

Abstract
Ameloblastoma is an odontogenic neoplasm whose molecular pathogenesis has only recently been elucidated. The discovery of recurrent activating mutations in FGFR2, BRAF, and RAS in a large majority of ameloblastomas has implicated dysregulation of MAPK pathway signaling as a critical step in the pathogenesis of this tumor. Some degree of controversy exists regarding the role of mutations affecting the sonic hedgehog (SHH) pathway, specifically Smoothened (SMO), which have been postulated to serve as either an alternative pathogenetic mechanism or secondary mutations. Here, we review recent advances in our understanding of the molecular pathogenesis of ameloblastoma as well as the diagnostic, prognostic, and therapeutic implications of these discoveries.
Keywords: ameloblastoma, MAPK, BRAF, RAS, odontogenic tumor, therapy
Introduction
Ameloblastoma is an uncommon, locally invasive odontogenic neoplasm arising in the jaw. The average age of diagnosis is 36 years, with equal incidence in men and women.1 Most ameloblastomas (up to 80%) occur in the posterior mandible, with fewer tumors arising in the maxilla.1 Patients typically present with painless swelling of the jaw, and the vast majority of ameloblastomas are slow-growing neoplasms without metastatic potential. However, ameloblastic tumors with cytologic atypia are classified as ameloblastic carcinoma and have a propensity for rapid growth and metastasis.2 Rarely, ameloblastic neoplasms metastasize in spite of a benign histologic appearance, and they are classified as metastasizing ameloblastoma. Current treatment options for ameloblastoma include both conservative treatment (enucleation or curettage) and resection. The former is associated with high rates of recurrence, while the latter results in significant facial deformity and morbidity.3
Ameloblastoma is thought to arise from cells of the dental lamina4 and resembles structures of the cap/bell stage of the developing tooth.5 In the 2005 World Health Organization (WHO) classification, ameloblastomas include four subtypes based on location and histopathology: solid/multicytic (91%), unicystic (6%), extra-osseous (2%), and desmoplastic (1%).6 Histologically, most tumors display a follicular pattern characterized by islands of epithelium within fibrous stroma that lacks inductive capacity to form hard tissue. The epithelium is comprised of columnar, preameloblast-like, palisaded cells with reverse polarization at the periphery, and loosely arranged cells resembling the stellate reticulum in the center (Fig. 1). However, some tumors display a plexiform pattern of epithelium with inconspicuous stellate reticulum.
Figure 1.
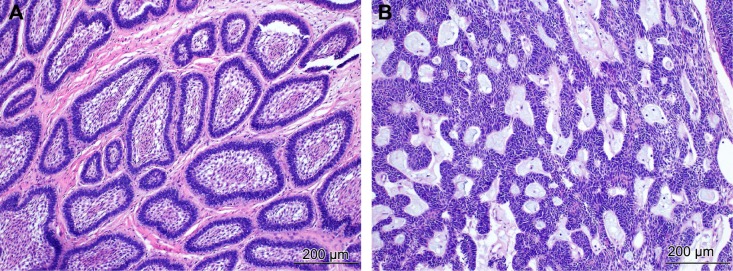
Histopathology of ameloblastoma. (A) The follicular pattern with islands of odontogenic epithelium within fibrous stroma. The epithelium consists of peripheral palisading cells showing reverse polarization and central loosely arranged cells resembling the stellate reticulum. H&E staining (×100). (B) The plexiform pattern with anastomosing strands of basal cells, delicate stroma, and inconspicuous stellate reticulum. H&E staining (×100).
