

Abstract
Amphiphilic block copolymers based on HPMA and ε-CL were synthesized by ring-opening polymerization of ε-CL followed by RAFT polymerization of HPMA. A copolymer composed of 34 kDa PHPMA and 8.5 kDa PCL associated into micelles with CMC of 5.4 μg · mL−1. A novel retinoid, 3-Cl-AHPC-OMe, was incorporated into micelles with 25 wt.-% loading by dialysis method. The effective diameter of drug loading micelles was 117 nm. Incubation of micelles in PBS at 37 °C indicated 86 wt.-% of the drug was released after 96 h. Cytotoxicity studies performed with C4-2 prostate cancer cells showed the IC50 dose was 1.96 μM after 72 h of incubation, whereas the micelles without drug showed no cytotoxicity.
Keywords: block copolymers, drug delivery systems, micelles, reversible addition fragmentation chain transfer (RAFT)
Introduction
Polymeric micelles are some of the most studied anticancer drug delivery systems.[1] They self-assemble from amphiphilic block copolymers in aqueous solution into nano-aggregates composed of a hydrophobic core and a hydrophilic shell.[2–4] Their advantages include simple preparation, high drug loading capacity, suitability for loading hydrophobic drugs, and accumulation in solid tumors due to enhanced permeability and retention (EPR) effect. Their main challenges are colloidal stability and an appropriate drug release profile.[1]
The micelle core is formed by the hydrophobic block. Its chemical structure and molecular weight are important factors that determine the stability, drug loading capacity and drug release profile of micelles.[2] Polyesters, such as poly(ε-caprolactone) (PCL)[5,6] and polylactide,[7] polyamino acids like poly(aspartic acid) and poly(β-benzyl-L-aspartate),[8] and poly(propylene oxide)[9] have been widely used as hydrophobic blocks. Poly(ethylene glycol) (PEG) has been the most frequently used hydrophilic shell-forming block,[3] while poly(N-vinyl-2-pyrrolidone) has also been repeatedly used.[10] PEG provides steric stabilization and minimization of non-specific protein interactions.[11] It is used in several products that have been approved by the Federal Drug Administration (FDA).[12] However, the lack of PEG functionality, the accelerated blood clearance (ABC) effect of PEGylated liposomes,[13] and the detection of anti-PEG antibodies[14] encouraged search for PEG alternatives.
Poly[N-(2-hydroxypropyl)methacrylamide] (PHPMA) is a hydrophilic, biocompatible and non-immunogenic water-soluble polymer intensively studied as drug carrier.[15] Like PEG, PHPMA modification has shown to increase blood-circulation times. Examples include semitelechelic PHPMA-modified nanospheres[16] and acetylcholinesterase modified at multiple points with PHPMA.[17] Recently, various micelles were designed with hydrophilic shells formed by PHPMA.[18–24] Interestingly, Hennink’s laboratory used lactate modified PHPMA as the hydrophobic block and PEG as the hydrophilic block to prepare thermosensitive, biodegradable micelles.[25] The multifunctionality of PHPMA demonstrated advantages in the design of different architectures.[2]
Lele and Leroux reported the synthesis of amphiphilic triblock[19] and star-shaped[20] copolymers containing PHPMA as a hydrophilic block and PCL, a biodegradable polymer, as the hydrophobic block. Doxorubicin and amphotericin B were loaded into the triblock based micelles (1–4 wt.-%), whereas indomethacin was incorporated into the star-shaped micelles (5–12 wt.-%).
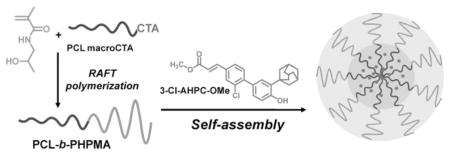
Researchers[20,21] used traditional free-radical polymerization to prepare PHPMA blocks. Consequently, the polydispersity of the PHPMA block will be >1.5 and might affect the control of size distribution of the final micelles. In this work, we explore the potential of a novel micellar drug carrier system based on a diblock copolymer of PCL and PHPMA. The block copolymer was synthesized by a combination of ring-opening polymerization (ROP) of ε-caprolactone (ε-CL) followed by “living” reversible addition-fragmentation chain transfer (RAFT) polymerization of HPMA. Using this approach, the molecular weight and polydispersity can be controlled. Polymeric micelles were formed by self-assembly of copolymers forming a hydrophobic PCL core and a hydrophilic PHPMA shell. To evaluate the feasibility of the micelles for drug delivery, a hydrophobic drug, (E)-4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorocinnamic methyl ester (3-Cl-AHPC-OMe; not used in polymeric micelle-based drug delivery systems before), was chosen and physically incorporated into the hydrophobic micelle core. The micelles were characterized by pyrene assay, dynamic light scattering (DLS), transmission electron microscopy (TEM), and stability measurements. In vitro cytotoxicity of the 3-Cl-AHPC-OMe delivery system was evaluated on the C4-2 prostate cancer cell line.
Experimental Section
Materials
ε-CL and tin(II) 2-ethylhexanoate (SnOct2) were purchased from Alfa Aesar (Ward Hill, MA). 2,2′-Azobis(2,4-dimethyl valeronitrile) (V-65) was purchased from Wako Chemicals (Richmond, VA). 4-(Dimethylamino)pyridine (DMAP), N,N′-dicyclohexylcarbodiimide (DCC), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and all other reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). Polyvinylidene fluoride (PVDF) filters (pore size 0.45 μm, ø 33 mm) were purchased from Millipore (Billerica, MA). 3-Cl-AHPC-OMe was synthesized according to literature[26] and purified by preparative RP-HPLC (Agilent Technologies 1100 series, VYDAC 218TP C18 column, 22 × 250 mm). HPMA[27] and 4-cyano-4-((thiobenzoyl)sulfanyl)pentanoic acid[28] were synthesized as previously described.
C4-2 prostate cancer cells were a gift from Dr. U. Elsässer-Beile, University of Freiburg, Germany.
Synthesis of PCL
ε-CL (5.02 g, 44.0 × 10−3 mol), benzyl alcohol (96 mg, 0.9 × 10−3 mol) and SnOct2 (45.8 mg, 0.1 × 10−3 mol) were dissolved in 10 mL of toluene. The polymerization was carried out at 115 °C for 24 h under a nitrogen atmosphere. The polymer was isolated by diluting the polymerized reaction mixture with 15 mL CH2Cl2, followed by precipitating the solution dropwise into 200 mL cold methanol under vigorous stirring. The solid was filtered off, washed with 150 mL cold methanol and dried under reduced pressure at room temperature. The yield of PCL was 4.25 g (84.7 wt.-%).
