

Abstract
An enantioselective synthesis of the indole diterpenoid natural product paspaline is disclosed. Critical to this approach was the implementation of stereoselective desymmetrization reactions to assemble key stereocenters of the molecule. The design and execution of these tactics are described in detail, and a thorough analysis of observed outcomes is presented, ultimately providing the title compound in high stereopurity. This synthesis provides a novel template for preparing key stereocenters in this family of molecules, and the reactions developed en route to paspaline present a series of new synthetic disconnections in preparing steroidal natural products.
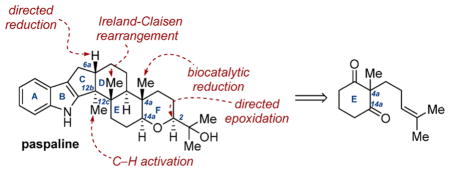
INTRODUCTION
Production of novel metabolites by the ergot fungus has been well-documented.1 Most notably, those produced by Claviceps purpurea have long been implicated in the contamination of various grains.2 Claviceps paspali, another species in this genus, has been linked to “paspalum stagger” poisoning in livestock,3 and it was from this fungus that Arigoni and co-workers isolated paspaline (1, Figure 1) and paspalicine (4), the first of a now extensive family of indole diterpene alkaloid natural products.4 A diverse range of related structures have since been reported including paspaline B (2),5 paspalinine (3),6 JBIR-03 (5),7 and paxilline (6).8
Figure 1.

Paspaline and related indole diterpenoid natural products.
The varied biological profiles of these compounds have rendered them particularly attractive to the chemical industry. The recently discovered JBIR-03 has displayed significant inhibition of Valsa ceratosperma (MIC = 128 μg/mL) while showing no cytotoxic effects to the human fibrosarcoma cell line HT-1080 at 100 μM.7 Moreover, paspalinine and its derivatives have demonstrated marked activity as Maxi-K channel antagonists and, as a result, are under examination as treatments for Alzheimer’s disease and other neurological disorders.9 Paxilline is currently under study for its properties as a BK channel antagonist toward the suppression of seizures in postnatal mammals.10 From a standpoint of structure–activity, prior work by Cole has underscored the significance of the axial tert-hydroxyl functionality (C4b, paspaline numbering) as an important source of activity for these structures, evidenced by the lack of tremorgenicity demonstrated by paspaline and paspalicine.11
The absolute structure of 1 was confirmed in 1980 by Springer and Clardy on the basis of X-ray diffraction studies.11a Paspaline and its related compounds are characterized by their unique indole and tetrahydropyran (or derivatives thereof) ring fusions. Furthermore, grafted onto the D/E decalin core, three all-carbon quaternary atoms are encountered (C4a, C12b, C12c). These salient features necessitate careful planning for endeavors in total synthesis. These challenges were first addressed by the Smith laboratory,12 whose body of work in this area has defined the state of the art for the synthesis of paspaline and its related structures. Subsequent partial13 and total14 synthetic studies of these molecules have since been disclosed, building on these advances. As an extension to previous work in our laboratory in developing total synthesis platforms for complex molecular frameworks,15 we sought to develop an expedient synthesis of 1, particularly of the key C4a, C12b, and C12c stereocenters, which could serve as a template for assembly of the remaining structures in this family. Our work toward this goal culminated in a highly stereocontrolled total synthesis of paspaline.16 Herein, we disclose the entirety of our efforts, ultimately leading to the conception and implementation of two critical stereoselective desymmetrization reactions for facile target assembly. These studies have laid the groundwork for future investigations in this family of natural products.
Our preliminary synthetic plan for 1 began with translation to hydroxyalkene 7 (Scheme 1). The decalin functionality (D and E rings) in 7 would be constructed via a transannular ketone addition/Friedel–Crafts alkylation cascade arising from cyclodecenone 8,17 establishing the vicinal C12b and C12c quaternary centers in a single operation. The tetrasubstituted (E)-alkene in 8 would be prepared via intramolecular coupling of the corresponding diene 9 or dicarbonyl 10 via a metathesis18 or McMurry process.19 Synthesis of this ketone would rely on the union of fragments 11 and 12 to assemble the C6a, C6, and C5 carbon–carbon bonds. Access to the tetrahydropyran 12 was envisioned via an alkylation/Michael addition cascade between dimethylmalonate and 13 inspired by methodology developed by Gharpure.20
Scheme 1.

Preliminary Synthesis Plan for Paspaline
RESULTS AND DISCUSSION
In accordance with the above strategy, initial focus was placed on synthesizing the tetrahydropyranyl F ring and C2/C14a stereodiad in 1 (Scheme 2). In a forward sense, tosylation of the previously reported diol 14 followed by oxy-Michael addition and iodination furnished the requisite iodoalkene 13 in 62% yield over three steps,21 setting the stage for the proposed annulation. Thus, treatment of 13 with CH2(CO2Me)2 and Cs2CO3 in DMF provided exclusively the desired 2,6-cis-pyran in 99% yield and >20:1 dr. Selective reduction of the ethyl ester in 17 proceeded smoothly to give alcohol 18 in 72% yield, and subsequent iodination and alkylation installed the requisite alkene in 12. With this compound in hand, we turned our attention to desymmetrization of the C4a gem-diester in 12 via nucleophilic addition. Experiments with this compound revealed a strong diastereotopic group bias for the equatorial ester, giving the desired relative stereochemistry at C4a.22 To enable maximum flexibility in the downstream strategy, the corresponding carboxylic acid 19, methyl ketone 20, and enone 21 were prepared.
Scheme 2. Synthesis of Tetrahydropyranyl F Ring and C4a Stereocentera.

aReagents and conditions: (a) TsCl, NEt3, DMAP (10 mol %), CH2Cl2, 0 °C; (b) N-methylmorpholine, ethyl propiolate, CH2Cl2, rt; (c) NaI, acetone, rt; (d) CH2(CO2Me)2, Cs2 CO3, DMF, rt; (e) DIBAL-H, THF, 0 °C; (f) (i) I2, PPh3, imidazole, CH2Cl2, 0 °C to rt; (ii) (isopropenyl)2CuLi, Et2O, −78 to 0 °C; (g) KOH, THF/MeOH (1.75:1), rt; (h) MeLi, THF, −78 °C; (i) (i) EtLi, THF, −78 °C; (ii) LDA, THF, −78 °C, then PhSeBr; (iii) H2O2(aq), CH2Cl2, 0 °C.
With the pyran subunit in place, the next challenge became introduction of the indole fragment bearing the atoms necessary for cyclodecenone synthesis (Scheme 3); however, we found this union to be significantly more challenging than first expected. In the first iteration, Michael addition of the enolate of 20 to the indole-derived enone 2223 using a variety of bases (LDA, LHMDS, NaOMe) showed no productive reactivity, presumably due to low reactivity of enone 22. Mukaiyama Michael addition to 22 using the enolsilane derived from 20 resulted in rapid desilylation prior to engaging 22 under all conditions examined. Methyl vinyl ketone also failed to react with 20 under these conditions. An alternative strategy explored reversal of the nucleophile/electrophile identities via the reaction of enolsilane 24 and pyranyl enone 21. However, exposure of these compounds to Lewis acidic conditions (BF3·OEt2, TiCl4, Cu(OTf)2, etc.) resulted only in desilylation of 24 and decomposition of enone 21. Finally, a Lewis acid promoted ene reaction was examined as a method for the union of enone 21 and nucleophilic alkene 25; unfortunately, the inherent instability of enone 21 remained problematic in this approach. These failed efforts led us to conclude that direct intermolecular coupling methodologies of these fragments to 1 from the C4a functionality were prohibitively challenging, and as a result, this approach was abandoned.
Scheme 3.

Unsuccessful Approaches to C5–C6a Bond Construction
In an effort to circumvent the issues associated with the above strategy, we postulated that an intramolecular approach to the critical bond disconnection might be more facile (Scheme 4). This process would be enabled via appendage of the appropriate functionality to the iodide 27 (which had been synthesized previously in the described route to alkene 12). We selected 2-methyl-1,3-cyclohexanedione 28 as this nucleophile, anticipating that Krapcho decarboxylation of the corresponding alkylation product 29 might initiate an intramolecular aldol addition process to assemble the D,E ring decalin moiety as well as the C12c and C4b stereocenters (33). In practice, alkylation of iodide 27 with 28 gave a ~1:2 mixture of diketone 29 and the undesired O-alkylation product 30 in 34 and 56% yields, respectively.23 While this issue of regiochemistry rendered material throughput challenging, we carried on in the interest of validating the proposed downstream reactivity. Operating first on small scale (15 mg), treatment of diketone 29 with NaCl in DMSO afforded a ~1:1 ratio of the Krapcho adduct 31 and the cyclization product 32 as a single diastereomer. However, a single-crystal X-ray diffraction study revealed 32 to be the undesired cis-decalinone product (e.g., epimeric at C12c). Fortunately, formation of 32 was suppressed when the reaction was further scaled (70 mg), giving exclusively the Krapcho adduct 31 in 43% yield. In hopes that a stepwise Krapcho/aldol process might proceed with selectivity orthogonal to 32, we began screening conditions for the conversion of 31 to 33. Toward this aim, treatment of 31 with Brønsted or Lewis acidic conditions gave either no reaction or starting material decomposition upon heating. Alternatively, exposure to basic conditions resulted in no reaction or retro-Dieckmann decomposition of the dione functionality.
Scheme 4. Decarboxylative Annulation Approach to Paspaline D,E Ringsa.

aReagents and conditions: (a) Cs2CO3, DMF, 65 °C; (b) NaCl, DMSO, 150 °C.
Having arrived at another critical impasse, we began to question the viability of this route in providing access to 1. While the alkylation/Michael cascade sequence (13 → 17) provided expedient access to the F ring tetrahydropyran stereochemistry and desymmetrization of the C4a stereocenter proceeded as planned, further elaboration of this material to 1 seemed an unlikely venture. At this critical stage in our studies, we began to examine alternative points of initiation for our synthesis (Scheme 5).
Scheme 5.

Revised Approach to 1 via Enantioselective Desymmetrization
Guided by our previous work in developing symmetry-breaking processes to enable rapid construction of complex natural products,15g,h we surmised that a synthesis beginning from desymmetrization of a paspaline E ring precursor might circumvent the problems associated with our initial strategy. It is important to note at this juncture that Smith’s synthesis of 1 also commences via a symmetry-breaking process;12a namely, the Wieland–Miescher ketone synthesis (28 → 34) assembles the D–E ring fusion of 1 concomitant with the C12c quaternary stereocenter. While this reaction is a classic “single stereocenter” desymmetrization, we envisioned an alternative E ring desymmetrization arising from stereoselective monoreduction of functionalized diketone 35. Reduction of this compound would establish the stereochemical identity of C4a and C14a in 36 in a single operation while supplying the needed functional handles for tetrahydropyran assembly and synthesis completion. Armed with this new hypothesis, we refocused our efforts in the synthesis of 1 via this approach.
The first challenge in our revised synthesis plan was preparation of the desymmetrization precursor 35 via alkylation of dione 28 or its derivatives (Scheme 6). In the event, deprotonation of 28 with NaH followed by addition of iodide 37 provided the desired cycloalkanone 35 in 7% yield along with 26% of the undesired O-alkylation product 38. This result was not entirely unexpected: challenges associated with regioselective C-alkylation of cyclic α-dicarbonyls have been well-documented.24 In hopes of enhancing C-nucleophilicity of this structure, we prepared hydrazone 39.24 Screening of conditions revealed that enolization with KH followed by addition of iodide 37 provided exclusively the corresponding C-alkylation adduct which, following hydrazone deprotection, afforded functionalized diketone 35 in 76% yield over two steps. Of particular importance is the scalability of this process: diketone 35 can be prepared in >10 g scale in a single batch. This reaction represents a useful advance over prior art in preparing this compound,25 and the scope of this method is currently under study.
Scheme 6. Desymmetrization Approach to 1: E,F Ring Synthesisa.
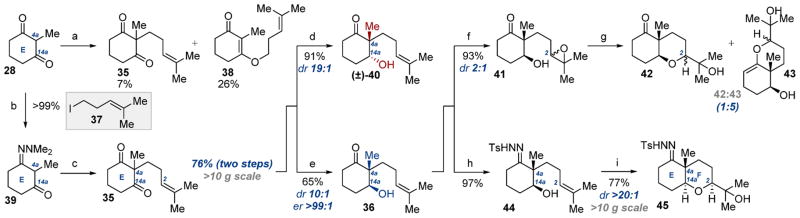
aReagents and conditions: (a) NaH, DMF, 0 °C, then 37, rt; (b) H2NNMe2, TsOH (3.0 mol %), C6H6, 100 °C; (c) (i) KH, THF, 0 °C, then 37, −78 °C to rt; (ii) Cu(OAc)2, THF/H2O (1:1), rt; (d) NaBH4, MeOH, 0 °C; (e) YSC-2, H2O/DMSO (10:1), 30 °C; (f) m-CPBA, CH2Cl2, 0 °C; (g) PPTS (20 mol %), CH2Cl2, rt; (h) TsHNNH2, C7H8, 70 °C; (i) m-CPBA, CH2Cl2, 0 °C, then PPTS.
With the critical desymmetrization precursor in our possession, we began investigating selective monoreduction of 35 to access the C4a–C14a stereodiad. Treatment of 35 with NaBH4 provided the racemic monoreduction product 40 with excellent yield and diastereoselectivity (19:1), albeit the opposite diastereomer to that desired. It is reasonable to expect formation of this diastereomer under strictly substrate-controlled conditions, although we were surprised by the magnitude of selectivity for this diastereomer. We were encouraged, however, by the recent reports of Nakada26 and Node27 which demonstrated access to the diastereomer needed for our synthesis on similar cyclic diketones using biocatalytic reducing conditions. In experimenting with our compound, we were pleased to find that monoreduction of 35 with yeast from Saccharomyces cerevisiae type 2 (YSC-2) proceeded with virtually complete reagent control, giving the desired alcohol diastereomer 36 in 65% yield, 10:1 dr, and >99:1 er. The success of this transformation provided encouragement to the viability of our revised synthesis plan and set the stage for further manipulation to 1.
From hydroxy olefin 36, we anticipated assembly of the tetrahydropyranyl F ring via an oxidative cyclization sequence. With this goal in mind, treating the alkene in 36 with m-CPBA provided the corresponding epoxide 41 in 93% yield and poor diastereoselectivity (2:1). While any number of asymmetric epoxidation methods could likely enhance this selectivity, of greater concern was that treatment of this diastereomeric mixture 41 with conditions requisite for ring closure (PPTS) gave an inseparable 5:1 mixture of products with the desired tetrahydropyran 42 as the minor product. The major material was identified as alcohol 43, the result of epoxide trapping by the enol tautomer of the ketone in 41. To circumvent this issue, we envisaged that masking the ketone in 36 would preclude this undesired mode of ring closure. Since it translated well to our downstream strategy for D ring construction, 36 was converted to the corresponding tosyl hydrazone 44 in 97% yield. To our surprise, the reaction of this compound with m-CPBA followed by PPTS initiated an epoxidation/cyclization cascade, providing the desired tetrahydropyran 45 directly in 77% yield and >20:1 dr. This reaction gave expedient preparation of the paspaline F ring in a single operation.
We were unaware of any previously reported directing effects of tosyl hydrazones on analogous systems (Scheme 7). To provide understanding to this difference in reactivity between hydroxyketone 36 and hydrazone 44, we carried out the following experiments. First, the alkene in hydrazone 44 was removed via hydrogenation to give alcohol 46. Treatment of 46 with the exact reaction conditions used in the epoxidation of 44 resulted in quantitative starting material recovery. This datum excluded the possibility of intramolecular oxygen delivery in the reaction via a transient oxazidirine such as 47. Concluding that the reactivity may be a consequence of underlying conformational differences between 36 and 44, we calculated both structures using density functional theory (DFT) at the level of B3LYP/6-311G(d).28 Interestingly, the optimized structures of 36 and 44 showed a significant difference in the dihedral angle about the C14a C–OH bond and the C4a C–CH2R bond (69° for 36 and 85° for 44). On the basis of these facts, we hypothesize that the observed selectivity is a consequence of the hydrazone in 44 imposing a favorable reactive conformation (48) on the cyclohexane such that the C14a hydroxyl is in close proximity to the alkene during the oxidation. It follows that this would enhance transfer of the substrate’s chiral information to C2 during the oxidation, giving the observed pyran 45 following ring closure. To the best of our knowledge, this reaction is the first example of an alkene epoxidation stereoselectivity being influenced by the presence of a tosyl hydrazone.29
Scheme 7. Mechanistic Investigations in the Conversion of 44 to 45a.

aReagents and conditions: (a) H2 (1 atm), Pd/C (1.50 mass equiv), MeOH, rt; (b) m-CPBA, CH2Cl2, 0 °C.
With assembly of the E and F rings complete, attention was directed to construction of the sterically congested D ring and C12c stereocenter (Scheme 8). We believed that the tosyl hydrazone in 45 would be engaged via the Shapiro reaction to produce a transient vinyllithium which, upon trapping with the appropriate electrophile, would provide the functionality required to meet these synthetic challenges.30
Scheme 8. Synthesis of Enone 53 and Attempts at D Ring Synthesisa.

aReagents and conditions: (a) TBSOTf, 2,6-lutidine, CH2Cl2, −50 °C; (b) n-BuLi, THF, −50 °C to rt, then DMF; (c) Ph3P=CH2, THF, 0 °C; (d) nitroethylene, CH2Cl2, 65 °C; (e) (i) KOH, MeOH, rt, then MsOH, 0 °C to rt; (ii) DBU, CH2Cl2, rt.
Thus, TBS protection of the tert-alcohol in 45 proceeded to give silyl ether 49 in 77% yield. Shapiro reaction of 49 followed by DMF trapping furnished unsaturated aldehyde 50 in 62% yield which, upon olefination, gave diene 51 poised for a Diels–Alder cycloaddition. Nitroethylene proved to be an effective dienophile in this reaction, giving the annulation product 52 in 94% yield and with complete regioselectivity under thermal conditions. Subsequent Nef reaction and alkene isomerization afforded the ketone 53, from which we envisioned manipulation of the alkene would complete D ring assembly to give 58. Accordingly, Birch reduction of 53 followed by electrophilic trapping with MeI furnished decalinone 54 in 67% yield and high stereoselectivity (>20:1). Unfortunately, this compound was identified as the undesired cis-decalinone (bearing the desired C4b stereochemistry and undesired C12c stereochemistry) via X-ray diffraction analysis of a derivative.31 After a screen of reducing metals, solvents, and addition methods showed no promise for over-riding this selectivity, we 2began exploring auxiliary methods for stereoselective introduction of the C12c methyl group. In the first iteration, Birch reduction of 53 followed by protic quenching and epimerization with DBU gave the trans-decalinone 55 as a single diastereomer. However, all attempts at thermodynamic methylation of this compound proved fruitless, giving either polymethylated products or starting material decomposition. We next examined whether the C12c methyl group could be introduced stereospecifically via an epoxidation/semipinacol reaction sequence. While epoxidation of 53 was achieved upon treatment with p-NPBA32 to give the desired oxirane 56 as a single diastereomer in 46% yield, the subsequent ketone methylation requisite for rearrangement consistently gave starting material recovery or decomposition under more forcing conditions. In a final case, the ketone in 53 was reduced upon treatment with LiAl(OtBu)3H to give alcohol 57 in 95% yield and 10:1 dr. From this compound, we pursued radical delivery of the C12c methyl group via tethering from the secondary hydroxyl.33 However, this approach also proved unsuccessful, as the alkene in 57 failed to engage all radical precursors bound to the alcohol.
Collectively, these reactions indicated that the inherent bias of enone 53 for the α-face of the D–E ring fusion (presumably influenced by the C4a angular methyl group) would preclude all attempts at late-stage introduction of the C12c methyl group. At this key juncture in our studies, we determined that if D ring assembly was preceded by introduction of this methyl group, then the subsequent annulation step might also proceed with α-face selectivity to give the requisite syn-diaxial methyl group relationship (Scheme 9). Thus, methylation of hydrazone 49 upon treatment with n-BuLi and MeI proceeded smoothly to give the monomethylated product 59 in excellent yield. In accordance with our Diels–Alder strategy, Shapiro reaction of 59 followed by trapping with DMF afforded aldehyde 60 in 61% yield, giving the diene 61 upon olefination. While we at first anticipated that the [4 + 2] annulation of 61 with nitroethylene would proceed in a manner similar to the previously described desmethyl cycloaddition (51 → 52), we quickly found the steric impact of the newly introduced methyl group to be much greater than expected. In our initial trials, the reaction of 61 with nitroethylene failed to produce cycloadduct 62 under both thermal and Lewis acidic conditions. An extensive screen of Diels–Alder dienophiles and promotors ensued, showing no further promise for D ring construction via this method. We then turned our attention to alternative annulation methods, making use of the flexibility of electrophile choice in the Shapiro reaction step and its subsequent intermediates. To bypass an intermolecular cycloaddition, we pursued an electrocyclization pathway to form the requisite D ring. Olefination of aldehyde 60 with the ylide derived from allyltriphenylphosphonium bromide gave the simplified triene 63 in 36% yield, and irradiation of 63 (Hg vapor lamp) gave complete conversion to a single product after 1 h. Unfortunately, this material was identified as the sigmatropic rearrangement product 65 and not the desired cyclization product 64. Suspecting that this rearrangement might predominate using any analogue of this triene, we abandoned this pathway in favor of alternative cyclization modes. Toward these aims, substrates 66–68 were prepared via modification of the electrophilic trap (and subsequent product manipulation) in the Shapiro reaction and examined for their viability in D ring synthesis. Electron-rich Diels–Alder diene 66 and Nazarov substrate 67 failed to participate in any productive reactivity, either giving no reaction or decomposing to complex mixtures. Iodide 68 was synthesized with the goal of completing D ring synthesis via cross-coupling; however, this approach also proved fruitless.
Scheme 9. Strategies Examined toward D Ring Synthesis via Methyl-Group-First Approacha.

aReagents and conditions: (a) n-BuLi, THF, −50 °C, then MeI; (b) n-BuLi, THF, −50 °C to rt, then DMF; (c) Ph3P=CH2, THF, 0 °C; (d) Ph3P=CHCHCH2, THF, 0 °C to rt; (e) hν, hexanes, rt.
Our options diminishing, we prepared primary alcohol 69 via trapping the Shapiro intermediate of 59 with (HCHO)n (Table 1). We surmised that the appropriately selected ester of 69 would participate in an Ireland–Claisen rearrangement,34 influenced by the C4a stereocenter, to install the C12c (and potentially C12b) quaternary methyl group(s) while providing functional handles for D ring construction. We then began screening esters of 69 compatible with our synthetic manifold. In the simplest cases, acetate 70a (entry 1) and propionate 70b (entry 2) did not undergo rearrangement as the corresponding silyl ketene acetals were labile at elevated reaction temperatures. Isobutyrate 70c (entry 3) performed exceptionally to give 71a (80% yield, 6:1 dr, 4 g scale), although a downstream C–H activation at C12b would be required for this product to be a viable intermediate toward 1. With the reaction’s viability demonstrated, functionalized esters 70d–h were probed. Indole ester 70d or protected analogues thereof failed to rearrange, presumably due to a steric impact of the indole on silyl ketene acetal generation. Esters 70e–g (entries 5–7) likewise suffered from the same issue. We were excited to find promising reactivity, however, in the case of silyl-functionalized isobutyrate 70h (entry 8, 52% yield, 6.6:1.1:1 dr). The stereochemistry at C12c of this compound was assigned by analogy to rearrangement product 71a (vide infra). The identity of the C12b stereocenter could not be identified.
Table 1.
Ireland–Claisen Screenings for D Ring Assembly
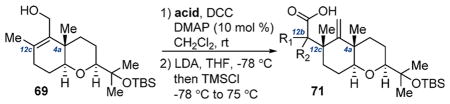
| |||
|---|---|---|---|
| entry | acid | ester (yield)a | acid (yield, dr)a |
| 1 | Ac2O | 70a (82%)b | -- |
| 2 |
|
70b (83%) | -- |
| 3 |
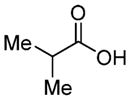
|
70c (73%) | 71a (80%, 6:1)c |
| 4 |
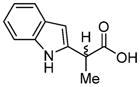
|
70d (93%) | -- |
| 5 |
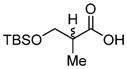
|
70e (63%) | -- |
| 6 |
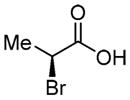
|
70f (74%) | -- |
| 7 |
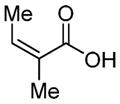
|
70g (54%) | -- |
| 8 |
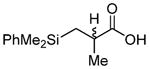
|
70h (87%) | 71b (57%, 6.6:1.1:1)c |
Isolated yields.
Conditions: Ac2O, NEt3, DMAP (10 mol %), CH2Cl2, rt.
Determined by 1H NMR analysis of crude mixtures.
The next portion of our strategy involved conversion of the rearrangement product to its methyl ketone for subsequent ring closure (Scheme 10). After first reoptimizing the Shapiro reaction step to facilitate one-pot conversion of desmethylhydrazone 49 to alcohol 69, we moved forward in this approach. Unfortunately, conversion of silyl-functionalized isobutyrate product 71b to its derived methyl ketone proved unfeasible due to a significant steric impact at the α-position. In contrast, early returns on the simpler isobutyrate rearrangement product 71a showed that the methyl ketone synthesis worked well, and as a result, we moved forward in our synthesis with this compound. Thus, esterification of acid 71a with TMSCHN2 followed by treatment with MeLi furnished ketone 72 in 84% yield. The C4b stereocenter was established via hydroboration/oxidation of 72 to give diol 73 in 74% yield and >20:1 dr. After some experimentation, bisoxidation of 73 was accomplished via Swern conditions to give ketoaldehyde 74 poised for intramolecular condensation. Exposure of 74 to basic conditions cleanly afforded enone 75 in 74% yield over two steps, thereby completing D ring synthesis. The resultant alkene was removed via hydrogenation to give the corresponding ketone, which was converted to oxime 76 in 82% yield.
Scheme 10. D Ring Synthesis Completion and Symmetry-Breaking C–H Activation of C12b Stereocentera.
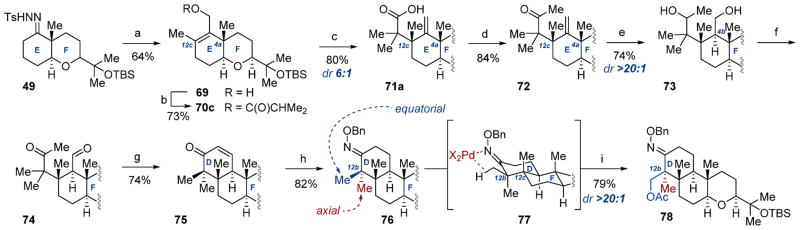
aReagents and conditions: (a) n-BuLi, THF, −50 °C, then MeI; n-BuLi, −50 °C to rt, then (HCHO)n; (b) isobutyric acid, DCC, DMAP (10 mol %), CH2Cl2, rt; (c) LDA, THF, −78 °C, then TMSCl, −78 to 75 °C; (d) (i) TMSCHN2, MeOH/C7H8 (2:1), rt; (ii) MeLi, Et2O, 0 °C to rt; (e) BH3T·HF, THF, 50 °C, then H2O2, NaOH, 0 °C to rt; (f) (COCl)2, DMSO, CH2Cl2, −78 °C, then DIPEA, −78 to 0 °C; (g) KOH(aq), THF/ MeOH (1:1), 0 °C to rt; (h) (i) H2 (1 atm), Pd/C (1.50 mass equiv), EtOAc, rt; (ii) NH2OBn·HCl, NaOAc, MeOH/H2O (5:1), 85 °C; (i) Pd(OAc)2 (15 mol %), PhI(OAc)2, AcOH/Ac2O (1:1), 100 °C.
With D ring synthesis concluded, desymmetrization of the nonstereogenic C12b dimethyl group in 76 became compulsory for synthesis completion. The success of this transformation would require a selective functionalization of the equatorial methyl group at C12b over its axial counterpart to provide the diastereomer needed; we were aware that the lowest energy conformer of 76 places the oxime C–N double bond in the same plane as the equatorial methyl group and anticipated that the appropriate catalytic system would operate on 76 using the oxime as a directing group. We selected the catalytic C–H oxidation reaction developed by Sanford and co-workers,35 which had demonstrated applicability to substituted cyclohexanone oximes. In the event, treatment of oxime 76 with Sanford’s conditions provided acetate 78 in 79% yield (via 77) with complete diastereoselection, establishing the stereochemistry of the final quaternary center in 1 and providing the necessary functional handle for synthesis completion.