While the molecular pathogenesis of ameloblastoma was largely unknown prior to 2014, there was mounting evidence suggesting that activation of the mitogen-activated protein kinase (MAPK) pathway plays a prominent role. Several studies demonstrated activation of components of the MAPK pathway in an ameloblastoma cell line (AM-1) under various circumstances, including stimulation with tumor necrosis factor alpha (TNF α)7 and fibroblast growth factors 7 and 10.8 In addition, transgenic mice expressing v-Ha-Ras under the zeta-globin promoter were shown to develop odontogenic tumors resembling ameloblastoma.9
MAPK Pathway Mutations
In 2014, three separate reports identified recurrent MAPK mutations in ameloblastoma.10–12 The most common and first mutation identified was BRAF V600E. Two of these reports found BRAF mutations at a similar frequency (64% and 63%; 54/84 and 15/24),10,12 while a third demonstrated a lower frequency (46%; 13/28).11 A more recent study reported BRAF mutations in 82% (14/17) of cases.13 The combined incidence from all four studies is 62.7% (96/153). BRAF is a serine-threonine kinase within the MAPK pathway. The V600E mutation is present in numerous neoplasms including melanoma,14 hairy cell leukemia,15 papillary thyroid carcinoma,16 Langerhans cell histiocytosis,17 and colorectal cancer.18 This mutation results in constitutive activation of the BRAF protein and downstream MEK and ERK signaling, enhancing cell proliferation, survival, and ultimately neoplastic transformation.19 Both Brown et al12 and Sweeney et al11 also identified the BRAF V600E mutation in the ameloblastoma cell line AM-1, and demonstrated evidence of in vitro activation of MAPK signaling that was blocked by BRAF inhibition.
In addition to BRAF, two studies identified mutations affecting other genes in the MAPK pathway upstream of BRAF (Fig. 2).11,12 The BRAF protein is normally activated by the G-protein RAS. RAS mutations were identified in up to 20% of ameloblastomas, including KRAS, NRAS, and HRAS.12 All RAS mutations occurred at sites commonly mutated in other neoplasms (codons 12 and 61) and are known to lead to constitutive activation of RAS signaling. The activation of RAS and the remainder of the MAPK pathway is normally triggered by the activation of a growth factor receptor in response to a growth factor. Fibroblast growth factor receptor 2 (FGFR2) is one of several receptors that activate MAPK signaling. FGFR2 mutations were identified in 6%–18% of ameloblastomas,11,12 occurring in either the transmembrane (C382R and V395D) or kinase domain (N549K) of the receptor. These mutations have been described in both endometrial carcinoma and craniosynostosis and are known to result in constitutive MAPK pathway activation that is abrogated by treatment with FGFR inhibitors.20–23 Together, FGFR2, RAS, and BRAF mutations are present in 78%–88% of ameloblastomas. Importantly, mutations affecting these genes were mutually exclusive in all 65 cases described except one (Fig. 3). This case from Sweeney et al11 demonstrated concomitant mutations of BRAF and FGFR2. The high prevalence and near-complete mutual exclusivity of these mutations suggests that activation of the MAPK pathway likely represents a critical event that occurs early in the pathogenesis of ameloblastoma.
Figure 2.

Schematic of the mitogen activated protein kinase (MAPK) pathway with mutation frequencies in ameloblastoma based on all studies in which each gene was evaluated.10–13
Figure 3.
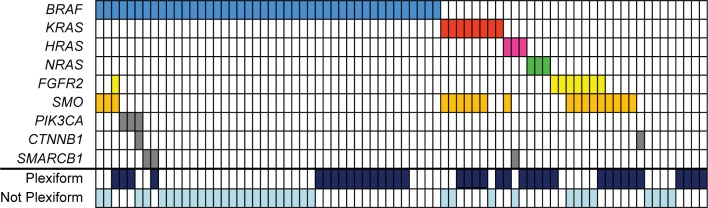
Summary of BRAF, KRAS, HRAS, NRAS, FGFR2, SMO, PIK3CA, CTNNB1, and SMARCB1 mutations in ameloblastoma based on two studies in which all of these genes were evaluated.11,12 Colored boxes indicate the presence of mutations in the indicated genes (rows) and samples (columns). The histologic pattern (plexiform versus non-plexiform) is also indicated (if known).