Synthesis of PCL Modified Chain Transfer Agent (PCL-CTA)
PCL (0.505 g, 0.060 × 10−3 mol), 4-cyano-4-((thiobenzoyl)sulfanyl)-pentanoic acid (0.109 g, 0.390 × 10−3 mol) and DMAP (12 mg, 0.098 × 10−3 mol) were dissolved in 5 mL CH2Cl2 and cooled to 0 °C. Then, 93 mg (0.451 × 10−3 mol) DCC were dissolved in 3 mL CH2Cl2 and added dropwise into the reaction mixture. The mixture was stirred in the dark at room temperature overnight. Dicyclohexylurea was removed by filtration, the filtrate was concentrated and the polymer was precipitated under vigorous stirring into 350 mL cold methanol. The pink solid was filtered and dried under reduced pressure. The yield of PCL-CTA was 0.442 g (87.5 wt.-%). The structure of the product was confirmed by 1H NMR.
Synthesis of Amphiphilic PCL-b-PHPMA Diblock Copolymer
HPMA (809 mg, 5.650 × 10−3 mol), PCL-CTA (195.8 mg, 0.023 × 10−3 mol), and V-65 (10.9 mg, 0.044 × 10−3 mol) were dissolved in a methanol-acetone (2/3) mixture. The solution was bubbled with N2 for 30 min, flame sealed and polymerized at 40 °C for 48 h. The pink reaction product was precipitated into diethyl ether, washed three times with diethyl ether, and then dried under reduced pressure. The yield was 697 mg (69.4 wt.-%).
Characterization of PCL-b-PHPMA
The composition of PCL-b-PHPMA was determined with 1H NMR on a Mercury 400 spectrometer using DMSO-d6 as solvent. Chemical shifts were reported in ppm (δ) relative to DMSO-d6 (2.49 ppm). The feature peaks of PHPMA (δ = 3.66, –CH(OH)–CH3) and PCL (δ = 4.04–CH2–OOC–) were chosen to calculate the molar ratio of the two blocks.
The molecular weights of the PCL polymer and PCL-b-PHPMA copolymer were calculated from 1H NMR measurement results. The molecular weight of the copolymer was further confirmed on a PL-GPC 50 plus gel permeation chromatography (GPC) instrument from Polymer Laboratories, Varian Inc. with a Polypore column 300 × 7.5 mm and dual detectors (PL-RI and PL-BV400RT). DMF with 0.1 vol.-% LiBr was used as eluent (1 mL · min−1).
Preparation of PCL-b-PHPMA Micelles
A solution of 19.8 mg PCL-b-PHPMA copolymer in 5 mL DMF was shaken overnight for equilibration and then dialyzed against deionized (DI) water for 24 h using Spectra/Por dialysis membrane 12–14 kDa molecular weight cut-off. The water was exchanged every hour for the first 3 h and every 2 h for the next 6 h. After dialysis, the aqueous solution was lyophilized. A slightly pink product was obtained. Yield: 18 mg (91.0 wt.-%).
Determination of the Critical Micelle Concentration (CMC)
The CMC was determined via fluorescence spectroscopy using pyrene as a fluorescence probe.[29–31] Acetone solution (0.2 mL) of pyrene (3.75 × 10−6 mol · L−1) was transferred into 2 mL Eppendorf vials. After the acetone was evaporated, 1.5 mL of aqueous solutions of PCL-b-PHPMA micelles with concentrations ranging from 10−0.9 mg · mL−1 to 10−3.0 mg · mL−1 were added. The final pyrene concentration was 5.0 × 10−7 mol · L−1 in every sample. The samples were heated up at 65 °C for 3 h followed by cooling down at room temperature overnight in the dark. Emission spectra (360–460 nm, λex = 339 nm) and excitation spectra (300–360 nm, λem = 390 nm) were recorded with a Perkin Elmer LS 55 luminescence spectrometer.
Preparation of 3-Cl-AHPC-OMe-Loaded Micelles and Determination of Drug Content
A solution of 20.6 mg PCL-b-PHPMA and 4.7 mg 3-Cl-AHPC-OMe in 5 mL DMF was equilibrated for 1 h and dialyzed against DI water for 24 h as described above. After dialysis, the precipitated 3-Cl-AHPC-OMe was removed by filtration with a PVDF filter. The filtrate was lyophilized for 24 h. Yield: 21.0 mg.
The content of 3-Cl-AHPC-OMe incorporated in the micelle core was determined by RP-HPLC after dissolving the freeze-dried drug-loaded micelles in pure ethanol.
Determination of Micelle Size by DLS
The effective mean diameter and the size distribution of PCL-b-PHPMA micelles and 3-Cl-AHPC-OMe-loaded PCL-b-PHPMA micelles were analysed by DLS using a Brookhaven BI-200SM goniometer and BI-9000AT digital correlator equipped with a He-Ne laser (λ = 633 nm) at room temperature in phosphate-buffered saline (PBS) (pH = 7.4). The scattering angel was 90°. All samples were measured four times to get the final average effective diameter and size distribution. Samples were filtered through a PVDF filter before measurement.
Morphology of Micelles by TEM
The morphology of PCL-b-PHPMA micelles with/without 3-Cl-AHPC-OMe loading were observed using a Philips Tecnai TEM at 100 kV accelerating voltage. The micelles were suspended in DI water (1 mg · mL−1) and 1 drop of micelle solution was put onto the carbon side of the copper specimen. Negatively staining was performed by adding 1 drop of 4 wt.-% uranyl acetate in distilled water. The specimen was dried at room temperature before measurement.
Release of 3-Cl-AHPC-OMe from PCL-b-PHPMA Micelles
3-Cl-AHPC-OMe-loaded micelles (11.0 mg) were dissolved in PBS pH 7.4 (1 mg · mL−1) and filtered through a PVDF filter. The clear solution was then incubated in a glass vial at 37 °C and 50 rpm in an Innova 4080 incubator shaker from New Brunswick Scientific. Samples (100 μL) were withdrawn at pre-determined time intervals. To separate free drug from drug-loaded micelles, every sample was filtered through a PVDF filter. The filter was first washed with 4 mL PBS, followed by washing with 4 mL pure ethanol. A PBS fraction and an ethanol fraction were collected, the drug content in both fractions was analysed by RP-HPLC.
The content and release of 3-Cl-AHPC-OMe from the micelles were measured using RP-HPLC (Agilent Technologies 1100 series, Zorbax C8 column 4.6 × 150 mm) with gradient elution from 40–90 vol.-% of Buffer B within 20 min and flow rate 1.0 mL · min−1 (Buffer A: DI H2O containing 0.1 vol.-% TFA; Buffer B: acetonitrile containing 0.1 vol.-% TFA). The injection volumes of all samples were 80 μL. 3-Cl-AHPC-OMe was detected with a diode array detector at wavelength λ = 325 nm.
