieved a large number of high coat color contribution male chimeras (10+) using ROSA26-targeted JM8A3 ES cells as described here, but this will also vary depending on the expertise of the individual laboratory.
Finally, we have bred our conditional ROSA26 line to several different Cre recombinases, including MMTV-cre, Mx1-cre, and Vav1-cre, and have found that the STOP sequence between the loxP sites is successfully excised and the GOI is ubiquitously transcribed and translated in cell types with active Cre recombinase activity, which is the ultimate desired output of this protocol. Using the abovementioned Cre lines to drive hematopoietic expression of the GOI, we have developed a model that develops fully penetrant and rapid onset acute lymphoblastic leukemia (Carofino et al., 2013). However, the inducible ROSA26 model can also be adapted to create tumor models in a variety of cell types. Multiple Cre lines may need to be tested and analyzed until the desired phenotype is achieved. For instance, some Cre lines are expressed in early stem cells, while others are expressed in more differentiated cells. Our GOI caused leukemia only when expression was initiated in stem cells, a consideration that may also be critical for the success of other investigator’s experiments.
It is also important to note that for cancer studies using a conditional ROSA26 allele, mice should not develop cancer until they are mated to a Cre transgenic line. We have not observed any leakiness in the expression of the downstream GOI in any of our crosses, but ROSA26-GOI and Cre-only control littermates should be aged and analyzed in parallel to ensure that leakiness does not occur and that Cre recombinase expression does not result in an independent phenotype. Expression of the GOI should also be verified using qRT-PCR, and a lack of expression after mating to different Cre lines may indicate a problem with the original ROSA26 targeting construct, further reinforcing the importance of verifying targeting construct integrity prior to generating the mouse model.
Time Considerations
The time needed to complete the protocol will depend on the cloning and molecular biology expertise of the individual investigator. For Basic Protocol 1, one day will be needed for primer design (step 2), and another for optimizing the first PCR (step 3). The In-Fusion reaction and transformation can be performed in one day (steps 4 – 6). The plates will need to be incubated overnight, colonies picked and cultures incubated over another night, and mini-preps and test digests can be performed on the third day. Sequencing of clones may take several days. Therefore, the generation of pENTR-GOI will take approximately one week (or less). Once the integrity of pENTR-GOI is verified, the LR reaction and transformation can be performed in one day (steps 7 – 9). Once again, picking colonies, performing mini-preps, test digests, and sequencing can take 3–5 days (step 10). Performing a maxi prep of correct clones, digestion, and purification of DNA (steps 11 – 14) can take an additional 2 – 3 days. Therefore, Basic Protocol 1 can be performed in approximately two weeks or less, depending on the investigator.
Between Basic Protocol 1 and Basic Protocol 2, ES cells must be electroporated, selected with neomycin, individual clones must be picked, and clones must be expanded in 96-well plates. Using core facilities, this process typically takes one month; however, this will vary depending on your expertise or resources available to your laboratory. Once clones in 96-well plates are obtained, the lysis of ES cells (Basic Protocol step 1) must be performed overnight, but all other DNA extraction (steps 2 – 6) and PCR (steps 7 – 8) reaction steps can be performed in one to two days. For Southern blotting, DNA extraction (step 10) can be performed in one day, and restriction digestion (step 11) should be performed overnight. The running of the gel and preparation of the gel for transfer can be performed in one or two days (steps 12 – 16). The transfer must be performed overnight (step 17). All other Southern blot steps can be performed in one day (steps 18 – 23), but hybridization should be performed overnight (step 24). Washing and exposing to film should be done the next day (steps 25 – 27). The length of exposure will vary, but can range from one day to several weeks depending on the specific activity of the probe and stringency of washing. Together, Basic Protocol 2 can be performed in approximately a week and half at the shortest, and several weeks at the longest, depending on necessary exposure time.
Acknowledgments
The authors would like to thank Isabel Lorenzo of the Mouse Embryonic Stem Cell Core at Baylor College of Medicine for performing all ES cell manipulations, and Dr. Franco DeMayo of the Genetically Engineered Mouse Core at Baylor College of Medicine for ES cell microinjection and chimera generation. This work was also supported by NIH R01 CA163849, awarded to MJJ.
