

Abstract
Unintended consequences of secondary prevention include potential introduction of bias into epidemiologic studies estimating genotype-disease associations. To better understand such bias, we simulated a family-based study of colorectal cancer (CRC), which can be prevented by resecting screen-detected polyps. We simulated genes related to CRC development through risk of polyps (G1), risk of CRC but not polyps (G2), and progression from polyp to CRC (G3). Then, we examined 4 analytical strategies for studying diseases subject to secondary prevention, comparing the following: 1) CRC cases with all controls, without adjusting for polyp history; 2) CRC cases with controls, adjusting for polyp history; 3) CRC cases with only polyp-free controls; and 4) cases with either CRC or polyps with controls having neither. Strategy 1 yielded estimates of association between CRC and each G that were not substantially biased. Strategies 2–4 yielded biased estimates varying in direction according to analysis strategy and gene type. Type I errors were correct, but strategy 1 provided greater power for estimating associations with G2 and G3. We also applied each strategy to case-control data from the Colon Cancer Family Registry (1997–2007). Generally, the best analytical option balancing bias and power is to compare all CRC cases with all controls, ignoring polyps.
Keywords: candidate gene, colorectal cancer, genetic association, polymorphisms, polyps, precursor, screening, secondary prevention
Precursors of disease, definable pathological states that frequently progress to disease without passing through a recognized intermediate state (1), have been described for a variety of diseases (e.g., low cluster of differentiation 4 count for acquired immunodeficiency syndrome, atherosclerosis for myocardial infarction, and precancers for several invasive malignancies). Numerous precursors studied as intermediate endpoints in epidemiologic research have provided valuable insight into pathophysiological mechanisms, and precancers have facilitated secondary prevention of several malignancies by serving as targets for screening followed by intervention to eliminate cells from which cancer may arise. Longstanding screening tests, the Papanicolaou (Pap) smear and endoscopy, are the basis of secondary prevention that led to notable reductions in the incidence of cervical cancer and colorectal cancer (CRC), respectively. In light of the substantial economic and humanitarian benefits of secondary prevention, rapid advances in molecular diagnostic techniques have motivated revitalized efforts to develop secondary prevention of additional malignancies and other disease types.
An unintended consequence of secondary prevention is the potential for identification of a disease precursor—rather than frank disease—to present complications for the design and analysis of epidemiologic research seeking to estimate associations between putative risk factors and disease incidence. In the context of genetic association studies, for example, epidemiologic practitioners have often wondered whether precancers should be taken into account when estimating the association between genetic variants and the risk of malignancy (1). The causal pathways leading from exposure (here, genetic variation) to cancer may be direct (no known precancer) or indirect (exposure causing a precancerous lesion that may develop subsequently into cancer) (1). Estimates of association between genetic polymorphisms and cancer may be biased if precancers are detected and removed, thereby reducing cancer incidence and converting some who would have been cases to potential controls. The potential for bias is exacerbated for studies of genetic determinants because an individual's screening behavior may be influenced by detection of cancer or its precursors in family members. In observational studies, investigators do not always have the opportunity to fully control for subtle influences of secondary prevention because complete or unbiased information about screening behaviors or detection of precursor lesions is often unavailable. Further, family data raise deeper issues of ascertainment bias (2).
A number of analytical strategies have been proposed to account for precancers in genetic association studies. One approach is to view individuals who develop precursors as “latent cases” and to exclude them from the control group during the analysis phase (3). However, this strategy has been shown to yield an estimator of the incidence rate ratio (relative risk estimated by the odds ratio in case-control studies under incidence density sampling (4)) that is biased away from the null (5, 6). An intuitive explanation for this bias is that the incidence rate is defined as the number of cases divided by the person-time at risk, including all time up to the diagnosis of the disease itself. Because controls in an incidence density case-control study should represent the person-time distribution in the source population for cases, it is appropriate that any individual should be eligible to serve as a control until he or she develops the disease itself. This approach yields an unbiased estimator only if 2 assumptions are met: 1) Detection of a precancer is independent of the risk factor under study, and 2) detection of the precancer does not influence the future course of the disease. Because neither assumption applies in this situation, ignoring precursor detection risks introducing bias. Others have suggested that excluding precancers specifically from the control group can increase power (7), but the resulting estimator reflects neither risk in the base population nor strength of the risk factor-disease association (8). An alternative possibility is to include as cases subjects who have been found to have the precancer, but this risks introducing substantial misclassification of the outcome if the precancer and cancer have different etiologies or if some precancers are of no clinical consequence.
These analytical strategies and, more broadly, the methodological issues associated with precursor lesions have been debated in the context of many genetic epidemiology studies involving cancers and other diseases with described precursors. Our own questions about appropriate epidemiologic methods in this scenario were motivated by a study focused on genetic variation related to folate metabolism and risk of CRC in the Colon Cancer Family Registry (CCFR) (9, 10). A particularly challenging situation arises with the methylenetetrahydrofolate reductase gene (MTHFR), for example, that encodes an enzyme in the folate metabolic pathway, which plays a key role in colorectal carcinogenesis. The T/T genotype of the frequently studied MTHFR polymorphism 677C>T (Ala222Val) is associated with a 65%–70% reduction in enzyme activity in vitro (11, 12) and an approximately 20% decrease in CRC risk among folate-replete individuals (13, 14). However, the MTHFR 677 T/T genotype is not associated with development of adenomas (13, 14), precursor lesions to CRC, suggesting that MTHFR is involved instead in progression of adenoma to CRC. The questions arise, therefore: Should an investigator seeking to characterize associations of genes to the risk of CRC exclude patients known to have had a history of polyps, treat them as cases, or include only cases and controls with a polyp history? How does any bias depend on screening, detection, and prevention? Is such bias introduced only when examining genes involved in polyp development or also genes involved in other pathways to cancer or the transition from polyps to carcinoma? Can the potential biases due to familial influences on screening behavior be adequately controlled by adjusting for family history of CRC or polyps, personal history of polyps, or screening behavior?