In Vitro Cytotoxicity Assay of Free and Micellar 3-Cl-AHPC-OMe
The cytotoxicity of 3-Cl-AHPC-OMe-loaded PCL-b-PHPMA micelles was measured using C4-2 prostate cancer cells. Free 3-Cl-AHPC-OMe dissolved in cell culture media containing ≤0.81 vol.-% dimethylsulfoxide (DMSO) and drug free PCL-b-PHPMA micelles dissolved in cell culture media were used as controls. C4-2 cells were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10 vol.-% fetal bovine serum (FBS) at 37 °C in an incubator with 5 vol.-% CO2. All experiments were performed in the presence of 10 vol.-% FBS. Tested concentrations of free and micelles incorporated 3-Cl-AHPC-OMe were prepared by serial dilution within a range of 0.313–80 μM. C4-2 prostate cancer cells harvested in the logarithmic growth phase were seeded into 96-well plates at a density of 7500 cells/well and incubated in 100 μL RPMA 1640 media containing 10vol.-% FBS. All plates were pre-cultured for 24 h in a humidified atmosphere with 5 vol.-% CO2 at 37 °C. Subsequently, media were replaced by 100 μL media containing free 3-Cl-AHPC-OMe, micelles loaded with 3-Cl-AHPC-OMe and drug free PCL-b-PHPMA micelles, respectively. After incubation for different time periods (12 h, 24 h, 48 h and 72 h), the drug containing media were replaced by 100 μL fresh media without drug. The plates were incubated for additional 24 h followed by MTT cell viability assay.[32] Microplates were analyzed on a BIO-RAD microplate reader Model 680.
Results and Discussion
Micelle-forming block copolymers containing PHPMA have been synthesized by varying synthetic approaches. Lele and Leroux[19] synthesized PHPMA-b-PCL-b-PHPMA copolymers by (traditional) free radical polymerization of HPMA in the presence of a biodegradable chain transfer agent, α,ω-poly(ε-caprolactone) dithiol. They used a similar synthetic scheme for the synthesis of star-shaped PCL-b-PHPMA.[20] Starting with pentaerythritol, they prepared a star-poly(ε-caprolactone)-tetrakis-thiol. Using the latter as a CTA during polymerization of HPMA produced the star-shaped block copolymer. Koňák et al. first used living atom transfer radical polymerization (ATRP) for the synthesis of micelle-forming diblock PHPMA containing copolymers, PHPMA-b-poly(n-butyl acrylate).[18] Here we synthesized PCL-b-PHPMA in three steps: first, PCL by ROP of ε-CL, followed by the conversion of PCL into a RAFT macro CTA, and in the third step the PHPMA block was formed by RAFT polymerization of HPMA.
Synthesis and Characterization of PCL-b-PHPMA Block Copolymer
The synthesis of the block copolymer PCL-b-PHPMA is shown in Scheme 1. PCL was synthesized by ROP of ε-CL using benzyl alcohol as the initiator and SnOct2 as the catalyst. This approach resulted in the formation of PCL having a hydroxyl group at one end and benzyloxy group at the other end. The molecular weight of PCL was thus determined by 1H NMR based on the integration ratio between the signal of the phenyl end moiety at δ = 7. 33 ppm (intensity 4.66) and the signal of the ε-methylene group in CL repeating units at δ = 4.0 ppm(intensity 136.64), giving rise to M̄n of 8.5 kDa (Figure 1A).
Scheme 1.
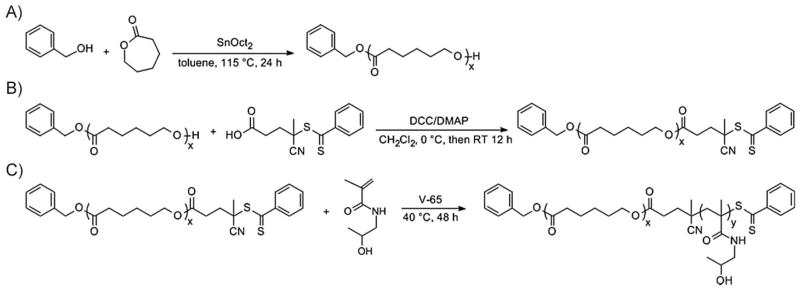
Synthesis of A) PCL, B) macromolecular chain transfer agent PCL-CTA and C) diblock copolymer PCL-b-PHPMA.
Figure 1.

A) 1H NMR spectrum of PCL for chain length determination. B) 1H NMR spectrum of PCL-b-PHPMA diblock copolymer for PHPMA chain length determination.
PCL is a semicrystalline polymer and more hydrophobic than poly(α-hydroxy acid)s, other commonly used core-forming blocks. Therefore, it has a higher loading capacity for most of the hydrophobic drugs.[33] To enhance drug loading in micelles, a long sequence of PCL may be favourable. Molecular weights between 2 kDa and 25 kDa of PCL were mostly used in research.[19,20,34] However, no micelles will be formed if the overall property of diblock copolymers is too hydrophobic.[35] It was shown that amphiphilic diblock copolymers can form spherical micelles when the length of a hydrophilic block exceeds the hydrophobic block to some degree.[36] We have tried different molecular weights of PCL and chosen 8.5 kDa; such macromolecules can form micelles with suitable size and have considerably stability in PBS. From a practical point, the molecular weight of the hydrophilic PHPMA block should not exceed 45 kDa, the renal filtration limit.
To prepare the PCL-b-PHPMA diblock copolymer, PCL was further modified with 4-cyano-4-((thiobenzoyl)sulfanyl)-pentanoic acid and functioned as macro-CTA to mediate RAFT polymerization of HPMA using V-65 as the initiator.
The composition of the diblock copolymer was estimated using 1H NMR. Figure 1B shows a typical 1H NMR spectrum of PCL-b-PHPMA block copolymer. From peak intensities of the ε-methylene group in PCL repeating units at δ = 3.96 ppm and the CH protons of the methine group in PHPMA repeating unit at δ = 3.66 ppm (intensity ratio of 0.62/1.00), the molar ratio of PCL block to PHPMA block was 0.31:1. The number-average molecular weight of the PHPMA block and the whole copolymer were thus calculated as 33.9 kDa and 42.3 kDa, respectively.
The molecular weight of PCL-b-PHPMA was further confirmed by GPC. The calibration curve was obtained by using poly(methyl methacrylate) as standard (molecular weights: 30, 74, 133.5, and 350 kDa; Polysciences, USA). As shown in Figure 2, the GPC shows only one sharp peak with M̄w/M̄n of 1.27, which suggests a well-controlled copolymerization. The weight-average molecular weight measured by GPC was 38.3 kDa, similar to an almost identical value to the result of 1H NMR measurements.