SMO and Other Mutations
Several mutations were identified within genes not involved in the MAPK pathway. These included SMO, CTNNB1, PIK3CA, and SMARCB1. Of these, SMO mutations were the most frequent, occurring in 16%–39% of cases.11,12 SMO mutations included W535L and L412F, which have been previously described in basal cell carcinoma24,25 and meningioma,26,27 as well as a novel mutation G416E. The Smoothened (SMO) protein is a nonclassical G-protein-coupled receptor that mediates sonic hedgehog (SHH) signaling and is normally repressed by patched (PTCH1) in the absence of the Hedgehog ligand.28 Polymorphisms and deleterious germline mutations within PTCH1 have been shown to affect the risk of ameloblastoma.29,30 Sweeney et al11 demonstrated increased sonic hedgehog signaling activity in Smo−/− mouse embryonic fibroblasts carrying SMO L412F. Furthermore, the effect of this mutation was inhibited by phar-macologic inhibitors of SHH signaling, including KAAD-cyclopamine and arsenic trioxide.
It is unclear whether MAPK and the Hedgehog pathway mutations represent two molecular subclasses of ameloblastoma, as suggested by Sweeney et al,11 or whether SMO mutations function as secondary events with MAPK pathway activation being the essential driver of pathogenesis, as suggested by Brown et al.12 BRAF and SMO were the two most frequently mutated genes in both studies, and mutations in these genes were mutually exclusive with one another in all but three instances (16% of SMO mutated cases). However, SMO mutations frequently co-occurred with RAS mutations (37% of SMO mutated cases) and FGFR2 mutations (32% of SMO mutated cases). Sixteen percent of SMO mutations occurred in the absence of any MAPK pathway mutations, accounting for 4% of ameloblastomas overall.
Brown et al12 also identified mutations in several other genes at a lower frequency. These included CTNNB1, PIK3CA, and SMARCB1 present in 4%, 6%, and 6% of cases, respectively. These mutations were not mutually exclusive with one another or with MAPK pathway or SMO mutations. All mutations have previously been described in other neoplasms. It is unclear precisely what role these mutations play in the pathogenesis of ameloblastoma.
MAPK Mutations in Other Odontogenic Tumors
Two studies investigated the pathogenetic specificity of MAPK pathway mutations, particularly BRAF V600E, by evaluating other odontogenic tumors. In one study, BRAF mutations were identified in 2 ameloblastic fibromas and 1 ameloblastic fibrodentinoma but were not identified in 37 other odontogenic tumors. These included ameloblastic carcinoma, odontoameloblastoma, clear cell odontogenic carcinomas, adenomatoid odontogenic tumor, keratocystic odontogenic tumor, calcifying cystic odontogenic tumor, calcifying epithelial odontogenic tumor, odontogenic fibroma, and odontogenic myxoma.12 A subsequent study identified BRAF V600E mutations in 3/8 (38%) ameloblastic carcinomas and 1/1 clear cell odontogenic tumor, but found no mutations in either of the two ghost cell odontogenic carcinomas.13 The presence of BRAF mutations in ameloblastic carcinoma and ameloblastic fibroma/fibrodentinoma suggests that these tumors may be pathogenetically related to ameloblastoma. Some ameloblastic carcinomas appear to arise from a pre-existing, benign ameloblastoma and are therefore designated dedifferentiated ameloblastic carcinoma.2 While ameloblastic fibromas and fibrodentinomastend to occur in younger individuals and exhibit less locally aggressive behavior, these tumors are histologically very similar to classic ameloblastoma, differing primarily in the appearance of the stroma surrounding odontogenic epithelium.31 Overall, these findings suggest that ameloblastic tumors are a distinct group of odontogenic tumors with characteristic genetic abnormalities. These findings also implicate the BRAF V600E mutation as a potential diagnostic marker in the work-up of odontogenic tumors that is specific for ameloblastic tumors. Notably, Brown et al10 observed perfect concordance between VE1 immunohistochemistry and the molecular detection of BRAF V600E mutations, demonstrating that both techniques may be useful in the diagnosis of ameloblastic tumors.