The yield and selectivity of this transformation are noteworthy; examples for the successful execution of this reaction as a platform for desymmetrization of achiral quaternary centers are scarce in recent literature (Scheme 11). In 2008, Yu and co-workers reported a stoichiometric desymmetrization of dimethyl oxime 79, proceeding in 72% yield and complete selectivity (assisted by the conformational rigidity of 79) en route to the synthesis of lobatoside E.36 Six years later, the Sorenson laboratory described the first symmetry-breaking implementation of Sanford’s catalytic reaction in their synthesis of jiadifenolide.37 In this reaction, treating oxime 81 with Pd(OAc)2 and PhI(OAc)2 afforded the desired acetate 82 in 22% yield and 1:1 dr. The poor selectivity in this transformation may be attributed to the oxime in 81 bisecting the two methyl groups. In our case, exposure of oxime 76 to Sanford’s conditions provided the desired acetate diastereomer 78 in 79% yield and >20:1 dr (presumably aided by the coplanar oxime and equatorial methyl group). That this reaction (76 → 78) provided the desired product diastereomer in such high yield illustrates the viability of this and related transformations in the late-stage pursuit of challenging quaternary stereocenters, particularly scenarios in which inherent structural biases may lend a degree of stereochemical predictability.
Scheme 11.

Recent Examples of Substrate-Directed sp3 C–H Oxidation/Desymmetrization
With acetate 78 in hand, we faced the remaining challenges of C ring installation, C6a reduction, and indolization to complete our synthesis (Scheme 12). Acetate 78 was subjected to global hydrolysis to remove the acetate, oxime, and silyl ether functionalities. The resulting primary alcohol was oxidized with Dess-Martin periodinane (DMP) to give ketoaldehyde 83 in 70% yield over two steps. From 83, we envisioned that bisvinylation followed by ring-closing metathesis (RCM) would install the needed carbon skeleton. Unfortunately, treatment of 83 with vinylmagnesium bromide at −78 °C gave predominantly retro-aldol decomposition products with only small amounts of 84. After some experimentation, we found that the CeCl3·2LiCl complex recently reported by Knochel aided in suppressing the retro-aldol product completely,38 giving diol 84 in 95% yield. Treatment of 84 with Grubb’s second generation catalyst provided allylic alcohol 85 in 71% yield. While an alcohol oxidation/hydroxyl elimination pathway was first pursued for the conversion of diol 85 to enone 86, we found that simply subjecting 85 to acidic conditions (TFA) resulted in direct elimination of the tert-hydroxyl to give nonconjugated enone 86 in 71% yield. This set the stage for hydrogenation of the resultant alkene to install the final stereocenter found in 1. In the event, catalytic hydrogenation of alkene 86 with Pd/C provided ketone 87 in 87% yield and >20:1 dr. However, 1H NMR spectral data of this compound were not consistent with that of the desired compound previously synthesized by Smith and co-workers,12d leading to the conclusion that this hydrogenation had delivered the opposite diastereomer to that required. In order to rationalize this result, we calculated the structure of nonconjugated enone 86. As anticipated, the DFT-optimized structure of 86 revealed a marked puckering of the C–D ring fusion; catalytic hydrogenation of this alkene to give the desired diastereomer at C6a would necessitate approach of H2 to the concave Re face of 86. This result is in accord with prior studies on similar steroidal systems39 which also describe convex surface hydrogenation on related enones.
Scheme 12. Paspaline C Ring Construction and Synthesis of C6a Epimeric Ketonea.

aReagents and conditions: (a) (i) HCl, H2O/MeOH/THF/acetone (10:10:10:1), 85 °C; (ii) DMP, CH2Cl2, rt; (b) vinylmagnesium bromide, CeCl3·2LiCl, THF, −78 °C; (c) Grubbs second generation catalyst (20 mol %), CH2Cl2, rt; (d) TFA, CH2Cl2, 0 °C to rt; (e) H2 (1 atm), Pd/C (1.50 mass equiv), EtOH, rt.
Upon assessing our available functional handles, we surmised that selective reduction of the ketone in 86 might alter the outcome of the ensuing alkene hydrogenation by virtue of the hydroxyl’s function as a directing group (Scheme 13). The use of Crabtree’s catalyst in alcohol-directed alkene hydrogenations has been well-documented40 and would presumably engage the alkene on the same face as the hydroxyl. To this end, treatment of ketone 86 with LiAlH4 afforded the desired (S)-alcohol 88 in 60% yield and >20:1 dr over two steps from diol 85. The steric impact of the C12c methyl group on the outcome of this reaction cannot be overstated; ketone reduction in analogous steroidal systems not bearing this methyl group generally proceed with the opposite sense of selectivity.39,41
Scheme 13. Substrate-Directed Control of the C6a Stereocenter and Completion of the Total Synthesis of Paspalinea.

aReagents and conditions: (a) LiAlH4, THF, 0 °C; (b) H2 (1 atm), C8H12IrP(C6H11)3C5H5N]PF6 (15 mol %), CH2Cl2, rt; (c) DMP, CH2Cl2, rt; (d) (i) LDA, THF, 0 °C, then HMPA, Me2S2; (ii) Nchloroaniline, CH2Cl2, −78 °C, then NEt3; (iii) Raney Ni, EtOH, rt; (iv) TsOH (66 mol %), CH2Cl2, 50 °C.
With this alcohol in hand, catalytic hydrogenation of 88 using Crabtree’s catalyst completely over-rode the inherent substrate bias, giving the corresponding alcohol 90 (via 89) in >20:1 dr and subsequently the ketone 91 in 86% over two steps after reoxidation of the alcohol. The stereochemistry of 91 was confirmed via 1H NMR comparison with Smith’s intermediate and an X-ray diffraction study.12d This left only indolization to complete our total synthesis of 1. The Gassman indolization utilized previously by Smith proved to be the method of choice in affording paspaline (1) in 46% yield from 91.12a,42 Synthetic 1 matched the reported analytical data for paspaline, and single-crystal X-ray analysis of this sample was in agreement with the reported structure.11a
CONCLUSIONS
In conclusion, we have described the entirety of our efforts toward the synthesis of paspaline. The final route totals 28 steps from commercially available 28 in 0.4% yield (Smith synthesis: 24 steps from 28, 0.2% yield).12a Of particular note is the stereoselectivity of the described route: the least stereoselective reactions in our synthesis are the Ireland–Claisen rearrangement (70c → 71a, 6:1 dr) and the biocatalytic reduction (35 → 36, >99:1 er, 10:1 dr). All other stereodetermining transformations occur in >20:1 dr (Scheme 14). After initial approaches for the assembly of 1 via a cationic transannular cyclization were unsuccessful, a symmetry-breaking approach to paspaline was developed to complete construction of the E,F ring fusion within the first four steps of the synthesis. A novel tosyl hydrazone influenced epoxidation enabled excellent control of the C2 stereocenter (>20:1), and the Ireland–Claisen rearrangement provided access to the D ring and C12c stereocenter of 1. A substrate-directed symmetry-breaking C–H acetoxylation inspired by Sanford and co-workers provided control of the C12b stereocenter (>20:1). To override the inherent facial bias in the hydrogenation of enone 86, stereoselective reduction of the ketone followed by hydrogenation with Crabtree’s catalyst provided the final stereocenter in 1 with excellent selectivity (>20:1). Emphasis was placed throughout on expedient assembly of the critical C4a, C12b, and C12c quaternary methyl groups toward facile preparation of the remaining structures in this family of molecules. The route and methods described in this work present a number of complementary conceptual disconnections in the preparation of “steroid-like” natural products. Work in our laboratory in preparing these and related compounds is ongoing and will be reported in due course.
Scheme 14. Summary of Paspaline Total Synthesisa.
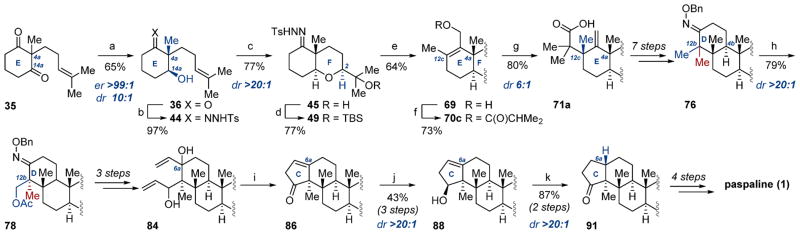
aReagents and conditions: (a) YSC-2, H2O/DMSO (10:1), 30 °C; (b) TsHNNH2, C7H8, 70 °C; (c) m-CPBA, CH2Cl2, 0 °C, then PPTS; (d) TBSOTf, 2,6-lutidine, CH2Cl2, −50 °C; (e) n-BuLi, THF, −50 °C, then MeI; n-BuLi, −50 °C to rt, then (HCHO)n; (f) isobutyric acid, DCC, DMAP (10 mol %), CH2Cl2, rt; (g) LDA, THF, −78 °C, then TMSCl, −78 to 75 °C; (h) Pd(OAc)2 (15 mol %), PhI(OAc)2, AcOH/Ac2O (1:1), 100 °C; (i) (i) Grubbs second generation catalyst (20 mol %), CH2Cl2, rt; (ii) TFA, CH2Cl2, 0 °C to rt; (j) LiAlH4, THF, 0 °C; (k) (i) H2 (1 atm), C8H12IrP(C6H11)3C5H5N]PF6 (15 mol %), CH2Cl2, rt; (ii) DMP, CH2Cl2, rt.
EXPERIMENTAL SECTION
Materials and Methods
General
Tetrahydrofuran (THF), diethyl ether (Et2O), dichloromethane (CH2Cl2), and toluene (C7H8) were dried by passage through a column of neutral alumina under nitrogen prior to use. Aniline, hexamethylphosphoramide (HMPA), and diisopropylamine were freshly distilled from calcium hydride prior to use. Compounds 14,43 37,44 and 3924 were prepared according to known procedures. All other reagents were purchased from commercial sources and were used as received unless otherwise noted. Proton and carbon magnetic resonance spectra (1H NMR and 13C NMR) were recorded with solvent resonance as the internal standard (1H NMR: CDCl3 at 7.26 ppm and C6D6 at 7.16 ppm; 13C NMR: CDCl3 at 77.0 ppm). 1H NMR data are reported as follows: chemical shift, multiplicity (s = singlet, br s = broad singlet, d = doublet, br d = broad doublet, t = triplet, q = quartet, m = multiplet), coupling constants (Hz), and integration. Mass spectra were obtained via Fourier transform mass spectromtetry (FTMS) with electrospray introduction (ESI) and external calibration in positive ion mode. All samples were prepared in methanol. Visualization for thin layer chromatography (TLC) was accomplished with UV light, KMnO4, and/or Seebach’s stain followed by heating. Purification of the reaction products was carried out by flash chromatography on silica gel. Unless otherwise noted, all reactions were carried out under an atmosphere of dry nitrogen in flame-dried glassware with magnetic stirring. Yield refers to isolated yield of analytically pure material unless otherwise noted. Yields are reported for a specific experiment and as a result may differ slightly from those found in figures, which are averages of at least two experiments.
Computation Analysis
High-level DFT calculations using the B3LYP28a,b approximate exchange-correlation energy density functional were performed with the standard Pople triple-ζ basis set 6-311G(d)28c,d for all elements when stable structures are optimized. Calculations were performed in the gas phase at 0 K with tight SCF convergence and ultrafine integration grids. All calculations were performed with the package of Gaussian 09 version D01.45 Cartesian coordinates of the studied systems are provided in the Supporting Information.
3-Hydroxy-4-methylpent-4-en-1-yl 4-Methylbenzenesulfonate (15)
A flame-dried, 1000 mL round-bottomed flask was charged with diol 14 (4.67 g, 40.2 mmol, 1.00 equiv) and CH2Cl2 (300 mL) under an atmosphere of N2. The solution was cooled to 0 °C, and NEt3 (14.0 mL, 100.5 mmol, 2.50 equiv), DMAP (0.49 g, 4.00 mmol, 0.10 equiv), and TsCl (8.43 g, 44.2 mmol, 1.10 equiv) were added sequentially. The resulting mixture was allowed to stir at this temperature until complete conversion of the starting material was observed by TLC analysis, typically 12 h. The mixture was then diluted with H2O (150 mL) and partitioned in a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 40 mL). The combined organic extracts were dried with sodium sulfate and concentrated in vacuo. The product was purified via flash chromatography (70:30 to 60:40 hexanes/EtOAc) to afford the tosylate 15 (8.75 g, 81% yield) as a pale yellow oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 7.79 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 4.91 (s, 1H), 4.82 (s, 1H), 4.22 (m, 1H), 4.16 (m, 1H), 4.09 (m, 1H), 2.44 (s, 3H), 1.91 (m, 1H), 1.79 (m, 1H), 1.75 (br s, 1H), 1.68 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 146.4, 144.8, 132.9, 129.8, 127.9, 111.4, 71.5, 67.6, 34.1, 21.6, 17.6; HRMS (ESI+) calcd for C13H18O4S+Na, 293.0824; found 293.0815; IR (thin film, cm−1) 3545, 3055, 2984, 2686, 1652, 1616, 1456, 1360, 1266, 1189; TLC (80:20 hexanes/EtOAc) Rf = 0.14.
Ethyl (E)-3-((2-Methyl-5-(tosyloxy)pent-1-en-3-yl)oxy)acrylate (16)
A flame-dried, 500 mL round-bottomed flask was charged with alcohol 15 (8.75 g, 32.0 mmol, 1.00 equiv) and CH2Cl2 (160 mL) under an atmosphere of N2 at rt. N-Methylmorpholine (3.60 mL, 35.7 mmol, 1.10 equiv) and ethyl propiolate (3.92 mL, 35.7 mmol, 1.10 equiv) were added sequentially, and the mixture was allowed to stir until complete conversion of the starting material was observed by TLC analysis, typically 4 h. The reaction mixture was concentrated on a rotary evaporator, and the crude product was purified via flash chromatography (80:20 to 70:30 hexanes/EtOAc) to give the vinyl ether 16 (11.4 g, 97% yield) as a clear oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 7.77 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 12.6 Hz, 1H), 5.14 (d, J = 12.6 Hz, 1H), 4.97 (s, 1H), 4.93 (s, 1H), 4.31 (dd, J = 4.8, 4.2 Hz, 1H), 4.16–4.06 (m, 4H), 2.43 (s, 3H), 1.97 (m, 2H), 1.61 (s, 3H), 1.26 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 167.6, 160.5, 145.0, 141.5, 132.6, 129.9, 127.9, 115.4, 98.6, 81.5, 66.2, 59.8, 32.7, 21.6, 16.7, 14.3; HRMS (ESI+) calcd for C18H24O6S+Na, 391.1191; found 391.1181; IR (thin film, cm−1) 2980, 2916, 2849, 1706, 1644, 1488, 1362, 1189, 1097, 923; TLC (80:20 hexanes/EtOAc) Rf = 0.32.
Ethyl (E)-3-((5-Iodo-2-methylpent-1-en-3-yl)oxy)acrylate (13)
To a solution of tosylate 16 (11.4 g, 30.8 mmol, 1.00 equiv) in acetone (300 mL) at rt was added NaI (40.0 g, 308.0 mmol, 10.0 equiv) portionwise with vigorous stirring. The resulting suspension was allowed to stir 12 h at which point TLC analysis confirmed complete consumption of the starting material. The reaction mixture was diluted with brine (150 mL) and transferred to a separatory funnel. The aqueous layer was extracted with EtOAc (3 × 60 mL), and the combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (90:10 to 80:20 hexanes/EtOAc) to afford the alkyl iodide 13 (8.67 g, 87% yield) as a pale yellow oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 7.46 (d, J = 12.6 Hz, 1H), 5.27 (d, J = 12.6 Hz, 1H), 5.05 (s, 1H), 5.04 (s, 1H), 4.39 (dd, J = 4.8, 3.0 Hz, 1H), 4.14 (m, 2H), 3.17 (m, 2H), 2.21 (m, 1H), 2.07 (m, 1H), 1.67 (s, 3H), 1.25 (t, J = 7.2 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 167.7, 160.8, 141.5, 115.3, 98.6, 85.5, 59.8, 36.7, 17.0, 14.3, 0.9; HRMS (ESI+) calcd for C11H17IO3+Na, 347.0120; found 347.0111; IR (thin film, cm−1) 3078, 2978, 2916, 1707, 1644, 1456, 1322, 1171, 1006, 834; TLC (80:20 hexanes/EtOAc) Rf = 0.64.
Dimethyl 2-(2-Ethoxy-2-oxoethyl)-6-(prop-1-en-2-yl)dihydro-2Hpyran-3,3(4H)-dicarboxylate (17)
A 500 mL round-bottomed flask was charged with the iodide 13 (8.75 g, 27.00 mmol, 1.00 equiv) and DMF (130 mL) at rt. Dimethyl malonate (6.20 mL, 54.0 mmol, 2.00 equiv) and Cs2CO3 (26.4 g, 81.0 mmol, 3.00 equiv) were added sequentially, whereupon a bright orange color was observed. The resulting mixture was allowed to stir for 14 h and was subsequently diluted with H2O (50 mL) and Et2O (50 mL). The layers were partitioned in a separatory funnel, and the aqueous layer was extracted with Et2O (3 × 30 mL). The combined organic extracts were washed with brine (40 mL), dried with magnesium sulfate, and concentrated in vacuo to give the crude pyran as a single diastereomer (as determined by 1H NMR spectroscopic analysis of the crude mixture, which revealed a single compound). The product was purified via flash chromatography (90:10 to 80:20 hexanes/EtOAc) to afford tetrahydropyran 17 (8.85 g, 99% yield) as a clear, viscous oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 4.86 (s, 1H), 4.75 (s, 1H), 4.31 (dd, J = 9.0, 6.0 Hz, 1H), 4.11 (m, 2H), 3.83 (d, J = 12.0 Hz, 1H), 3.72 (s, 3H), 3.67 (s, 3H), 2.77 (m, 2H), 2.54 (m, 1H), 1.92 (m, 1H), 1.82 (m, 1H), 1.67 (br s, 1H), 1.65 (s, 3H), 1.20 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 171.4, 170.7, 169.0, 144.6, 110.7, 81.1, 77.3, 60.3, 55.8, 52.5, 52.2, 38.0, 31.7, 26.0, 18.8, 14.1; HRMS (ESI+) calcd for C16H24O7+Na, 351.1420; found 351.1409; IR (thin film, cm−1) 3446, 2955, 2849, 1733, 1652, 1455, 1267, 1186, 1072, 904; TLC (80:20 hexanes/EtOAc) Rf = 0.43.
Dimethyl-2-(2-hydroxyethyl)-6-(prop-1-en-2-yl)dihydro-2Hpyran-3,3(4H)-dicarboxylate (18)
A flame-dried, 500 mL roundbottomed flask was charged with ester 17 (6.00 g, 18.3 mmol, 1.00 equiv) and THF (150 mL) under an atmosphere of N2. The solution was cooled to 0 °C, and DIBAL-H (1 M solution in hexane, 18.3 mL, 18.3 mmol, 1.00 equiv) was added slowly. The reaction was then analyzed for reaction completion via TLC analysis, which indicated incomplete starting material conversion. Another 1.00 equiv of DIBAL-H was added, whereupon TLC analysis indicated complete conversion of the starting material. The reaction mixture was quenched via addition of acetone (30 mL), and the mixture was stirred 5 min at 0 °C. Saturated Rochelle’s salt(aq) (40 mL) was then added, and the mixture was transferred to a separatory funnel. The aqueous layer was extracted with Et2O (3 × 40 mL), and the combined organic extracts were washed with 1 M HCl(aq) (40 mL) and brine (40 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (60:40 to 50:50 to 40:60 hexanes/EtOAc) to afford alcohol 18 (3.78 g, 72% yield) as a clear, viscous oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 4.92 (s, 1H), 4.81 (s, 1H), 4.01 (dd, J = 8.4, 1.8 Hz, 1H), 3.88 (m, 1H), 3.79–3.76 (m, 5H), 3.71 (s, 3H), 2.54 (m, 1H), 2.42 (d, J = 5.4 Hz, 1H), 2.14 (m, 1H), 1.93–1.88 (m, 3H), 1.70 (s, 3H), 1.67 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 171.2, 169.3, 144.7, 111.1, 81.5, 81.1, 62.1, 56.2, 52.6, 52.1, 34.7, 31.9, 26.6, 18.7; HRMS (ESI+) calcd for C14H22O6+Na, 309.1314; found 309.1305; IR (thin film, cm−1) 3446, 3055, 2954, 2883, 1731, 1455, 1266, 1078, 906, 737; TLC (75:25 hexanes/EtOAc) Rf = 0.05.
Dimethyl-2-(2-iodoethyl)-6-(prop-1-en-2-yl)dihydro-2H-pyran-3,3(4H)-dicarboxylate (27)
A 500 mL round-bottomed flask was charged with CH2Cl2 (96 mL), and the solution was cooled to 0 °C. Imidazole (3.22 g, 47.4 mmol, 4.96 equiv) and PPh3 (5.14 g, 19.0 mmol, 2.05 equiv) were added followed by I2 (4.83 g, 19.0 mmol, 2.00 equiv). The mixture was allowed to stir at 0 °C for 10 min, whereupon a pale yellow suspension was observed. The alcohol 18 (2.73 g, 9.55 mmol, 1.00 equiv) was then added as a solution in CH2Cl2 (20 mL), and the mixture was allowed to warm to rt and stirred until complete consumption of the starting material was observed by TLC analysis, typically 12 h. The mixture was then quenched via addition of saturated Na2S2O3(aq) (50 mL) and transferred to a separatory funnel. The aqueous layer was extracted with CH2Cl2 (3 × 30 mL), and the combined organic extracts were washed with brine (30 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (95:5 to 90:10 hexanes/EtOAc) to afford primary iodide 27 (2.64 g, 70% yield) as a white solid. Analytical data: mp 61–65 °C; 1H NMR (600 MHz, CDCl3) δ 4.93 (s, 1H), 4.83 (s, 1H), 3.88 (d, J = 10.2 Hz, 1H), 3.84 (d, J = 11.4 Hz, 1H), 3.75 (s, 3H), 3.72 (s, 3H), 3.36 (m, 1H), 3.28 (m, 1H), 2.56 (dt, J = 6.6, 3.0 Hz, 1H), 2.37 (m, 1H), 2.13 (m, 1H), 1.95 (m, 1H), 1.84 (m, 1H), 1.73 (s, 3H), 1.69 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 171.0, 169.3, 144.8, 110.9, 81.4, 80.9, 56.4, 52.6, 52.2, 35.8, 32.0, 26.3, 19.0, 4.3; HRMS (ESI+) calcd for C14H21IO5+Na, 419.0326; found 419.0320; IR (thin film, cm−1) 2917, 2849, 1731, 1652, 1540, 1455, 1265, 1083, 905; TLC (75:25 hexanes/EtOAc) Rf = 0.50.
Dimethyl-2-(3-methylbut-3-en-1-yl)-6-(prop-1-en-2-yl)dihydro-2H-pyran-3,3(4H)-dicarboxylate (12)
A flame-dried, 50 mL roundbottomed flask was charged with 2-bromopropene (0.67 mL, 7.57 mmol, 3.00 equiv) and Et2O (13 mL) under an atmosphere of N2. The mixture was cooled to −78 °C, and tBuLi (1.70 M solution in pentane, 8.91 mL, 15.14 mmol, 6.00 equiv) was added dropwise. The reaction mixture was allowed to stir 30 min at −78 °C, then warmed to rt and stirred for 1 h. During this time period, a second flame-dried, 100 mL round-bottomed flask was charged with CuI (0.72 g, 3.79 mmol, 1.50 equiv) and Et2O (12 mL) and was cooled to −78 °C. The isopropenyllithium solution was then cooled to −78 °C and transferred via cannula to the CuI suspension over a period of ~1 min. The resulting suspension was then warmed to −45 °C and stirred 1 h, upon which a color change from pale brown to dark gray to dark yellow-green was observed. The mixture was cooled to −78 °C, and a solution of iodide 27 (1.00 g, 2.52 mmol, 1.00 equiv) in Et2O (5 mL) was added. The reaction was then warmed to 0 °C and stirred until complete conversion of the starting material was observed by TLC analysis, typically 30 min. The reaction was then quenched via addition of saturated NH4Cl(aq) (20 mL), and the mixture was transferred to a separatory funnel. The aqueous layer was extracted with Et2O (3 × 20 mL), and the combined organic extracts were washed with saturated NH4Cl(aq) (20 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 95:5 to 90:10 hexanes/EtOAc) to afford the alkene 12 (0.77 g, 99% yield) as a clear oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 4.93 (s, 1H), 4.81 (s, 1H), 4.72 (s, 1H), 4.69 (s, 1H), 3.75 (br s, 4H), 3.70 (br s, 4H), 2.53 (m, 1H), 2.21 (m, 1H), 2.12 (m, 1H), 1.94–1.78 (m, 4H), 1.74 (s, 3H), 1,72 (s, 3H), 1.67 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 171.6, 169.7, 154.4, 145.1, 110.6, 110.3, 81.0, 80.5, 56.6, 52.4, 52.0, 34.9, 32.2, 30.1, 26.2, 22.2, 19.2; HRMS (ESI+) calcd for C17H26O5+Na, 333.1678; found 333.1669; IR (thin film, cm−1) 3446, 3056, 2953, 2849, 1731, 1669, 1636, 1520, 1455, 1203, 1266; TLC (75:25 hexanes/EtOAc) Rf = 0.52.
3-(Methoxycarbonyl)-2-(3-methylbut-3-en-1-yl)-6-(prop-1-en-2-yl)tetrahydro-2H-pyran-3-carboxylic Acid (19)
A 20 mL scintillation vial was charged with diester 12 (0.10 g, 0.32 mmol, 1.00 equiv) and THF (3 mL) with stirring at rt. KOH (1 M in MeOH, 1.70 mL, 1.70 mmol, 5.27 equiv) was added, and the resulting mixture was allowed to stir at rt until complete consumption of the starting material was observed by TLC analysis. This time period varied widely for each experiment (from 12 h to 6 days dependent on scale; in this iteration, 5 days were required to reach complete conversion). Once complete, the reaction mixture was concentrated on a rotary evaporator. The residue was diluted with H2O (10 mL), transferred to a separatory funnel, and extracted with Et2O (2 × 5 mL). The aqueous layer was acidified to pH = 1 with 1 M HCl(aq) and extracted with EtOAc (3 × 5 mL). The combined EtOAc extracts were dried with magnesium sulfate and concentrated in vacuo to afford the crude monoacid 19 (0.094 g, >99% crude yield) as a pale yellow, viscous oil. The diastereomeric ratio was determined via 1H NMR spectroscopic analysis of this crude material, which revealed a single compound. Analytical data: 1H NMR (600 MHz, C6D6) δ 10.56 (br s, 1H), 5.02 (s, 1H), 4.90 (s, 1H), 4.84 (s, 1H), 4.80 (s, 1H), 3.84 (dd, J = 7.2, 1.8 Hz, 1H), 3.58 (d, J = 11.4 Hz, 1H), 3.31 (s, 3H), 2.56 (d, J = 13.2 Hz, 1H), 2.33 (m, 2H), 2.17 (m, 2H), 2.08 (m, 1H), 1.71 (s, 3H), 1.68–1.67 (m, 4H), 1.34 (d, J = 1.8 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 177.1, 169.6, 145.3, 145.0, 110.8, 110.4, 81.1, 80.3, 56.6, 52.2, 34.8, 32.2, 30.1, 26.1, 22.2, 19.2; HRMS (ESI+) calcd for C16H24O5+Na, 319.1521; found 319.1513; IR (thin film, cm−1) 3566, 3074, 2952, 2857, 2633, 1732, 1650, 1438, 1268, 1080, 891; TLC (75:25 hexanes/ EtOAc) Rf = 0.32.