Figure 2.
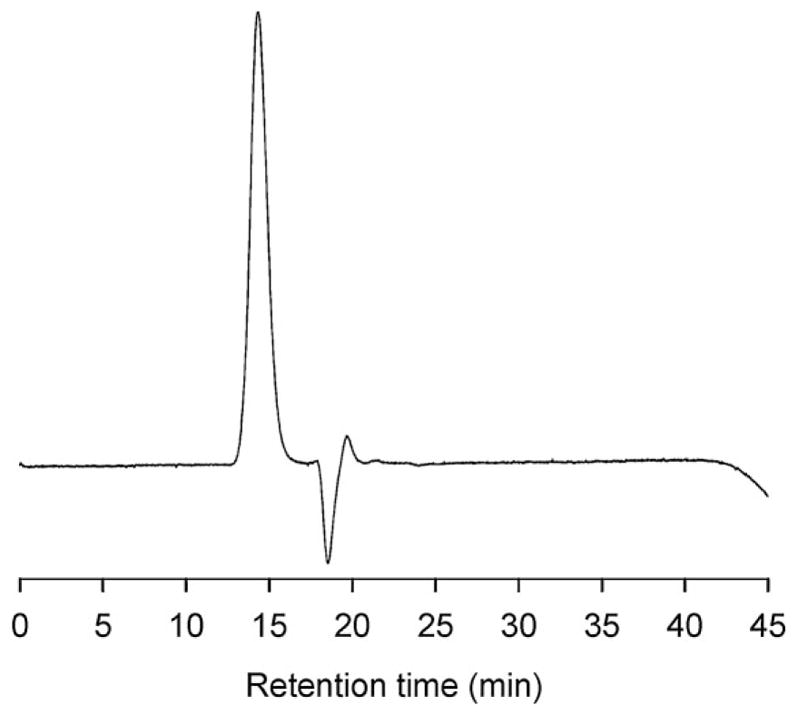
GPC diagram of PCL-b-PHPMA for molecular weight determination.
Preparation of PCL-b-PHPMA Micelles and CMC Determination
The dialysis method was used for the preparation of polymeric micelles.[5] The copolymer was dissolved in DMF and the micelle formation was then induced by the slow replacement of DMF by DI water (Scheme 2). After dialysis, the solution was completely transparent. Furthermore, there was no visible precipitation during incubation of PCL-b-PHPMA micelles over a period of one week at a concentration of 1 mg · mL−1 (V = 10 mL PBS).
Scheme 2.

Micelle formation of amphiphilic PCL-b-PHPMA diblock copolymers with entrapment of 3-Cl-AHPC-OMe, a novel drug effective in prostate cancer treatment.
The CMC of PCL-b-PHPMA micelles was determined by fluorescence spectroscopy with pyrene as a hydrophobic probe. This method is considered to be the most sensitive and precise method for CMC detection.[37,38] Pyrene changes its fluorescence intensity when the polarity of the surrounding environment changes.[39] Below the CMC, the fluorescence intensity of pyrene is low due to lack of hydrophobic environment. When the copolymer concentration surpasses the CMC, micelles are formed and hydrophobic pyrene molecules preferentially incorporate into the hydrophobic micelle core. Therefore, a fast increase of the pyrene fluorescence is detectable.[29]
A pyrene fluorescence excitation spectrum (excitation wavelength λex = 300–360 nm, emission wavelength measured at λem = 390 nm) was measured for PCL-b-PHPMA micelle preparations with copolymer concentrations ranging from 10−0.9 mg · mL−1 to 10−3.0 mg · mL−1 (diagram not shown). The pyrene concentration of each preparation was 5.0 × 10−7 mol · L−1. The detected fluorescence intensity quickly increased with PCL-b-PHPMA copolymer concentrations ≥10−2.2 mg · mL−1. Furthermore, increasing copolymer concentration lead to a red shift of the fluorescence excitation curves. For a copolymer concentration of 10−0.9 mg · mL−1, the maximum of the highest peak was detected at λex = 337.5 nm. In contrast, for a PCL-b-PHPMA concentration of 10−3.0 mg · mL−1, the maximum of the highest peak was detected at an excitation wavelength λex = 334 nm. For CMC calculation, the fluorescence intensity ratio of the peaks at λex = 337.5 nm to the peaks at λex = 334 nm (I337.5/I334) was plotted against the logarithm of the PHPMA-b-PCL copolymer concentration.[29] The plot of all measured concentrations is shown in Figure 3. The CMC was obtained from the crossover point of the 2 straight lines, which gave a CMC of 5.42 μg · mL−1.
Figure 3.
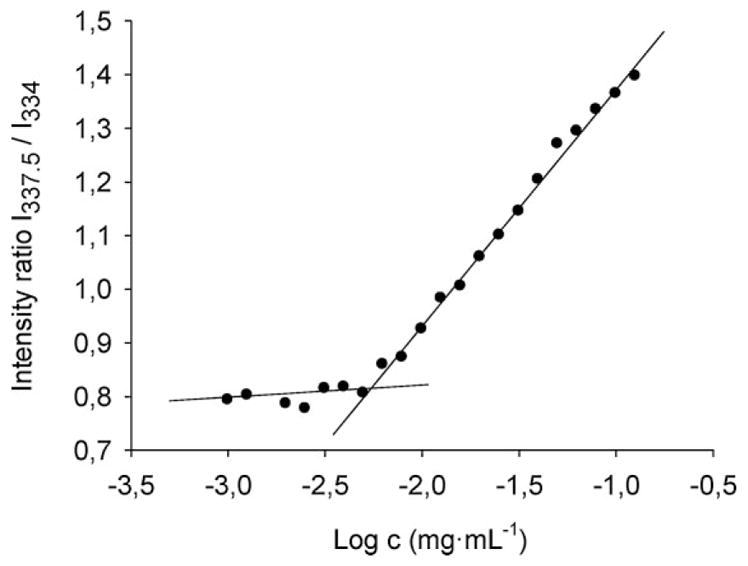
Determination of CMC of PCL-b-PHPMA micelles. Plot of the pyrene fluorescence intensity ratio I337.5/I334 vs. the logarithm of the copolymer concentration. The crossover point of the two straight lines describes the CMC.
According to these results, the developed PCL-b-PHPMA micelles had a low CMC value compared to values reported earlier in literature.[19,40,41] Thus, the micelles possessed a high thermodynamic stability. This might be the result of two factors: a) the relatively high molecular weight of the PCL block, which resulted in strong hydrophobic interaction within the hydrophobic core; and b) the presence of hydroxyl groups in the PHPMA block which can form H-bonds within the hydrophilic micellar shell.