To explore these questions relevant to genetic association studies of CRC, with the objective of understanding the most appropriate analytical choices more generally in situations involving disease precursors, we designed a simulation experiment and applied a variety of analytical strategies using data from the CCFR folate metabolism candidate gene study. Although for simplicity of illustration we focus our application on candidate genes, the same issues are relevant to genome-wide association studies.
METHODS
Simulation study
The simulation study was based on a simple discordant sibship design. We simulated 1,000 sibships of size 4, each having at least 1 CRC case and at least 1 member unaffected by CRC. For each sibship, we first generated genotypes at 3 unlinked loci (Figure 1). These simulated genes related to CRC development through risk of polyps, risk of CRC but not polyps, and progression from polyps to CRC were designated G1–G3, respectively. We also generated 2 correlated γ “frailties” (1 for polyps and 1 for cancer), representing residual familial dependencies due to other genes or shared environmental factors. We then simulated times to the development of the first polyp for each subject, times to cancer with or without a polyp, and censoring times. All of these times were assumed to be independently exponentially distributed, with relative rates being a multiplicative function of the corresponding genotype (a log-additive model) and frailty (except for censoring, which was assumed to occur at the same rate for all simulated subjects).
Figure 1.
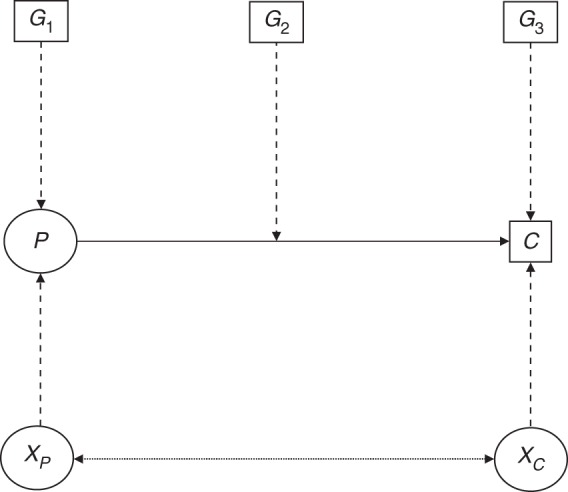
Schematic representation of the biological model for colorectal polyps and cancer for a single individual, where solid arrows represent biological progression; dashed arrows represent genetic determinants; and dotted, double-headed arrows represent correlations in frailties (XP for polyps and XC for cancer). C, [colorectal] cancer; G1–G3, simulated genes related to colorectal cancer (CRC) development through risk of polyps, risk of CRC but not polyps, and progression from polyps to CRC, respectively; P, polyp.
Each individual was assigned a random screening time, and if a polyp was present, it was designated as “detected” at that time. Then, subsequent screening times were simulated for each member of the sibship based on the family history of randomly detected polyps or cancer, and again, each subject was designated as having a detected polyp accordingly. If a polyp was detected, the subsequent risk of cancer was reduced by 50%. Figure 2 represents the simulation of the screening, detection, and prevention process for a single sib pair. Details of these simulation procedures are described in the Web Appendix, available at http://aje.oxfordjournals.org/.
Figure 2.
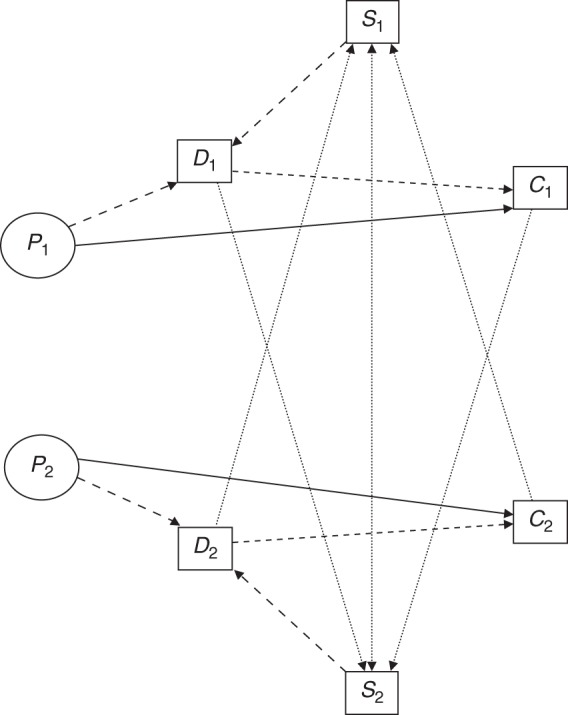
Schematic representation of the model for screening, polyp detection, and cancer prevention for a pair of siblings (subscripts 1 and 2, respectively), where solid arrows denote biological progression; dotted arrows represent screening uptake; large dashed arrows represent polyp detection; and small dashed arrows represent cancer prevention. C, colorectal cancer; D, detected polyp; P, polyp; S, screening (colonoscopy).