Methyl-3-acetyl-2-(3-methylbut-3-en-1-yl)-6-(prop-1-en-2-yl)-tetrahydro-2H-pyran-3-carboxylate (20)
A flame-dried, 25 mL round-bottomed flask was charged with diester 12 (0.35 g, 1.13 mmol, 1.00 equiv) and THF (11 mL) under an atmosphere of N2. The solution was cooled to −78 °C, and MeLi (1.60 M in Et2O, 0.6 mL, 0.97 mmol, 2.00 equiv) was added over 5 s. The reaction was then checked via TLC analysis, which showed incomplete conversion of the starting material. Another 1.00 equiv of MeLi was added, whereupon TLC analysis showed complete conversion of the starting material. The reaction mixture was then quenched via addition of saturated NH4Cl(aq) (5 mL) and subsequently warmed to rt. The mixture was transferred to a separatory funnel, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo to give the crude ketone as a single diastereomer (as determined via 1H NMR spectroscopic analysis of the crude product residue, which revealed a single stereoisomer in combination with overaddition products). The product was purified via flash chromatography (100:0 to 98:2 to 95:5 to 90:10 hexanes/EtOAc) to afford ketone 20 (0.22 g, 65% yield) as a clear, viscous oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 4.94 (s, 1H), 4.82 (s, 1H), 4.71 (s, 1H), 4.69 (s, 1H), 3.78–3.75 (m, 4H), 3.71 (d, J = 11.4 Hz, 1H), 2.45 (m, 1H), 2.22 (m, 1H), 2.12 (br s, 4H), 1.99 (m, 1H), 1.75 (br s, 4H), 1.73 (br s, 4H), 1.68 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 205.1, 171.0, 145.6, 145.2, 110.6, 110.3, 80.8, 80.3, 62.3, 52.0, 34.9, 31.4, 30.2, 27.1, 26.4, 22.3, 19.3; HRMS (ESI+) calcd for C17H26O4+Na, 317.1729; found 317.1720; IR (thin film, cm−1) 3445, 3072, 2969, 2857, 1708, 1649, 1436, 1356, 1221, 1081; TLC (75:25 hexanes/EtOAc) Rf = 0.45.
Synthesis of Unsaturated Ketone 21
Methyl-2-(3-methylbut-3-en-1-yl)-6-(prop-1-en-2-yl)-3-propionyltetrahydro-2H-pyran-3-carboxylate (S1)
A flame-dried, 20 mL scintillation vial was charged with bromoethane (0.13 mL, 1.69 mmol, 3.50 equiv) and THF (5 mL) under an atmosphere of N2. The solution was cooled to −78 °C, and tBuLi (1.70 M in pentane, 1.99 mL, 3.38 mmol, 7.00 equiv) was added dropwise. The mixture was allowed to stir 30 min at −78 °C, whereupon a solution of the diester 12 (0.15 g, 0.48 mmol, 1.00 equiv) was added over ~10 s. The reaction progress was immediately checked via TLC analysis, which confirmed complete consumption of the starting material. The reaction was then quenched via addition of saturated NH4Cl(aq) (5 mL) and warmed to rt. The mixture was transferred to a separatory funnel, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo to afford the crude ketone as a single diastereomer (as determined via 1H NMR spectroscopic analysis of the crude product residue, which revealed a single stereoisomer in combination with overaddition products). The product was purified via flash chromatography (100:0 to 98:2 to 95:5 to 90:10 hexanes/EtOAc) to afford ketone S1 (0.13 g, 89% yield) as a clear, viscous oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 4.93 (s, 1H), 4.82 (s, 1H), 4.71 (s, 1H), 4.68 (s, 1H), 3.79–3.77 (m, 4H), 3.71 (d, J = 3.6 Hz, 1H), 2.42 (m, 3H), 2.21 (m, 1H), 2.12 (m, 1H), 1.95 (m, 1H), 1.78 (m, 1H), 1.74 (s, 3H), 1.73 (s, 3H), 1.69 (m, 2H), 1.60 (br s, 1H), 1.03 (t, J = 7.2 Hz, H); 13C NMR (150 MHz, CDCl3) δ 208.0, 171.2, 145.6, 145.2, 110.6, 110.3, 80.8, 80.5, 62.4, 51.9, 34.9, 32.6, 31.7, 30.2, 26.4, 22.3, 19.3, 7.9; HRMS (ESI+) calcd for C18H28O4+Na, 331.1885; found 331.1876; IR (thin film, cm−1) 3446, 3073, 2970, 2855, 1739, 1650, 1455, 1342, 1159, 892; TLC (75:25 hexanes/EtOAc) Rf = 0.47.
Methyl 3-Acryloyl-2-(3-methylbut-3-en-1-yl)-6-(prop-1-en-2-yl)-tetrahydro-2H-pyran-3-carboxylate (21)
A flame-dried, 20 mL scintillation vial was charged with THF (4 mL) and diisopropylamine (0.08 mL, 0.55 mmol, 1.30 equiv) under an atmosphere of N2. The mixture was cooled to 0 °C, and nBuLi (1.74 M in hexanes, 0.32 mL, 0.55 mmol, 1.30 equiv) was added dropwise. After being stirred 30 min, the mixture was cooled to −78 °C, and a solution of ketone S1 (0.13 g, 0.42 mmol, 1.00 equiv) in THF (1 mL) was added. After being stirred 45 min at −78 °C, PhSeBr (0.11 g, 0.51 mmol, 1.10 equiv) was added, and the mixture was allowed to stir until complete consumption of the starting material was observed by TLC analysis, typically 45 min. The reaction mixture was diluted with H2O (10 mL), warmed to rt, and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo to give the crude α-selenide, which was used in the next step without further purification.
The intermediate selenide was dissolved in CH2Cl2 (2 mL), and the mixture was cooled to 0 °C. H2O2 (30% w/w in H2O, 0.80 mL) was added dropwise, and the mixture was stirred at 0 °C until complete consumption of the starting material was observed by TLC analysis, typically 15 min. The reaction mixture was diluted with H2O (7 mL) and transferred to a separatory funnel. The aqueous layer was extracted with EtOAc (3 × 7 mL), and the combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 98:2 to 95:5 hexanes/EtOAc) to afford unsaturated ketone 21 (0.079 g, 56%) as a pale yellow, viscous oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 6.39 (d, J = 3.0 Hz, 1H), 6.38 (s, 1H), 5.71 (dd, J = 4.2, 3.0 Hz, 1H), 4.95 (s, 1H), 4.84 (s, 1H), 4.72 (s, 1H), 4.70 (s, 1H), 3.84 (d, J = 10.2 Hz, 1H), 3.76 (s, 3H), 3.72 (d, J = 11.4 Hz, 1H), 2.43 (m, 1H), 2.22 (m, 1H), 2.17 (m, 1H), 2.08 (m, 1H), 1.81 (m, 1H), 1.77 (s, 3H), 1.74 (s, 3H), 1.72–1.66 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 195.6, 170.9, 145.6, 145.2, 131.7, 129.7, 110.7, 110.4, 80.7, 79.9, 60.8, 52.0, 34.8, 31.0, 30.2, 26.2, 22.3, 19.4; HRMS (ESI+) calcd for C18H26O4+Na, 329.1729; found 329.1720; IR (thin film, cm−1) 3420, 3054, 2952, 2852, 1740, 1636, 1455, 1265, 1049, 894; TLC (75:25 hexanes/EtOAc) Rf = 0.63.
Synthesis of Unsaturated Ketone 22
tert-Butyl 3-(3-oxopropyl)-1H-indole-1-carboxylate (S2)
A flame-dried, 50 mL round-bottomed flask was charged with 3-(1H-indol-3-yl)propanal46 (0.37 g, 2.10 mmol, 1.00 equiv), CH2Cl2 (14 mL), NEt3 (0.44 mL, 3.15 mmol, 1.50 equiv), and DMAP (0.005 g, 0.21 mmol, 0.10 equiv) at rt under an atmosphere of N2. Boc2O (0.55 g, 2.52 mmol, 1.20 equiv) was added in one porition, and the resulting mixture was allowed to stir until complete consumption of the starting material was observed by TLC analysis, typically 5 h. The mixture was then diluted with H2O (10 mL) and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 90:10 to 80:20 hexanes/ EtOAc) to afford the protected indole S2 (0.24 g, 42% yield) as a clear, viscous oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 9.87 (s, 1H), 8.13 (br s, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.38 (br s, 1H), 7.33 (t, J = 7.2 Hz, 1H), 7.25 (t, J = 7.2 Hz, 1H), 3.04 (t, J = 7.2 Hz, 2H), 2.87 (t, J = 7.8 Hz, 2H), 1.67 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 201.5, 124.5, 122.6, 122.4, 119.1, 118.7, 115.3, 43.1, 28.2, 17.4; HRMS (ESI+) calcd for C16H19NO3+Na, 296.1263; found 296.1256; IR (thin film, cm−1) 3446, 2977, 2916, 1731, 1670, 1636, 1455, 1373, 1256, 1158, 1018, 746; TLC (80:20 hexanes/EtOAc) Rf = 0.53.
tert-Butyl 3-(2-formylallyl)-1H-indole-1-carboxylate (S3)
A flame-dried, 50 mL round-bottomed flask was charged with aldehyde S2 (0.16 g, 0.60 mmol, 1.00 equiv) and CH2Cl2 (12 mL) at rt under an atmosphere of N2. NEt3 (0.84 mL, 6.00 mmol, 10.0 equiv) was added followed last by dimethylmethylideneiminium iodide (0.33 g, 1.8 mmol, 3.00 equiv). The mixture was allowed to stir at rt until complete conversion of the starting material was observed by TLC analysis, typically 12 h. The reaction was then concentrated on a rotary evaporator and purified via flash chromatography (95:5 to 90:10 hexanes/EtOAc) to afford unsaturated aldehyde S3 (0.08 g, 45% yield) as a yellow, viscous oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 9.67 (s, 1H), 8.13 (br s, 1H), 7.42 (br s, 1H), 7.39 (d, J = 7.8 Hz, 1H), 7.32 (t, J = 8.4 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 3.65 (s, 2H), 1.67 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 194.0, 149.7, 147.9, 135.3, 130.1, 124.4, 124.1, 122.5, 119.1, 116.8, 115.3, 83.6, 28.2, 23.3; HRMS (ESI+) calcd for C17H19NO3+Na, 308.1263; found 308.1255; IR (thin film, cm−1) 3446, 2916, 1732, 1685, 1488, 1455, 1370, 1255, 1158, 1083, 959; TLC (80:20 hexanes/EtOAc) Rf = 0.60.
tert-Butyl 3-(2-methylene-3-oxobutyl)-1H-indole-1-carboxylate (22)
A flame-dried, 20 mL scintillation vial was charged with aldehyde S3 (0.04 g, 0.12 mmol, 1.00 equiv) and THF (2 mL) under an atmosphere of N2. The solution was cooled to 0 °C, and MeMgBr (3 M in Et2O, 0.12 mL, 0.37 mmol, 3.00 equiv) was added over a period of ~1 min. The mixture was allowed to stir until complete consumption of the starting material was observed by TLC analysis, typically 30 min. The reaction was then quenched via addition of saturated NH4Cl(aq) (5 mL), and the mixture was transferred to a separatory funnel. The aqueous layer was extracted with Et2O (3 × 10 mL), and the combined organic extracts were dried with magnesium sulfate and concentrated in vacuo to give the crude alcohol, which was used in the next step without further purification.
The crude residue was dissolved in CH2Cl2 (2 mL) and transferred to a 20 mL scintillation vial. Dess-Martin periodinane (0.10 g, 0.25 mmol, 2.00 equiv) was added to the vial, and the resulting mixture was allowed to stir until complete consumption of the starting material was observed by TLC analysis, typically 20 min. The reaction mixture was then quenched via a 1:1 mixture of saturated NaHCO3(aq) and saturated Na2S2O3(aq) (5 mL) and allowed to stir 5 min. The mixture was then transferred to a separatory funnel, and the aqueous layer was extracted with Et2O (3 × 5 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (95:5 to 90:10 hexanes/EtOAc) to afford enone 22 (0.026 g, 71% yield) as a yellow viscous oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 8.12 (br s, 1H), 7.41 (d, J = 9.0 Hz, 1H), 7.39 (br s, 1H), 7.31 (t, J = 9.0 Hz, 1H), 7.21 (t, J = 9.0 Hz, 1H), 6.10 (s, 1H), 5.72(s, 1H), 3.67 (s, 2H), 2.39 (s, 3H), 1.67 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 199.4, 146.8, 126.5, 124.3, 124.0, 122.4, 119.2, 117.8, 115.2, 36.6, 28.2, 25.9; HRMS (ESI+) calcd for C18H21NO3+Na, 322.1419; found 322.1411; IR (thin film, cm−1) 3445, 3054, 2980, 2930, 1731, 1680, 1628, 1454, 1368, 1256, 1158, 1082; TLC (80:20 hexanes/EtOAc) Rf = 0.60.
Synthesis of Enol Silane 24
4-(1-(2,2,2-Trifluoroacetyl)-1Hindol-3-yl)butan-2-one (S4)
A flame-dried, 100 mL round-bottomed flask was charged with TFAA (1.51 mL, 10.7 mmol, 4.00 equiv) and CH2Cl2 (25 mL) under an atmosphere of N2. 4-(1H-Indol-3-yl)butan-2-one47 (0.50 g, 2.67 mmol, 1.00 equiv) was dissolved in CH2Cl2 (2 mL) and added dropwise to the TFAA solution. Once the addition was complete, the mixture was allowed to stir at rt until complete consumption of the starting material was observed by TLC analysis, typically 12 h. The reaction was quenched via addition of saturated NaHCO3(aq) (10 mL) and transferred to a separatory funnel. The aqueous layer was extracted with EtOAc (3 × 10 mL), and the combined organic extracts were dried with sodium sulfate and concentrated in vacuo. The product was purified via flash chromatography (90:10 to 80:20 hexanes/EtOAc) to afford TFAprotected indole S4 (0.54 g, 71% yield) as a pale yellow solid. Analytical data: mp 55–58 °C; 1H NMR (600 MHz, CDCl3) δ 8.43 (d, J = 7.8 Hz, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.42 (m, 2H), 7.25 (br s, 1H), 2.99 (t, J = 7.8 Hz, 2H), 2.87 (t, J = 7.8 Hz, 2H), 2.20 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 207.0, 136.2, 130.5, 126.4, 125.5, 125.2, 120.3, 120.2, 119.2, 117.0, 42.2, 30.0, 18.6; HRMS (ESI+) calcd for C14H12F3NO2+Na, 306.0718; found 306.0709; IR (thin film, cm−1) 2917, 1717, 1459, 1419, 1292, 1207, 1155, 880; TLC (80:20 hexanes/ EtOAc) Rf = 0.48.
2,2,2-Trifluoro-1-(3-(3-((trimethylsilyl)oxy)but-2-en-1-yl)-1Hindol-1-yl)ethan-1-one (24)
A flame-dried, 20 mL scintillation vial was charged with ketone S4 (0.05 g, 0.267 mmol, 1.00 equiv) and CH2Cl2 (3 mL) under an atmosphere of N2. The mixture was cooled to −10 °C, and HMDS (0.17 mL, 0.801 mmol, 3.00 equiv) was added followed by TMSI (0.02 mL, 0.267 mmol, 1.00 equiv) dropwise. The reaction mixture was warmed to rt and stirred until TLC analysis confirmed complete consumption of the starting material, typically 45 min. The reaction mixture was then quenched via addition of saturated NaHCO3(aq) (5 mL) and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 5 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo to afford the crude enol silane as a ~3:1 mixture of alkene isomers as determined by 1H NMR analysis. This material was unstable to further purification and was used directly in reaction screenings. The crude 1H NMR spectrum is included in the Supporting Information.
tert-Butyl 3-(3-Methylbut-2-en-1-yl)-1H-indole-1-carboxylate (25)
A flame-dried, 20 mL scintillation vial was charged with 3-(3-methylbut-2-en-1-yl)-1H-indole48 (0.05 g, 0.27 mmol, 1.00 equiv), NEt3 (0.06 mL, 0.41 mmol, 1.50 equiv), DMAP (0.003 g, 0.027 mmol, 0.10 equiv), and CH2Cl2 (3 mL) at rt under an atmosphere of N2. Boc2O (0.07 mL, 0.32 mmol, 1.20 equiv) was added, and the mixture was allowed to stir at rt until TLC analysis confirmed complete consumption of the starting material, typically 12 h. The mixture was diluted with H2O (5 mL) and transferred to a separatory funnel. The aqueous layer was extracted with CH2Cl2 (3 × 5 mL), and the combined organic extracts were washed with H2O (5 mL), dried with sodium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 98:2 hexanes/EtOAc) to afford protected indole 25 (0.06 g, 73% yield) as a yellow viscous oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 8.11 (d, J = 9.0 Hz, 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.35–7.31 (m, 2H), 7.25 (t, J = 7.8 Hz, 1H), 5,41 (t, J = 7.2 Hz, 1H), 3.39 (d, J = 7.2 Hz, 2H), 1.78 (br s, 6H), 1.68 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 133.0, 124.2, 123.1, 122.3, 122.2, 121.5, 120.6, 120.5, 119.1, 115.2, 107.1, 28.2, 25.7, 23.9, 17.8; HRMS (ESI+) calcd for C18H23NO2+Na, 308.1626; found 308.1619; IR (thin film, cm−1) 3421, 3053, 2980, 2931, 1730, 1454, 1371, 1265, 1158, 855; TLC (80:20 hexanes/EtOAc) Rf = 0.95.
Dimethyl 2-(2-(1-Methyl-2,6-dioxocyclohexyl)ethyl)-6-(prop-1-en-2-yl)dihydro-2H-pyran-3,3(4H)-dicarboxylate (29)
A flame-dried, 20 mL scintillation vial was charged with iodide 27 (0.60 g, 1.51 mmol, 1.00 equiv), 2-methyl-1,3-cyclohexanedione (0.27 g, 2.12 mmol, 1.4 equiv), and DMF (3 mL) at rt under an atmosphere of N2. Cs2CO3 (0.74 g, 2.27 mmol, 1.50 equiv) was added, and the mixture was warmed to 65 °C. The reaction was allowed to stir at this temperature until complete consumption of the starting material was observed by TLC analysis, typically 5 h. The reaction mixture was cooled to rt, diluted with H2O (6 mL) and Et2O (5 mL), and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were washed with brine (10 mL), dried with magnesium sulfate, and concentrated in vacuo. The mixture was purified via flash chromatography (70:30 to 60:40 to 50:50 hexanes/ EtOAc) to afford diketone 29 (0.20 g, 34% yield) as a clear, viscous oil and enol ether 30 (0.34 g, 56% yield) as a clear, viscous oil. Analytical data: O-alkylation product 30: 1H NMR (600 MHz, CDCl3) δ 4.90 (s, 1H), 4.79 (s, 1H), 4.09 (m, 2H), 3.92 (d, J = 10.8 Hz, 1H), 3.77 (d, J = 11.4 Hz, 1H), 3.74 (s, 3H), 3.69 (s, 3H), 2.55–2.51 (m, 3H), 2.31 (t, J = 6.6 Hz, 2H), 2.21 (m, 1H), 2.09 (m, 1H), 1.96–1.90 (m, 3H), 1.79 (m, 1H), 1.70 (s, 3H), 1.68 (s, 3H), 1.66 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 198.8, 171.5, 110.9, 169.3, 144.8, 115.0, 110.8, 81.3, 77.1, 64.6, 56.4, 52.6, 52.1, 36.2, 32.7, 31.9, 26.4, 25.3, 20.9, 18.8, 7.3; HRMS (ESI+) calcd for C21H30O7+Na, 417.1889; found 417.1879; IR (thin film, cm−1) 2953, 1731, 1635, 1455, 1377, 1355, 1262, 1095, 921; TLC (75:25 hexanes/EtOAc) Rf = 0.10. C-alkylation product 29: 1H NMR (600 MHz, CDCl3) δ 4.90 (s, 1H), 4.79 (s, 1H), 3.73–3.70 (m, 4H), 3.68 (s, 3H), 3.61 (m, 1H), 2.76 (m, 2H), 2.56–2.48 (m, 3H), 2.12 (m, 1H), 2.04 (m, 1H), 1.85–1.74 (m, 4H), 1.69 (s, 3H), 1.63 (m, 1H), 1.59 (m, 2H), 1.18 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 210.0, 209.8, 171.3, 169.2, 145.0, 110.6, 81.1, 80.9, 66.1, 56.2, 52.5, 52.0, 37.5, 35.5, 32.0, 27.6, 26.4, 18.8, 17.8, 17.0; HRMS (ESI+) calcd for C21H30O7+Na, 417.1889; found 417.1879; IR (thin film, cm−1) 3403, 3057, 2954, 2872, 1729, 1696, 1455, 1266, 1084, 905; TLC (75:25 hexanes/EtOAc) Rf = 0.13.
Methyl 10a-Hydroxy-6a-methyl-7-oxo-3-(prop-1-en-2-yl)-decahydro-1H-benzo[f]chromene-10b(4aH)-carboxylate (32)
A 5 mL dram vial was charged with diketone 29 (0.015 g, 0.04 mmol, 1.00 equiv) and DMSO (2 mL), and NaCl (0.02 g, 0.38 mmol, 10.0 equiv) was added in one portion. The vial was sealed with a screw-cap, and the mixture was warmed to 150 °C and stirred 9 h. The mixture was cooled to rt, diluted with Et2O (2 mL), and transferred to a separatory funnel containing H2O (10 mL). The aqueous layer was extracted with Et2O (3 × 5 mL), and the combined organic extracts were washed with brine (5 mL), dried with magnesium sulfate, and concentrated in vacuo. Crude 1H NMR analysis revealed a ~1:1 mixture of the diastereomeric decarboxylation product 31 and annulation product 32. This mixture was purified via flash chromatography (70:30 to 60:40 hexanes/EtOAc) to afford annulation product 32 (0.006 g, 47% yield) as a clear, viscous oil and Krapcho adduct 31 (0.005 g, 39% yield) as a clear, viscous oil. Slow evaporation of 32 from acetone and hexanes provided crystals suitable for X-ray crystallographic analysis. (Note: when this reaction was conducted on 0.07 g, scale, only the Krapcho adduct 31 was isolated in 43% yield. No cyclization product 32 was detected on this scale.) Analytical data: Decarboxylation product 31: 1H NMR (600 MHz, CDCl3) δ 4.94 (s, 2H), 4.82–4.81 (m, 2H), 3.73–3.70 (m, 2H), 3.68–6.67 (m, 3H), 3.47–3.41 (m, 2H), 2.80–2.69 (m, 4H), 2.60–2.54 (m, 4H), 2.24 (m, 1H), 2.14–2.12 (m, 2H), 2.07–1.98 (m, 5H), 1.85–1.77 (m, 3H), 1.74–1.73 (m, 5H), 1.51–1.39 (m, 5H), 1.21 (s, 3H), 1.18 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 210.3, 210.1, 210.0, 209.9, 174.3, 172.8, 145.8, 145.5, 110.6, 110.2, 81.5, 80.0, 78.0, 77.3, 66.1, 65.5, 51.7, 51.3, 46.7, 37.8, 37.7, 37.5, 34.0, 33.5, 29.7, 29.1, 28.9, 28.8, 27.7, 26.2, 25.7, 19.0, 18.8, 18.3, 17.9, 17.7, 16.4; HRMS (ESI+) calcd for C19H28O5+Na, 359.1834; found 359.1825; IR (thin film, cm−1) 3446, 2917, 2849, 1731, 1652, 1540, 1456, 1200, 901; TLC (75:25 hexanes/EtOAc) Rf = 0.17. Annulation product 32: 1H NMR (600 MHz, CDCl3) δ 4.91 (s, 1H), 4.78 (s, 1H), 3.86 (d, J = 12.0 Hz, 1H), 3.66 (dd, J = 7.8, 4.8 Hz, 1H), 3.59 (s, 3H), 2.47 (m, 2H), 2.31 (m, 2H), 2.16 (dd, J = 10.8, 6.0 Hz, 1H), 2.10–2.00 (m, 3H), 1.77 (m, 2H), 1.68 (s, 3H), 1.62 (m, 1H), 1.52 (br s, 1H), 1.45 (m, 1H), 1.35 (m, 1H), 1.18 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 210.0, 172.5, 145.7, 110.9, 82.0, 80.0, 78.2, 53.5, 53.2, 50.5, 34.1, 29.1, 28.1, 27.5, 26.9, 25.9, 25.4, 18.4, 18.1; HRMS (ESI+) calcd for C19H28O5+Na, 359.1834; found 359.1825; IR (thin film, cm−1) 3446, 3055, 2950, 1718, 1456, 1339, 1265, 1073, 899; TLC (75:25 hexanes/EtOAc) Rf = 0.07.
2-Methyl-3-((4-methylpent-3-en-1-yl)oxy)cyclohex-2-en-1-one (38)
A flame-dried, 25 mL round-bottomed flask was charged with 2-methyl-1,3-cyclohexanedione (1.00 g, 7.93 mmol, 100 equiv) and DMF (8 mL) under an atmosphere of N2. The mixture was cooled to 0 °C, and NaH (60% dispersion in oil, 0.39 g, 10.3 mmol, 1.30 equiv) was added portionwise. The mixture was warmed to rt and stirred 10 min, whereupon the iodide 37 (2.16 g, 10.3 mmol, 1.30 equiv) was added. The mixture was allowed to stir 12 h, and the reaction mixture was poured into a separatory funnel containing H2O (20 mL). CH2Cl2 (20 mL) was added, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine (20 mL), dried with magnesium sulfate, and concentrated in vacuo. The products were purified via flash chromatography (90:10 to 80:20 to 60:40 hexanes/EtOAc) to afford cycloalkanedione 35 (0.12 g, 7% yield) as a yellow oil and vinyl ether 38 (0.43 g, 26% yield) as a clear, viscous oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 5.11 (m, 1H), 3.93 (t, J = 6.6 Hz, 2H), 2.51 (m, 2H), 2.36 (q, J = 7.2 Hz, 2H), 2.30 (t, J = 6.6 Hz, 2H), 1.93 (m, 2H), 1.68 (s, 3H), 1.66 (t, J = 1.2 Hz, 3H), 1.61 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 198.8, 171.4, 134.8, 118.9, 115.0, 67.4, 36.2, 28.7, 25.7, 25.4, 20.9, 17.7, 7.29; HRMS (ESI+) calcd for C13H20O2+Na, 231.1361; found 231.1354; IR (thin film, cm−1) 3446, 2926, 1732, 1646, 1472, 1376, 1238, 1096; TLC (70:30 hexanes/EtOAc) Rf = 0.26.
(E)-3-(2,2-Dimethylhydrazono)-2-methylcyclohexan-1-one (39)
A 250 mL round-bottomed flask was charged with 2-methyl-1,3-cyclohexanedione (12.0 g, 95.1 mmol, 1.00 equiv), C6H6 (150 mL), H2NNMe2 (8.70 mL, 114.2 mmol, 1.20 equiv), and TsOH (0.50 g, 2.63 mmol, 0.03 equiv). A Dean–Stark apparatus was connected to the flask, and the mixture was heated to 100 °C with vigorous stirring for 6 h. The mixture was cooled to rt and concentrated on a rotary evaporator. The crude residue was then recrystallized from C7H8 to afford ketohydrazone 39 (16.00 g, 99% yield) as a yellow powder. Analytical data for this compound matched that reported in the literature:24 1H NMR (600 MHz, CDCl3) δ 5.05 (br s, 1H), 2.64 (m, 2H), 2.53 (s, 6H), 2.32 (t, J = 7.2 Hz, 2H), 1.90 (m, 2H), 1.66 (s, 3H).
2-Methyl-2-(4-methylpent-3-en-1-yl)cyclohexane-1,3-dione(35)
A flame-dried, 500 mL round-bottomed flask was charged with THF (250 mL) under an atmosphere of N2. KH (10.40 g, 30% dispersion in oil, 78.50 mmol, 1.20 equiv) was washed free of oil three times with petroleum ether, suspended in THF (20 mL), and added to the flask with stirring. The reaction mixture was cooled to −78 °C, and a solution of ketohydrazone 39 (11.00 g, 65.42 mmol, 1.00 equiv) in THF (25 mL) was slowly added. The reaction was warmed to 0 °C and allowed to stir 4.5 h. The resulting dark-brown mixture was recooled to −78 °C, and iodide S2 (17.3 g, 78.50 mmol, 1.20 equiv) was added. The reaction mixture was allowed to stir while slowly warming to rt overnight, producing a cream-white suspension. The reaction was then quenched with saturated NH4Cl(aq) (50 mL), and the resulting mixture was partitioned in a separatory funnel. The aqueous layer was extracted with Et2O (3 × 50 mL), and the combined organic extracts were washed with brine (40 mL), dried with magnesium sulfate, and concentrated in vacuo to give the intermediate alkylation product, which was used in the next step without further purification.