Preparation of 3-Cl-AHPC-OMe-Loaded Micelles
Targeting of the Nur77/Bcl-2 pathway has emerged as an attractive anticancer therapeutic approach based on targeting of mitochondria and induction of apoptosis, due to the simultaneous suppression of antiapoptotic effects and activation of proapoptotic effects.[42–44] Novel retinoids, such as 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalenecarboxylic acid (AHPN), have been identified that activate this pathway and initiate apoptosis.[45] A series of AHPN analogs has been synthesized and tested, of which (E)-4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorocinnamic acid (3-Cl-AHPC) is well characterized.[46–49] Consequently, its ester was used in this study.
The physical entrapment of 3-Cl-AHPC-OMe was performed by a dialysis technique.[5,30,40] Polymer and drug were both dissolved in DMF and dialysed against DI water. The gradual replacement of DMF by water triggered the micellation of the copolymer and the incorporation of drug in the hydrophobic micelle core. Drug, which was not incorporated in the micelle core was removed by filtration through a PVDF filter. The clear filtrate was then lyophilized. The drug concentration incorporated into the micelle core was analysed by RP-HPLC using a calibration curve, and gave a drug loading content of 25.1 wt.-%. The higher content of drug loading, when compared with published data,[19] may be caused by high hydrophobicity of 3-Cl-AHPC-OMe and longer PCL block.
Characterization of PCL-b-PHPMA Micellar Systems by DLS and TEM
Figure 4A shows the particle size distribution of PCL-b-PHPMA micelles measured by DLS. The mean effective diameter was 62.45 ± 2.97 nm with a polydispersity (PD) of 0.243 ± 0.029. Figure 4B shows the particle size distribution of 3-Cl-AHPC-OMe-loaded PCL-b-PHPMA micelles measured by DLS. The mean effective diameter was 116.58 ± 2.68 nm with a PD of 0.227 ± 0.026. Incorporation of 3-Cl-AHPC-OMe into the hydrophobic micelle core resulted in an almost doubled effective diameter. The DLS diagram of 3-Cl-AHPC-OMe-loaded PCL-b-PHPMA micelles showed a unimodal size distribution without secondary micelle aggregates. Dialysis is an equilibration method to prepare micelles. At comparable preparation conditions, the size of the micelles will be affected by the hydrophobic/hydrophilic balance. Addition of a free hydrophobic drug will shift the balance to higher hydrophobicity, resulting in increased size of micelles. The size and shape of both drug free and drug-loaded micelles was analysed by TEM. Both micelle types were spherical. Figure 5 shows a TEM image of 3-Cl-AHPC-OMe-loaded PCL-b-PHPMA micelles. The diameter detected by TEM was slightly lower than the diameter measured by DLS, because the TEM measurements were performed in dry state. Therefore, these micelles had no hydrated shell in contrast to the micelles in the DLS measurements. In general, the result from the DLS measurement was in agreement with the result from TEM measurement.
Figure 4.

Size distribution of micelles as determined by DLS. A) Empty micelles: effective diameter 62.5 nm, polydispersity 0.243; B) 3-Cl-AHPC-OMe-loaded PCL-b-PHPMA micelles: effective diameter 116.6 nm, polydispersity 0.227.
Figure 5.
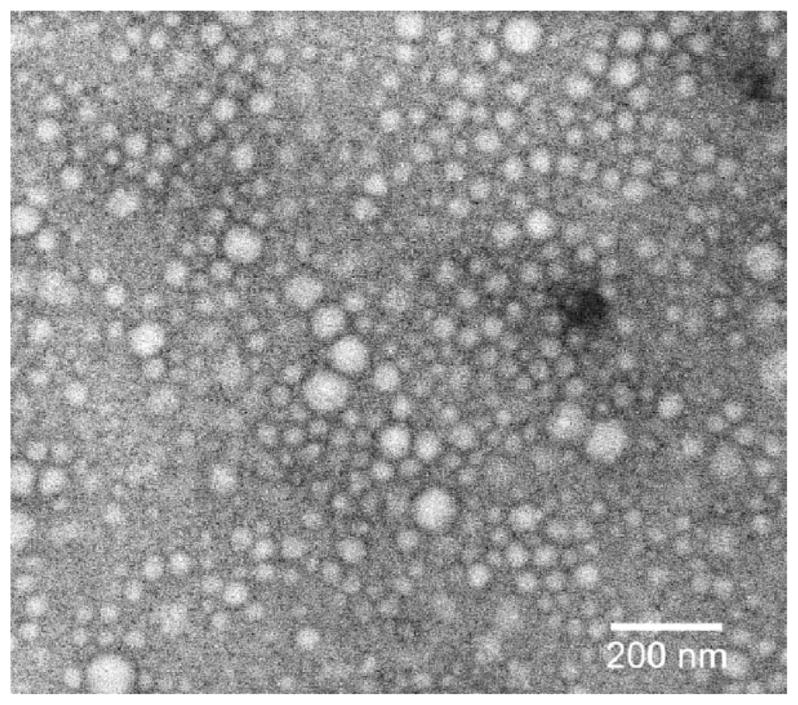
TEM image of 3-Cl-AHPC-OMe-loaded PCL-b-PHPMA micelles prepared in DI water (1.0 mg · mL−1) at room temperature.
In vitro 3-Cl-AHPC-OMe Release
The drug release from polymeric micelles was frequently studied by a method where drug-loaded micelles were entrapped inside a dialysis bag and only the released drug could pass the dialysis membrane. This method was used for several model drugs, e.g. for indomethacin,[30] doxorubicin,[40] paclitaxel,[41] and docetaxel.[50] However, because of the very hydrophobic nature of 3-Cl-AHPC-OMe, it was not possible to detect any free drug outside the dialysis bag after incubation of 3-Cl-AHPC-OMe-loaded PCL-b-PHPMA micelles. Thus, a new method was developed and validated for the release measurements. Here, free drug and drug-loaded micelles were separated by a 0.45 μm PVDF filter and washed with 2 different eluents: first PBS, followed by ethanol.
Samples (100 μL) were withdrawn from the incubation vial that contained drug-loaded micelles incubated in PBS with an Eppendorf pipette, diluted with 1 mL PBS and injected into a syringe and filtered through a PVDF filter. The filter was washed with PBS (3 × 1 mL). In doing so, drug-loaded micelles passed the filter membrane with PBS. However, the released free drug was sticking to the filter membrane. In order to elute the attached drug from the filter membrane, the filter was washed with ethanol (4 × 1 mL), which dissolved the drug. The drug content in the 4 mL PBS fraction and the 4 mL ethanol fraction were then measured by RP-HPLC and the amount of released drug was calculated. It is worth mentioning that the total drug concentration (drug content in PBS fraction + ethanol fraction) was constant, but the drug concentration in the PBS fraction decreased whereas the drug concentration in the ethanol fraction increased over time.