Each of 1,000 replicate data sets was analyzed for association between variants in 3 candidate genes and CRC by using conditional logistic regression for sib-matched case-control data using 4 strategies:
Cases were individuals with CRC; controls were individuals without CRC before their censoring times, with no adjustment for the individual's polyp history.
The same comparison as strategy 1 was used, except with adjustment for history of screen-detected polyps.
Cases were individuals with CRC; controls were individuals with neither CRC nor polyps detected before their censoring times.
Cases were individuals with either CRC or a detected polyp; controls were individuals with neither CRC nor polyps detected before their censoring times.
Only sibships with at least 1 case and 1 control were included in the corresponding analysis. For the first definition of case-control status, all ascertained sibships were included, but for the latter 2, sibships with no controls were uninformative.
The mean and variance of the estimated natural log of relative risk (ln RR) parameters for each gene and the test size and power for testing the null hypothesis were tabulated across 1,000 replicates. For test size, the relative risks for all 3 genes were set to 1.0, and the proportion of replicates that produced univariate Wald χ2 > 1.96 (2-sided α < 0.05) for each gene was tabulated. To investigate the bias due to polyp-based screening for CRC, we first estimated the “true” relative risk for CRC (compared with controls with no CRC) for each gene from a large sample (n = 100,000) with no screening or intervention (i.e., the marginal associations of each gene and CRC risk in the presence of the other pathway and residual frailties; refer to the Web Appendix). For a 2-fold causal association of each of the genes on their respective pathway, these marginal relative risks for CRC were 1.27 for G1, 1.39 for G2, and 1.42 for G3.
Application to the Colon Cancer Family Registry
The application used data from a large family-based case-control association study of genes involved in folate metabolism nested within the CCFR. This study included a total of 1,237 population-based and 410 clinic-based cases of pathologically confirmed CRC diagnosed from 1997 to 2002 and their unaffected siblings or first cousins. Families were ascertained from 6 sites in the United States (Seattle, Washington; Mayo Clinic, Minnesota; Hawaii; and the University of Southern California Consortium comprising Los Angeles, California; North Carolina; Colorado; Arizona; Minnesota; Dartmouth Medical School; and the Cleveland Clinic); Canada (Ontario); and Australia (Melbourne). Some of these families were systematically ascertained through probands from population-based cancer registries with various criteria based on age, race, specific family history, or microsatellite instability status (described below as “population-based families”), and some were ascertained through genetic clinics, which would tend to overrepresent strong family histories (“clinic-based families”). All aspects of this study received institutional review board approval under the policies of the CCFR, and all study participants provided written, informed consent.
At the time of study enrollment, a core questionnaire collected information on personal and family histories of polyps, CRC and other cancer types, and other cancer risk factors. Specific questions focused on screening behavior (colonoscopy and sigmoidoscopy) and age at and reasons for screening, including previous family history and self-reported personal history of polyps including type (benign vs. adenomatous), age at detection, and removal dates.
For illustration purposes, we considered 6 nonsynonymous single-nucleotide polymorphisms (SNPs) in 5 folate-related genes: MTHFR; 5-methyltetrahydrofolate-homocysteine methyltransferase gene (MTR); 5-methyltetrahydrofolate-homocysteine methyltransferase reductase gene (MTRR); cystathionine-β-synthase gene (CBS); and solute carrier family 19 (folate transporter), member 1, gene (SLC19A1). Full details of the substantive results for these associations in which polyps were not incorporated into case or control inclusion/exclusion criteria have been reported elsewhere (15–18). In the present research, multivariable conditional logistic regression with family as the matching factor was conducted to estimate associations between genetic variants and risk of CRC using each of the 4 analysis strategies detailed above. Because genotyped SNPs may not be the causal variants, we used a robust variance estimator to prevent biased estimates that could otherwise result from testing association in the presence of linkage (19). For the majority of SNPs, we grouped heterozygous genotypes with the common homozygous genotypes in agreement with findings from previous studies. However, for 2 SNPs, we assumed a dominant model of risk, either because this mode of genetic inheritance had been indicated by prior studies or because the number of homozygotes for the minor allele was too small. Multivariable models were also adjusted for alcohol consumption and use of folic acid and multivitamins, but addition of these other variables did not substantially change the estimates of risk, so results from the more parsimonious models were reported. Population-based and clinic-based families were analyzed separately because we anticipated that the latter could be affected by additional biases related to ascertainment. All statistical analyses were performed by using R, version 2.6.2 (R Foundation for Statistical Computing, Vienna, Austria) (http://www.r-project.org/).
RESULTS
Simulation study
Table 1 presents the percent bias in ln RR estimates for associations of the 3 gene types (G1–G3) and risk of CRC, with entries that exceed 25% of the true values denoted by footnote. Estimates for the comparison of CRC cases against all others without cancer (unadjusted for polyp history, strategy 1) were not substantially biased under the various secondary prevention models illustrated by the rows of the table. However, the analysis strategies that took polyps into account in case or control definitions (strategies 3 and 4) led to quite substantial biases, with direction and extent of the bias varying across gene types and analysis approach. Bias was most severe for estimates of association with G1. Adjusting for polyps in a case-control comparison of CRC cases against all controls (strategy 2) produced results similar to those from analyses in which we excluded individuals with detected polyps from the control group (strategy 3). Additional adjustment for personal history of colonoscopy yielded results that were not appreciably different from adjustment for polyps alone (data not shown). Excluding individuals with detected polyps led to substantial losses of informative sibships, resulting in larger standard errors than with the other 2 strategies.