Clinicopathologic Associations
The mutation profile of ameloblastomas correlates with histopathology, location (Fig. 4), age at diagnosis, and prognosis (Fig. 5). As stated earlier, Sweeney et al11 postulated that BRAF-mutated and SMO-mutated tumors represent two distinct molecular subtypes with ameloblastoma with different clinicopathologic features including location, histologic pattern (follicular versus plexiform), and possibly prognosis. The former two showed statistically significant associations with genotype (BRAF-mutated vs SMO-mutated). However, in the larger series from Brown et al,12 these associations correlated better with the presence or absence of the BRAF mutation rather than the presence of SMO mutations, which were found in only a minority of BRAF wild-type tumors (37%). BRAF mutations were shown to occur much more frequently in the mandible and only rarely in the maxilla (5.6%), while 43% of BRAF wild-type tumors arose in the maxilla. This trend was not specific for SMO-mutated tumors; indeed, 64% of BRAF wild-type, SMO wild-type tumors also arose in the maxilla.
Figure 4.

Relationship between the anatomic location and mutation frequency in ameloblastoma based on all studies in which BRAF, RAS, FGFR2, and SMO were evaluated.10–13,40
Figure 5.
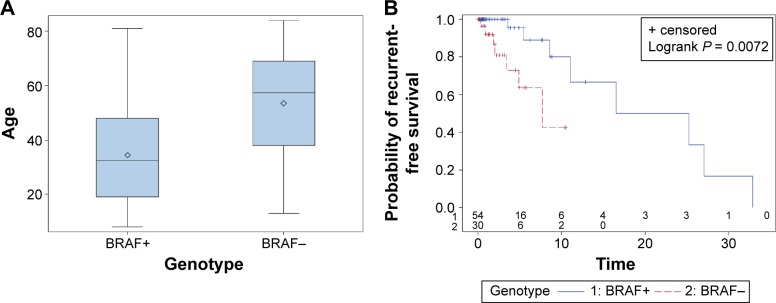
Clinical and prognostic significance of the BRAF V600E mutation in ameloblastoma adapted from Brown et al.12 (A) Box plot showing a statistically significant difference in age distribution for BRAF V600E-mutated and BRAF wild-type ameloblastomas (P = 0.0007). Diamond indicates the mean, middle horizontal line indicates the median, box indicates 25th and 75th percentiles, and whiskers indicate minimum and maximum. (B) Recurrence-free survival (in years) for BRAF V600E-mutated and BRAF wild-type ameloblastomas using the Kaplan–Meier method.
Sweeney et al11 observed that 80% of ameloblastomas with the plexiform histologic pattern were BRAF wild-type, SMO mutant (P < 0.02). Brown et al10 did not comment on the relationship of follicular/plexiform pattern and genotype. However, a review of their data shows that, while there was no statistically significant association between plexiform histology and SMO mutations (50% frequency in SMO-mutated compared to 43% frequency in SMO wild-type), plexiform histology was significantly more common among BRAF wild-type tumors (62%) compared with BRAF-mutated tumors (35%; P < 0.02). Similarly, when data from these two studies are combined (Fig. 3), plexiform histology remains significantly more common among BRAF wild-type tumors (62% versus 36%; P = 0.026), while SMO status shows no significant association.
Brown et al12 also found that BRAF mutations occurred in younger patients with a mean age at diagnosis of 34.5 years compared to 53.6 in BRAF wild-type cases (P < 0.0001). Similarly, the mean age at diagnosis among BRAF wild-type, SMO wild-type cases was 57.2 years. In addition, this study showed that BRAF V600E is an independent predictor of recurrence-free survival with BRAF wild-type tumors recurring earlier (P = 0.046). No statistically significant association was observed between recurrence and SMO mutation status.
Overall, several clinicopathologic features correlate with the presence or absence of BRAF mutations and are not specific for SMO mutations. These findings are analogous to BRAF V600E mutations in melanoma, which also occur in younger patients and have a different anatomic distribution compared with NRAS and other mutations.32–35 In melanoma, different anatomic distributions are thought to result from differences in ultraviolet light exposure. It is unclear why the anatomic distribution differs between BRAF V600E and BRAF wild-type ameloblastomas.