Cu(OAc)2·H2O (26.00 g, 130.9 mmol, 2.00 equiv) was dissolved in H2O (300 mL) in a 1000 mL round-bottomed flask with vigorous stirring. The crude hydrazone was then dissolved in THF (300 mL) and added to the Cu(OAc)2·H2O solution, and the reaction mixture was allowed to stir until TLC analysis confirmed complete conversion of the starting material, typically 12 h. The resulting mixture was concentrated on a rotary evaporator to remove the THF, and the solution was then diluted with saturated NH4Cl(aq) (100 mL) and CH2Cl2 (100 mL). This mixture was transferred to a separatory funnel and extracted with CH2Cl2 (3 × 50 mL). The combined organic extracts were washed with brine (2 × 50 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (90:10 to 80:20 hexanes/EtOAc) to afford diketone 35 (10.34 g, 76% yield) as an orange, viscous oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 4.99 (br s, 1H), 2.70 (m, 2H), 2.60 (m, 2H), 2.01 (m, 1H), 1.86–1.80 (m, 5H), 1.64 (s, 3H), 1.55 (s, 3H), 1.23 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 210.3, 132.9, 122.9, 65.6, 37.9, 37.5, 25.6, 23.3, 18.9, 17.7, 17.6; HRMS (ESI+) calcd for C13H20O2+H, 209.1542; found 209.1537; IR (thin film, cm−1) 3400, 2967, 2929, 1725, 1695, 1602, 1451, 1280, 1169, 1026; TLC (80:20 hexanes/EtOAc) Rf = 0.40.
3-Hydroxy-2-methyl-2-(4-methylpent-3-en-1-yl)cyclohexan-1-one (40)
A 20 mL scintillation vial was charged with diketone 35 (0.1 g, 0.48 mmol, 1.00 equiv) and MeOH (10 mL), and the solution was cooled to 0 °C. NaBH4 (0.005 g, 0.12 mmol, 0.25 equiv) was added, and the mixture was allowed to stir at this temperature until complete consumption of the starting material was observed by TLC analysis, typically 10 min. The reaction was diluted with brine (5 mL) and CH2Cl2 (5 mL), and the mixture was transferred to a separatory funnel. The aqueous layer was extracted with CH2Cl2 (3 × 5 mL), and the combined organic extracts were dried with sodium sulfate and concentrated in vacuo to give the crude alcohol as a 19.4:1 mixture of diastereomers. The diastereomeric ratio was determined by 1H NMR spectroscopic analysis of the crude reaction mixture by comparison of the integration of the resonances at δ 1.14 (major diastereomer) and δ 1.09 (minor diastereomer). The product was purified via flash chromatography (80:20 to 70:30 hexanes/EtOAc) to afford hydroxyketone 40 (0.093 g, 93% yield) as a clear, viscous oil. Analytical data: 1H NMR (600 MHz, CDCl3) δ 5.05 (t, J = 6.0 Hz, 1H), 3.65 (d, J = 7.8 Hz, 1H), 2.39 (m, 1H), 2.32 (m, 1H), 1.99–1.88 (m, 5H), 1.73 (m, 1H), 1.66–1.63 (m, 4H), 1.55 (br s, 4H), 1.15 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 214.1, 132.1, 123.9, 77.5, 54.7, 37.6, 31.5, 28.7, 25.6, 21.9, 20.7, 18.7, 17.6; HRMS (ESI+) calcd for C13H22O2+Na, 233.1518; found 233.1510; IR (thin film, cm−1) 3420, 2939, 2871, 1698, 1455, 1375, 1161, 1059, 993, 831; TLC (70:30 hexanes/EtOAc) Rf = 0.32.
(2R,3S)-3-Hydroxy-2-methyl-2-(4-methylpent-3-en-1-yl)-cyclohexan-1-one (36)
A 1000 mL round-bottomed flask was charged with H2O (320 mL), and YSC-2 (77 g, purchased from Sigma-Aldrich) was added portionwise with vigorous stirring. Diketone 35 (2.00 g, 9.60 mmol, 1.00 equiv) was dissolved in DMSO (32 mL) and added to the YSC-2 suspension, and the mixture was warmed to 30 °C and vigorously stirred for 24 h. The reaction mixture was then cooled to rt, diluted with Et2O (50 mL), and Celite (10 g) was added. The stirring was stopped, and the mixture was allowed to let stand at rt for 12 h. The resulting mixture was then filtered through a pad of Celite in a Buchner funnel. Once the filter cake was dry, the Celite pad was then washed with Et2O (100 mL), CH2Cl2 (100 mL), acetone (100 mL), Et2O (100 mL), and EtOAc (100 mL), ensuring that the filter cake was loosened with a spatula between each wash. The filtrate was transferred to a separatory funnel, and the organic layer was separated. The aqueous layer was extracted with EtOAc (50 mL), and the combined organic extracts were dried with sodium sulfate and concentrated in vacuo, giving crude alcohol 36 as a 10:1 mixture of diastereomers. The diastereomeric ratio was determined by 1H NMR spectroscopic analysis of the crude reaction mixture by comparison of the integration of the resonances at δ 1.15 (minor diastereomer) and δ 1.10 (major diastereomer). The product was purified via flash chromatography (80:20 to 70:30 hexanes/ EtOAc) to afford alcohol 36 (1.32 g, 67% yield) as a yellow, viscous oil. (Note: for purposes of material throughput, the crude residue may be stored indefinitely with no deleterious effects to yield. In practice, up to 8 iterations of this procedure were carried out, and the crude residues were combined and purified simultaneously.) The enantioselectivity (>99:1) was determined via 19F NMR analysis of the resulting Mosher ester S8 (vide infra). Analytical data: [α]D 28 −74.7 (c = 0.30, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.04 (m, 1H), 3.89 (dd, J = 3.0, 2.4 Hz, 1H), 2.41 (m, 1H), 2.31 (m, 1H), 2.08 (m, 1H), 2.02 (m, 1H), 1.93 (m, 1H), 1.87–1.79 (m, 4H), 1.65 (br s, 4H), 1.56 (s, 3H), 1.53 (m, 1H), 1.10 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 214.4, 132.2, 123.7, 76.3, 54.3, 37.8, 36.2, 28.1, 25.6, 22.6, 20.7, 17.6, 17.3; HRMS (ESI+) calcd for C13H22O2+Na, 233.1518; found 233.1514; IR (thin film, cm−1) 3434, 3054, 2985, 2305, 1703, 1630, 1442, 1265, 738; TLC (80:20 hexanes/EtOAc) Rf = 0.23.
(1S,2R)-2-Methyl-2-(4-methylpent-3-en-1-yl)-3-oxocyclohexyl-(R)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoate (Mosher Ester of 36)
A flame-dried, 20 mL scintillation vial was charged with (R)-(+)-α-methoxy-α-trifluoromethylphenylacetic acid (0.45 g, 1.90 mmol, 2.00 equiv) and CH2Cl2 (8 mL) with magnetic stirring at rt under an atmosphere of N2. DCC (0.39 g, 1.90 mmol, 2.00 equiv) was added followed by DMAP (0.01 g, 0.10 mmol, 0.10 equiv) and a 10:1 diastereomeric mixture of alcohol 36 (0.20 g, 0.95 mmol, 1.00 equiv) in CH2Cl2 (2 mL). The reaction mixture was allowed to stir at rt until complete conversion of the starting material was observed by TLC analysis, typically 12 h. The resulting mixture was filtered through cotton and concentrated in vacuo. The product was purified via flash chromatography (95:5 to 90:10 hexanes/EtOAc) to provide the Mosher ester (0.40 g, 99% yield) as an inseparable 10:1 mixture of diastereomers (as determined by integration of the resonances at δ 5.33 (major diastereomer) and δ 5.06 (minor diastereomer)). 19F NMR analysis revealed only a 10:1 mixture of diastereomers at δ –71.1 ppm (minor diastereomer) and δ –71.2 ppm (major diastereomer). Analytical data: [α]D 28 +22.6 (c = 0.50, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.50 (m, 2H), 7.39 (m, 3H), 5.33 (dd, J = 3.0, 3.0 Hz, 1H), 3.50 (s, 3H), 2.45 (m, 1H), 2.35 (m, 1H), 2.22 (m, 1H), 1.96–1.74 (m, 5H), 1.66 (s, 3H), 1.57 (s, 3H), 1.54 (m, 2H), 0.96 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 211.5, 165.8, 132.5, 131.9, 129.6, 128.4, 127.2, 123.2, 80.3, 55.3, 52.6, 37.4, 35.9, 25.6, 25.5, 22.4, 20.4, 17.8, 17.6; HRMS (ESI+) calcd for C23H29F3O4+Na, 449.1916; found 449.1923; IR (thin film, cm−1) 3423, 2949, 2855, 1746, 1713, 1451, 1270, 1168, 1019, 807, 721; TLC (80:20 hexanes/ EtOAc) Rf = 0.51.
(2R,3S)-2-(2-(3,3-Dimethyloxiran-2-yl)ethyl)-3-hydroxy-2-methylcyclohexan-1-one (41)
A 20 mL scintillation vial was charged with hydroxyketone 36 (0.10 g, 0.48 mmol, 1.00 equiv) and CH2Cl2 (5 mL), and the mixture was cooled to 0 °C. m-CPBA (70% dispersion in H2O, 0.19 g, 0.76 mmol, 1.60 equiv) was added in one portion, and the mixture was stirred until complete consumption of the starting material was observed by TLC analysis, typically 20 min. The reaction was quenched via saturated Na2S2O3 (5 mL), and the mixture was transferred to a separatory funnel. The aqueous layer was extracted with CH2Cl2 (3 × 5 mL), and the combined organic extracts were dried with sodium sulfate and concentrated in vacuo to give the crude epoxide as a 2:1 mixture of diastereomers. The diastereomeric ratio was determined by 1H NMR spectroscopic analysis of the crude reaction mixture by comparison of the integration of the resonances at δ 1.13 (major diastereomer) and δ 1.12 (minor diastereomer). The product was purified via flash chromatography (60:40 to 50:50 to 40:60 hexanes/EtOAc) to afford epoxide 41 (0.10 g, 93% yield) as a clear oil in an inseparable mixture of diastereomers. Analytical data: [α]D 25 +1.9 (c = 1.25, CHCl3); 1H NMR (600 MHz, CDCl3) δ 3.83 (dd, J = 4.2, 3.0 Hz, 1H), 2.68 (m, 1H), 2.33 (m, 2H), 2.01 (m, 2H), 1.84–1.54 (m, 5H), 1.48–1.40 (m, 1H), 1.27 (m, 3H), 1.23 (m, 3H), 1.09 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 214.2, 214.1, 75.6, 74.4, 64.7, 64.3, 59.1, 58.7, 54.3, 54.0, 37.6, 37.5, 32.0, 31.7, 28.4, 28.3, 24.8, 23.6, 23.5, 20.4, 20.3, 18.6, 18.5, 18.0, 17.1; HRMS (ESI+) calcd for C13H22O3+Na, 249.1467; found 249.1459; IR (thin film, cm−1) 3446, 3054, 2982, 2874, 1732, 1702, 1497, 1422, 1266, 1156, 1016, 895; TLC (80:20 hexanes/EtOAc) Rf = 0.07.
(4aR,8aS)-2-(2-Hydroxypropan-2-yl)-4a-methyloctahydro-5Hchromen-5-one (42) and (4aR,5S)-2-(2-hydroxypropan-2-yl)-4amethyloctahydro-2H-chromen-5-ol (43)
A 20 mL scintillation vial was charged with keto-epoxide 41 (0.05 g, 0.22 mmol, 1.00 equiv) and CH2Cl2 (2 mL), and PPTS (0.01 g, 0.04 mmol, 0.20 equiv) was added. The mixture was allowed to stir at rt until TLC analysis indicated complete consumption of the starting material, typically 30 min. The reaction mixture was diluted with saturated NaHCO3(aq) (5 mL) and transferred to a separatory funnel. The aqueous layer was extracted with CH2Cl2 (3 × 5 mL), and the combined organic extracts were dried with sodium sulfate and concentrated in vacuo. Crude 1H NMR analysis revealed an inseparable ~1:5 mixture of diastereomeric tetrahydropyrans 42 and diastereomeric vinyl ethers 43. The crude 1H NMR spectrum is included in the Supporting Information: HRMS (ESI+) calcd for +Na, 249.1467; found 249.1459.
N′-((2S,3S,E)-3-Hydroxy-2-methyl-2-(4-methylpent-3-en-1-yl)-cyclohexylidene)-4-methylbenzenesulfonohydrazide (44)
The alcohol 40 (8.20 g, 38.99 mmol, 1.00 equiv) was dissolved in wet C7H8 (195 mL) in a 500 mL round-bottomed flask, and p-toluenesulfonylhydrazine (8.71 g, 46.79 mmol, 1.20 equiv) was added with magnetic stirring. The mixture was placed in a preheated oil bath at 70 °C and allowed to stir for 50 min. (Note: product decomposition was observed if the reaction was allowed to stir for longer than this time period.) The resulting mixture was cooled to rt and concentrated on a rotary evaporator. The product was purified via flash chromatography (70:30 to 60:40 to 50:50 hexanes/EtOAc) to provide the hydrazone 44 (14.75 g, > 99% yield) as a pale yellow, viscous foam. Analytical data: [α]D28 −144.6 (c = 0.50, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.82 (d, J = 8.4 Hz, 2H), 7.64 (br s, 1H), 7.28 (d, J = 7.8 Hz, 2H), 4.93 (t, J = 6.6 Hz, 1H), 3.63 (dd, J = 3.0, 2.4 Hz, 1H), 2.39 (s, 3H), 2.35 (m, 1H), 2.00 (m, 1H), 1.90 (m, 1H), 1.75–1.66 (m, 3H), 1.64 (s, 3H), 1.57–1.49 (m, 2H), 1.46 (s, 3H), 1.37 (m, 2H), 1.04 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 163.3, 143.9, 135.1, 131.5, 129.3, 128.2, 124.1, 75.4, 47.6, 36.5, 25.6, 22.0, 21.5, 19.8, 19.1, 17.5; HRMS (ESI+) calcd for C20H30N2O3S+Na, 401.1875; found 401.1892; IR (thin film, cm−1) 3516, 3212, 2933, 2872, 1914, 1725, 1598, 1447, 1329, 1185, 1165, 1091, 736; TLC (80:20 hexanes/EtOAc) Rf = 0.17.
N′-((2S,4aS,8aS,E)-2-(2-Hydroxypropan-2-yl)-4a-methyloctahydro-5H-chromen-5-ylidene)-4-methylbenzenesulfonohydrazide(45)
Hydrazone 44 (14.76 g, 38.99 mmol, 1.00 equiv) was dissolved in CH2Cl2 (320 mL) in a 1000 mL round-bottomed flask with stirring. The mixture was cooled to 0 °C, and m-CPBA (14.42 g, 70% dispersion in H2O, 58.49 mmol, 1.50 equiv) was added. The reaction was allowed to stir at this temperature until TLC analysis showed full conversion of the starting material, typically 10 min. The reaction was quenched via addition of saturated Na2S2O3(aq) (70 mL), and the mixture was partitioned in a separatory funnel. The mixture was extracted with CH2Cl2 (3 × 50 mL), and the combined organic extracts were washed with brine (50 mL), dried with magnesium sulfate, and concentrated to a volume of ~300 mL on a rotary evaporator. A stir bar was added followed by PPTS (0.98 g, 3.90 mmol, 0.10 equiv), and the mixture was allowed to stir 12 h at rt. The reaction mixture was then concentrated in vacuo to give the crude tetrahydropyran 45 as a single diastereomer (as determined by 1H NMR spectroscopic analysis of the crude reaction mixture, which revealed a single stereoisomer). The product was purified via flash chromatography (60:40 to 50:50 to 40:60 hexanes/EtOAc) to afford pyran 45 (11.63 g, 76% yield) as a pale yellow, viscous foam. Analytical data: [α]D28 −63.2 (c = 0.40, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.82 (d, J = 7.8 Hz, 2H), 7.73 (br s, 1H), 7.30 (d, J = 7.8 Hz, 2H), 3.11 (t, J = 3.6 Hz, 1H), 3.09 (t, J = 2.4 Hz, 1H), 2.52 (dd, J = 12.0, 3.0 Hz, 1H), 2.45–2.40 (m, 4H), 1.94 (m, 1H), 1.82 (m, 2H), 1.67 (m, 2H), 1.59–1.50 (m, 3H), 1.33–1.26 (m, 2H), 1.15 (s, 3H), 1.14 (s, 3H), 0.96 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 164.5, 143.9, 135.1, 129.3, 128.1, 84.5, 82.0, 71.8, 42.4, 32.1, 26.3, 21.6, 17.2; HRMS (ESI+) calcd for C20H30N2O4S+Na, 417.1824; found 417.1840; IR (thin film, cm−1) 3451, 3216, 2946, 2870, 1630, 1598, 1450, 1333, 1166, 1089, 925; TLC (80:20 hexanes/EtOAc) Rf = 0.11.
N′-((2S,3S,E)-3-Hydroxy-2-methyl-2-(4-methylpentyl)-cyclohexylidene)-4-methylbenzenesulfonohydrazide (46)
A 20 mL scintillation vial was charged with alkene 44 (0.05 g, 0.13 mmol, 1.00 equiv) and MeOH (4 mL). Pd/C (0.025 g, 0.50 mass equiv) was added, and the resulting suspension was placed under 1 atm H2 (balloon) and allowed to stir 1 h, whereupon TLC analysis indicated complete consumption of the starting material. The suspension was filtered through a pad of Celite and concentrated on a rotary evaporator to afford hydrazone 46 (0.05 g, > 99% crude yield) as a single diastereomer (as determined by 1H NMR analysis of the crude mixture, which revealed a single stereoisomer). When this material was subjected to the reaction conditions used in the conversion of 44 to 45, no reaction was observed, and the starting material was recovered quantitatively. Analytical data: [α]D28 −51.9 (c = 1.25, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.83 (d, J = 8.2 Hz, 2H), 7.65 (br s, 1H), 7.28 (d, J = 7.8 Hz, 2H), 3.63 (dd, J = 3.0, 1.8 Hz, 1H), 2.40 (s, 3H), 2.36 (m, 1H), 1.93 (m, 2H), 1.79–1.64 (m, 3H), 1.56 (m, 1H), 1.46 (m, 1H), 1.34 (m, 1H), 1.27 (m, 1H), 1.02 (s, 3H), 1.00–0.98 (m, 3H), 0.77 (d, J = 6.6 Hz, 3H), 0.76 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 163.5, 143.8, 135.2, 129.3, 128.2, 75.6, 47.6, 39.5, 36.8, 27.7, 27.6, 22.6, 22.5, 21.5, 21.1, 19.7, 19.2; HRMS (ESI+) calcd for C20H32N2O3S+Na, 403.2031; found 403.2022; IR (thin film, cm−1) 3503. 3214, 2951, 2868, 1670, 1470, 1329, 1165, 1092, 1001, 924; TLC (80:20 hexanes/EtOAc) Rf = 0.07.
N′-((2S,4aS,8aS,E)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a-methyloctahydro-5H-chromen-5-ylidene)-4-methylbenzenesulfonohydrazide(49)
A flame-dried, 150 mL round-bottomed flask was charged with pyran 45 (9.41 g, 23.88 mmol, 1.00 equiv) and CH2Cl2 (120 mL) under an atmosphere of N2. The reaction mixture was cooled to −50 °C (CO2(s)/acetonitrile bath), and 2,6-lutidine (5.50 mL, 47.46 mmol, 2.00 equiv) and TBSOTf (9.87 mL, 42.99 mmol, 1.8 equiv) were added sequentially. The reaction was allowed to stir at this temperature until TLC analysis confirmed complete consumption of the starting material, typically 30 min. The reaction was quenched via addition of saturated NaHCO3(aq) (40 mL), and the mixture was warmed to rt and partitioned in a separatory funnel. The aqueous layer was extracted with EtOAc (3 × 30 mL), and the combined organic extracts were washed with brine (40 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 95:5 to 90:10 to 80:20 hexanes/EtOAc) to remove silanol byproducts then purified a second time (90:10 to 80:20 hexanes/EtOAc) to afford silyl ether 49 (9.46 g, 79% yield) as a pale yellow, viscous foam. Analytical data: [α]D28 −75.5 (c = 0.35, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.84 (d, J = 8.4 Hz, 2H), 7.57 (br s, 1H), 7.31 (d, J = 7.8 Hz, 2H), 3.04 (dd, J = 7.8, 3.6 Hz, 1H), 2.99 (d, J = 11.4 Hz, 1H), 2.50 (d, J = 14.4 Hz, 1H), 2.43 (s, 3H), 1.93 (m, 1H), 1.83–1.76 (m, 2H), 1.68–1.50 (m, 6H), 1.19 (s, 3H), 1.15 (s, 3H), 0.95 (s, 3H), 0.84 (s, 9H), 0.07 (s, 3H), 0.04 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 165.0, 143.8, 135.2, 129.3, 128.1, 85.3, 82.0, 76.8, 74.7, 42.5, 32.4, 27.2, 25.1, 21.6, 21.3, 17.3, −2.1, −2.2; HRMS (ESI+) calcd for C26H44N2O4SSi+Na, 531.2689; found 531.2704; IR (thin film, cm−1) 3433, 3054, 2985, 2855, 2305, 1630, 1422, 1167, 1092, 835, 739; TLC (80:20 hexanes/ EtOAc) Rf = 0.37.
(2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a-methyl-3,4,4a,7,8,8a-hexahydro-2H-chromene-5-carbaldehyde (50)
A flame-dried, 100 mL round-bottomed flask was charged with hydrazone 49 (2.00 g, 3.93 mmol, 1.00 equiv) and THF (39 mL) under an atmosphere of N2. The mixture was cooled to −50 °C, and nBuLi (1.64 M in hexane, 12.0 mL, 19.7 mmol, 5.00 equiv) was added dropwise, producing a dark orange color. The mixture was allowed to stir 30 min at −50 °C. The flask was fitted with a venting needle, and the mixture was warmed to 0 °C and stirred 5 min, then warmed to rt and stirred until complete consumption of the starting material was observed by TLC analysis, typically 20 min (scale dependent). The venting needle was removed, and DMF (3.02 mL, 39.3 mmol, 10.0 equiv) was added. Following this addition, the reaction was stirred 20 min, diluted with H2O (20 mL) and Et2O (20 mL) and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 20 mL). The combined organic extracts were washed with brine (20 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 95:5 to 90:10 hexanes/EtOAc) to afford unsaturated aldehyde 50 (0.92 g, 66% yield) as a yellow, viscous oil. Analytical data: [α]D28 −138.0 (c = 0.55, CHCl3); 1H NMR (600 MHz, CDCl3) δ 9.38 (s, 1H), 6.55 (t, J = 3.0 Hz, 1H), 3.22 (dd, J = 8.4, 3.6 Hz, 1H), 3.15 (dd, J = 9.0, 3.0 Hz, 1H), 2.70 (m, 1H), 2.46 (m, 2H), 1.68 (m, 2H), 1.57 (m, 2H), 1.28 (m, 1H), 1.23 (s, 3H), 1.18 (s, 3H), 1.14 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 193.8, 151.0, 148.3, 85.9, 80.9, 74.9, 35.4, 32.6, 27.2, 26.4, 25.8, 25.1, 23.2, 21.3, 17.9, −2.2; HRMS (ESI+) calcd for C20H36O3Si+Na, 375.2331; found 375.2323; IR (thin film, cm−1) 3435, 2955, 2855, 1692, 1635, 1472, 1376, 1251, 1173, 1042; TLC (90:10 hexanes/EtOAc) Rf = 0.49.
tert-Butyldimethyl-((2-((2S,4aS,8aS)-4a-methyl-5-vinyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-2-yl)propan-2-yl)oxy)silane (51)
A flame-dried, 100 mL round-bottomed flask was charged with methyltriphenylphosphonium bromide (4.90 g, 13.7 mmol, 6.00 equiv) and THF (20 mL) under an atmosphere of N2. The mixture was cooled to 0 °C and nBuLi (1.65 M in hexanes, 7.63 mL, 12.6 mmol, 5.50 equiv) was added dropwise. The deep yellow mixture was allowed to stir 1 h at 0 °C upon which the aldehyde 50 (0.81 g, 2.29 mmol, 1.00 equiv) was added as a solution in THF (3 mL). The reaction was allowed to stir until complete consumption of the starting material was observed by TLC analysis, typically 15 min. The reaction was diluted with H2O (15 mL) and transferred to a separatory funnel. The layers were separated, and the aqueous layer was extracted with Et2O (3 × 15 mL). The combined organic extracts were washed with brine (15 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 99:1 to 97.5:2.5 hexanes/EtOAc) to afford diene 51 (0.69 g, 86% yield) as a clear oil. Analytical data: [α]D28 −167.4 (c = 0.35, CHCl3); 1H NMR (600 MHz, CDCl3) δ 6.24 (dd, J = 10.8, 6.0 Hz, 1H), 5.61 (t, J = 3.6 Hz, 1H), 5.26 (d, J = 17.4 Hz, 1H), 4.93 (d, J = 10.8 Hz, 1H), 3.23 (dd, J = 5.4, 4.8 Hz, 1H), 3.12 (m, 1H), 2.20 (m, 1H), 1.93 (dt, J = 6.0, 3.0 Hz, 1H), 1.66 (m, 2H), 1.60 (m, 2H), 1.35 (m, 1H), 1.24 (s, 3H), 1.18 (s, 3H), 1.07 (s, 3H), 0.85 (s, 9H), 0.09 (s, 3H), 0.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 144.5, 135.4, 121.6, 113.5, 85.5, 81.5, 74.9, 36.1, 34.3, 27.4, 25.9, 25.0, 23.8, 21.8, 18.9, 18.2, −2.1, −2.2; HRMS (ESI+) calcd for C21H38O2Si+Na, 373.2539; found 373.2529; IR (thin film, cm−1) 3053, 2985, 2956, 2854, 2685, 1716, 1636, 1456, 1265, 1143; TLC (90:10 hexanes/EtOAc) Rf = 0.91.
tert-Butyldimethyl-((2-((3S,4aS,10bS)-10b-methyl-7-nitro-2,3,4a,5,6,6a,7,8,9,10b-decahydro-1H-benzo[f]chromen-3-yl)-propan-2-yl)oxy)silane (52)
A 20 mL scintillation vial was charged with diene 51 (0.66 g, 1.88 mmol, 1.00 equiv) and CH2Cl2 (9 mL). Nitroethylene49 (10 M solution in CH2Cl2, 0.75 mL, 7.50 mmol, 4.00 equiv) was added, and the vial was sealed with a screw-cap. The mixture was heated to 65 °C and stirred until complete conversion of the starting material was observed by TLC analysis, typically 12 h. The mixture was cooled to rt and concentrated on a rotary evaporator. The product was purified via flash chromatography (100:0 to 97.5:2.5 to 95:5 to 90:10 hexanes/EtOAc) to afford alkene 52 (0.75 g, 95% yield) as a clear, viscous oil in an inseparable mixture of diastereomers. Analytical data: [α]D28 −4.7 (c = 0.75, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.51 (br s, 1H), 5.45 (d, J = 4.8 Hz, 1H), 4.79–4.66 (m, 1H), 4.32–4.20 (m, 1H), 3.45 (dd, J = 7.8, 3.0 Hz, 1H), 3.06–3.01 (m, 4H), 2.96–2.87 (m, 3H), 2.27–1.89 (m, 13H), 1.76–1.72 (m, 3H), 1.66–1.37 (m, 17H), 1.25 (m, 2H), 1.21–1.19 (m, 8H), 1.17–1.15 (m, 3H), 1.05–1.03 (m, 8H), 0.84 (br s, 25H), 0.07 (s, 8H), 0.05 (s, 8H); 13C NMR (150 MHz, CDCl3) δ 144.8, 143.9, 143.2, 118.4, 117.9, 117.7, 90.6, 89.8, 85.6, 85.4, 85.1, 84.9, 83.4, 82.2, 74.8, 39.6, 37.5, 36.8, 36.4, 36.1, 34.4, 28.0, 27.3, 27.1, 27.0, 25.5, 25.2, 25.0, 24.4, 24.0, 23.0, 22.7, 21.9, 21.8, 21.6, 21.5, 18.1, 17.0, −2.2; HRMS (ESI+) calcd for C23H41NO4Si+Na, 446.2703; found 446.2692; IR (thin film, cm−1) 3054, 2954, 2930, 2855, 1732, 1670, 1546, 1488, 1362, 1265, 1167, 1046; TLC (90:10 hexanes/EtOAc) Rf = 0.66.