Table 1.
Percent Bias and Standard Error of Estimates of Log Relative Risk for the Association Between 3 Gene Types and the Risk of Colorectal Cancer Among Population-Based Families in the Colon Cancer Family Registry, 1997–2007
| Pr(Screen | FH Polyps) | Pr(Screen | FH CRC) | Pr(CRC Prevented | Polyp Detected) |
G1 Bias |
G2 Bias |
G3 Bias |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CRC vs. No CRC |
CRC vs. Neither CRC Nor Detected Polypc | CRC or Detected Polyp vs. Neitherd | CRC vs. No CRC |
CRC vs. Neither CRC Nor Detected Polypc | CRC or Detected Polyp vs. Neitherd | CRC vs. No CRC |
CRC vs. Neither CRC Nor Detected Polypc | CRC or Detected Polyp vs. Neitherd | ||||||
| Unadjusteda | Adjusted for Polypsb | Unadjusteda | Adjusted for Polypsb | Unadjusteda | Adjusted for Polypsb | |||||||||
| 0.00 | 0.00 | 0.00 | −3 | −61e | −41e | 55e | 0 | 8 | 17 | −13 | −4 | 2 | −16 | −30e |
| 0.25 | 0.00 | 0.00 | −2 | −65e | −43e | 61e | −1 | 7 | 16 | −14 | 7 | 15 | −8 | −25 |
| 0.00 | 0.75 | 0.00 | −4 | −93e | −64e | 73e | 2 | 18 | 36e | −18 | 2 | 18 | −17 | −41e |
| 0.00 | 0.00 | 0.50 | −10 | −51e | −23 | 98e | −3 | −1 | 7 | −17 | 6 | 7 | −8 | −20 |
| 0.25 | 0.75 | 0.50 | 2 | −91e | −47e | 147e | −2 | 4 | 24 | −21 | −2 | 3 | −23 | −41e |
Abbreviations: CRC, colorectal cancer; FH, family history; G1–G3, simulated genes related to CRC development through risk of polyps, risk of CRC but not polyps, and progression from polyps to CRC, respectively; ln RR, natural log of relative risk; Pr, probability.
a Strategy 1: standard error of ln RR = 0.086.
b Strategy 2: standard error of ln RR = 0.091.
c Strategy 3: standard error of ln RR = 0.115.
d Strategy 4: standard error of ln RR = 0.086.
e Absolute bias is greater than 25% of the true value.
Web Table 1 summarizes the power and test size for these same combinations, and Figure 3 shows power curves across a range of causal relative risks for the first and last of these combinations. There was no evidence that any strategy led to significantly increased or decreased type I error rates. Power varied substantially across the 3 analysis methods; however, power for G1 was particularly low when detected polyps were excluded from the analysis in every situation. The comparison of CRC cases against all non-CRC controls (strategy 1) was consistently more powerful than strategies 3 or 4 for G2 and G3. For G1, treating detected polyps as cases was consistently the most powerful method.
Figure 3.

Power curves for the 4 analyses: solid curves, colorectal cancer (CRC) versus no CRC (black, unadjusted; gray, adjusted for polyps); small dashed curves, CRC versus neither CRC nor detected polyp; large dashed curves, CRC or detected polyp versus neither. Left (A, C, E): Pr(screening | family history of polyps) = 0, Pr(screening | family history of CRC) = 0, and Pr(CRC prevented | polyp detected) = 0; right (B, D, F): Pr(screening | family history of polyps) = 0.25, Pr(screening | family history of CRC) = 0.75, and Pr(CRC prevented | polyp detected) = 0.50. Top (A and B), G1; middle (C and D), G2; bottom (E and F), G3. PR, probability; G1–G3, simulated genes related to CRC development through risk of polyps, risk of CRC but not polyps, and progression from polyps to CRC, respectively.
Application to the Colon Cancer Family Registry
Tables 2 and 3 provide the odds ratios and corresponding 95% confidence intervals for 6 SNPs among population-based and clinic-based families, respectively. There is insufficient biological knowledge to predict whether any of these SNPs can be defined as G1, G2, or G3, except perhaps MTHFR 677C>T, which the literature suggests is more likely of type G2 or G3.
Table 2.