Implications for Therapy
Several small-molecule inhibitors targeting BRAF and MEK are FDA-approved or in clinical trials for the treatment of neoplasms with activating MAPK pathway mutations, principally BRAF V600E-mutated melanoma. Two separate studies showed that ameloblastoma cells harboring the BRAF V600E mutation are sensitive to the BRAF inhibitor vemurafenib in vitro.11,12 More recently, Kaye et al36 reported a patient with multiply recurrent ameloblastoma in the mandible and metastatic ameloblastoma in the lung that was found to harbor a BRAF V600E mutation. This patient was treated with a combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) and achieved a dramatic response after 8 weeks of therapy.
These findings suggest a potential role for BRAF and MEK inhibitors in ameloblastoma treatment. While ameloblastoma is typically treated surgically, surgical resection often results in significant facial deformity and recurrences are common. In addition, pharmacological treatment may be particularly useful in metastatic and locally aggressive cases and in patients who are poor surgical candidates. A clinical trial investigating the utility of dabrafenib in BRAF-mutated melanoma is currently under way.
Other mutations identified in ameloblastoma may also be targetable. MEK inhibitors are currently being investigated as potential treatment for NRAS-mutated melanoma and may therefore also be useful in ameloblastomas with RAS mutations.37 The utility of FGFR inhibitors is also under investigation,38 as is SHH pathway inhibition with medications such as itraconazole and arsenic trioxide.39
Conclusion
Great strides have recently been made in our understanding of the underlying molecular pathogenesis of ameloblastoma. Mutations affecting several genes within the MAPK pathway are now known to occur in a large majority of cases. The biologic importance of these mutations is highlighted by their high frequency and pattern of mutual exclusivity. The BRAF V600E mutation is the most common mutation, occurring in approximately two-thirds of cases. The presence or absence of this mutation correlates with several clinicopathologic features including location, age at diagnosis, histology, and prognosis. This mutation has also been shown to be specific for ameloblastic tumors, suggesting a potential role as a diagnostic marker. Somatic mutations affecting the Hedgehog pathway, specifically SMO, are also fairly common. It is currently unclear whether MAPK and Hedgehog pathway mutations represent two molecular subclasses of ameloblastoma, or whether SMO mutations function as secondary events with MAPK pathway mutations being the essential driver of pathogenesis. However, the higher frequency of MAPK mutations, the lack of mutual exclusivity of Hedgehog with MAPK pathway mutations, and the lack of clinicopathologic associations with SMO that are independent of BRAF status would argue against viewing SMO-mutated tumors as a truly distinct subclass of ameloblastoma. Finally, both in vitro and anecdotal clinical data implicate MAPK pathway inhibition as a promising future treatment option for ameloblastoma.
Acknowledgments
The authors would like to thank Katherine Betz for her assistance with the artwork.
Footnotes
ACADEMIC EDITOR: Barbara Guinn, Editor in Chief
PEER REVIEW: Two peer reviewers contributed to the peer review report. Reviewers’ reports totaled 577 words, excluding any confidential comments to the academic editor.