(3S,4aS,10bS)-3-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-10b-methyl-1,2,3,4a,5,6,8,9,10,10b-decahydro-7H-benzo[f]-chromen-7-one (53)
A 100 mL round-bottomed flask was charged with alkene 52 (0.753 g, 1.78 mmol, 1.00 equiv) and a 1:1 mixture of THF/MeOH (35 mL). The solution was cooled to 0 °C, and KOH (1 M in H2O, 5.34 mL, 5.34 mmol, 3.00 equiv) was added dropwise, subsequently warming to rt. The mixture was stirred until complete conversion of the starting material was observed by TLC analysis, typically 45 min. The mixture was cooled to 0 °C, and MsOH was added drop-by-drop until the reaction pH reached <1 (scaledependent, ~2 mL was required in this iteration), resulting in the formation of a white suspension. The resulting mixture was warmed to rt and stirred vigorously for 1 h, whereupon the mixture was neutralized with saturated NaHCO3(aq) (20 mL). The mixture was transferred to a separatory funnel, the layers were separated, and the aqueous layer was extracted with Et2O (3 × 15 mL). The combined organic extracts were washed with brine (15 mL), dried with magnesium sulfate, and concentrated in vacuo to give the crude nonconjugated enone, which was used in the next step without further purification.
The crude ketone was transferred to a flame-dried, 50 mL roundbottomed flask and dissolved in CH2Cl2 (18 mL) under an atmosphere of N2. DBU (0.52 mL, 3.60 mmol, 2.00 equiv) was added, and the mixture was allowed to stir at rt until complete conversion of the starting material was observed by TLC analysis, typically 3 h. The reaction was diluted with H2O (15 mL) and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried with sodium sulfate and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 97.5:2.5 to 95:5 to 90:10 hexanes/ EtOAc) to afford conjugated enone 53 (0.38 g, 54% yield) as a yellow solid. Analytical data: mp 85–89 °C; [α]D28 −118.8 (c = 0.85, CHCl3); 1H NMR (600 MHz, CDCl3) δ 3.19 (dd, J = 9.6, 3.0 Hz, 1H), 3.08 (dd, J = 4.2, 4.2 Hz, 1H), 2.44 (m, 2H), 2.34–2.20 (m, 4H), 2.01 (m, 1H), 1.93 (dt, J = 6.0, 3.0 Hz, 1H), 1.84 (m, 1H), 1.76 (m, 1H), 1.65–1.59 (m, 3H), 1.40 (m, 1H), 1.24 (s, 3H), 1.19 (s, 3H), 1.10 (s, 3H), 0.84 (s, 9H), 0.09 (s, 3H), 0.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 199.8, 162.7, 129.8, 85.1, 80.4, 74.8, 38.0, 37.7, 33.3, 27.5, 25.8, 25.2, 24.9, 23.3, 22.9, 22.4, 21.4, 18.1, 18.0, −2.1, −2.2; HRMS (ESI+) calcd for C23H40O3Si+Na, 415.2644; found 415.2636; IR (thin film, cm−1) 3053, 2954, 2887, 2855, 1683, 1616, 1576, 1472, 1362, 1265, 1172, 1045; TLC (90:10 hexanes/EtOAc) Rf = 0.34.
(3S,4aS,6aR,10aS,10bS)-3-(2-((tert-Butyldimethylsilyl)oxy)-propan-2-yl)-6a,10b-dimethyldodecahydro-7H-benzo[f]chromen-7-one (54)
An oven-dried, 50 mL two-neck round-bottomed flask was fitted with a stir bar and an oven-dried coldfinger condenser and placed under an atmosphere of Ar. The flask and condenser were cooled to −78 °C, and liq. NH3 (5 mL) was allowed to condense into the flask. Freshly cut Li0 (0.01 g, 1.43 mmol, 14.3 equiv) was washed with hexanes and added to the flask, resulting in the formation of a dark blue color. After being stirred 5 min at −78 °C, a solution of ketone 53 (0.04 g, 0.10 mmol, 1.00 equiv) in THF (3 mL) was added, and the reaction was warmed to −33 °C and stirred 15 min. The reaction was the cooled to −78 °C, diluted with THF (5 mL), and a solution of MeI (0.38 mL, 6.0 mmol, 60.0 equiv) in THF (2 mL) was added dropwise. The mixture was allowed to warm to rt and stirred until liq. NH3 had completely evaporated. The residue was quenched with saturated NH4Cl(aq) (10 mL), diluted with Et2O (10 mL) and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo to give the crude ketone 54 as a single diastereomer (as determined by 1H NMR spectroscopic analysis of the crude reaction mixture, which revealed a single compound). The product was purified via flash chromatography (100:0 to 98:2 to 95:5 to 90:10 hexanes/EtOAc) to afford ketone 54 (0.025 g, 61% yield) as a clear, viscous oil. Analytical data: [α]D28 −38.2 (c = 0.75, CHCl3); 1H NMR (600 MHz, CDCl3) δ 2.98 (dd, J = 8.4, 3.0 Hz, 1H), 2.84 (dd, J = 7.8, 3.6 Hz, 1H), 2.64 (m, 1H), 2.44 (dt, J = 7.2, 3.0 Hz, 1H), 2.25 (dd, J = 10.2, 6.0 Hz, 1H), 2.08 (m, 2H), 1.95–1.87 (m, 3H), 1.52–1.44 (m, 5H), 1.25 (m, 1H), 1.24 (s, 3H), 1.20 (s, 3H), 1.14 (s, 3H), 1.06 (m, 1H), 0.83 (s, 9H), 0.82 (s, 3H) 0.06 (s, 3H), 0.04 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 216.0, 84.9, 84.3, 74.8, 54.4, 47.9, 37.9, 37.3, 36.4, 32.6, 29.9, 27.3, 25.8, 25.1, 25.1, 23.8, 21.5, 19.1, 18.2, 16.0, −2.1, −2.2; HRMS (ESI+) calcd for C24H44O3Si+Na, 431.2957; found 431.2949; IR (thin film, cm−1) 3421, 2954, 2855, 1792, 1698, 1377, 1265, 1215, 1058; TLC (90:10 hexanes/EtOAc) Rf = 0.54.
(3S,4aS,6aS,10aR,10bS)-3-(2-((tert-Butyldimethylsilyl)oxy)-propan-2-yl)-10b-methyldodecahydro-7H-benzo[f]chromen-7-one (55)
An oven-dried, 50 mL two-neck round-bottomed flask was fitted with a stir bar and an oven-dried coldfinger condenser and placed under an atmosphere of Ar. The flask and condenser were cooled to −78 °C, and liq. NH3 (5 mL) was allowed to condense into the flask. Freshly cut Li0 (0.005 g, 0.714 mmol, 14.3 equiv) was washed with hexanes and added to the flask, resulting in the formation of a dark blue color. After being stirred 5 min at −78 °C, a solution of ketone 53 (0.02 g, 0.05 mmol, 1.00 equiv) in THF (2 mL) was added, and the reaction was warmed to −33 °C and stirred 15 min. The reaction was carefully quenched via portionwise addition of NH4Cl(s), and the mixture was allowed to warm to rt and stirred until liq. NH3 had completely evaporated. The residue was diluted with H2O (10 mL) and Et2O (10 mL) and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo to afford the crude ketone as a 1:1 mixture of diastereomers, which was taken on directly to the next step without further purification. A crude 1H NMR spectrum of this reaction is included in the Supporting Information.
This crude residue was transferred to a flame-dried, 20 mL scintillation vial and dissolved in C7H8 under an atmosphere of N2. DBU (0.01 mL, 0.05 mmol, 1.00 equiv) was added, and the mixture was warmed to 65 °C and stirred 12 h. The reaction was cooled to rt, diluted with H2O (10 mL) and CH2Cl2 (5 mL) and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 5 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. At this juncture, crude 1H NMR analysis revealed complete epimerization to a single diastereomer. The product was purified via flash chromatography (100:0 to 97.5:2.5 to 90:10 hexanes/EtOAc) to afford ketone 55 (0.015 g, 75% yield) as a clear, viscous oil. Analytical data: [α]D28 −72.0 (c = 0.75, CHCl3); 1H NMR (600 MHz, CDCl3) δ 3.03 (dd, J = 7.2, 3.6 Hz, 1H), 2.87 (dd, J = 7.8, 3.6 Hz, 1H), 2.36 (m, 1H), 2.26 (m, 2H), 2.10 (m, 1H), 1.91–1.83 (m, 3H), 1.63 (m, 1H), 1.57–1.52 (m, 4H), 1.43–1.36 (m, 3H), 1.21 (s, 3H), 1.16 (br s, 4H), 0.91 (s, 3H), 0.84 (s, 9H), 0.07 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 213.2, 85.1, 83.2, 74.8, 52.3, 49.2, 41.8, 36.7, 36.6, 27.4, 26.5, 26.2, 25.8, 24.9, 24.3, 23.6, 21.8, 18.1, 12.1, −2.1, −2.2; HRMS (ESI+) calcd for C23H42O3Si+Na, 417.2801; found 417.2793; IR (thin film, cm−1) 3420, 2951, 2854, 1715, 1652, 1472, 1376, 1251, 1155, 1051, 835; TLC (90:10 hexanes/EtOAc) Rf = 0.40.
(3S,4aS,6aS,10aR,10bR)-3-(2-((Butyldimethylsilyl)oxy)propan-2-yl)-10b-methyloctahydro-1H-6a,10a-epoxybenzo[f]chromen-7(8H)-one (56)
A 20 mL scintillation vial was charged with enone 53 (0.10 g, 0.26 mmol, 1.00 equiv) and (CH2Cl)2 (5 mL). p-NPBA32 (0.19 g, 0.89 mmol, 3.50 equiv) was added, and the vial was sealed with a screw-cap. The mixture was warmed to 65 °C and stirred until complete consumption of the starting material was observed by TLC analysis, typically 3 h. The reaction mixture was warmed to rt, quenched via saturated Na2S2O3(aq) (5 mL), and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 7 mL). The combined organic extracts were dried with sodium sulfate and concentrated in vacuo to afford the crude epoxide as a single diastereomer (as determined by 1H NMR spectroscopic analysis of the crude reaction mixture, which revealed a single compound). The product was purified via flash chromatography (100:0 to 97.5:2.5 to 95:5 hexanes/EtOAc) to afford keto-epoxide 56 (0.05 g, 47% yield) as a clear, viscous oil. Slow evaporation of 56 from HPLC grade methanol afforded crystals suitable for X-ray crystallographic analysis. Analytical data: [α]D28 −105.2 (c = 0.70, CHCl3); 1H NMR (600 MHz, CDCl3) δ 3.43 (dd, J = 8.4, 4.2 Hz, 1H), 3.06 (m, 1H), 2.58 (m, 1H), 2.08 (m, 2H), 1.91–1.85 (m, 3H), 1.64 (m, 3H), 1.55–1.49 (m, 3H), 1.37 (m, 1H), 1.20 (s, 3H), 1.15 (s, 3H), 1.03 (s, 3H), 0.84 (s, 9H), 0.07 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 207.2, 84.8, 75.4, 74.7, 64.3, 36.4, 36.2, 32.0, 27.4, 25.8, 24.9, 22.3, 21.6, 21.3,18.9, 18.8, 18.1, 15.9, −2.1, −2.2; HRMS (ESI+) calcd for C23H40O4Si+Na, 431.2594; found 431.2585; IR (thin film, cm−1) 3420, 2955, 2856, 1704, 1646, 1488, 1396, 1265, 1173, 1072, 835, 739; TLC (90:10 hexanes/EtOAc) Rf = 0.25.
(3S,4aS,7S,10bS)-3-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-10b-methyl-2,3,4a,5,6,7,8,9,10,10b-decahydro-1H-benzo[f]-chromen-7-ol (57)
A flame-dried, 20 mL scintillation vial was charged with ketone 53 (0.06 g, 0.15 mmol, 1.0 equiv) and THF (2 mL) under an atmosphere of N2. The reaction mixture was cooled to −78 °C, and LiAl(OtBu)3H (1 M solution in THF, 0.31 mL, 0.31 mmol, 2.00 equiv) was added in one portion. The reaction mixture was allowed to stir for 12 h, slowly warming to rt during this time period at which point TLC analysis confirmed complete consumption of the starting material. The reaction was quenched via saturated NH4Cl(aq) (5 mL) and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 7 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo to give the crude alcohol as a 10:1 mixture of diastereomers. The diastereomeric ratio was determined by 1H NMR spectroscopic analysis of the crude reaction mixture by comparison of the integration of the resonances at δ 3.99 (major diastereomer) and δ 3.82 (minor diastereomer). The product was purified via flash chromatography (90:10 to 80:20 hexanes/EtOAc) to afford alcohol 57 (0.054 g, 90% yield) as a clear, viscous oil. Analytical data: [α]D28 −92.7 (c = 1.00, CHCl3); 1H NMR (600 MHz, CDCl3) δ 3.99 (m, 1H), 3.20 (dd, J = 8.4, 3.6 Hz, 1H), 3.08 (dd, J = 7.2, 3.6 Hz, 1H), 2.49 (m, 1H), 1.98 (m, 2H), 1.88 (m, 2H), 1.82 (m, 1H), 1.71 (m, 2H), 1.65 (m, 2H), 1.57 (m, 2H), 1.52 (m, 2H), 1.29 (m, 1H), 1.23 (s, 3H), 1.17 (s, 3H), 0.99 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 139.7, 128.2, 85.1, 81.2, 74.9, 70.6, 34.5, 34.0, 32.6, 27.3, 26.8, 25.9, 25.1, 24.0, 23.8, 21.8, 19.8, 18.4, 18.2, −2.1, −2.2; HRMS (ESI+) calcd for C23H42O3Si+Na, 417.2801; found 417.2791; IR (thin film, cm−1) 3420, 2930, 2855, 1683, 1636, 1507, 1456, 1361, 1264, 1046, 835; TLC (90:10 hexanes/EtOAc) Rf = 0.25.
N′-((2S,4aS,8aS,E)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyloctahydro-5H-chromen-5-ylidene)-4-methylbenzene-sulfonohydrazide (59)
A flame-dried, 500 mL round-bottomed flask was charged with hydrazone 49 (6.21 g, 12.2 mmol, 1.00 equiv) and THF (122 mL) under an atmosphere of N2. The mixture was cooled to −50 °C, and nBuLi (2.60 M in hexanes, 16.4 mL, 42.7 mmol, 3.50 equiv) was added over a period of ~2 min, producing a dark orange color. The reaction mixture was allowed to stir 40 min, whereupon MeI (1.90 mL, 30.5 mmol, 2.50 equiv) was added, resulting in a color change from orange to yellow. The reaction was allowed to stir until complete consumption of the starting material was observed by TLC analysis, typically 20 min. The reaction was quenched via saturated NH4Cl(aq) (40 mL) and allowed to warm to rt. The mixture was transferred to a separatory funnel, the organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 40 mL). The combined organic extracts were washed with brine (40 mL), dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (90:10 to 80:20 hexanes/EtOAc) to afford hydrazone 59 (6.37 g, 98% yield) as a white foam in a 7:1 diastereomeric ratio. Analytical data: [α]D28 −121.0 (c = 0.60, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.83 (d, J = 7.2 Hz, 2H), 7.72 (br s, 1H), 7.30 (d, J = 7.8 Hz, 2H), 3.05 (m, 1H), 3.00 (s, 1H), 2.73 (q, J = 7.8 Hz, 1H), 2.43 (s, 3H), 2.01 (d, J = 13.2 Hz, 1H), 1.69 (m, 1H), 1.57–1.54 (m, 5H), 1.45 (m, 1H), 1.34 (m, 1H), 1.20 (s, 3H), 1.15 (s, 3H), 1.07 (d, J = 7.2 Hz, 3H), 0.95 (s, 3H), 0.84 (s, 9H), 0.07 (s, 3H), 0.04 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 167.1, 143.8, 135.3, 129.3, 128.0, 127.9, 85.2, 82.0, 74.7, 41.9, 33.3, 28.3, 27.7, 27.2, 25.8, 25.0, 22.8, 21.6, 21.2, 19.1, 18.3, 18.1, −2.1, −2.2; HRMS (ESI+) calcd for C27H46N2O4SSi+Na, 545.2845; found 545.2840; IR (thin film, cm−1) 3225, 2954, 2855, 1472, 1396, 1265, 1168, 1090, 1038, 812, 773; TLC (90:10 hexanes/EtOAc) Rf = 0.35.
(2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromene-5-carbaldehyde (60)
A flame-dried, 25 mL round-bottomed flask was charged with hydrazone 59 (0.48 g, 0.92 mmol, 1.00 equiv) and THF (9.5 mL) under an atmosphere of N2. The solution was cooled to −50 °C, and nBuLi (1.70 M in hexanes, 3.25 mL, 5.52 mmol, 6.00 equiv) was added over a period of ~2 min, producing a dark orange color. The reaction was allowed to stir 30 min, whereupon a venting needle was added, and the mixture was warmed to 0 °C and stirred 5 min. The reaction was then warmed to rt and stirred until complete consumption of the starting material was observed by TLC analysis, typically 20 min. The venting needle was removed, DMF (0.71 mL, 9.2 mmol, 10.0 equiv) was added, and the reaction was stirred 20 min. The mixture was diluted with H2O (15 mL) and Et2O (10 mL) and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 15 mL). The combined organic extracts were washed with brine (15 mL), dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (95:5 to 90:10 hexanes/EtOAc) to afford aldehyde 60 (0.21 g, 62% yield) as a yellow, viscous oil. Analytical data: [α]D28 −151.8 (c = 0.80, CHCl3); 1H NMR (600 MHz, CDCl3) δ 10.05 (br s, 1H), 3.16 (m, 1H), 3.11 (dd, J = 9.0, 3.0 Hz, 1H), 2.65 (m, 1H), 2.38 (m, 1H), 2.28 (m, 1H), 2.06 (s, 3H), 1.69 (m, 2H), 1.62–1.53 (m, 3H), 1.22 (s, 3H), 1.17 (s, 3H), 1.14 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 191.9, 153.9, 140.3, 85.8, 80.6, 74.9, 35.7, 34.3, 33.5, 27.1, 25.8, 25.1, 23.7, 21.6, 18.8, 18.2, 18.1, −2.1, −2.2; HRMS (ESI+) calcd for C21H38O3Si+Na, 389.2488; found 389.2481; IR (thin film, cm−1) 2954, 2928, 2855, 1733, 1674, 1472, 1376, 1251, 1095, 1005, 835; TLC (90:10 hexanes/EtOAc) Rf = 0.50.
tert-Butyl((2-((2S,4aS,8aS)-4a,6-dimethyl-5-vinyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-2-yl)propan-2-yl)oxy)dimethylsilane (61)
A flame-dried, 25 mL round-bottomed flask was charged with methyltriphenylphosphonium bromide (1.90 g, 5.28 mmol, 8.00 equiv) and THF (7 mL) under an atmosphere of N2. The mixture was cooled to 0 °C and nBuLi (1.69 M in hexanes, 2.94 mL, 4.95 mmol, 7.50 equiv) was added dropwise. The deep yellow mixture was allowed to stir 1 h at 0 °C upon which the aldehyde 60 (0.24 g, 0.66 mmol, 1.00 equiv) was added as a solution in THF (2 mL). The reaction was allowed to stir until complete consumption of the starting material was observed by TLC analysis, typically 15 min. The reaction was diluted with H2O (15 mL) and transferred to a separatory funnel. The layers were separated, and the aqueous layer was extracted with Et2O (3 × 15 mL). The combined organic extracts were washed with brine (15 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 99:1 to 97.5:2.5 hexanes/EtOAc) to afford diene 61 (0.20 g, 82% yield) as a clear oil. Analytical data: [α]D28 −94.4 (c = 1.50, CHCl3); 1H NMR (600 MHz, CDCl3) δ 6.13 (dd, J = 12.0, 6.0 Hz, 1H), 5.23 (dd, J = 8.4, 3.0 Hz, 1H), 4.96 (dd, J = 15.6, 2.4 Hz, 1H), 3.19 (dd, J = 7.2, 4.8 Hz, 1H), 3.08 (dd, J = 6.6, 4.2 Hz, 1H), 2.18 (m, 1H), 2.08 (dd, J = 11.4, 6.6 Hz, 1H), 1.84 (dt, J = 6.0, 4.2 Hz, 1H), 1.66 (br s, 4H), 1.55 (br s, 3H), 1.28 (m, 1H), 1.23 (s, 3H), 1.17 (s, 3H), 1.02 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 138.1, 134.3, 127.5, 118.0, 85.2, 81.4, 75.0, 36.2, 35.2, 31.6, 27.3, 25.9, 25.1, 24.3, 21.8, 20.5, 18.7, 18.2; HRMS (ESI+) calcd for C22H40O2Si+Na, 387.2695; found 387.2688; IR (thin film, cm−1) 2954, 2855, 1717, 1471, 1376, 1253, 1167, 1039, 880, 741; TLC (90:10 hexanes/EtOAc) Rf = 0.93.
((2-((2S,4aS,8aS)-5-((Z)-Buta-1,3-dien-1-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-2-yl)propan-2-yl)oxy)(tert-butyl) dimethylsilane (63)
A flame-dried, 20 mL scintillation vial was charged with allyltriphenylphosphonium bromide (1.31 g, 3.43 mmol, 8.00 equiv) and THF (5 mL) under an atmosphere of N2. The mixture was cooled to 0 °C and nBuLi (2.64 M in hexanes, 1.22 mL, 3.21 mmol, 7.50 equiv) was added dropwise. The deep yellow mixture was allowed to stir 1 h at 0 °C, whereupon the aldehyde 60 (0.16 g, 0.43 mmol, 1.00 equiv) was added as a solution in THF (2 mL). The reaction was allowed to stir until complete consumption of the starting material was observed by TLC analysis, typically 12 h. The reaction was diluted with H2O (15 mL) and transferred to a separatory funnel. The layers were separated, and the aqueous layer was extracted with Et2O (3 × 15 mL). The combined organic extracts were washed with brine (15 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 99:1 to 97.5:2.5 hexanes/EtOAc) to afford triene 63 (0.06 g, 36% yield) as a clear oil. Analytical data: [α]D28 −49.8 (c = 1.25, CHCl3); 1H NMR (600 MHz, CDCl3) δ 6.37 (m, 1H), 6.05 (m, 2H), 5.15 (d, J = 16.8 Hz, 1H), 5.03 (d, J = 10.2 Hz, 1H), 3.19 (dd, J = 7.2, 4.8 Hz, 1H), 2.20 (m, 1H), 2.11 (dd, J = 12.6, 5.4 Hz, 1H), 1.88 (dt, J = 6.0, 3.0 Hz, 1H), 1.68 (br s, 5H), 1.56 (m, 2H), 1.27 (m, 1H), 1.23 (s, 3H), 1.17 (s, 3H), 1.04 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 137.7, 137.1, 133.9, 130.7, 128.7, 115.4, 85.2, 81.4, 74.9, 36.6, 35.3, 31.8, 27.3, 25.8, 25.0, 24.2, 21.8, 20.8, 18.9, 18.2, −2.1, −2.2; HRMS (ESI+) calcd for C24H42O2Si +Na, 413.2852; found 413.2843; IR (thin film, cm−1) 3420, 2929, 2855, 1670, 1497, 1457, 1387, 1265, 1165, 1040, 835; TLC (90:10 hexanes/EtOAc) Rf = 0.94.
((2-((2S,4aS,8aS,E)-5-(But-3-en-1-ylidene)-4a-methyl-6-methyle-neoctahydro-2H-chromen-2-yl)propan-2-yl)oxy)(tert-butyl)-dimethylsilane (65)
The triene 63 (0.017 g, 0.043 mmol, 1.00 equiv) was taken up into hexanes and transferred to a toroidal photochemical reactor equipped with a water-cooled Pyrex immersion well. A 450 W Hanovia medium pressure mercury vapor lamp was lowered inside the immersion well, and the triene solution was irradiated for 1 h. The solution was subsequently concentrated in vacuo. The product was purified via flash chromatography to give rearrangement product 65 (0.009 g, 53% yield) as a clear, viscous oil. Analytical data: [α]D28 −11.8 (c = 0.10, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.83 (m, 1H), 5.20 (t, J = 7.8 Hz, 1H), 5.01 (m, 1H), 4.97 (m, 1H), 4.66 (t, J = 1.8 Hz, 1H), 3.10 (dd, J = 7.8, 4.2 Hz, 1H), 3.05 (m, 1H), 2.90 (m, 2H), 2.33 (m, 1H), 2.06 (m, 1H), 1.70 (m, 2H), 1.66–1.55 (m, 6H), 1.22 (s, 3H, 1.17 (s, 3H), 0.94 (s, 3H), 0.85 (s, 9H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 148.0, 144.1, 138.3, 119.1, 114.3, 112.8, 85.2, 82.5, 74.9, 39.8, 34.5, 33.8, 33.2, 28.5, 27.2, 25.8, 25.1, 21.9, 18.2, 17.9, −2.1, −2.2; HRMS (ESI+) calcd for C24H42O2Si +Na, 413.2852; found 413.2843; IR (thin film, cm−1) 3053, 2956, 2855, 1749, 1670, 1540, 1456, 1265, 1046, 835; TLC (90:10 hexanes/EtOAc) Rf = 0.97.
Synthesis of Enol Silane 66
1-((2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)ethan-1-one (S4)
A flame-dried, 25 mL round-bottomed flask was charged with hydrazone 59 (0.30 g, 0.57 mmol, 1.00 equiv) and THF (6 mL) under an atmosphere of N2. The solution was cooled to −50 °C, and nBuLi (2.64 M in hexanes, 1.30 mL, 3.44 mmol, 6.00 equiv) was added over a period of ~2 min, producing a dark orange color. The reaction was allowed to stir 30 min, whereupon a venting needle was added, and the mixture was warmed to 0 °C and stirred 5 min. The reaction was then warmed to rt and stirred until complete consumption of the starting material was observed by TLC analysis, typically 20 min. The venting needle was removed, the mixture was cooled to −78 °C, and acetaldehyde (0.32 mL, 5.74 mmol, 10.0 equiv) was added dropwise. The reaction was allowed to stir 25 min, whereupon H2O (5 mL) and Et2O (5 mL) were added, and the mixture was warmed to rt and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were washed with brine (10 mL), dried with magnesium sulfate, and concentrated in vacuo to give the crude alcohol, which was taken on to the next step without further purification.
The crude residue was taken up into CH2Cl2 (5 mL) and transferred to a 20 mL scintillation vial. Dess-Martin periodinane (0.29 g, 0.68 mmol, 2.00 equiv) was added to the vial, and the mixture was allowed to stir until TLC analysis indicated complete consumption of the starting material, typically 15 min. The mixture was then quenched via a 1:1 solution of saturated NaHCO3(aq) and saturated Na2S2O3(aq) (5 mL), and the mixture was stirred 5 min. The reaction mixture was then diluted with Et2O (10 mL) and partitioned in a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 97.5:2.5 to 95:5 hexanes/EtOAc) to afford ketone S4 (0.09 g, 43% yield) as a yellow, viscous oil. Analytical data: [α]D28 −31.6 (c = 0.50, CHCl3); 1H NMR (600 MHz, CDCl3) δ 3.21 (dd, J = 6.0, 3.6 Hz, 1H), 3.09 (dd, J = 5.4, 3.6 Hz, 1H), 2.25 (s, 3H), 2.16 (m, 1H), 2.08 (m, 1H), 1.67 (m, 2H), 1.56–1.54 (m, 6H), 1.44 (m, 1H), 1.21 (s, 3H), 1.17–1.15 (m, 6H), 0.83 (s, 9H), 0.07 (s, 3H), 0.04 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 208.2, 143.5, 128.5, 85.4, 80.4, 74.8, 35.4, 34.4, 33.3, 30.6, 27.3, 25.8, 25.0, 23.8, 21.3, 20.1, 19.6, 18.1, −2.1, −2.2; HRMS (ESI+) calcd for C22H40O3Si+Na, 403.2644; found 403.2636; IR (thin film, cm−1) 2955, 2854, 1829, 1686, 1488, 1361, 1249, 1095, 835, 739; TLC (90:10 hexanes/EtOAc) Rf = 0.38.
tert-Butyl((1-((2S,4aS,8aS)-2-(2-((tert-butyldimethylsilyl)oxy)-propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)vinyl)oxy)dimethylsilane (66)
A flame-dried, 20 mL scintillation vial was charged with ketone S4 (0.06 g, 0.16 mmol, 1.00 equiv) and THF (2 mL) under an atmosphere of N2. The reaction was cooled to 0 °C, and NEt3 (0.07 mL, 0.47 mmol, 3.00 equiv) and TBSOTf (0.075 mL, 0.32 mmol, 2.00 equiv) were added sequentially. The reaction mixture was warmed to rt and stirred until TLC analysis showed complete consumption of the starting material, typically 3 h. The reaction was quenched via addition of saturated NaHCO3(aq) (2 mL) and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with pentane (3 × 5 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 98:2 to 97.5:2.5 hexanes/EtOAc) to afford silyloxydiene 66 (0.077 g, 99% yield) as a clear, viscous oil. Analytical data: [α]D28 −20.8 (c = 0.33, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.27 (s, 1H), 3.90 (s, 1H), 3.15 (dd, J = 7.2, 4.8 Hz, 1H), 3.08 (dd, J = 7.8, 3.0 Hz, 1H), 2.15 (m, 1H), 2.06 (dd, J = 11.4, 6.6 Hz, 1H), 1.81 (d, J = 13.2 Hz, 1H), 1.66 (br s, 5H), 1.54 (m, 2H), 1.39 (m, 1H), 1.23 (s, 3H), 1.16 (s, 3H), 1.07 (s, 3H), 0.92 (s, 9H), 0.84 (s, 9H), 0.20 (s, 3H), 0.17 (s, 3H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 155.6, 138.9, 128.6, 85.3, 81.1, 75.0, 34.7, 30.5, 27.2, 25.9, 25.8, 25.7, 25.2, 24.1, 21.9, 20.8, 18.2, 18.1, −2.1, −2.2, −4.5, −4.6; HRMS (ESI+) calcd for C28H54O3Si2+Na, 517.3509; found 517.3499; IR (thin film, cm−1) 2930, 2896, 1611, 1497, 1376, 1265, 1165, 1038, 835, 775; TLC (90:10 hexanes/EtOAc) Rf = 0.94.