Footnotes
Conflict of Interest
The authors have declared no conflicts of interest for this article.
Internet Resources
http://tools.lifetechnologies.com/content/sfs/manuals/pentrdualselectionvectors_man.pdf Selection guide and manual for choosing Gateway pENTR vectors
http://bioinfo.clontech.com/infusion/convertPcrPrimersInit.do
Tool for converting PCR primers into In-Fusion primers
http://www.informatics.jax.org/searchtool/Search.do?query=”gt%28rosa%2926sor”
Listing of gene trapped ROSA26 alleles
Literature Cited
- Abe T, Kiyonari H, Shioi G, Inoue KI, Nakao K, Aizawa S, Fujimori T. Establishment of conditional reporter mouse lines at ROSA26 locus for live cell imaging. Genesis (New York, NY : 2000) 2011;49:579–590. doi: 10.1002/dvg.20753. [DOI] [PubMed] [Google Scholar]
- Bäckman CM, Zhang Y, Malik N, Shan L, Hoffer BJ, Westphal H, Tomac AC. Generalized tetracycline induced Cre recombinase expression through the ROSA26 locus of recombinant mice. Journal of Neuroscience Methods. 2009;176:16–23. doi: 10.1016/j.jneumeth.2008.08.024. [DOI] [PubMed] [Google Scholar]
- Bernard P, Couturier M. Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. Journal of Molecular Biology. 1992;226:735–745. doi: 10.1016/0022-2836(92)90629-x. [DOI] [PubMed] [Google Scholar]
- Bernard P, Kézdy KE, Van Melderen L, Steyaert J, Wyns L, Pato ML, Higgins PN, Couturier M. The F plasmid CcdB protein induces efficient ATP-dependent DNA cleavage by gyrase. Journal of Molecular Biology. 1993;234:534–541. doi: 10.1006/jmbi.1993.1609. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Elefanty AG, Fisher SJ, Patient R, Schlaeger T, Snyder EY, editors. Current Protocols in Stem Cell Biology. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2007. [Accessed June 21, 2014]. Available at: http://www.currentprotocols.com/WileyCDA/CPUnit/refId-sc01c04.html. [Google Scholar]
- Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE Mouse Genome Database Group. The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Research. 2014;42:D810–817. doi: 10.1093/nar/gkt1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown T. Current Protocols in Immunology. John Wiley & Sons, Inc; 2001. [Accessed June 21, 2014]. Southern Blotting. Available at: http://onlinelibrary.wiley.com/doi/10.1002/0471142735.im1006as06/abstract. [Google Scholar]
- Carofino BL, Ayanga B, Justice MJ. A mouse model for inducible overexpression of Prdm14 results in rapid-onset and highly penetrant T-cell acute lymphoblastic leukemia (T-ALL) Disease models & mechanisms. 2013;6:1494–1506. doi: 10.1242/dmm.012575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola S. Mouse models for miRNA expression: the ROSA26 locus. Methods in Molecular Biology (Clifton, NJ ) 2010;667:145–163. doi: 10.1007/978-1-60761-811-9_10. [DOI] [PubMed] [Google Scholar]
- Frese KK, Tuveson DA. Maximizing mouse cancer models. Nature Reviews Cancer. 2007;7:654–658. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- Friedrich G, Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes & Development. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Daitoku Y, Sekiguchi K, Tanimoto Y, Mizuno-Iijima S, Mizuno S, Kajiwara N, Ema M, Miwa Y, Mekada K, et al. Novel ROSA26 Cre-reporter knock-in C57BL/6N mice exhibiting green emission before and red emission after Cre-mediated recombination. Experimental Animals / Japanese Association for Laboratory Animal Science. 2013;62:295–304. doi: 10.1538/expanim.62.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenstein P, Slight J, Ozdemir D, Burn S, Berry R, Hastie N. High-efficiency Rosa26 knock-in vector construction for Cre-regulated overexpression and RNAi. PathoGenetics. 2008;1:3. doi: 10.1186/1755-8417-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I, Hirano K, Sakamoto M, Miyoshi G, Imura T, Kitano S, Miyachi H, Kageyama R. A multifunctional teal-fluorescent Rosa26 reporter mouse line for Cre- and Flp-mediated recombination. Neuroscience Research. 2012;73:85–91. doi: 10.1016/j.neures.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Jullien N, Goddard I, Selmi-Ruby S, Fina JL, Cremer H, Herman JP. Use of ERT2-iCre-ERT2 for conditional transgenesis. Genesis (New York, NY : 2000) 2008;46:193–199. doi: 10.1002/dvg.20383. [DOI] [PubMed] [Google Scholar]