Odds Ratio Estimates of the Association Between Selected Polymorphisms and Risk of Colorectal Cancer Among Population-Based Families in the Colon Cancer Family Registry, 1997–2007
| Gene-rsID | Nucleotide Substitution | Amino Acid Substitution | CRC vs. No CRC |
CRC vs. Neither CRC Nor Detected Polyp, Adjusted for Sex and Agea |
CRC or Detected Polyp vs. Neither, Adjusted for Sex and Ageb |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adjusted for Sex and Agec |
Adjusted for Sex, Age, and Polypsd |
Adjusted for Sex, Age, Polyps, and Colonoscopyd |
||||||||||
| OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | |||
| MTHFR-rs1801133e | 677C>T | V222A | 0.75 | 0.55, 1.03 | 0.78 | 0.57, 1.07 | 0.77 | 0.56, 1.06 | 0.74 | 0.53, 1.04 | 0.87 | 0.66, 1.14 |
| MTHFR-rs1801131e | 1298C>T | A429E | 0.80 | 0.59, 1.09 | 0.76 | 0.55, 1.03 | 0.75 | 0.54, 1.03 | 0.74 | 0.52, 1.06 | 0.89 | 0.67, 1.17 |
| MTR-rs1805087e | 2756A>G | D919G | 1.10 | 0.70, 1.70 | 1.05 | 0.67, 1.65 | 1.04 | 0.67, 1.61 | 1.13 | 0.70, 1.83 | 0.99 | 0.67, 1.45 |
| MTRR-rs1801394f | 66A>G | I22M | 0.95 | 0.76, 1.19 | 0.93 | 0.74, 1.17 | 0.93 | 0.73, 1.18 | 1.05 | 0.81, 1.36 | 1.10 | 0.90, 1.35 |
| CBS-rs234706f | 699C>T | Y233Y | 0.98 | 0.80, 1.21 | 1.01 | 0.81, 1.25 | 1.03 | 0.83, 1.28 | 0.90 | 0.70, 1.14 | 0.96 | 0.79, 1.18 |
| SLC19A1-rs1051266e | 80G>A | R27H | 0.85 | 0.65, 1.09 | 0.80 | 0.61, 1.05 | 0.81 | 0.62, 1.06 | 0.85 | 0.62, 1.15 | 0.96 | 0.76, 1.22 |
Abbreviations: CBS, cystathionine-β-synthase gene; CI, confidence interval; CRC, colorectal cancer; MTHFR, methylenetetrahydrofolate reductase gene; MTR, 5-methyltetrahydrofolate-homocysteine methyltransferase gene; MTRR, 5-methyltetrahydrofolate-homocysteine methyltransferase reductase gene; OR, odds ratio; rsID, related sequence identifier; SLC19A1, solute carrier family 19 (folate transporter), member 1, gene.
a Strategy 3: n (total) = 2,173; 924 discordant sibships; 953 cases of CRC, 1,220 controls (no CRC or polyps).
b Strategy 4: n (total) = 2,736; 968 discordant sibships; 1,233 cases of CRC or polyps, 1,503 controls (no CRC or polyps).
c Strategy 1: n (total) = 2,935; 1,186 discordant sibships; 1,237 cases of CRC, 1,698 controls (no CRC).
d Strategy 2: n (total) = 2,935; 1,186 discordant sibships; 1,237 cases of CRC, 1,698 controls (no CRC). Adjusted for 1) sex, age, and polyps detected prior to baseline or 2) sex, age, polyps detected prior to baseline, and at least 1 screening colonoscopy prior to baseline.
e Recessive model.
f Dominant model.
Table 3.
Odds Ratio Estimates of the Association Between Selected Polymorphisms and Risk of Colorectal Cancer Among Clinic-Based Families in the Colon Cancer Family Registry, 1997–2007
| Gene-rsID | Nucleotide Substitution | Amino Acid Substitution | CRC vs. No CRC |
CRC vs. Neither CRC Nor Detected Polyp, Adjusted for Sex and Agea |
CRC or Detected Polyp vs. Neither, Adjusted for Sex and Ageb |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adjusted for Sex and Agec |
Adjusted for Sex, Age, and Polypsd |
Adjusted for Sex, Age, Polyps, and Colonoscopyd |
||||||||||
| OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | |||
| MTHFR-rs1801133e | 677C>T | V222A | 0.81 | 0.55, 1.37 | 0.70 | 0.42, 1.15 | 0.67 | 0.40, 1.11 | 1.08 | 0.53, 2.21 | 1.15 | 0.72, 1.82 |
| MTHFR-rs1801131e | 1298C>T | A429E | 0.87 | 0.50, 1.51 | 0.91 | 0.52, 1.59 | 0.91 | 0.52, 1.59 | 0.60 | 0.33, 1.10 | 0.65 | 0.40, 1.05 |
| MTR-rs1805087e | 2756A>G | D919G | 1.00 | 0.30, 3.38 | 0.82 | 0.25, 2.75 | 0.82 | 0.25, 2.74 | 2.39 | 0.86, 6.61 | 1.64 | 0.83, 3.25 |
| MTRR-rs1801394f | 66A>G | I22M | 0.76 | 0.54, 1.08 | 0.70 | 0.49, 1.00 | 0.70 | 0.49, 1.00 | 0.88 | 0.58, 1.33 | 0.96 | 0.71, 1.30 |
| CBS-rs234706f | 699C>T | Y233Y | 1.10 | 0.79, 1.53 | 1.17 | 0.83, 1.65 | 1.18 | 0.84, 1.67 | 1.07 | 0.75, 1.54 | 1.07 | 0.83, 1.38 |
| SLC19A1-rs1051266e | 80G>A | R27H | 1.13 | 0.75, 1.69 | 1.09 | 0.72, 1.64 | 1.11 | 0.73, 1.67 | 1.11 | 0.71, 1.75 | 1.03 | 0.74, 1.45 |
Abbreviations: CBS, cystathionine-β-synthase gene; CI, confidence interval; CRC, colorectal cancer; MTHFR, methylenetetrahydrofolate reductase gene; MTR, 5-methyltetrahydrofolate-homocysteine methyltransferase gene; MTRR, 5-methyltetrahydrofolate-homocysteine methyltransferase reductase gene; OR, odds ratio; rsID, related sequence identifier; SLC19A1, solute carrier family 19 (folate transporter), member 1, gene.
a Strategy 3: n (total) = 2,173; 924 discordant sibships; 953 cases of CRC, 1,220 controls (no CRC or polyps).
b Strategy 4: n (total) = 2,736; 968 discordant sibships; 1,233 cases of CRC or polyps, 1,503 controls (no CRC or polyps).
c Strategy 1: n (total) = 2,935; 1,186 discordant sibships; 1,237 cases of CRC, 1,698 controls (no CRC).
d Strategy 2: n (total) = 2,935; 1,186 discordant sibships; 1,237 cases of CRC, 1,698 controls (no CRC). Adjusted for 1) sex, age, and polyps detected prior to baseline or 2) sex, age, polyps detected prior to baseline, and at least 1 screening colonoscopy prior to baseline.
e Recessive model.
f Dominant model.