(E)-1-((2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)but-2-en-1-one (67)
A flame-dried, 25 mL round-bottomed flask was charged with hydrazone 59 (0.30 g, 0.57 mmol, 1.00 equiv) and THF (6 mL) under an atmosphere of N2. The solution was cooled to −50 °C, and nBuLi (2.64 M in hexanes, 1.30 mL, 3.44 mmol, 6.00 equiv) was added over a period of ~2 min, producing a dark orange color. The reaction was allowed to stir 30 min, whereupon a venting needle was added, and the mixture was warmed to 0 °C and stirred 5 min. The reaction was then warmed to rt and stirred until complete consumption of the starting material was observed by TLC analysis, typically 20 min. The venting needle was removed, the mixture was cooled to −78 °C, and (E)-crotonaldehyde (0.48 mL, 5.74 mmol, 10.0 equiv) was added dropwise. The reaction was allowed to stir 25 min, whereupon H2O (5 mL) and Et2O (5 mL) were added, and the mixture was warmed to rt and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were washed with brine (10 mL), dried with magnesium sulfate, and concentrated in vacuo to give the crude alcohol, which was taken on to the next step without further purification
The crude residue was taken up into CH2Cl2 (5 mL) and transferred to a 20 mL scintillation vial. Dess-Martin periodinane (0.29 g, 0.68 mmol, 2.00 equiv) was added to the vial, and the mixture was allowed to stir until TLC analysis indicated complete consumption of the starting material, typically 15 min. The mixture was then quenched via a 1:1 solution of saturated NaHCO3(aq) and saturated Na2S2O3(aq) (5 mL), and the mixture was stirred 5 min. The reaction mixture was then diluted with Et2O (10 mL) and partitioned in a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 97.5:2.5 to 95:5 hexanes/EtOAc) to afford ketone 67 (0.10 g, 46% yield) as a yellow, viscous oil. Analytical data: [α]D28 −72.2 (c = 0.48, CHCl3); 1H NMR (600 MHz, CDCl3) δ 6.73 (m, 1H), 6.14 (dd, J = 13.8, 1.8 Hz, 1H), 3.26 (dd, J = 6.6, 5.4 Hz, 1H), 3.09 (dd, J = 7.8, 1.8 Hz, 1H), 2.21 (m, 1H), 2.11 (dd, J = 11.4, 6.6 Hz, 1H), 1.93 (dd, J = 5.4, 1.8 Hz, 3H), 1.74–1.70 (m, 2H), 1.51–1.47 (m, 6H), 1.38 (m, 1H), 1.22 (s, 3H), 1.15 (s, 3H), 1.14 (s, 3H), 0.83 (s, 9H), 0.07 (s, 3H), 0.04 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 200.7, 146.4, 140.2, 134.6, 130.2, 85.4, 80.4, 74.9, 35.7, 34.5, 30.5, 27.2, 25.8, 25.0, 23.9, 21.4, 20.7, 19.7, 18.4, 18.1, −2.1, −2.2; HRMS (ESI+) calcd for C24H42O3Si+Na, 429.2801; found 429.2792; IR (thin film, cm−1) 2955, 2855, 1671, 1472, 1361, 1265, 1165, 1041, 835, 739; TLC (90:10 hexanes/EtOAc) Rf = 0.56.
tert-Butyl((2-((2S,4aR,8aS)-5-iodo-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-2-yl)propan-2-yl)oxy)dimethylsilane (68)
A flame-dried, 20 mL scintillation vial was charged with hydrazone 59 (0.30 g, 0.57 mmol, 1.00 equiv) and THF (6 mL) under an atmosphere of N2. The solution was cooled to −50 °C, and nBuLi (1.70 M in hexanes, 2.00 mL, 3.42 mmol, 6.00 equiv) was added over a period of ~2 min, producing a dark orange color. The reaction was allowed to stir 30 min, whereupon a venting needle was added, and the mixture was warmed to 0 °C and stirred 5 min. The reaction was then warmed to rt and stirred until complete consumption of the starting material was observed by TLC analysis, typically 20 min. The venting needle was removed, the mixture was cooled to 0 °C, and I2 (0.43 g, 1.71 mmol, 3.00 equiv) was added portionwise. The reaction was allowed to stir 20 min, whereupon H2O (5 mL) and Et2O (5 mL) were added, and the mixture was warmed to rt and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were washed with brine (10 mL) and saturated Na2S2O3(aq), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 99:1 to 98:2) to afford iodide 68 (0.18 g, 67% yield) containing 17% of the inseparable vinyl C–H compound (arising from protic quenching of the transient vinyllithium) by 1H NMR analysis. Analytical data: [α]D28 −248.0 (c = 1.00, CHCl3); 1H NMR (600 MHz, CDCl3) δ 3.32 (dd, J = 7.8, 4.2 Hz, 1H), 3.07 (dd, J = 9.0, 3.0 Hz, 1H), 2.30 (m, 1H), 2.22 (dd, J = 11.4, 6.0 Hz, 1H), 1.90 (m, 1H), 185 (s, 3H), 1.70 (m, 1H), 1.60 (br s, 1H), 1.55 (m, 2H), 1.28 (m, 1H), 1.24 (s, 3H), 1.18 (s, 3H), 0.99 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 136.2, 131.2, 114.8, 85.5, 81.1, 74.6, 41.5, 41.3, 32.3, 29.8, 27.4, 25.8, 25.0, 24.2, 22.7, 18.5, 18.1, −2.1, −2.2; HRMS (ESI+) calcd for C20H37IO2Si+Na, 487.1505; found 487.1497; IR (thin film, cm−1) 2954, 2854, 1771, 1670, 1488, 1376, 1264, 1162, 1040, 834; TLC (90:10 hexanes/EtOAc) Rf = 0.91.
((2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)methanol (69)
A flame-dried, 50 mL round-bottomed flask was charged with hydrazone 59 (0.58 g, 1.10 mmol, 1.00 equiv) and THF (11 mL) under an atmosphere of N2. The solution was cooled to −50 °C, and nBuLi (1.55 M in hexanes, 4.27 mL, 6.62 mmol, 6.00 equiv) was added over a period of ~2 min, producing a dark orange color. The reaction was allowed to stir 30 min, whereupon a venting needle was added, and the mixture was warmed to 0 °C and stirred 5 min. The reaction was then warmed to rt and stirred until complete consumption of the starting material was observed by TLC analysis, typically 20 min. The venting needle was removed, (HCHO)n (0.35 g, 11.0 mmol, 10.0 equiv) was added to the mixture in one portion, and the reaction was allowed to stir 40 min at rt. H2O (10 mL) and Et2O (5 mL) were added, and the mixture was transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were washed with brine (10 mL), dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 95:5 to 90:10 to 80:20 hexanes/EtOAc) to afford alcohol 69 (0.26 g, 65% yield) as a yellow, viscous oil. Analytical data: [α]D28 −53.7 (c = 0.70, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.20 (d, J = 11.4 Hz, 1H), 4.07 (d, J = 11.4 Hz, 1H), 3.18 (dd, J = 6.0, 4.2 Hz, 1H), 3.09 (dd, J = 6.6, 3.6 Hz, 1H), 2.17 (m, 1H), 2.06 (m, 1H), 1.98 (dt, J = 6.0, 3.6 Hz, 1H), 1.71 (s, 3H), 1.66 (m, 2H), 1.60 (m, 2H), 1.44 (m, 1H), 1.23 (s, 3H), 1.17 (s, 3H), 1.00 (s, 3H), 0.84 (s, 9H), 0.80 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 137.4, 132.2, 85.2, 81.3, 74.9, 58.2, 31.5, 25.8, 25.0, 24.2, 21.7, 19.4, 19.0, 18.1, −2.16, −2.21; HRMS (ESI+) calcd for C21H40O3Si+Na, 391.2645; found 391.2652; IR (thin film, cm−1) 3409, 2953, 2855, 1641, 1461, 1377, 1252, 1168, 1092, 834; TLC (85:15 hexanes/EtOAc) Rf = 0.29.
Procedure for One-Pot Synthesis of 69 from Alcohol 49
((2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)methanol (69)
A flame-dried, 250 mL round-bottomed flask was charged with hydrazone 49 (1.50 g, 2.95 mmol, 1.00 equiv) and THF (30 mL) under an atmosphere of N2. The solution was cooled to −50 °C, and nBuLi (3.97 mL, 2.6 M in hexanes, 10.32 mmol, 3.50 equiv) was added dropwise, producing a dark orange color. The reaction mixture was allowed to stir 40 min at this temperature, then MeI (0.46 mL, 7.37 mmol, 2.50 equiv) was added. The reaction was allowed to stir at −50 °C until TLC analysis confirmed complete conversion of 49, typically 20 min. An additional charge of nBuLi (9.07 mL, 2.6 M in hexanes, 23.6 mmol, 8.00 equiv) was added to the reaction, and the resulting mixture was stirred 30 min. The flask was fitted with a venting needle, and the reaction mixture was then warmed to 0 °C, stirred 5 min, then warmed to rt and stirred until complete consumption of the intermediate hydrazone was observed by TLC analysis, typically 15–25 min (scale dependent). The septum was partially removed, and (HCHO)n (0.89 g, 29.5 mmol, 10.0 equiv) was added in one portion with vigorous stirring. The reaction was allowed to stir 30 min at rt, at which time the mixture was diluted with H2O (25 mL) and Et2O (20 mL) and transferred to a separatory funnel. The organic layer was separated and the aqueous layer was extracted with Et2O (3 × 20 mL). The combined organic extracts were washed with brine (30 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 95:5 to 90:10 to 80:20 hexanes/EtOAc) to afford alcohol 69 (0.76 g, 66% yield).
((2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)methyl acetate (70a)
A flame-dried, 20 mL scintillation vial was charged with alcohol 69 (0.05 g, 0.14 mmol, 1.00 equiv) and CH2Cl2 under an atmosphere of N2. The mixture was cooled to 0 °C, and NEt3 (0.04 mL, 0.27 mmol, 2.00 equiv), DMAP (0.002 g, 0.014 mmol, 0.1 equiv), and last Ac2O (0.03 mL, 0.27 mmol, 2.00 equiv) were added sequentially. The mixture was allowed to stir at this temperature until TLC analysis showed complete consumption of the starting material, typically 3 h. The mixture was diluted with H2O (7 mL) and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 7 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 98:2 to 95:5 to 90:10 hexanes/EtOAc) to afford acetate 70a (0.046 g, 83% yield) as a clear, viscous oil. Analytical data: [α]D28 −59.0 (c = 1.35, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.59 (dd, J = 12.0, 5.4 Hz, 2H), 3.19 (dd, J = 6.0, 4.8 Hz, 1H), 3.08 (dd, J = 6.0, 4.2 Hz, 1H), 2.20 (m, 1H), 2.11 (m, 1H), 2.05 (s, 3H), 1.82 (m, 1H), 1.67 (br s, 5H), 1.57 (m, 1H), 1.36 (m, 1H), 1.23 (s, 3H), 1.17 (s, 3H), 0.99 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 171.3, 134.8, 132.2, 85.1, 81.0, 74.9, 60.4, 36.3, 33.9, 31.6, 27.3, 25.8, 25.0, 24.1, 21.6, 21.2, 19.3, 19.2, 18.1, −2.1, −2.2; HRMS (ESI+) calcd for C23H42O4Si+Na, 433.2750; found 433.2741; IR (thin film, cm−1) 2955, 2856, 1771, 1730, 1472, 1377, 1249, 1092, 1039, 835, 759; TLC (90:10 hexanes/EtOAc) Rf = 0.54.
((2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)methyl propionate (70b)
A flame-dried, 20 mL scintillation vial was charged with CH2Cl2 (3 mL) and propionic acid (0.02 g, 0.27 mmol, 2.00 equiv) at rt under an atmosphere of N2. DCC (0.06 g, 0.27 mmol, 2.00 equiv) and DMAP (0.002 g, 0.014 mmol, 0.10 equiv) were added followed last by a solution of alcohol 69 (0.05 g, 0.14 mmol, 1.00 equiv) in CH2Cl2 (1 mL), and the reaction was allowed to stir at rt until TLC analysis confirmed complete conversion of the starting material, typically 3.5 h. The reaction mixture was filtered through cotton into a separatory funnel, and H2O (10 mL) and EtOAc (10 mL) were added. The mixture was extracted with EtOAc (3 × 10 mL), and the combined organic extracts were washed with saturated NaHCO3(aq) (10 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 98:2 to 95:5 hexanes/EtOAc) to afford ester 70b (0.05 g, 86% yield) as a clear, viscous oil. Analytical data: [α]D28 −51.4 (c = 1.25, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.60 (dd, J = 12.0, 7.8 Hz, 2H), 3.19 (dd, J = 5.4, 5.4 Hz, 1H), 3.08 (dd, J = 6.0, 4.2 Hz, 1H), 2.32 (q, J = 7.2 Hz, 2H), 2.19 (m, 1H), 2.09 (dd, J = 12.6, 4.8 Hz, 1H), 1.82 (dt, J = 6.0, 3.6 Hz, 1H), 1.67 (br s, 5H), 1.56 (m, 1H), 1.37 (m, 1H), 1.23 (s, 3H), 1.17 (s, 3H), 1.40 (t, J = 7.2 Hz, 3H), 0.99 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 174.6, 134.6, 132.3, 85.1, 81.0, 74.9, 60.3, 36.3, 34.0, 27.7, 27.3, 25.8, 25.0, 24.2, 21.7, 19.3, 19.2, 18.2, 9.2, −2.1, −2.2; HRMS (ESI+) calcd for C24H44O4Si+Na, 447.2907; found 447.2897; IR (thin film, cm−1) 3053, 2955, 2855, 1731, 1540, 1472, 1322, 1265, 1179, 1071, 835; TLC (90:10 hexanes/EtOAc) Rf = 0.68.
((2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)methyl 2-(1H-indol-2-yl)propanoate (70d)
A 20 mL scintillation vial was charged with ethyl 2-(1H-indol-2-yl)propanoate50 (0.2 g, 0.92 mmol, 1.00 equiv) and a 3:1 mixture of MeOH/THF (5 mL). LiOH (4 M in H2O, 0.7 mL, 2.76 mmol, 3.00 equiv) was added, and the mixture was allowed to stir at rt until complete consumption of the starting material was observed by TLC analysis, typically 6 h. The reaction mixture was concentrated on a rotary evaporator, and the residue was diluted with H2O (10 mL) and transferred to a separatory funnel. The aqueous layer was extracted with EtOAc (2 × 10 mL), and the aqueous layer was then acidified to pH = 0 with 1 M HCl(aq) and extracted with CH2Cl2 (3 × 10 mL). The combined CH2Cl2 extracts were dried with magnesium sulfate and concentrated in vacuo to give the crude carboxylic acid. This material could not be isolated due to spontaneous decarboxylation, but could be carried forward directly to the next step without further purification.
The crude acid (~4.00 equiv) was dissolved in CH2Cl2 (3 mL) and transferred to a flame-dried, 20 mL scintillation vial under an atmosphere of N2. DCC (0.095 g, 0.46 mmol, 2.00 equiv) was added followed by DMAP (0.003 g, 0.023 mmol, 0.10 equiv) and last a solution of alcohol 69 (0.085 g, 0.23 mmol, 1.00 equiv) in CH2Cl2 (1 mL). The reaction was allowed to stir until TLC analysis confirmed complete consumption of the starting material, typically 20 min. The reaction mixture was filtered through cotton into a separatory funnel, and H2O (10 mL) and EtOAc (10 mL) were added. The mixture was extracted with EtOAc (3 × 10 mL), and the combined organic extracts were washed with saturated NaHCO3(aq) (10 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 95:5 to 90:10 hexanes/ EtOAc) to afford an inseparable mixture of diastereomeric esters 70d (0.14 g, 99% yield) as a brown, viscous oil. Analytical data: [α]D28 −68.4 (c = 0.43, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.59 (m, 1H), 7.56 (m, 1H), 7.32 (m, 1H), 7.15 (m, 1H), 7.09 (m, 1H), 6.37 (br s, 1H), 4.66 (m, 2H), 3.95 (m, 1H), 3.17 (m, 1H), 3.06–2.98 (m, 1H), 2.21 (m, 1H), 2.10 (m, 1H), 1.68–1.66 (m, 4H), 1.64–1.62 (m, 4H), 1.46 (m, 2H), 1.32 (m, 2H), 1.23–1.21 (m, 3H), 1.16–1.14 (m, 3H), 0.97–0.96 (m, 3H), 0.86 (s, 9H), 0.10–0.09 (m, 3H), 0.07 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 173.6, 136.7, 136.6, 136.0, 135.4, 131.9, 128.0, 121.7, 120.2, 119.7, 110.6, 100.1, 85.1, 85.0, 80.9, 74.8, 61.3, 61.2, 41.5, 39.3, 39.2, 36.2, 33.9, 31.6, 27.2, 27.1, 26.1, 25.8, 25.2, 25.1, 24.1, 23.3, 21.5, 19.2, 18.1, 17.4, 17.2, 14.1, −2.2, −2.3; HRMS (ESI+) calcd for C32H49NO4Si+Na, 562.3329; found 562.3320; IR (thin film, cm−1) 3392, 2954, 2855, 1716, 1471, 1377, 1250, 1172, 1069, 835; TLC (90:10 hexanes/EtOAc) Rf = 0.41.
((2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)methyl 3-((tert-butyldimethylsilyl)oxy)-2-methylpropanoate (70e)
A flamedried, 20 mL scintillation vial was charged with CH2Cl2 (3 mL) and 3-((tert-butyldimethylsilyl)oxy)-2-methylpropanoic acid51 (0.05 g, 0.22 mmol, 2.00 equiv) at rt under an atmosphere of N2. DCC (0.04 g, 0.22 mmol, 2.00 equiv) and DMAP (0.002 g, 0.014 mmol, 0.10 equiv) were added followed last by a solution of alcohol 69 (0.04 g, 0.11 mmol, 1.00 equiv) in CH2Cl2 (1 mL), and the reaction was allowed to stir at rt until TLC analysis confirmed complete conversion of the starting material, typically 3.5 h. The reaction mixture was filtered through cotton into a separatory funnel, and H2O (10 mL) and EtOAc (10 mL) were added. The mixture was extracted with EtOAc (3 × 10 mL), and the combined organic extracts were washed with saturated NaHCO3(aq) (10 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 98:2 to 95:5 hexanes/EtOAc) to afford an inseparable mixture of diastereomeric esters 70e (0.047 g, 76% yield) as a clear, viscous oil. Analytical data: [α]D28 −42.1 (c = 1.20, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.58 (m, 2H), 3.79 (m, 1H), 3.64 (m, 1H), 3.18 (m, 1H), 3.08 (dd, J = 7.2, 3.6 Hz, 1H), 2.62 (m, 1H), 2.19 (m, 1H), 2.09 (m, 1H), 1.82 (m, 1H), 1.67–1.66 (m, 5H), 1.57 (m, 2H), 1.37 (m, 1H), 1.23 (s, 3H), 1.17 (s, 3H), 1.14–1.12 (m, 3H), 0.99 (m, 3H), 0.87 (s, 9H), 0.84 (s, 9H), 0.08 (s, 3H), 0.05 (s, 3H), 0.03 (br s, 6H), 13C NMR (150 MHz, CDCl3) δ 175.1, 134.6, 134.5, 132.3, 85.1, 81.0, 74.9, 65.3, 65.2, 60.4, 60.3, 42.7, 36.3, 34.1, 34.0, 31.7, 27.4, 27.3, 25.9, 25.8, 25.0, 24.2, 21.7, 21.6, 19.3, 19.2, 18.2, 13.6, −2.1, −2.2, −5.5; HRMS (ESI+) calcd for C31H60O5Si2+Na, 591.3877; found 591.3867; IR (thin film, cm−1) 3053, 2955, 2884, 2857, 1727, 1471, 1377, 1265, 1179, 1049, 836; TLC (90:10 hexanes/EtOAc) Rf = 0.73.
((2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)methyl (S)-2-Bromopropanoate (70f)
A flame-dried, 20 mL scintillation vial was charged with CH2Cl2 (3 mL) and (S)-2-bromopropanoic acid52 (0.04 g, 0.27 mmol, 2.00 equiv) at rt under an atmosphere of N2. DCC (0.06 g, 0.27 mmol, 2.00 equiv) and DMAP (0.002 g, 0.014 mmol, 0.10 equiv) were added followed last by a solution of alcohol 69 (0.05 g, 0.14 mmol, 1.00 equiv) in CH2Cl2 (1 mL), and the reaction was allowed to stir at rt until TLC analysis confirmed complete conversion of the starting material, typically 3.5 h. The reaction mixture was filtered through cotton into a separatory funnel, and H2O (10 mL) and EtOAc (10 mL) were added. The mixture was extracted with EtOAc (3 × 10 mL), and the combined organic extracts were washed with saturated NaHCO3(aq) (10 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 98:2 to 95:5 hexanes/EtOAc) to afford ester 70f (0.062 g, 90% yield) as a clear, viscous oil. Analytical data: [α]D28 −49.2 (c = 1.50, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.68 (br s, 1H), 4.36 (q, J = 6.6 Hz, 1H), 3.19 (dd, J = 6.0, 4.2 Hz, 1H), 3.08 (dd, J = 6.6, 3.6 Hz, 1H), 2.20 (m, 1H), 2.10 (m, 1H), 1.83–1.81 (m, 5H), 1.69–1.66 (m, 5H), 1.58 (m, 1H), 1.38 (m, 1H), 1.23 (s, 3H), 1.17 (s, 3H), 1.01 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 170.4, 135.7, 131.6, 85.1, 80.9, 74.9, 62.0, 40.3, 36.2, 34.1, 31.7, 27.3, 25.8, 25.0, 24.1, 21.7, 21.6, 19.4, 19.3, 18.1, −2.1, −2.2; HRMS (ESI+) calcd for C24H43BrO4Si+Na, 525.2012; found 525.2004; IR (thin film, cm−1) 2929, 2856, 1732, 1472, 1378, 1329, 1217, 1159, 1070, 835; TLC (90:10 hexanes/ EtOAc) Rf = 0.62.
((2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)methyl (Z)-2-Methylbut-2-enoate (70g)
A flame-dried, 20 mL scintillation vial was charged with CH2Cl2 (3 mL) and angelic acid (0.03 g, 0.27 mmol, 2.00 equiv) at rt under an atmosphere of N2. DCC (0.06 g, 0.27 mmol, 2.00 equiv) and DMAP (0.002 g, 0.014 mmol, 0.10 equiv) were added followed last by a solution of alcohol 69 (0.05 g, 0.14 mmol, 1.00 equiv) in CH2Cl2 (1 mL), and the reaction was allowed to stir at rt until TLC analysis confirmed complete conversion of the starting material, 30 h. In some cases, an additional 2.00 equiv of angelic acid and DCC were added after 12 h to aide starting material conversion. The reaction mixture was filtered through cotton into a separatory funnel, and H2O (10 mL) and EtOAc (10 mL) were added. The aqueous layer was extracted with EtOAc (3 × 10 mL), and the combined organic extracts were washed with saturated NaHCO3(aq) (10 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 98:2 to 95:5 hexanes/EtOAc) to afford ester 70g (0.040 g, 59% yield) as a pale yellow, viscous oil. Analytical data: [α]D28 −53.1 (c = 1.00, CHCl3); 1H NMR (600 MHz, CDCl3) δ 6.83 (q, J = 6.6 Hz, 1H), 4.65 (br s, 2H), 3.19 (dd, J = 5.4, 5.4 Hz, 1H), 3.09 (dd, J = 6.0, 3.6 Hz, 1H), 2.19 (m, 1H), 2.10 (m, 1H), 1.97 (m, 1H), 1.88–1.83 (m, 4H), 1.79 (d, J = 6.6 Hz, 3H), 1.67 (br s, 4H), 1.56 (m, 2H), 1.37 (m, 1H), 1.23 (s, 3H), 1.16 (s, 3H), 1.01 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 168.3, 137.4, 136.9, 134.3, 132.4, 128.8, 85.1, 81.0, 74.9, 60.4, 36.2, 34.1, 31.7, 31.6, 27.2, 25.8, 25.0, 24.2, 21.7, 19.4, 19.2, 14.4, 12.1, −2.1, −2.2; HRMS (ESI+) calcd for C26H46O4Si+Na, 473.3063; found 473.3055; IR (thin film, cm−1) 2955, 2855, 1731, 1703, 1636, 1487, 1361, 1263, 1070, 835, 758; TLC (90:10 hexanes/EtOAc) Rf = 0.65.
((2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)methyl 3-(dimethyl(phenyl)silyl)-2-methylpropanoate (70h)
A flame-dried, 20 mL scintillation vial was charged with CH2Cl2 (5 mL) and 3-(dimethyl(phenyl)silyl)-2-methylpropanoic acid53 (0.18 g, 0.81 mmol, 2.00 equiv) at rt under an atmosphere of N2. DCC (0.17 g, 0.81 mmol, 2.00 equiv) and DMAP (0.005 g, 0.04 mmol, 0.10 equiv) were added followed last by a solution of alcohol 69 (0.15 g, 0.41 mmol, 1.00 equiv) in CH2Cl2 (2 mL), and the reaction was allowed to stir at rt until TLC analysis confirmed complete conversion of the starting material, 5 h. The reaction mixture was filtered through cotton into a separatory funnel, and H2O (10 mL) and EtOAc (10 mL) were added. The mixture was extracted with EtOAc (3 × 10 mL), and the combined organic extracts were washed with saturated NaHCO3(aq) (10 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 98:2 to 98:2 to 95:5 hexanes/EtOAc) to afford ester 70h (0.21 g, 91% yield) as a clear, viscous oil. Analytical data: [α]D28 −42.5 (c = 1.30, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.51 (br s, 2H), 7.36 (br s, 3H), 4.56 (dd, J = 7.8, 4.2 Hz, 1H), 4.50 (d, J = 12.0 Hz, 1H), 3.20 (dd, J = 6.0, 4.2 Hz, 1H), 3.10 (dd, J = 6.0, 3.6 Hz, 1H), 2.54 (m, 1H), 2.20 (m, 1H), 2.10 (m, 1H), 1.79 (d, J = 12.6 Hz, 1H), 1.68–1.66 (m, 5H), 1.58 (m, 2H), 1.32 (m, 2H), 1.25 (s, 3H), 1.19 (s, 3H), 1.15 (d, J = 6.6 Hz, 3H), 1.00 (s, 3H), 0.94–0.89 (m, 2H), 0.87 (s, 9H), 0.31 (br s, 6H), 0.10 (s, 3H), 0.08 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 177.6, 138.8, 134.5, 133.5, 132.3, 129.1, 128.9, 127.9, 127.785.1, 81.0, 74.9, 60.3, 36.6, 36.3, 36.2, 34.1, 31.6, 27.3, 27.2, 25.8, 25.1, 24.1, 21.7, 20.7, 20.6, 20.5, 19.8, 19.3, 19.2, 19.2, 18.1, −2.1, −2.2, −2.3, −2.4, −2.6; HRMS (ESI+) calcd for C33H56O4Si2+Na, 595.3615; found 595.3604; IR (thin film, cm−1) 3052, 2956, 2856, 1809, 1718, 1487, 1457, 1361, 1265, 1198, 1047, 835; TLC (90:10 hexanes/EtOAc) Rf = 0.78.