Before release measurements were performed, the freeze-dried drug-loaded micelles were dissolved in PBS and directly filtered through a PVDF filter in order to completely remove all free drug molecules, which may be released during the freeze-drying process. The same was done before the cytotoxicity assay. The exact drug concentration of the micelle solution was measured by RP-HPLC immediately after filtration.
Several experiments were performed in order to validate the separation of free 3-Cl-AHPC-OMe from 3-Cl-AHPC-OMe-loaded PCL-b-PHPMA micelles by a PVDF filter. For instance, free drug was dissolved in DMF and dialyzed against DI water before filtration in order to get comparable conditions. After dialysis, it was possible to filter out 99.91 wt.-% of the drug from the dialysate, composed of drug saturated solution and drug suspension, at a concentration range similar to that used for the release experiments. Moreover, it was shown that drug-loaded micelles totally passed the filter in the first 3 mL of the PBS eluent (no drug-loaded micelles were eluted after the first 3 mL of PBS) and the free drug was washed out with the first 2 mL of the pure ethanol eluent (no free drug was eluted after first 2 mL of ethanol). In addition, freshly prepared drug-loaded micelle solution easily passed the PVDF filter membrane with PBS as eluent without any drug release induced by the filter membrane. After the micelles passed the membrane with PBS, the membrane was washed with ethanol. No drug was detected in the ethanol; this means that the freshly prepared drug-loaded micelles can pass through the filter quantitatively.
As shown in Figure 6, a fast release of 3-Cl-AHPC-OMe occurred during the first hour, which plateaued between 12–14 wt.-% released drug during the next several hours. After 24 h, 14.69 wt.-% of the incorporated drug was released. The stability during this time interval is important for passive targeting by the EPR effect. The release is expected to occur via diffusion within 24 h. After incubation for 24 h, the drug release quickly increased but slowed down after 72 h when 73.09 wt.-% drug was released. After 168 h, 93.23 wt.-% drug was released from the PCL-b-PHPMA micelle core. Apparently, after 24 h, the micelle becomes destabilized and releases 3-Cl-AHPC-OMe faster (Figure 6). The destabilization may be caused by hydrolysis. Although the degradation of bulk hydrophobic PCL is slow, the PHPMA block is linked to PCL block via an ester bond. Due to the hydrophilicity of PHPMA, the link point ester bond is accessible to water, and thus susceptible to hydrolysis. A loss of PHPMA blocks may destabilize the micelles. The release profile can be manipulated by the change in the molecular weight of the hydrophilic and hydrophobic blocks.[34,51,52] However, the release profile resulted in reasonable cytotoxicity in vitro (see below). The drug can sustain release for 7 d with a fast release on day 2 and day 3 (Figure 6), which may ensure stability for passive targeting and keep a higher drug concentration after reaching the target site. However, only in vivo experiments on an animal model will decide if adjustments of molecular weight of the blocks will be needed.
Figure 6.
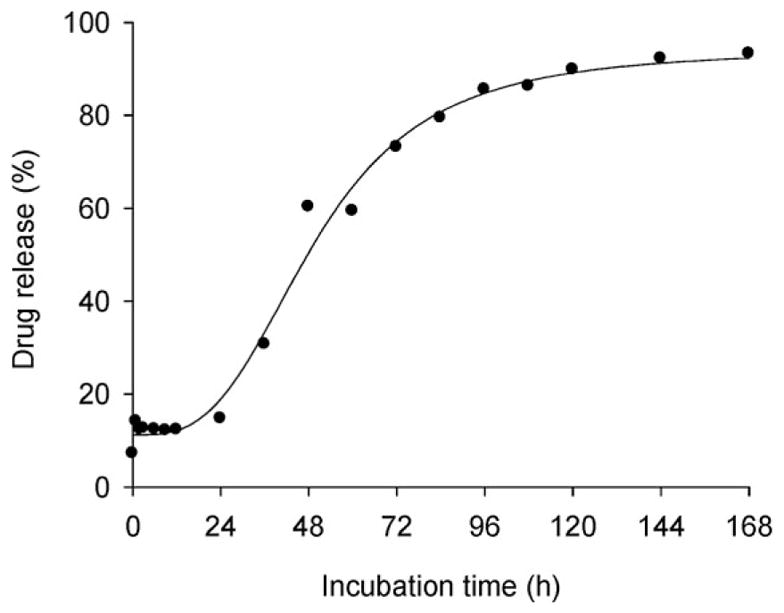
In vitro release of 3-Cl-AHPC-OMe from PCL-b-PHPMA polymeric micelles in PBS, pH 7.4 at 37 °C.
In vitro Cytotoxicity Assay
The cytotoxicity of 3-Cl-AHPC-OMe-loaded PCL-b-PHPMA micelles and free 3-Cl-AHPC-OMe was tested on C4-2 prostate carcinoma cell line, a subline of LNCap that is one of the most commonly used prostate cancer cells.[53] Figure 7 shows the C4-2 cell viability curves of free drug and drug-loaded micelles following incubation times of 24 h, 48 h, and 72 h. As expected, the IC50 dose decreased with incubation time for both formulations (Table 1), in agreement with published data.[32] The difference between the IC50 dose of free and micellar drug decreased with increasing incubation time. This reflects two mechanisms of cell entry, diffusion and endocytosis. All free drug molecules could cross the plasma membrane by diffusion. On the other hand, the micellar drug shared two pathways: a) endocytosis, followed by release of drug from the micelles within the subcellular organelles and diffusion via the endosomal/lysosomal membrane into the cytoplasm; and b) the fraction of drug that was released from micelles into the incubation media before internalization could participate in the diffusion-type of cell entry. The pure PCL-b-PHPMA micelles without drug did not show cytotoxicity at any concentration evaluated.
Figure 7.

Cytotoxicity of free (empty squares) and micellar (full circles) 3-Cl-AHPC-OMe to human prostate cancer C4-2 cells following different times of incubation: A) 24 h; B) 48 h; C) 72 h.
Table 1.