- Kleinhammer A, Wurst W, Kühn R. Target validation in mice by constitutive and conditional RNAi. Methods in Molecular Biology (Clifton, NJ ) 2013;986:307–323. doi: 10.1007/978-1-62703-311-4_19. [DOI] [PubMed] [Google Scholar]
- Luche H, Weber O, Nageswara Rao T, Blum C, Fehling HJ. Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. European Journal of Immunology. 2007;37:43–53. doi: 10.1002/eji.200636745. [DOI] [PubMed] [Google Scholar]
- Mao X, Fujiwara Y, Chapdelaine A, Yang H, Orkin SH. Activation of EGFP expression by Cre-mediated excision in a new ROSA26 reporter mouse strain. Blood. 2001;97:324–326. doi: 10.1182/blood.v97.1.324. [DOI] [PubMed] [Google Scholar]
- Mao X, Fujiwara Y, Orkin SH. Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:5037–5042. doi: 10.1073/pnas.96.9.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nagy A, Mar L, Watts G. Creation and use of a cre recombinase transgenic database. Methods in molecular biology. 2009;530:365–378. doi: 10.1007/978-1-59745-471-1_19. [DOI] [PubMed] [Google Scholar]
- Pettitt SJ, Liang Q, Rairdan XY, Moran JL, Prosser HM, Beier DR, Lloyd KC, Bradley A, Skarnes WC. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nature Methods. 2009;6:493–495. doi: 10.1038/nmeth.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safran M, Kim WY, Kung AL, Horner JW, DePinho RA, Kaelin WG. Mouse reporter strain for noninvasive bioluminescent imaging of cells that have undergone Cre-mediated recombination. Molecular Imaging. 2003;2:297–302. doi: 10.1162/15353500200303154. [DOI] [PubMed] [Google Scholar]
- Shioi G, Kiyonari H, Abe T, Nakao K, Fujimori T, Jang CW, Huang CC, Akiyama H, Behringer RR, Aizawa S. A mouse reporter line to conditionally mark nuclei and cell membranes for in vivo live-imaging. Genesis (New York, NY : 2000) 2011;49:570–578. doi: 10.1002/dvg.20758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature Genetics. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Developmental Biology. 2001;1:4–4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukup-Jackson MR, Kiraly O, Kay JE, Na L, Rowland EA, Winther KE, Chow DN, Kimoto T, Matsuguchi T, Jonnalagadda VS, et al. Rosa26-GFP Direct Repeat (RaDR-GFP) Mice Reveal Tissue- and Age-Dependence of Homologous Recombination in Mammals In Vivo. PLoS genetics. 2014;10:e1004299. doi: 10.1371/journal.pgen.1004299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompers DM, Labosky PA. Electroporation of Murine Embryonic Stem Cells: A Step-by-Step Guide. STEM CELLS. 2004;22:243–249. doi: 10.1634/stemcells.22-3-243. [DOI] [PubMed] [Google Scholar]
- Ueda J, Maehara K, Mashiko D, Ichinose T, Yao T, Hori M, Sato Y, Kimura H, Ohkawa Y, Yamagata K. Heterochromatin Dynamics during the Differentiation Process Revealed by the DNA Methylation Reporter Mouse, MethylRO. Stem Cell Reports. 2014;2:910–924. doi: 10.1016/j.stemcr.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Shook NA, Kanisicak O, Yamamoto S, Wosczyna MN, Camp JR, Goldhamer DJ. A multifunctional reporter mouse line for Cre- and FLP-dependent lineage analysis. Genesis (New York, NY : 2000) 2009;47:107–114. doi: 10.1002/dvg.20474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrowicz BP, Imamoto A, Fiering S, Herzenberg LA, Kerr WG, Soriano P. Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proceedings of the National Academy of Sciences. 1997;94:3789–3794. doi: 10.1073/pnas.94.8.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]