2-((2S,4aS,6S,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-5-methyleneoctahydro-2H-chromen-6-yl)-3-(dimethyl(phenyl)silyl)-2-methylpropanoic acid (71b)
A flamedried, 20 mL scintillation vial was charged with THF (2 mL) under an atmosphere of N2. The mixture was cooled to −78 °C, and a premade solution of LDA (0.5 M in THF/hexanes, 0.52 mL, 0.26 mmol, 3.00 equiv) was added followed by a solution of ester 70h (0.05 g, 0.087 mmol, 1.00 equiv) in THF (1 mL). The reaction was allowed to stir 45 min at this temperature at which point TMSCl (0.04 mL, 0.26 mmol, 3.00 equiv) was added, and the mixture was warmed to rt and stirred 5 min. The septum was replaced with a screw cap, the vial was sealed, and the mixture was warmed to 75 °C and stirred until TLC analysis indicated complete consumption of the starting material, typically 12 h. The mixture was cooled to rt and quenched via addition of 1 M HCl(aq) (4 mL). The mixture was transferred to a separatory funnel and diluted with Et2O (10 mL). The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic extracts were washed with brine (10 mL) and concentrated in vacuo to give the crude rearrangement product in a 6.6:1.1:1 diastereomeric ratio. The diastereomeric ratio was determined by 1H NMR spectroscopic analysis of the crude reaction mixture by comparison of the integration of the resonances at δ 5.25 (minor diastereomer), δ 5.10 (major diastereomer), and δ 5.04 (minor diastereomer, overlapping signals). The product was purified via flash chromatography (100:0 to 95:5 to 90:10 hexanes/EtOAc) to afford carboxylic acid 71b (0.032 g, 62% yield) as a clear viscous oil. Analytical data: [α]D28 −18.3 (c = 1.25, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.51 (m, 2H), 7.34 (m, 3H), 5.10 (s, 1H), 5.01 (s, 1H), 3.11 (dd, J = 6.0, 4.8 Hz, 1H), 3.01 (m, 1H), 1.97–1.90 (m, 2H), 1.74 (m, 1H), 1.66–1.54 (m, 6H), 1.38 (s, 3H), 1.27 (m, 2H), 1.22 (s, 3H), 1.17 (s, 3H), 1.16 (s, 3H), 1.15 (m, 1H), 1.07 (s, 3H), 1.05 (m, 1H), 0.84 (s, 9H), 0.37 (s, 3H), 0.29 (s, 3H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 184.0, 160.8, 140.3, 133.5, 128.8, 127.7, 111.7, 84.7, 80.4, 74.8, 52.5, 46.5, 39.3, 36.8, 36.6, 32.7, 30.2, 27.3, 25.9, 25.0, 24.7, 24.5, 23.3, 23.1, 22.1, 22.1, 18.2, −1.2, −1.4, −2.1, −2.2; HRMS (ESI+) calcd for C33H56O4Si2+Na, 595.3615; found 595.3605; IR (thin film, cm−1) 3420, 3053, 2956, 2956, 2855, 1716, 1689, 1487, 1377, 1265, 1093, 896, 835; TLC (90:10 hexanes/EtOAc) Rf = 0.46.
((2S,4aS,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-3,4,4a,7,8,8a-hexahydro-2H-chromen-5-yl)methyl Isobutyrate (70c)
A flame-dried, 500 mL round-bottomed flask was charged with CH2Cl2 (110 mL) and isobutyric acid (2.22 mL, 24.47 mmol, 2.00 equiv) at rt under an atmosphere of N2. DCC (5.05 g, 24.47 mmol, 2.00 equiv) and DMAP (0.15 g, 1.22 mmol, 0.10 equiv) were added followed last by a solution of alcohol 69 (4.51 g, 12.23 mmol, 1.00 equiv) in CH2Cl2 (10 mL), and the reaction was allowed to stir at rt until TLC analysis confirmed complete conversion of the starting material, typically 2.5 h. The reaction mixture was filtered through cotton into a separatory funnel, and H2O (40 mL) and EtOAc (100 mL) were added. The mixture was extracted with EtOAc (3 × 30 mL), and the combined organic extracts were washed with saturated NaHCO3(aq) (2 × 30 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 97.5:2.5 to 95:5 hexanes/EtOAc) to afford ester 70c (4.01 g, 75%) as a clear, viscous oil. Analytical data: [α]D28 −73.0 (c = 0.75, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.58 (br s, 2H), 3.19 (dd, J = 5.4, 4.8 Hz, 1H), 3.08 (dd, J = 5.4, 4.2 Hz, 1H), 2.54 (m, 1H), 2.19 (m, 1H), 2.09 (m, 1H), 1.82 (dt, J = 6.0, 3.0 Hz, 1H), 1.67–1.65 (m, 5H), 1.57 (m, 2H), 1.37 (m, 1H), 1.23 (s, 3H), 1.17 (br s, 6H), 1.15 (d, J = 2.4 Hz, 3H), 0.99 (s, 3H), 0.84 (s, 3H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 177.3, 134.5, 132.3, 85.1, 81.0, 74.9, 60.3, 25.8, 24.2, 21.7, 19.3, 19.2, 19.1, 19.0, 18.2, −2.1, −2.2; HRMS (ESI+) calcd for C25H46O4Si+Na, 461.3063; found 461.3062; IR (thin film, cm−1) 2955, 2856, 1721, 1470, 1378, 1215, 1092, 835, 756; TLC (85:15 hexanes/EtOAc) Rf = 0.66.
2-((2S,4aS,6S,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-5-methyleneoctahydro-2H-chromen-6-yl)-2-methylpropanoic acid (71a)
A flame-dried, 250 mL round-bottomed flask was charged with THF (80 mL) and diisopropylamine (3.84 mL, 27.42 mmol, 3.00 equiv) under an atmosphere of N2. The mixture was cooled to 0 °C and nBuLi (1.85 M solution in hexanes, 14.82 mL, 27.42 mmol, 3.00 equiv) was added slowly. After being stirred for 30 min at 0 °C, the mixture was cooled to −78 °C, and isobutyrate 70c (4.01 g, 9.14 mmol, 1.00 equiv) was added as a solution in THF (15 mL). The mixture was allowed to stir for 45 min at which time TMSCl (3.52 mL, 27.42 mmol, 3.00 equiv) was added. The reaction mixture was then allowed to warm to rt, stirred for 5 min, and subsequently warmed to 75 °C and stirred until TLC analysis indicated complete conversion of the starting material, typically 12h. The reaction mixture was cooled to rt and quenched via 1 M HCl(aq) (25 mL). The mixture was then partitioned in a separatory funnel and extracted with Et2O (3 × 20 mL). The combined organic extracts were washed with 6 M HCl (2 × 30 mL), dried with magnesium sulfate, and concentrated in vacuo to provide the crude acid as a 6:1 mixture of diastereomers. The diastereomeric ratio was determined by 1H NMR spectroscopic analysis of the crude reaction mixture by comparison of the integration of the resonances at δ 5.13 (minor diastereomer) and δ 5.12 (major diastereomer). The product was purified via flash chromatography (100:0 to 90:10 to 80:20 hexanes/EtOAc) to afford acid 71a (3.14 g, 78% yield) as a clear, viscous oil in an inseparable 6:1 diastereomeric ratio. Analytical data: [α]D28 −43.5 (c = 0.70, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.12 (s, 1H), 5.04 (s, 1H), 3.12 (dd, J = 6.6, 2.4 Hz, 1H), 3.04 (m, 1H), 2.09 (m, 1H), 1.95 (m, 1H), 1.66 (m, 2H), 1.59 (m, 2H), 1.54 (m, 1H), 1.41 (m, 1H), 1.33 (s, 3H), 1.31 (s, 3H), 1.25 (s, 3H), 1.23 (s, 3H), 1.17 (s, 3H), 1.09 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 184.5, 161.1, 110.4, 84.7, 81.0, 74.8, 50.2, 44.4, 39.5, 36.9, 33.2, 28.3, 27.4, 25.6, 25.0, 24.6, 23.7, 23.6, 22.4, 22.1, 18.2, −2.2; HRMS (ESI+) calcd for C25H46O4Si+Na, 461.3063; found 461.3063; IR (thin film, cm−1) 3406, 2955, 2856, 1693, 1641, 1471, 1378, 1252, 1170, 1094, 1042, 835, 760; TLC (85:15 hexanes/EtOAc) Rf = 0.40.
Synthesis of Ketone 72
Methyl 2-((2S,4aS,6S,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-5-methyleneoctahydro-2H-chromen-6-yl)-2-methylpropanoate (S5)
The acid 71a (3.14 g, 7.16 mmol, 1.00 equiv) was dissolved in MeOH/C7H8 (2:1, 75 mL) in a 250 mL round-bottomed flask with magnetic stirring at rt. TMSCHN2 (2 M in Et2O, 10.00 mL, 20 mmol, 2.79 equiv) was added dropwise until the yellow color of excess TMSCHN2 in solution persisted. AcOH (1.50 g, 24.98 mmol, 3.50 mmol) was added dropwise, giving a clear solution. The resulting mixture was concentrated in vacuo and purified via flash chromatography (100:0 to 97.5:2.5 to 95:5 hexanes/EtOAc) to afford ester S5 (3.06 g, 94% yield) as a clear, viscous oil in an inseparable 6.3:1 diastereomeric ratio (as determined by integration of the resonances at δ 3.64 (minor diastereomer) and δ 3.62 (major diastereomer)). Analytical data: [α]D28 −89.7 (c = 0.60, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.01 (s, 1H), 5.00 (s, 1H), 3.62 (s, 3H), 3.08 (m, 1H), 3.03 (dd, J = 6.0, 2.4 Hz, 1H), 2.08 (m, 1H), 1.95 (dt, J = 5.4, 3.6 Hz, 1H), 1.65 (m, 2H), 1.59 (m, 2H), 1.57 (br s, 1H), 1.50 (m, 1H), 1.39 (m, 1H), 1.29 (s, 3H), 1.28 (s, 3H), 1.22 (s, 3H), 1.21 (s, 3H), 1.16 (s, 3H), 1.08 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 178.7, 161.4, 110.0, 84.8, 81.0, 74.9, 51.4, 50.3, 44.3, 39.5, 36.9, 33.1, 28.5, 27.3, 25.6, 25.0, 24.6, 23.9, 23.7, 22.4, 22.1, 18.2, −2.1, −2.2; HRMS (ESI+) calcd for C26H48O4Si+Na, 475.3220; found 475.3221; IR (thin film, cm−1) 2954, 2855, 1722, 1601, 1451, 1378, 1169, 1051, 835, 741; TLC (85:15 hexanes/EtOAc) Rf = 0.66.
3-((2S,4aS,6S,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-4a,6-dimethyl-5-methyleneoctahydro-2H-chromen-6-yl)-3-methylbutan-2-one (72)
A flame-dried, 500 mL round-bottomed flask was charged with ester S5 (3.82 g, 8.44 mmol, 1.00 equiv) and Et2O (84 mL) under an atmosphere of N2. The mixture was cooled to 0 °C, and MeLi (1.6 M in Et2O, 21.09 mL, 33.75 mmol, 4.00 equiv) was added. The mixture was warmed to rt, whereupon TLC analysis showed incomplete conversion of the starting material. A second addition of MeLi (4.00 equiv) was carried out, upon which TLC analysis showed remaining starting material. A third addition of MeLi (4.00 equiv) was carried out, upon which TLC analysis showed complete conversion of the starting material. The reaction mixture was cooled to 0 °C and quenched carefully with saturated NH4Cl(aq) (25 mL). The mixture was partitioned in a separatory funnel, and the aqueous layer was extracted with Et2O (3 × 20 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 97.5:2.5 to 95:5 hexanes/EtOAc) to afford ketone 72 (3.52 g, 86% yield) as a clear, viscous oil in an inseparable 7:1 ratio of diastereomers (as determined by integration of the resonances at δ 5.05 (major diastereomer) and δ 5.03 (minor diastereomer)). Analytical data: [α]D28 −92.2 (c = 0.60, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.05 (s, 1H), 4.90 (s, 1H), 3.15 (dd, J = 5.4, 4.8 Hz, 1H), 3.04 (m, 1H), 2.18 (s, 3H), 1.96 (m, 1H), 1.66 (m, 1H), 1.61–1.59 (m, 3H), 1.53 (m, 1H), 1.44 (m, 1H), 1.30 (s, 3H), 1.23 (s, 3H), 1.22 (s, 3H), 1.16 (br s, 6H), 1.08 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 215.1, 161.4, 111.1, 84.7, 80.5, 74.8, 54.7, 44.9, 39.4, 36.8, 33.0, 29.7, 29.4, 27.4, 25.8, 25.0, 24.6, 23.6, 23.5, 22.7, 22.0, 18.1, −2.1, −2.2; HRMS (ESI+) calcd for C26H48O3Si+Na, 459.3271; found 459.3267; IR (thin film, cm−1) 2955, 2856, 1694, 1620, 1470, 1377, 1251, 1094, 835; TLC (85:15 hexanes/EtOAc) Rf = 0.54.
3-((2S,4aS,5R,6S,8aS)-2-(2-((tert-Butyldimethylsilyl)oxy)propan-2-yl)-5-(hydroxymethyl)-4a,6-dimethyloctahydro-2H-chromen-6-yl)-3-methylbutan-2-ol (73)
A flame-dried, 250 mL round-bottomed flask was charged with ketone 72 (1.63 g, 3.74 mmol, 1.00 equiv) and THF (70 mL) under an atmosphere of N2. BH3·THF (1 M in THF, 16.82 mL, 4.50 equiv) was added, and the mixture was warmed to 50 °C and stirred until complete conversion of the starting material was observed by TLC analysis, typically 12 h. The reaction mixture was then cooled to 0 °C, and 3 M NaOH(aq) (7.5 mL) was added slowly followed by H2O2 (30% w/w in H2O, 7.5 mL). The resulting mixture was warmed to rt and stirred for 2.5 h, upon which the mixture was partitioned in a separatory funnel, diluted with H2O (30 mL), and extracted with Et2O (3 × 20 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo to afford the crude diol as an inseparable mixture of diastereomers at C12c and C6a. The diastereoselection of this reaction at C4b was determined via 1H NMR analysis of the subsequent intermediate 74. The product was purified via flash chromatography (80:20 to 70:30 hexanes/EtOAc) to afford diol 73 (1.27 g, 74% yield) as a white, viscous foam. This diastereomeric mixture was carried on to the next step without further separation. Analytical data: [α]D28 −83.9 (c = 0.60, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.18 (m, 2H), 3.88 (t, J = 12.6 Hz, 2H), 3.68 (dd, J = 9.0, 3.0 Hz, 1H), 3.56 (d, J = 12.0 Hz, 1H), 3.06 (m, 2H), 2.93 (dd, J = 6.0, 4.8 Hz, 1H), 2.86 (dd, J = 7.2, 4.2 Hz, 1H), 1.98 (m, 3H), 1.76 (s, 1H), 1.59–1.49 (m, 11H), 1.42–1.36 (m, 3H), 1.25 (d, J = 6.0 Hz, 5H), 1.21 (s, 6H), 1.15 (s, 7H), 1.01 (s, 2H), 0.95 (br s, 9H), 0.90 (br s, 4H), 0.89 (s, 3H), 0.86 (s, 2H), 0.83 (br s, 22H), 0.07 (s, 7H), 0.05 (s, 7H); 13C NMR (150 MHz, CDCl3) δ 85.1, 84.9, 84.2, 83.7, 74.9, 68.7, 61.5, 61.0, 54.2, 52.9, 45.8, 45.1, 42.5, 42.4, 39.0, 38.5, 37.9, 37.8, 34.0, 33.5, 27.4, 27.3, 25.8, 25.2, 25.0, 24.9, 24.6, 21.5, 21.4, 21.2, 19.8, 18.1, 17.8, 17.5, 14.7, 14.2, −2.1, −2.2; HRMS (ESI+) calcd for C26H52O4Si+Na, 479.3533; found 479.3549; IR (thin film, cm−1) 3320, 2955, 2855, 1471, 1379, 1251, 1172, 1100, 834, 759; TLC (85:15 hexanes/EtOAc) Rf = 0.14.
(3S,4aS,6aS,10aR,10bS)-3-(2-((tert-Butyldimethylsilyl)oxy)-propan-2-yl)-6a,7,7,10b-tetramethyl-2,3,5,6,6a,7,10a,10b-octahydro-1H-benzo[f]chromen-8(4aH)-one (75)
A flame-dried, 250 mL round-bottomed flask was charged with CH2Cl2 (70 mL) and (COCl)2 (1.71 mL, 19.92 mmol, 5.00 equiv) under an atmosphere of N2. The mixture was cooled to −78 °C, and DMSO (2.83 mL, 39.84 mmol, 10.00 equiv) was added slowly. The mixture was allowed to stir 30 min at −78 °C then the diol 73 (1.82 g, 3.98 mmol, 1.00 equiv) was added as a solution in CH2Cl2 (10 mL). The reaction mixture was stirred at this temperature for 2 h then DIPEA (13.88 mL, 79.69 mL, 20.0 equiv) was added. The reaction was stirred 30 min at −78 °C then warmed to 0 °C and stirred 15 min. At this time TLC analysis confirmed complete conversion of the starting material. The reaction was quenched with saturated NH4Cl(aq) (25 mL), and the mixture was partitioned in a separatory funnel. The mixture was extracted with CH2Cl2 (3 × 20 mL), and the combined organic extracts were washed with brine (20 mL), dried with magnesium sulfate and concentrated in vacuo to afford the crude ketoaldehyde 74, which was carried to the next step without further purification. (Note: at this stage, a single diastereomer was observed in the 1H NMR spectrum of the crude aldehyde, thereby establishing complete control of the C4b methine stereocenter in the hydroboration/oxidation step. This crude spectrum is provided in the Supporting Information.)
The crude ketoaldehyde 74 was dissolved in MeOH/THF (1:1, 80 mL) in a 250 mL round-bottomed flask and cooled to 0 °C with magnetic stirring. KOH(aq) (2 M, 8 mL) was added, and the reaction was warmed to rt and stirred for 12 h. The resulting mixture was concentrated on a rotary evaporator and partitioned with EtOAc (30 mL) and H2O (30 mL) in a separatory funnel. The mixture was extracted with EtOAc (3 × 20 mL), and the combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 97.5:2.5 to 95:5 to 90:10 hexanes/EtOAc) to afford the enone 75 (1.29 g, 75% yield) as a yellow, viscous oil. Analytical data: [α]D28 −109.0 (c = 0.85, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.73 (d, J = 12.6 Hz, 1H), 5.99 (dd, J = 7.0, 3.5 Hz, 1H), 3.14 (dd, J = 9.0, 3.0 Hz, 1H), 2.97 (dd, J = 5.0, 5.0 Hz, 1H), 2.29 (br s, 1H), 1.96 (d, J = 9.0 Hz, 1H), 1.70–1.58 (m, 6H), 1.43 (m, 1H), 1.23 (s, 3H), 1.17 (s, 3H), 1.07 (s, 3H), 1.00 (s, 3H), 0.95 (s, 3H), 0.93 (s, 3H), 0.84 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 204.9, 146.2, 129.0, 85.6, 85.0, 74.8, 51.5, 49.7, 43.8, 37.3, 35.5, 30.5, 27.4, 25.8, 24.9, 23.8, 21.2, 20.3, 18.1, 16.9, 16.6, 14.7, −2.1, −2.2; HRMS (ESI+) calcd for C26H46O3Si+Na, 457.3114; found 457.3129; IR (thin film, cm−1) 2954, 2855, 1677, 1461, 1389, 1251, 1174, 1103, 1041, 834, 756; TLC (85:15 hexanes/EtOAc) Rf = 0.43.
(3S,4aS,6aS,10aR,10bS,E)-3-(2-((tert-Butyldimethylsilyl)oxy)-propan-2-yl)-6a,7,7,10b-tetramethyldecahydro-1H-benzo[f]-chromen-8(4aH)-one O-Benzyl Oxime (76)
The enone 75 (1.61 g, 3.70 mmol, 1.00 equiv) was dissolved in EtOAc (60 mL) in a 250 mL round-bottomed flask and charged with Pd/C (2.40 g, 1.50 mass equiv). The reaction mixture was placed under 1 atm (balloon) of H2 and stirred until full conversion of the starting material was observed by TLC analysis, typically 30 min. The mixture was then filtered through a pad of Celite, and the filter cake was washed with two 20 mL portions of EtOAc. The solution was then concentrated in vacuo to afford the crude ketone, which was carried to the next step without further purification.
The residue was dissolved in MeOH/H2O (5:1, 80 mL) in a 250 mL round-bottomed flask. BnONH3Cl (11.84 g, 74.19 mmol, 20.00 equiv) and NaOAc (4.56 g, 55.64 mmol, 15.00 equiv) were added, and the resulting suspension was fitted with a reflux condenser and heated to 85 °C with stirring until TLC analysis confirmed complete consumption of the starting material, typically 16 h. The reaction mixture was cooled to rt and concentrated on a rotary evaporator. The residue was taken up into H2O (30 mL) and CH2Cl2 (30 mL), and the mixture was partitioned in a separatory funnel and extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with H2O (30 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (100:0 to 98:2 to 97.5:2.5 to 95:5 hexanes/EtOAc) to afford oxime 76 (1.66 g, 83% yield) as a clear, viscous oil. Analytical data: [α]D28 −112.8 (c = 0.45, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.37–7.28 (m, 5H), 5.08 (br s, 2H), 3.35 (dd, J = 9.6, 4.2 Hz, 1H), 3.10 (dd, J = 8.4, 3.0 Hz, 1H), 2.88 (dd, J = 6.0, 4.2 Hz, 1H), 1.82 (m, 2H), 1.66–1.45 (m, 7H), 1.36 (m, 1H), 1.23 (s, 3H), 1.16 (s, 3H), 1.13 (s, 3H), 1.01 (s, 3H), 0.85 (s, 9H), 0.09 (s, 3H), 0.07 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 165.2, 138.7, 128.1, 128.0, 127.4, 85.4, 85.3, 75.1, 74.9, 45.9, 45.5, 41.1, 38.2, 36.2, 31.3, 27.3, 25.8, 25.0, 24.5, 23.3, 21.4, 20.9, 20.0, 19.0, 18.1, 16.8, 13.3, −2.2; HRMS (ESI+) calcd for C33H55NO3Si+Na, 564.3849; found 564.3862; IR (thin film, cm−1) 2951, 2855, 1626, 1470, 1378, 1250, 1173, 1040, 898, 835, 757; TLC (85:15 hexanes/EtOAc) Rf = 0.77.
((3S,4aS,6aS,7R,10aR,10bS,E)-8-((Benzyloxy)imino)-3-(2-((tert-butyldimethylsilyl) oxy)propan-2-yl)-6a,7,10b-trimethyldodecahydro-1H-benzo[f]chromen-7-yl)methyl acetate (78)
A 100 mL round-bottomed flask was charged with oxime 76 (1.66 g, 3.06 mmol, 1.00 equiv) and AcOH:Ac2O (1:1, 31 mL) with magnetic stirring at rt. Pd(OAc)2 (0.10 g, 0.46 mmol, 0.15 equiv) and PhI(OAc)2 (1.48 g, 4.60 mmol, 1.50 equiv) were added sequentially, and the reaction mixture was warmed to 100 °C. This temperature was maintained until TLC analysis showed complete conversion of the starting material, typically 1 h. The mixture was cooled to rt, diluted with pentane (30 mL) and H2O (20 mL), and transferred to a separatory funnel. Saturated NaHCO3(aq) (30 mL) was added dropwise into the separatory funnel, and the mixture was allowed to stand 10 min upon completion of the addition. The layers were separated, and the aqueous layer was extracted with pentane (3 × 20 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo to afford the crude acetate 78 as a single diastereomer (as determined by 1H NMR spectroscopic analysis of the crude reaction mixture, which revealed a single compound). The product was purified via flash chromatography (100:0 to 95:5 to 90:10 hexanes/EtOAc) to afford the acetate 78 (1.49 g, 81% yield) as a reddish-brown, viscous oil. Analytical data: [α]D28 −66.2 (c = 0.70, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.34–7.27 (m, 5H), 5.04 (br s, 2H), 4.55 (d, J = 10.8 Hz, 1H), 4.03 (d, J = 11.4 Hz, 1H), 3.36 (dd, J = 10.8, 13.6 Hz, 1H), 3.09 (dd, J = 8.4, 3.0 Hz, 1H), 2.88 (dd, J = 5.4, 4.8 Hz, 1H),1.94 (s, 3H), 1.82–1.74 (m, 2H), 1.63–1.52 (m, 8H), 1.35 (m, 1H), 1.22 (s, 3H), 1.21 (s, 3H), 1.15 (s, 3H), 0.91 (s, 3H), 0.84 (s, 12H), 0.08 (s, 3H), 0.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 171.1, 161.6, 128.2, 128.0, 127.4, 85.3, 85.1, 75.4, 74.8, 65.6, 48.4, 46.0, 42.1, 38.2, 36.3, 32.0, 27.3, 25.8, 25.0, 24.4, 21.4, 21.1, 20.8, 20.1, 18.1, 17.3, 17.0, 13.5, −2.2; HRMS (ESI+) calcd for C35H57NO5Si+Na, 622.3904; found 622.3908; IR (thin film, cm−1) 2953, 2884, 1732, 1470, 1380, 1249, 1038, 835, 756; TLC (60:40 hexanes/EtOAc) Rf = 0.80.
Synthesis of Ketoaldehyde 83
(3S,4aS,6aS,7S,10aR,10bS)-7-(Hydroxymethyl)-3-(2-hydroxypropan-2-yl)-6a,7,10b-trimethyldecahydro-1H-benzo[f]chromen-8(4aH)-one (S6)
A 50 mL roundbottomed flask was charged with acetate 78 (0.71 g, 1.18 mmol, 1.00 equiv) and 2 M HCl(aq)/MeOH/THF/acetone (10:10:10:1, 12 mL). The mixture was warmed to 85 °C and stirred until full convergence to a single product was observed by TLC analysis, typically 5 h. The mixture was cooled to rt and concentrated on a rotary evaporator, and the residue was taken up into H2O (15 mL) and CH2Cl2 (15 mL) and partitioned in a separatory funnel. The mixture was extracted with CH2Cl2 (3 × 10 mL), and the combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (60:40 EtOAc:hexanes) to afford hydroxy ketone S6 (0.28 g, 71% yield) as a reddish-brown, viscous oil. Analytical data: [α]D28 −159.6 (c = 0.30, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.12 (dd, J = 8.4, 3.0 Hz, 1H), 3.22 (m, 2H), 3.02 (dd, J = 6.0, 3.0 Hz, 1H), 2.64 (dd, J = 7.2, 3.0 Hz, 1H), 2.59 (br s, 1H), 2.55 (m, 1H), 2.29 (m, 1H), 1.89–1.86 (m, 2H), 1.80 (m, 1H), 1.70–1.60 (m, 6H), 1.46 (m, 1H), 1.40 (dt, J = 6.0, 3.6 Hz, 1H), 1.31 (s, 3H), 1.23 (m, 1H), 1.18 (s, 3H), 1.15 (s, 3H), 0.99 (s, 3H), 0.91 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 219.3, 85.0, 84.6, 71.8, 63.6, 57.4, 45.3, 42.1, 37.9, 37.6. 36.4, 30.7, 26.1, 23.9, 23.7, 21.7, 21.2, 18.2, 16.9, 13.5; HRMS (ESI+) calcd for C20H34O4+Na, 361.2355; found 361.2360; IR (thin film, cm−1) 3450, 2950, 1692, 1425, 1166, 1102, 735, 685; TLC (60:40 hexanes/EtOAc) Rf = 0.12.