Cytotoxicity of free 3-Cl-AHPC-OMe and PCL-b-PHPMA micelle-loaded 3-Cl-AHPC-OMe for different incubation times.
| Formulation | IC50a) [μM] (mean ± SD)
|
|||
|---|---|---|---|---|
| 12 h | 24 h | 48 h | 72 h | |
| Free 3-Cl-AHPC-OMe | 45.02 ± 6.47 | 28.52 ± 4.55 | 3.71 ± 1.09 | 1.71 ± 0.17 |
| 3-Cl-AHPC-OMe-loaded PCL-b-PHPMA micelles | –b) | 58.92 ± 10.94 | 6.78 ± 0.62 | 1.96 ± 0.23 |
Values are expressed as mean 50% inhibition concentration (IC50) values with standard deviation (SD) calculated from 3 replicas (2 for the 72 h points);
Cytotoxicity was too low for IC50 determination.
In the cytotoxicity assay, the concentration of drug-loaded micelles may reach values lower than the CMC. But the stability of polymeric micelles is controlled by both thermodynamic stability and kinetic stability. Hydrogen bonds in PHPMA shell and crystallinity in PCL core will largely improve the kinetic stability. Accordingly, the micelles, even at concentration lower than CMC, will not immediately dissociate.
The higher IC50 dose of a drug delivery system when compared to free drug is the result of different mechanisms of cell entry and was generally observed with polymer-based systems. However, when the cytotoxicity of free and polymer-bound drugs was compared based on intracellular concentration of the drug a similar cytotoxicity was observed.[54] More importantly, a higher efficiency of polymer-bound drugs than free drugs in vivo was observed.[55] Thus the advantages of drug-loaded micelles for an in vivo situation will be: increased solubility of the hydrophobic drug; enhanced accumulation in solid tumors due to EPR effect; and different subcellular pharmacokinetics and decrease in non-specific side-effects due to participation of endocytosis in the internalization.
Conclusion
In summary, the studied PCL-b-PHPMA micelles appear to be promising biocompatible, non-immunogenic vehicles for drug delivery. The favorable hydrophobic/hydrophilic balance of the designed amphiphilic block copolymer provided a novel micelle drug delivery system with improved solubility for the hydrophobic 3-Cl-AHPC-OMe and controlled drug release. The cytotoxicity of 3-Cl-AHPC-OMe towards prostate cancer cells via the Nur77/Bcl-2 apoptosis induction pathway is a promising apoptosis induction mechanism for the development of new anticancer agents with higher potency.
Acknowledgments
The research was supported in part by NIH grant CA132831. C4-2 prostate cancer cells were kindly provided by Prof. Ursula Elsässer-Beile, University of Freiburg, Germany. S. K. was an exchange student from the Philipps University Marburg, Germany. His visit was within the framework of the Global Pharmaceutical Education Network (GPEN).
Contributor Information
Stefan G. Krimmer, Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, Utah 84112, USA
Huaizhong Pan, Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, Utah 84112, USA.
Jihua Liu, Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, Utah 84112, USA.
Jiyuan Yang, Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, Utah 84112, USA.
Jindřich Kopeček, Email: jindrich.kopecek@utah.edu, Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, Utah 84112, USA. Department of Bioengineering, University of Utah, Salt Lake City, Utah 84112, USA.
References
- 1.Maeda H. Bioconjugate Chem. 2010;21:797. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- 2.Talleli M, Rikken CJF, van Nostrum CF, Storm G, Hennink WE. Adv Drug Delivery Rev. 2010;62:231. doi: 10.1016/j.addr.2009.11.029. [DOI] [PubMed] [Google Scholar]
- 3.Nishiyama N, Kataoka K. Adv Polym Sci. 2006;193:67. [Google Scholar]
- 4.Riess G. Progr Polym Sci. 2003;28:1107. [Google Scholar]
- 5.Wang F, Bronich TK, Kabanov AV, Rauh RD, Roovers J. Bioconjugate Chem. 2005;16:397. doi: 10.1021/bc049784m. [DOI] [PubMed] [Google Scholar]
- 6.Vakil R, Kwon GS. Langmuir. 2006;22:9723. doi: 10.1021/la061408y. [DOI] [PubMed] [Google Scholar]
- 7.Hagan SA, Combes AGA, Carnett MC, Dunn SE, Davies MC, Illum L, Davis SS, Harding SE, Purkiss S, Gellert PR. Langmuir. 1996;12:2153. [Google Scholar]
- 8.Kwon GS, Naito M, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Langmuir. 1993;9:945. [Google Scholar]
- 9.Batrakova EV, Kabanov AV. J Controlled Release. 2008;130:98. doi: 10.1016/j.jconrel.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benahmed A, Ranger M, Leroux J. Pharm Res. 2001;18:323. doi: 10.1023/a:1011054930439. [DOI] [PubMed] [Google Scholar]
- 11.Molineux G. Cancer Treat Rev. 2002;28:13. doi: 10.1016/s0305-7372(02)80004-4. [DOI] [PubMed] [Google Scholar]
- 12.Harris JM, Chess RB. Nat Rev Drug Disc. 2003;2:214. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 13.Laverman P, Brouwers AH, Dams ETM, Oyen WJG, Storm G, van Rooijen N, Corstens FHM, Boerman OC. J Pharmacol Exp Ther. 2000;293:996. [PubMed] [Google Scholar]
- 14.Ishida T, Kiwada H. Int J Pharm. 2008;354:56. doi: 10.1016/j.ijpharm.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Kopeček J, Kopečková P. Adv Drug Delivery Rev. 2010;62:122. doi: 10.1016/j.addr.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamei S, Kopeček J. Pharm Res. 1995;12:663. doi: 10.1023/a:1016247206531. [DOI] [PubMed] [Google Scholar]
- 17.Lääne A, Aaviksaar A, Haga M, Chytrý V, Kopeček J. Makromol Chem Suppl. 1985;9:35. [Google Scholar]
- 18.Koňák Č, Ganchev B, Teodorescu M, Matyjaszewski K, Kopečková P, Kopeček J. Polymer. 2002;43:3735. [Google Scholar]
- 19.Lele BS, Leroux JC. Polymer. 2002;43:5595. [Google Scholar]