There were 1,186 discordant families in our analyses with strategies 1 and 2 comparing population-based individuals who had CRC with siblings who did not have CRC, irrespective of history of colorectal polyps. Considering strategy 1 with all CRC cases and all controls with adjustment for sex and age as the base analysis, the estimates across all 6 SNPs did not substantially differ with adjustment for personal history of polyps and/or screening colonoscopy (Table 2), although none were significantly different from the null, despite being strong a priori candidates. With exclusion of controls with polyps using strategy 3 (924 discordant sibships), the odds ratio estimates also did not differ substantially from the base analysis, except possibly for MTRR 66A>G and less so for CBS 699C>T. When treating CRC or polyps as cases using strategy 4 (968 discordant sibships), the odds ratio estimates appeared to be biased toward the null for MTHFR 667C>T, MTHFR 1298C>T, MTR 2756A>G, and SLC19A1 80G>A.
The results among clinic-based families differed from those for the population-based families (Table 3), in part because of increased variability from the smaller sample of clinic-based families. There were 349 discordant families for the analysis with strategy 1, 272 discordant families for strategy 2, and 291 families for strategy 3. Comparing CRC cases with controls without CRC, we found that the estimates differed by more than 10% between models with (strategy 2) and without (strategy 1) adjustment for polyps and colonoscopy screening for the MTHFR 677C>T and MTR 919D>G polymorphisms, but not for the other SNPs. For strategies excluding individuals with polyps from controls (3) or including individuals with polyps as cases (4), the results differed substantially from strategy 1 except for CBS 699C>T and SLC19A1 80G>A.
DISCUSSION
This study illustrated the potential for bias to arise in family-based association studies of genetic variation and risk of cancer resulting from screening behaviors and the existence of removable disease precursors. In our simulation, we generally observed a negative bias for variants in genes associated with polyp development (G1) in analyses treating only those with CRC as cases (strategies 1–3) and bias in the opposite direction in analyses that included polyps in the case definition (strategy 4). The latter phenomenon is particularly striking in scenarios of screening wherein polyp detection leads to reduction of CRC risk. For variants in genes directly associated with CRC risk (G2), biases were more modest but generally in a positive direction when adjusted for polyps (strategy 2) or excluding polyps from the controls (strategy 3). For genes associated with progression from polyps to CRC (G3), we observed substantial bias only for strategy 4 that included polyps as cases. In most epidemiologic studies of genetic variants, the biological role of the measured genotypic variants in development of outcomes of interest is unknown. Therefore, it would be difficult to make predictions about whether specific genes act as G1, G2, or G3 and to thereby identify the most appropriate analysis strategy based on the simulation results. This becomes even more challenging in the genome-wide association study setting, where millions of variants are examined in a single analysis based on a tag SNP approach. It is therefore comforting that, while all approaches lead to some bias, regardless of the gene type (G1, G2, or G3), a single strategy is generally the least biased and the most powerful: strategy 1 that compares all CRC cases with all controls, including among controls those without CRC but with a history of polyps.
In our applied example, we focused on 6 SNPs in folate metabolism-relevant genes hypothesized to play a role in colon carcinogenesis. We found that the potential biases identified in our simulation resulted in only minor differences in estimation across analytical strategies when considering population-based ascertainment (no differences greater than 15%). For the clinic-based families, results were more variable across approaches, which may reflect complex biases inherent in the ascertainment process, as well as the smaller sample size. MTHFR 677C>T may be considered a SNP involved in progression from polyps to carcinoma; this polymorphism has been associated with a decreased risk of CRC among folate-replete individuals but not the risk of developing adenomas (13, 14). Based on results from our simulation study for G3 genes, for this SNP, one should neither exclude patients known to have had a history of polyps from controls nor treat them as cases.
Our results apply broadly to other types of cancer and complex diseases for which precursors are detectable. We conclude that, for the purpose of identifying whether a gene has a role—directly or indirectly—in the etiology of disease, the best option is generally an analysis that compares all cases with those not known to have the disease at that point in time, without specifically accounting for precursors by either inclusion among cases, exclusion from controls, or adjustment. Adjusting for history of precursor lesions has an influence similar to that of excluding from controls individuals with the lesion, albeit with less inflation of the variance. Only if a gene's function is causally related to developing a disease precursor is an analysis of the joint phenotype warranted (and only for discovery, not estimation), and this would require the collection of systematic and unbiased data on that precursor. A better analysis in that circumstance would be some form of multivariate time-to-event analysis, assuming cohort data were available, but such an analysis is beyond the scope of this paper.