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: NAB, BLB. Analyzed the data: NAB, BLB. Wrote the first draft of the manuscript: NAB, BLB. Contributed to the writing of the manuscript: NAB, BLB. Agree with manuscript results and conclusions: NAB, BLB. Jointly developed the structure and arguments for the paper: NAB, BLB. Made critical revisions and approved final version: NAB, BLB. Both authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Reichart PA, Philipsen HP, Sonner S. Ameloblastoma: biological profile of 3677 cases. Eur J Cancer B Oral Oncol. 1995;31B(2):86–99. doi: 10.1016/0964-1955(94)00037-5. [DOI] [PubMed] [Google Scholar]
- 2.Sciubba JJ, Eversole LR, Slootweg PJ. Odontogenic/ameloblastic carinomas. In: Barnes L, Eveson JW, Reichart P, Sidransky D, editors. World Health Organization Classification Head and Neck Tumours. Lyon: IARC Press; 2005. pp. 287–289. [Google Scholar]
- 3.Mendenhall WM, Werning JW, Fernandes R, Malyapa RS, Mendenhall NP. Ameloblastoma. Am J Clin Oncol. 2007;30(6):645–648. doi: 10.1097/COC.0b013e3181573e59. [DOI] [PubMed] [Google Scholar]
- 4.Heikinheimo K, Kurppa KJ, Laiho A, et al. Early dental epithelial transcription factors distinguish ameloblastoma from keratocystic odontogenic tumor. J Dent Res. 2015;94(1):101–111. doi: 10.1177/0022034514556815. [DOI] [PubMed] [Google Scholar]
- 5.Melrose RJ. Benign epithelial odontogenic tumors. Semin Diagn Pathol. 1999;16(4):271–287. [PubMed] [Google Scholar]
- 6.Gardner DG, Heikinheimo K, Shear M, Philipsen HP, Coleman H. Ameloblastomas. In: Barnes L, Eveson JW, Reichart P, Sidransky D, editors. World Health Organization Classification Head and Neck Tumours. Lyon: IARC Press; 2005. pp. 287–289. [Google Scholar]
- 7.Hendarmin L, Sandra F, Nakao Y, Ohishi M, Nakamura N. TNFalpha played a role in induction of Akt and MAPK signals in ameloblastoma. Oral Oncol. 2005;41(4):375–382. doi: 10.1016/j.oraloncology.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 8.Nakao Y, Mitsuyasu T, Kawano S, Nakamura N, Kanda S, Nakamura S. Fibroblast growth factors 7 and 10 are involved in ameloblastoma proliferation via the mitogenactivated protein kinase pathway. Int J Oncol. 2013;43(5):1377–1384. doi: 10.3892/ijo.2013.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardiff RD, Leder A, Kuo A, Pattengale PK, Leder P. Multiple tumor types appear in a transgenic mouse with the ras oncogene. Am J Pathol. 1993;142(4):1199–1207. [PMC free article] [PubMed] [Google Scholar]
- 10.Kurppa KJ, Catón J, Morgan PR, et al. High frequency of BRAF V600E mutations in ameloblastoma. J Pathol. 2014;232(5):492–498. doi: 10.1002/path.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sweeney RT, McClary AC, Myers BR, et al. Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nat Genet. 2014;46(7):722–725. doi: 10.1038/ng.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown NA, Rolland D, McHugh JB, et al. Activating FGFR2-RAS-BRAF mutations in ameloblastoma. Clin Cancer Res. 2014;20(21):5517–5526. doi: 10.1158/1078-0432.CCR-14-1069. [DOI] [PubMed] [Google Scholar]
- 13.Diniz MG, Gomes CC, Guimarães BV, et al. Assessment of BRAFV600E and SMOF412E mutations in epithelial odontogenic tumours. Tumour Biol. 2015;36(7):5649–5653. doi: 10.1007/s13277-015-3238-0. [DOI] [PubMed] [Google Scholar]
- 14.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353(20):2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 15.Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364(24):2305–2315. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puxeddu E, Moretti S, Elisei R, et al. BRAF(V599E) mutation is the leading genetic event in adult sporadic papillary thyroid carcinomas. J Clin Endocrinol Metab. 2004;89(5):2414–2420. doi: 10.1210/jc.2003-031425. [DOI] [PubMed] [Google Scholar]
- 17.Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in langerhans cell histiocytosis. Blood. 2010;116(11):1919–1923. doi: 10.1182/blood-2010-04-279083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418(6901):934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 19.Niault TS, Baccarini M. Targets of Raf in tumorigenesis. Carcinogenesis. 2010;31(7):1165–1174. doi: 10.1093/carcin/bgp337. [DOI] [PubMed] [Google Scholar]