- 20.Lele BS, Leroux J. Macromolecules. 2002;35:6714. [Google Scholar]
- 21.Chytil P, Etrych T, Koňák Č, Šírová M, Mrkvan T, Říhová B, Ulbrich K. J Controlled Release. 2006;115:26. doi: 10.1016/j.jconrel.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 22.Chytil P, Etrych T, Koňák Č, Šírová M, Mrkvan T, Bouček J, Říhová B, Ulbrich K. J Controlled Release. 2008;127:121. doi: 10.1016/j.jconrel.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 23.Hrubý M, Koňák Č, Kučka J, Větřík M, Filippov SK, Vetvička D, Macková H, Karlsson G, Edwards K, Říhová B, Ulbrich K. Macromol Biosci. 2009;9:1016. doi: 10.1002/mabi.200900083. [DOI] [PubMed] [Google Scholar]
- 24.Jia Z, Wong L, Davis TP, Bulmus V. Biomacromolecules. 2008;9:3106. doi: 10.1021/bm800657e. [DOI] [PubMed] [Google Scholar]
- 25.Soga O, van Nostrum CF, Fens M, Rijcken CJF, Schiffelers RM, Hennink WE. J Controlled Release. 2005;103:341. doi: 10.1016/j.jconrel.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 26.Cincinelli R, Dallavalle S, Nannei R, Carella S, De Zani D, Merlini L, Penco S, Garattini E, Giannini G, Pisano C, Vesci L, Carminati P, Zuco V, Zanchi C, Zunino F. J Med Chem. 2005;48:4931. doi: 10.1021/jm049440h. [DOI] [PubMed] [Google Scholar]
- 27.Kopeček J, Bažilová H. Eur Polym J. 1973;9:7. [Google Scholar]
- 28.Mitsukami Y, Donovan MS, Lowe AB, McCormick CL. Macromolecules. 2001;34:2248. [Google Scholar]
- 29.Wilhelm M, Zhao CL, Wang Y, Xu R, Winnik MA, Mura JL, Riess G, Croucher MD. Macromolecules. 1991;24:1033. [Google Scholar]
- 30.La SB, Okano T, Kataoka K. J Pharm Sci. 1996;85:85. doi: 10.1021/js950204r. [DOI] [PubMed] [Google Scholar]
- 31.Kalyanasundaram K, Thomas JK. J Am Chem Soc. 1977;99:2039. [Google Scholar]
- 32.Cuchelkar V, Kopečková P, Kopeček J. Mol Pharm. 2008;5:776. doi: 10.1021/mp800019g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen C, Yu Y, Maysinger D, Eisenberg A. Bioconjugate Chem. 1998;9:564. doi: 10.1021/bc9702157. [DOI] [PubMed] [Google Scholar]
- 34.Shuai X, Ai H, Nasongkla N, Kim S, Gao J. J Controlled Release. 2004;98:415. doi: 10.1016/j.jconrel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Lee H, Zeng F, Dunne M, Allen C. Biomacromolecules. 2005;6:3119. doi: 10.1021/bm050451h. [DOI] [PubMed] [Google Scholar]
- 36.Torchilin VP. Medical Applications, editor. In: Polymer-based Nanostructures. Broz P, editor. Royal Society of Chemistry; 2010. p. 261. [Google Scholar]
- 37.Torchilin VP. J Controlled Release. 2001;73:137. doi: 10.1016/s0168-3659(01)00299-1. [DOI] [PubMed] [Google Scholar]
- 38.Astafieva I, Zhong XF, Eisenberg A. Macromolecules. 1993;26:7339. [Google Scholar]
- 39.Jones M, Leroux J. Eur J Pharm Biopharm. 1999;48:101. doi: 10.1016/s0939-6411(99)00039-9. [DOI] [PubMed] [Google Scholar]
- 40.Xu X, Li L, Zhou J, Lu S, Yang J, Yin X, Ren J. Colloids Surf B: Biointerfaces. 2007;55:222. [Google Scholar]
- 41.Zhang C, Qineng P, Zhang H. Colloids Surf B: Biointerfaces. 2004;39:69. doi: 10.1016/j.colsurfb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 42.Zhang X. Exp Opin Therap Targets. 2007;11:69. doi: 10.1517/14728222.11.1.69. [DOI] [PubMed] [Google Scholar]
- 43.Lin B, Kolluri SK, Lin F, Liu W, Han YH, Cao X, Dawson MI, Reed JC, Zhang XK. Cell. 2004;116:527. doi: 10.1016/s0092-8674(04)00162-x. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Kolluri SK, Gu J, Dawson MI, Cao X, Hobbs PD, Lin B, Chen G, Lu J, Lin F, Xie Z, Fontana JA, Reed JC, Zhang X. Science. 2000;289:1159. doi: 10.1126/science.289.5482.1159. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, Lin B, Agadir A, Liu R, Dawson MI, Reed JC, Fontana JA, Bost F, Hobbs PD, Zheng Y, Chen GQ, Shroot B, Mercola D, Zhang XK. Mol Cell Biol. 1998;18:4719. doi: 10.1128/mcb.18.8.4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Dawson MI, Ning Y, Polin L, Parchment RE, Corbett T, Mohamed AN, Feng KC, Farhana L, Rishi AK, Hogge D, Leid M, Peterson VJ, Zhang XK, Mohammad R, Lu JS, Willman C, VanBuren E, Biggar S, Edelstein M, Eilender D, Fontana JA. Blood. 2003;102:3743. doi: 10.1182/blood-2003-01-0108. [DOI] [PubMed] [Google Scholar]
- 47.Farhana L, Dawson MI, Huang Y, Zhang Y, Rishi AK, Reddy KB, Freeman RS, Fontana JA. Oncogene. 2004;23:1874. doi: 10.1038/sj.onc.1207311. [DOI] [PubMed] [Google Scholar]
- 48.Dawson MI, Harris DL, Liu G, Hobbs PD, Lange CW, Jong L, Bruey-Sedano N, James SY, Zhang XK, Peterson VJ, Leid M, Farhana L, Rishi AK, Fontana JA. J Med Chem. 2004;47:3518. doi: 10.1021/jm030524k. [DOI] [PubMed] [Google Scholar]
- 49.Dawson MI, Hobbs PD, Peterson VJ, Leid M, Lange CW, Feng K-C, Chen G, Gu J, Li H, Kumar Kolluri S, Zhang X-K, Zhang Y, Fontana JA. Cancer Res. 2001;61:4723. [PubMed] [Google Scholar]
- 50.Yang M, Ding Y, Zhang L, Qian X, Jiang X, Liu B. J Biomed Mater Res A. 2007;81:847. doi: 10.1002/jbm.a.31129. [DOI] [PubMed] [Google Scholar]
- 51.Kim SY, Shin IG, Lee YM, Cho CS, Sung YK. J Controlled Release. 1998;51:13. doi: 10.1016/s0168-3659(97)00124-7. [DOI] [PubMed] [Google Scholar]
- 52.Park EK, Kim SY, Lee SB, Lee YM. J Controlled Release. 2005;109:158. doi: 10.1016/j.jconrel.2005.09.039. [DOI] [PubMed] [Google Scholar]
- 53.Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Int J Cancer. 1994;57:406. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- 54.Minko T, Kopečková P, Kopeček J. Pharm Res. 1999;16:986. doi: 10.1023/a:1018959029186. [DOI] [PubMed] [Google Scholar]
- 55.Minko T, Kopečková P, Kopeček J. Int J Cancer. 2000;86:108. doi: 10.1002/(sici)1097-0215(20000401)86:1<108::aid-ijc17>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]