These methodological issues have been explored in a series of publications focusing on the apparent paradoxical observation that smoking is associated with the occurrence of adenomas but not with CRC. Terry and Neugut (7) argued that, because individuals with polyps typically form part of the control group in case-control studies of smoking and CRC and because polyps are associated with smoking, the estimated smoking-CRC associations are biased toward the null. Potter (20) framed this issue as a problem with misclassification of an outcome. Poole (8) disagreed with the recommendation to exclude individuals with polyps from the control group, arguing that because polyps are an intermediate step in a hypothetical causal pathway between the exposure and disease, individuals with polyps comprise part of the population at risk for CRC who should not be removed. He showed that if they were removed, the bias would be away from rather than toward the null (8). After these reports, substantive findings in the area of smoking, colorectal polyps, and CRC have evolved substantially (21–23), but the broader methodological questions related to precursor lesions remain relevant. These papers did not explicitly address the additional complications addressed in our study resulting from familial aggregation of polyps or cancer due to the specific genes under study.
Our study has several limitations. First, for simplicity, we have taken the parameter of interest to be the odds ratio estimated by conditional logistic regression. Under incidence density sampling, this is an exact estimator of the incidence rate ratio without the need to invoke a rare disease assumption (4). However, in family-based case-control studies, the test of association by conditional logistic regression is biased in the presence of linkage (24), but this can be overcome by the use of a robust variance estimator as done in our application with CCFR data (19). Also, when families are ascertained through incident cases during a fixed calendar time window, there can be bias toward the null unless the ascertainment is taken into account (2). In terms of other simulation parameters, we made several additional simplifying assumptions. For example, we did not consider polyp subtype even though the type of polyp detected (e.g., adenomatous, hyperplastic) could affect how much reduction in cancer risk is expected following removal of the polyp and influence the recommendations for frequency of subsequent screening.
Second, another potential source of bias concerns the inclusion of prevalent cases if the genes under study are related to survival from the cancer. In the population-based series from the CCFR, all probands were incident cases, but any relatives with a history of CRC by the time of the proband's enrollment were included as prevalent cases. We have not attempted to model this aspect in either the simulation or application studies reported here.
Third, the problem of detection bias can involve complicated family dynamics, with screening behavior depending on the family's history of both cancer and polyps, as well as on the influence of early detection on subsequent prognosis. Although our simulations could address these considerations in only a simplified manner, they nonetheless illustrate some of the biases that can result. In the CCFR, no systematic screening for polyps was done, relying only on participants' reports of their own and family members' polyp histories, so substantive analyses of polyps as an endpoint are unlikely to be reliable. We did not attempt to model the misclassification of polyp reports in our simulation, but this could have been an additional source of bias for all analysis strategies except the first. Although there was no major difference in estimates across strategies, at least for the population-based data, it was not possible to determine that any strategy was unbiased from the (unknown) truth. Further, it is important to note that cancer epidemiologists typically have incomplete or biased information about screening and detection of precancerous lesions and, thus, they may not have the opportunity to adjust for those variables in their analysis.
Finally, we have limited our study to family-based designs, where it is possible to observe and model the familial dependencies of disease incidence (polyps and cancer) and screening behavior directly. We assume that the same dynamics apply in population-based studies, although the information available on family members' histories would typically be available only by self-report of the cases and controls. The requirement that each sibship include at least 1 case and 1 control means that our subjects are not sampled from exactly the same populations of subjects as in a standard case-control design, but for rare diseases, the difference would be minor. The matched odds ratio from the family-based design estimates approximately the same population parameter as that from a case-control study with unrelated subjects.
In summary, genetic variation may be important in carcinoma development either directly or indirectly via the development of precancerous lesions. Untangling these complex interrelations is difficult given our limited understanding of the biological significance of particular genetic variants in relation to disease processes, and careful interpretation is needed. This becomes even more challenging in the genome-wide setting. Although bias from screening behaviors and the existence of precancers does occur, estimates of relative risk appeared to be only modestly affected in our real data application, at least where incident cases were recruited in a population-based setting. Further study is needed in larger samples where SNPs are associated with CRC to be able to reach a strong conclusion about whether the choice of analytical method matters, and if that choice makes a greater difference for population-based or clinic-based data.
ACKNOWLEDGMENTS
Author affiliations: USC Norris Comprehensive Cancer Center, Keck School of Medicine, University of Southern California, Los Angeles, California (Stephanie L. Schmit, Jane C. Figueiredo, Victoria K. Cortessis, Duncan C. Thomas); Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles, California (Stephanie L. Schmit, Jane C. Figueiredo, Victoria K. Cortessis, Duncan C. Thomas); and Department of Obstetrics and Gynecology, Keck School of Medicine, University of Southern California, Los Angeles, California (Victoria K. Cortessis).
This work was supported by the National Cancer Institute (grants U19 CA148107, R01 CA52862, UM1 CA167551, R01 CA112237) and the National Institute of Environmental Health Sciences (grant T32 ES013678). This work was also funded through cooperative agreements with the following Colon Cancer Family Registry centers: the Australasian Colorectal Cancer Family Registry (grants U01 CA074778 and U01/U24 CA097735); the Mayo Clinic Cooperative Family Registry for Colon Cancer Studies (grant U01/U24 CA074800); the Ontario Familial Colorectal Cancer Registry (grant U01/U24 CA074783); the Seattle Colorectal Cancer Family Registry (grant U01/U24 CA074794); the University of Hawaii Colorectal Cancer Family Registry (grant U01/U24 CA074806); and the University of Southern California Consortium Colorectal Cancer Family Registry (grant U01/U24 CA074799).
We thank Dr. Patricia Thompson for her insights and discussion on this paper.