- 20.Byron SA, Gartside M, Powell MA, et al. FGFR2 point mutations in 466 endometrioid endometrial tumors: relationship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological features. PLoS One. 2012;7(2):e30801. doi: 10.1371/journal.pone.0030801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konecny GE, Kolarova T, O’Brien NA, et al. Activity of the fibroblast growth factor receptor inhibitors dovitinib (TKI258) and NVP-BGJ398 in human endo-metrial cancer cells. Mol Cancer Ther. 2013;12(5):632–642. doi: 10.1158/1535-7163.MCT-12-0999. [DOI] [PubMed] [Google Scholar]
- 22.Dutt A, Salvesen HB, Chen TH, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A. 2008;105(25):8713–8717. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pollock PM, Gartside MG, Dejeza LC, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene. 2007;26(50):7158–7162. doi: 10.1038/sj.onc.1210529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie J, Murone M, Luoh SM, et al. Activating smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391(6662):90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 25.Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361(12):1164–1172. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 26.Clark VE, Erson-Omay EZ, Serin A, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339(6123):1077–1080. doi: 10.1126/science.1233009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brastianos PK, Horowitz PM, Santagata S, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet. 2013;45(3):285–289. doi: 10.1038/ng.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stone DM, Hynes M, Armanini M, et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384(6605):129–134. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- 29.Kawabata T, Takahashi K, Sugai M, et al. Polymorphisms in PTCH1 affect the risk of ameloblastoma. J Dent Res. 2005;84(9):812–816. doi: 10.1177/154405910508400906. [DOI] [PubMed] [Google Scholar]
- 30.Dalati T, Zhou H. Gorlin syndrome with ameloblastoma: a case report and review of literature. Cancer Invest. 2008;26(10):975–976. doi: 10.1080/07357900802039979. [DOI] [PubMed] [Google Scholar]
- 31.Slootweg PJ. Ameloblastic fibroma/fibrodentinoma. In: Barnes L, Eveson JW, Reichart P, Sidransky D, editors. World Health Organization Classification Head and Neck Tumours. Lyon: IARC Press; 2005. p. 308. [Google Scholar]
- 32.Edlundh-Rose E, Egyházi S, Omholt K, et al. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Res. 2006;16(6):471–478. doi: 10.1097/01.cmr.0000232300.22032.86. [DOI] [PubMed] [Google Scholar]
- 33.Bauer J, Büttner P, Murali R, et al. BRAF mutations in cutaneous melanoma are independently associated with age, anatomic site of the primary tumor, and the degree of solar elastosis at the primary tumor site. Pigment Cell Melanoma Res. 2011;24(2):345–351. doi: 10.1111/j.1755-148X.2011.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bucheit AD, Syklawer E, Jakob JA, et al. Clinical characteristics and outcomes with specific BRAF and NRAS mutations in patients with metastatic melanoma. Cancer. 2013;119(21):3821–3829. doi: 10.1002/cncr.28306. [DOI] [PubMed] [Google Scholar]
- 35.Platz A, Egyhazi S, Ringborg U, Hansson J. Human cutaneous melanoma; a review of NRAS and BRAF mutation frequencies in relation to histogenetic subclass and body site. Mol Oncol. 2008;1(4):395–405. doi: 10.1016/j.molonc.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaye FJ, Ivey AM, Drane WE, Mendenhall WM, Allan RW. Clinical and radiographic response with combined BRAF-targeted therapy in stage 4 amelo-blastoma. J Natl Cancer Inst. 2014;107(1):378. doi: 10.1093/jnci/dju378. [DOI] [PubMed] [Google Scholar]
- 37.Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249–256. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- 38.Powell MA, Sill MW, Goodfellow PJ, et al. A phase II trial of brivanib in recurrent or persistent endometrial cancer: an NRG Oncology/Gynecologic Oncology Group Study. Gynecol Oncol. 2014;135(1):38–43. doi: 10.1016/j.ygyno.2014.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim J, Aftab BT, Tang JY, et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23(1):23–34. doi: 10.1016/j.ccr.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McClary AC, West RB, McClary AC, et al. Ameloblastoma: a clinical review and trends in management. Eur Arch Otorhinolaryngol. 2015 doi: 10.1007/s00405-015-3631-8. [DOI] [PubMed] [Google Scholar]