Principal investigators of the Colon Cancer Family Registry sites include Dr. Robert W. Haile, Dr. Dennis J. Ahnen, Kristen Anton, Dr. Graham Casey, Dr. Iona Cheng, Dr. James M. Church, Dr. Timothy Church, Dr. Steven Gallinger, Dr. Mark A. Jenkins, Dr. Loic Le Marchand, Dr. Noralane M. Lindor, and Dr. Polly A. Newcomb.
The content of this report does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the CCFRs, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the CCFR.
Conflict of interest: none declared.
REFERENCES
- 1.Wacholder S. Precursors in cancer epidemiology: aligning definition and function. Cancer Epidemiol Biomarkers Prev. 2013;224:521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langholz B, Ziogas A, Thomas DC et al. Ascertainment bias in rate ratio estimation from case-sibling control studies of variable age-at-onset diseases. Biometrics. 1999;554:1129–1136. [DOI] [PubMed] [Google Scholar]
- 3.Hogue CJ, Gaylor DW, Schulz KF. Estimators of relative risk for case-control studies. Am J Epidemiol. 1983;1183:396–407. [DOI] [PubMed] [Google Scholar]
- 4.Greenland S, Thomas DC. On the need for the rare disease assumption in case-control studies. Am J Epidemiol. 1982;1163:547–553. [DOI] [PubMed] [Google Scholar]
- 5.Lubin JH, Gail MH. Biased selection of controls for case-control analyses of cohort studies. Biometrics. 1984;401:63–75. [PubMed] [Google Scholar]
- 6.Greenland S, Thomas DC, Morgenstern H. The rare-disease assumption revisited. A critique of “estimators of relative risk for case-control studies.” Am J Epidemiol. 1986;1246:869–883. [DOI] [PubMed] [Google Scholar]
- 7.Terry MB, Neugut AI. Cigarette smoking and the colorectal adenoma-carcinoma sequence: a hypothesis to explain the paradox. Am J Epidemiol. 1998;14710:903–910. [DOI] [PubMed] [Google Scholar]
- 8.Poole C. Controls who experienced hypothetical causal intermediates should not be excluded from case-control studies. Am J Epidemiol. 1999;1506:547–551. [DOI] [PubMed] [Google Scholar]
- 9.Haile RW, Siegmund KD, Gauderman WJ et al. Study-design issues in the development of the University of Southern California Consortium's Colorectal Cancer Family Registry. J Natl Cancer Inst Monogr. 1999;26:89–93. [DOI] [PubMed] [Google Scholar]
- 10.Newcomb PA, Baron J, Cotterchio M et al. Colon Cancer Family Registry: an international resource for studies of the genetic epidemiology of colon cancer. Cancer Epidemiol Biomarkers Prev. 2007;1611:2331–2343. [DOI] [PubMed] [Google Scholar]
- 11.Weisberg I, Tran P, Christensen B et al. A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol Genet Metab. 1998;643:169–172. [DOI] [PubMed] [Google Scholar]
- 12.Frosst P, Blom HJ, Milos R et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;101:111–113. [DOI] [PubMed] [Google Scholar]
- 13.Kono S, Chen K. Genetic polymorphisms of methylenetetrahydrofolate reductase and colorectal cancer and adenoma. Cancer Sci. 2005;969:535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Little J, Sharp L, Duthie S et al. Colon cancer and genetic variation in folate metabolism: the clinical bottom line. J Nutr. 2003;133(11 suppl 1):3758S–3766S. [DOI] [PubMed] [Google Scholar]
- 15.Figueiredo JC, Levine AJ, Lee WH et al. Genes involved with folate uptake and distribution and their association with colorectal cancer risk. Cancer Causes Control. 2010;214:597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levine AJ, Figueiredo JC, Lee W et al. A candidate gene study of folate-associated one carbon metabolism genes and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2010;197:1812–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levine AJ, Figueiredo JC, Lee W et al. Genetic variability in the MTHFR gene and colorectal cancer risk using the Colorectal Cancer Family Registry. Cancer Epidemiol Biomarkers Prev. 2010;191:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poynter JN, Haile RW, Siegmund KD et al. Associations between smoking, alcohol consumption, and colorectal cancer, overall and by tumor microsatellite instability status. Cancer Epidemiol Biomarkers Prev. 2009;1810:2745–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siegmund KD, Langholz B, Kraft P et al. Testing linkage disequilibrium in sibships. Am J Hum Genet. 2000;671:244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Potter JD. Invited commentary: Old problem, new wrinkles. Am J Epidemiol. 1998;14710:911–913. [DOI] [PubMed] [Google Scholar]
- 21.Morimoto LM, Newcomb PA, Ulrich CM et al. Risk factors for hyperplastic and adenomatous polyps: evidence for malignant potential? Cancer Epidemiol Biomarkers Prev. 2002;11(10 pt 1):1012–1018. [PubMed] [Google Scholar]
- 22.Botteri E, Iodice S, Raimondi S et al. Cigarette smoking and adenomatous polyps: a meta-analysis. Gastroenterology. 2008;1342:388–395. [DOI] [PubMed] [Google Scholar]
- 23.Gong J, Hutter C, Baron JA et al. A pooled analysis of smoking and colorectal cancer: timing of exposure and interactions with environmental factors. Cancer Epidemiol Biomarkers Prev. 2012;2111:1974–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curtis D. Use of siblings as controls in case-control association studies. Ann Hum Genet. 1997;61(pt 4):319–333. [DOI] [PubMed] [Google Scholar]