(3S,4aS,6aS,7S,10aR,10bS)-3-(2-Hydroxypropan-2-yl)-6a,7,10b-trimethyl-8-oxododecahydro-1H-benzo[f]chromene-7-carbaldehyde (83)
A 20 mL scintillation vial was charged with alcohol S6 (0.29 g, 0.84 mmol, 1.00 equiv) and CH2Cl2 (8 mL). Dess-Martin periodinane (0.71 g, 1.68 mmol, 2.00 equiv) was added at rt with stirring. The reaction mixture was allowed to stir at room temperature until TLC analysis confirmed complete conversion of the starting material, typically 20 min. The mixture was then quenched via a 1:1 solution of saturated NaHCO3(aq) and saturated Na2S2O3(aq) (10 mL), and the mixture was stirred 5 min. The reaction mixture was then diluted with Et2O (15 mL) and partitioned in a separatory funnel. The aqueous layer was extracted with Et2O (3 × 10 mL), and the combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (60:40 to 50:50 hexanes/EtOAc) to afford the ketoaldehyde 83 (0.28 g, 99% yield) as a pale white powder. Analytical data: mp 121–125 °C; [α]D28 −223.7 (c = 0.50, CHCl3); 1H NMR (600 MHz, CDCl3) δ 10.06 (s, 1H), 3.22 (dd, J = 9.0, 3.0 Hz, 1H), 3.02 (dd, J = 6.0, 2.4 Hz, 1H), 2.52–2.46 (m, 2H), 1.94–1.82 (m, 3H), 1.57–1.70 (m, 5H), 1.47 (m, 1H), 1.26 (s, 3H), 1.24 (s, 3H), 1.22 (m, 1H), 1.18 (s, 3H), 1.15 (s, 3H), 0.94 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 214.0, 204.2, 84.9, 84.6, 71.9, 64.7, 45.1, 43.4, 37.8, 37.6, 36.5, 31.6, 26.1, 23.7, 23.5, 21.6, 20.9, 19.5, 14.8, 13.6; HRMS (ESI+) calcd for C20H32O4+Na, 359.2199; found 359.2198; IR (thin film, cm−1) 3019, 2955, 2857, 2400, 1721, 1388, 1265, 1215, 1098; TLC (60:40 hexanes/EtOAc) Rf = 0.24.
(3S,4aS,6aS,7S,10aR,10bS)-7-(1-Hydroxyallyl)-3-(2-hydroxypropan-2-yl)-6a,7,10b-trimethyl-8-vinyldodecahydro-1H-benzo[f]-chromen-8-ol (84)
A flame-dried, 20 mL scintillation vial was charged with LiCl (0.30 g, 7.13 mmol, 20.00 equiv equiv), anhydrous CeCl3 (0.88 g, 3.57 mmol, 10.00 equiv), and a stir bar in a nitrogen-filled glovebox. The vial was removed from the glovebox and placed under an N2 atmosphere. THF (5 mL) was added, and this mixture was stirred at rt for 2.5 h. A separate flame-dried, 20 mL scintillation vial was charged with aldehyde 83 (0.12 g, 0.36 mmol, 1.00 equiv) and THF (2 mL) under an atmosphere of N2. The CeCl3·2LiCl suspension was added to the solution of 83 at rt, and the resulting mixture was stirred 2.5 h. The reaction was subsequently cooled to −78 °C, and vinylmagnesium bromide (1 M in THF, 3.57 mL, 3.57 mmol, 10 equiv) was added. The reaction mixture was allowed to stir at this temperature until TLC analysis confirmed complete consumption of the starting material, typically 20 min. The reaction was quenched with MeOH (3 mL), and the mixture was immediately warmed to rt upon which 5% AcOH(aq) (2 mL) and Et2O (2 mL) were added with stirring. Once the vial had reached rt, the solution was transferred to a separatory funnel, diluted with H2O (15 mL) and extracted with Et2O (3 × 15 mL). The combined organic extracts were washed with saturated NaHCO3(aq) (10 mL), dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (80:20 to 70:30 to 60:40 hexanes/EtOAc) to afford an inseparable 2.6:1 mixture of diol diastereomers 84 (0.14 g, 99% yield) as a pale white, viscous foam. Analytical data: [α]D28 −182.8 (c = 0.25, CHCl3); 1H NMR (600 MHz, CDCl3) δ 6.30 (dd, J = 10.8, 6.0 Hz, 1H), 6.14 (m, 1H), 5.24 (d, J = 17.4 Hz, 1H), 5.08 (d, J = 10.8 Hz, 1H), 5.03–4.99 (m, 2H), 4.40 (d, J = 8.4 Hz, 1H), 3.16 (m, 1H), 2.88 (m, 1H), 1.83 (m, 3H), 1.72 (m, 2H), 1.59 (m, 3H), 1.53 (s, 3H), 1.45–1.36 (m, 5H), 1.15 (s, 3H), 1.14 (s, 3H), 1.03 (m, 1H), 0.90 (s, 3H), 0.87 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 145.8, 140.7, 116.4, 112.6, 85.7, 84.5, 80.0, 79.6, 72.0, 49.4, 47.3, 43.1, 38.0, 36.3, 35.9, 32.4, 26.0, 24.3, 23.6, 21.9, 19.5, 18.6, 17.0, 13.3; HRMS (ESI+) calcd for C24H40O4+Na, 415.2825; found 415.2829; IR (thin film, cm−1) 3303, 2949, 2877, 1621, 1461, 1301, 1089, 920, 737; TLC (60:40 hexanes/EtOAc) Rf = 0.32.
(2S,4aS,4bR,9aS,9bS,11aS)-2-(2-Hydroxypropan-2-yl)-4a,9a,9b-trimethyl-3,4,4a,4b,5,6,9,9a,9b,10,11,11a-dodecahydroindeno[5,4-f]chromene-6a,9(2H)-diol (85)
A flame-dried, 20 mL scintillation vial was charged with Grubbs’ second generation catalyst (0.99 g, 0.12 mmol, 0.20 equiv) and a stir bar in a nitrogen-filled glovebox. The vial was removed from the glovebox and charged with CH2Cl2 (12 mL) under an atmosphere of N2. Diol 84 (0.23 g, 0.59 mmol, 1.00 equiv) was added as a solution in CH2Cl2 (3 mL), and the mixture was allowed to stir at rt until complete conversion of the starting material was observed by TLC analysis, typically 3 h. The reaction mixture was concentrated in vacuo, and the product was purified via flash chromatography (80:20 to 70:30 to 60:40 hexanes/EtOAc) to afford allylic alcohol 85 (0.16 g, 73% yield) as a pale-brown viscous foam. Analytical data: [α]D28 −62.8 (c = 0.75, CHCl3); 1H NMR (600 MHz, CDCl3) δ 6.22 (d, J = 5.4 Hz, 1H), 6.14 (dd, J = 3.0, 2.4 Hz, 1H), 4.42 (br s, 1H), 3.18 (dd, J = 9.0, 3.0 Hz, 1H), 2.95 (dd, J = 7.8, 3.6 Hz, 1H), 2.69 (br s, 1H), 2.26 (d, J = 5.4 Hz, 1H), 2.22 (br s, 1H), 1.83–1.75 (m, 8H), 1.63 (s, 3H), 1.56–1.54 (m, 3H), 1.42 (m, 2H), 1.17 (s, 3H), 1.16 (s, 3H), 0.94 (s, 3H), 0.92 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 143.2, 137.2, 87.3, 86.0, 84.6, 83.3, 71.9, 52.5, 47.9, 41.8, 38.3, 36.3, 32.9, 30.4, 26.8, 26.1, 23.9, 23.6, 21.9, 20.2, 17.7, 13.2; HRMS (ESI+) calcd for C22H36O4+Na, 387.2512; found 387.2519; IR (thin film, cm−1) 3400, 2951, 2675, 1729, 1449, 1384, 1256, 1097, 1023, 910, 754; TLC (60:40 hexanes/EtOAc) Rf = 0.25.
(2S,4aS,4bR,9aS,9bS,11aS)-2-(2-Hydroxypropan-2-yl)-4a,9a,9btrimethyl-3,4,4a,4b,5,6,8,9a,9b,10,11,11a-dodecahydroindeno[5,4-f]chromen-9(2H)-one (86)
A flame-dried, 20 mL scintillation vial was charged with diol 85 (0.15 g, 0.40 mmol, 1.00 equiv) and CH2Cl2 (9 mL) under and atmosphere of N2. The mixture was cooled to 0 °C, and TFA (0.15 mL, 2.02 mmol, 5.00 equiv) was added. The reaction mixture was warmed to rt and allowed to stir until complete conversion of the starting material was observed by TLC analysis, typically 30 min. The reaction was quenched with saturated NaHCO3(aq) (5 mL), and the mixture was partitioned in a separatory funnel. The aqueous layer was extracted with CH2Cl2 (3 × 5 mL), and the combined organic extracts were dried with Na2SO4 and concentrated in vacuo. The product was purified via flash chromatography (90:10 to 80:20 to 70:30 hexanes/EtOAc) to afford the nonconjugated enone 86 (0.10 g, 71% yield) as a pale brown, viscous oil. Analytical Data: [α]D28 −77.7 (c = 0.50, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.64 (m, 1H), 3.17 (dd, J = 9.0, 2.4 Hz, 1H), 2.95 (dd, J = 5.4, 4.8 Hz, 1H), 2.83 (m, 1H), 2.69 (m, 1H), 2.61 (br s,1H), 2.40 (m, 1H), 2.10 (br s, 1H), 1.84 (m, 2H), 1.64 (m, 3H), 1.55 (m, 2H), 1.43–1.33 (m, 3H), 1.16 (s, 3H), 1.13 (br s, 4H), 1.12 (s, 3H), 0.91 (s, 3H), 0.83 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 223.0, 148.2, 116.3, 85.5, 84.4, 71.8, 59.3, 46.6, 43.0, 41.1, 37.9, 36.5, 30.9, 27.6, 26.1, 24.0, 23.7, 21.8, 21.7, 17.6, 17.4, 13.4; HRMS (ESI+) calcd for C22H34O3+Na, 369.2406; found 369.2398; IR (thin film, cm−1) 3053, 2979, 2977, 1734, 1558, 1472, 1373, 1265, 1139, 1086, 971, 921, 704; TLC (80:20 hexanes/EtOAc) Rf = 0.23.
(2S,4aS,4bR,6aR,9aS,9bS,11aS)-2-(2-Hydroxypropan-2-yl)-4a,9a,9b-trimethyltetradecahydroindeno[5,4-f]chromen-9(2H)-one (87)
A 20 mL scintillation vial was charged with ketone 86 (0.008 g, 0.02 mmol, 1.00 equiv) and EtOH (2 mL), and Pd/C (0.013 g, 1.50 mass equiv) was added. The reaction mixture was placed under 1 atm H2 (balloon), and the mixture was allowed to stir overnight. The reaction was filtered through a Celite plug, and the filtrate was concentrated on a rotary evaporator to give the crude ketone as a single diastereomer (as determined by 1H NMR spectroscopic analysis of the crude reaction mixture, which revealed a single compound). The product was purified via flash chromatography (90:10 to 80:20 to 70:30 hexanes/EtOAc) to afford ketone 87 as a clear, viscous oil. Analytical data: [α]D28 −45.2 (c = 0.35, CHCl3); 1H NMR (600 MHz, CDCl3) δ 3.18 (dd, J = 9.0, 3.0 Hz, 1H), 2.92 (d, J = 6.6, 4.2 Hz, 1H), 2.60 (br s, 1H), 2.33 (m, 1H), 2.21 (m, 1H), 2.03 (m, 1H), 1.92–1.82 (m, 3H), 1.74 (m, 2H), 1.64–1.41 (m, 11H), 1.17 (s, 3H), 1.14 (s, 3H), 1.10 (s, 3H), 1.04 (s, 3H), 0.85 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 224.9, 85.4, 84.4, 71.8, 54.5, 47.5, 46.2, 39.3, 38.8, 37.8, 36.3, 32.5, 26.8, 26.2, 26.1, 23.7, 23.5, 21.8, 20.9, 19.5, 17.3, 13.5; HRMS (ESI+) calcd for C22H36O3+Na, 371.2562; found 371.2554; IR (thin film, cm−1) 3446, 2955, 2852, 1731, 1636, 1520, 1473, 1396, 1085, 754; TLC (80:20 hexanes/EtOAc) Rf = 0.20.
(2S,4aS,4bR,9S,9aS,9bS,11aS)-2-(2-Hydroxypropan-2-yl)-4a,9a,9b-trimethyl-2,3,4,4a,4b,5,6,8,9,9a,9b,10,11,11a-tetradecahydroindeno[ 5,4-f]chromen-9-ol (88)
A flame-dried, 20 mL scintillation vial was charged with diol 86 (0.16 g, 0.43 mmol, 1.00 equiv) and CH2Cl2 (9 mL) under and atmosphere of N2. The mixture was cooled to 0 °C, and TFA (0.17 mL, 2.14 mmol, 5.00 equiv) was added. The reaction mixture was warmed to rt and allowed to stir until complete conversion of the starting material was observed by TLC analysis, typically 30 min. The reaction was quenched with saturated NaHCO3(aq) (5 mL), and the mixture was partitioned in a separatory funnel. The aqueous layer was extracted with CH2Cl2 (3 × 5 mL), and the combined organic extracts were dried with Na2SO4 and concentrated in vacuo to afford the crude nonconjugated enone 86, which was carried to the next step without further purification.
A flame-dried, 20 mL scintillation vial was charged with the crude ketone 86 and THF (5 mL) under an atmosphere of N2. The reaction mixture was cooled to 0 °C, and LiAlH4 (1 M in THF, 2.00 mL, 2.00 mmol, 4.70 equiv) was added dropwise. The reaction mixture was allowed to stir at this temperature until TLC analysis indicated complete consumption of the starting material, typically 30 min. The reaction was then carefully quenched with saturated NH4Cl(aq) (4 mL) and stirred 5 min at rt. The resulting mixture was partitioned in a separatory funnel and extracted with Et2O (3 × 5 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo to afford the crude alcohol 88 as a single diastereomer (as determined by 1H NMR spectroscopic analysis of the crude reaction mixture, which revealed a single compound). The crude product was purified via flash chromatography (80:20 to 70:30 to 60:40 hexanes/ EtOAc) to afford alcohol 88 (0.90 g, 60% yield) as a pale yellow foam. Analytical data: [α]D28 −116.4 (c = 0.50, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.22 (s, 1H), 4.37 (t, J = 8.4 Hz, 1H), 3.18 (dd, J = 9.0, 3.0 Hz, 1H), 2.95 (m, 1H), 2.68 (br s, 1H), 2.54 (m, 1H), 2.25 (d, J = 10.8 Hz, 1H), 2.20 (m, 1H), 1.94 (br s, 1H), 1.80 (d, J = 2.4 Hz, 1H), 1.69–1.59 (m, 6H), 1.50 (m, 1H), 1.42–1.36 (m, 3H), 1.19 (s, 3H), 1.17 (s, 3H), 1.14 (s, 3H), 0.86 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 148.1, 117.5, 85.8, 85.2, 84.4, 71.9, 55.1, 48.1, 43.9, 40.9, 38.0, 36.7, 31.8, 27.0, 26.1, 24.6, 23.8, 23.6, 22.8, 21.9, 16.7, 13.5; HRMS (ESI+) calcd for C22H36O3+Na, 371.2562; found 371.2570; IR (thin film, cm−1) 3433, 2979, 2678, 2399, 1452, 1373, 1215, 1093, 955, 755, 668; TLC (60:40 hexanes/EtOAc) Rf = 0.36
(2S,4aS,4bR,6aS,9aS,9bS,11aS)-2-(2-Hydroxypropan-2-yl)-4a,9a,9b-trimethyltetradecahydroindeno[5,4-f]chromen-9(2H)-one (91)
A flame-dried, 20 mL scintillation vial was charged with Crabtree’s catalyst (0.01 g, 0.01 mmol, 0.15 equiv) in a nitrogen-filled glovebox. The vial was sealed with a rubber-septum, removed from the glovebox, and placed under an atmosphere of N2. CH2Cl2 (4 mL, freshly degassed via N2 bubbling for 30 min) was added followed by a solution of alcohol 88 (0.025 g, 0.07 mmol, 1.00 equiv) in degassed CH2Cl2 (2 mL), and the resulting mixture was placed under an atmosphere of H2 (balloon) and allowed to stir 36 h at rt. The resulting mixture was concentrated in vacuo to afford the crude alcohol 90, which was carried forward to the next step without purification. Although this material was not isolated, the diastereomeric ratio was determined by 1H NMR spectroscopic analysis of the crude reaction mixture, which revealed a single compound. This crude 1H NMR spectrum is included in the Supporting Information.
A 20 mL scintillation vial was charged with the crude alcohol 90 and CH2Cl2 (3 mL) with magnetic stirring. Dess-Martin periodinane (0.045 g, 0.11 mmol, 1.50 equiv) was added, and the reaction mixture was allowed to stir at rt until complete conversion of the starting materal was observed by TLC analysis, typically 20 min. The reaction was then quenched via a 1:1 solution of saturated NaHCO3(aq) and saturated Na2S2O3(aq) (3 mL), and the mixture was stirred 5 min. The reaction mixture was then diluted with Et2O (5 mL) and partitioned in a separatory funnel. The aqueous layer was extracted with Et2O (3 × 5 mL), and the combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (90:10 to 80:20 to 70:30 hexanes/EtOAc) to afford ketone 91 (0.022 g, 89% yield) as a clear semisolid. Slow evaporation from HPLC-grade hexanes provided crystals suitable for X-ray crystallographic analysis. Analytical data: mp 125–130 °C; [α]D27 −89.3 (c = 0.85, CHCl3); 1H NMR (500 MHz, CDCl3) δ 3.16 (dd, J = 9.0, 3.0 Hz, 1H), 2.93 (m, 1H), 2.65 (br s, 1H), 2.32 (dd, J = 11.0, 8.5 Hz, 1H), 2.19–2.14 (m, 2H), 2.00 (m, 1H), 1.78–1.23 (m, 15H), 1.16 (s, 3H), 1.14 (s, 3H), 1.02 (s, 3H), 0.92 (s, 3H), 0.83 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 221.2, 85.7, 84.4, 71.8, 56.1, 46.9, 40.2, 39.9, 37.8, 37.5, 36.5, 31.1, 26.1, 25.8, 24.2, 23.8, 23.7, 21.9, 21.2, 18.9, 12.9, 10.3; HRMS (ESI+) calcd for C22H36O3+Na, 371.2562; found 371.2560; IR (thin film, cm−1) 3566, 3446, 2946, 2876, 1772, 1731, 1472, 1385, 1259, 1158, 1098, 974, 735; TLC (70:30 hexanes/EtOAc) Rf = 0.60.
Note: The following sequence for conversion of 91 to paspaline was adapted from the previously published protocol by Smith and coworkers.12a,d
(2S,4aS,4bR,6aS,9aS,9bS,11aS)-2-(2-Hydroxypropan-2-yl)-4a,9a,9b-trimethyl-8-(methylthio)tetradecahydroindeno[5,4-f]-chromen-9(2H)-one (S7)
A flame-dried, 20 mL scintillation vial was cooled to 0 °C and charged with THF (1 mL) and a freshly prepared solution of lithium diisopropylamide (0.5 M in THF, 0.57 mL, 0.29 mmol, 5.00 equiv) under an atmosphere of N2. The resulting solution was then charged with a solution of ketone 91 (0.02 g, 0.06 mmol, 1.00 equiv) in THF (0.5 mL), and the reaction mixture was allowed to stir 15 min at 0 °C. HMPA (0.6 mL) was added followed by Me2S2 (0.031 mL, 0.34 mmol, 6.00 equiv), and the reaction was allowed to stir until TLC analysis showed complete conversion of the starting material, typically 10 min. The reaction was quenched via addition of H2O (5 mL). The resulting mixture was transferred to a separatory funnel, and the organic layer was separated. The aqueous layer was extracted with Et2O (3 × 5 mL), and the combined organic extracts were washed with brine (2 × 10 mL), dried with magnesium sulfate, and concentrated in vacuo. The product was purified via flash chromatography (90:10 to 80:20 to 70:30 hexanes/EtOAc) to afford an inseparable, diastereomeric mixture of thioethers S7 (0.019 g, 84% yield) as a yellow, viscous oil. Analytical data: [α]D27 −57.9 (c = 0.70, CHCl3); 1H NMR (600 MHz, CDCl3) δ 3.16 (dd, J = 9.0, 3.0 Hz, 1H), 2.94 (m, 2H), 2.61 (br s, 1H), 2.25 (br s, 3H), 2.22–2.13 (m, 3H), 1.63–1.57 (m, 11H), 1.47 (m, 10H), 1.17 (s, 3H), 1.14 (s, 3H), 1.02 (s, 3H), 1.01 (s, 3H), 0.83 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 218.4, 85.6, 84.4, 71.8, 56.6, 49.8, 46.6, 40.1, 38.1, 37.8, 36.4, 31.8, 31.1, 26.1, 25.2, 24.2, 23.7, 21.8, 21.1, 19.0, 15.4, 12.9, 11.0; HRMS (ESI+) calcd for C23H38O3S+Na, 417.2439; found 417.2438; IR (thin film, cm−1) 3446, 2946, 2874, 1732, 1652, 1519, 1456, 1386, 1232, 1152, 1086, 946; TLC (70:30 hexanes/EtOAc) Rf = 0.63.
(2S,4aS,4bR,6aS,9aS,9bS,11aS)-8-(2-Aminophenyl)-2-(2-hydroxypropan-2-yl)-4a,9a,9b-trimethyltetradecahydroindeno[5,4-f]-chromen-9(2H)-one (S8)
A flame-dried, 20 mL scintillation vial was charged with a solution of aniline (0.25 M in CH2Cl2, 0.26 mL, 0.07 mmol, 2.00 equiv) under an atmosphere of N2, and the resulting solution was cooled to −78 °C. The lights in the fume hood were turned off, and a solution of tBuOCl (0.25 M in CH2Cl2, 0.26 mL, 0.07 mmol, 2.00 equiv) was added dropwise. The reaction mixture was allowed to stir 15 min, upon which a solution of thioether S8 (0.013 g, 0.03 mmol, 1.00 equiv) in CH2Cl2 (1.5 mL) was added. The mixture was allowed to stir 50 min, upon which NEt3 (0.02 mL, 0.13 mmol, 4.00 equiv) was added. The reaction was then warmed to rt and allowed to stir until a bright orange color was observed, typically 5 min. The resulting solution was diluted with H2O (5 mL) and Et2O (10 mL) and partitioned in a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 5 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo to afford a crude mixture of diastereomeric keto-anilines, which was carried directly on to the next step without further purification.
The residue was taken up into EtOH (1 mL) in a 20 mL scintillation vial, and a slurry of Raney Ni in H2O (150 mg) was added. The reaction mixture was stirred vigorously at rt until complete conversion of the intermediate thioether was observed by TLC analysis, typically 1 h. The reaction mixture was filtered through a Celite plug, and the resulting solution was concentrated in vacuo. The crude product was purified via flash chromatography (90:10 to 80:20 to 70:30 to 60:40 hexanes/EtOAc) to afford ketoaniline S8 (0.009 g, 62% yield) as yellow, viscous oil. Analytical data: [α]D27 +26.6 (c = 0.45, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.06 (m, 2H), 6.77 (m, 2H), 4.21 (br s, 2H), 3.54 (t, J = 9.0 Hz, 1H), 3.16 (dd, J = 9.6, 2.4 Hz, 1H), 2.94 (m, 1H), 2.62 (br s, 1H), 2.35 (m, 1H), 2.14–2.04 (m, 3H), 1.84–1.37 (m, 16H), 1.17 (s, 3H), 1.15 (s, 1H), 1.11 (s, 3H), 0.98 (s, 3H), 0.86 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 221.0, 146.0, 127.6, 125.8, 125.4, 119.1, 117.5, 85.6, 84.5, 71.8, 57.0, 51.5, 46.8, 40.1, 38.0, 37.8, 36.5, 31.3, 28.9, 26.1, 25.4, 24.2, 23.7, 21.9, 21.2, 19.2, 12.9, 10.0; HRMS (ESI+) calcd for C28H41NO3+Na, 462.2984; found 462.2983; IR (thin film, cm−1) 3421, 3053, 2984, 2877, 2305, 1732, 1652, 1456, 1362, 1265, 738; TLC (70:30 hexanes/EtOAc) Rf = 0.30.
Paspaline (1)
A 1 mL dram vial was charged with ketone S8 (0.007 g, 0.02 mmol, 1.00 equiv), CH2Cl2 (1.2 mL), and PTSA (0.002 g, 0.01 mmol, 0.66 equiv). The vial was sealed, and the mixture was warmed to 50 °C and stirred for 16 h. The reaction mixture was cooled to rt, diluted with H2O (10 mL) and Et2O (10 mL), and transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was extracted with Et2O (3 × 5 mL). The combined organic extracts were dried with magnesium sulfate and concentrated in vacuo. The product was purified via flash chromatography (90:10 to 80:20 hexanes/EtOAc) to afford paspaline (0.006 g, 89% yield) as a yellow foam. Slow evaporation from HPLC-grade hexanes provided crystals suitable for X-ray crystallographic analysis. Analytical data: [α]D25 −16.4 (c = 0.30, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.72 (br s, 1H), 7.42 (m, 1H), 7.30 (m, 1H), 7.07 (m, 2H), 3.21 (dd, J = 9.6, 2.4 Hz, 1H), 3.03 (dd, J = 8.4, 3.6 Hz, 1H), 2.77–2.65 (m, 3H), 2.32 (dd, J = 10.8, 2.4 Hz, 1H), 1.96 (m, 1H), 1.84–1.77 (m, 3H), 1.70–1.56 (m, 6H), 1.49–1.37 (m, 3H), 1.19 (s, 3H), 1.17 (s, 3H), 1.13 (s, 3H), 1.03 (s, 3H), 0.88 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 150.8, 139.3, 125.1, 120.4, 119.5, 118.4, 118.2, 111.4, 85.7, 84.7, 71.9, 53.0, 48.7, 46.4, 40.0, 37.6, 36.5, 33.9, 27.5, 26.1, 25.2, 24.6, 23.7, 22.0, 21.9, 20.0, 14.6, 12.6; HRMS (ESI+) calcd for C28H39NO2+H, 422.3059; found 422.3056; IR (thin film, cm−1) 3565, 3467, 3053, 2982, 2930, 2855, 1455, 1386, 1375, 1331, 1265, 1158, 1087, 1037; TLC (70:30 hexanes/EtOAc) Rf = 0.42.
Supplementary Material
Acknowledgments
The project described was supported by Award No. R01 GM084927 from the National Institute of General Medical Sciences. R.J.S. acknowledges an NSF Graduate Research Fellowship and ACS Division of Organic Chemistry graduate fellowship. X-ray crystallography was performed by Dr. Peter White (UNC). The authors thank Dr. Shubin Liu (UNC) for assistance with DFT structure optimization calculations.
Footnotes
The authors declare no competing financial interest.
- NMR spectra, X-ray crystallographic data (x1504008, x1405008, x1501005, and x1502014), and computational details (PDF)
- X-ray data for 32 (CIF)
- X-ray data for 54 (CIF)
- X-ray data for 91 (CIF)
- X-ray data for 1 (CIF)
References
- 1.Sings H, Singh S. Alkaloids: Chemistry and Biology 2003. 60:51. doi: 10.1016/s0099-9598(03)60002-7. [DOI] [PubMed] [Google Scholar]
- 2.Miedaner T, Geiger HH. Toxins. 2015;7:659. doi: 10.3390/toxins7030659. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Steyn PS, Vleggar R. Progress in the Chemistry of Organic Natural Products. Springer-Verlag; New York: 1985. [Google Scholar]; (b) Mantle PG, Mortimer PB, White EP. Res Vet Sci. 1977;24:49. [PubMed] [Google Scholar]; (c) DiMenna ME, Mantle PB. Res Vet Sci. 1977;24:347. [Google Scholar]; (d) Mantle PG, Day JB, Haigh CR, Penny HC. Vet Rec. 1978;103:403. doi: 10.1136/vr.103.18.403-a. [DOI] [PubMed] [Google Scholar]; (e) Cole RJ, Dorner JW, Lansden JA, Cox RH, Pape C, Cunfer B, Nicholson SS, Bedell DM. J Agric Food Chem. 1977;25:1197. doi: 10.1021/jf60213a061. [DOI] [PubMed] [Google Scholar]
- 4.(a) Fehr T, Acklin W. Helv Chim Acta. 1966;49:1907. doi: 10.1002/hlca.19750580829. [DOI] [PubMed] [Google Scholar]; (b) Gysi RP. PhD Dissertation. 4990 Eidgenossiche Technische Hochschule; Zürich, Switzerland: 1973. [Google Scholar]; (c) Leutwiler A. Dissertation No 5163. Eidgenossiche Technische Hochschule; Zürich, Switzerland: 1973. [Google Scholar]
- 5.Munday-Finch SC, Wilkins AL, Miles CO. Phytochemistry. 1996;41:327. [Google Scholar]
- 6.(a) Cole RJ, Dorner JW, Springer JP, Cox RH. J Agric Food Chem. 1981;29:293. [Google Scholar]; (b) Gallagher RT, Finer J, Clardy J, Leutwiler A, Weibel F, Acklin W, Arigoni D. Tetrahedron Lett. 1980;21:231. and references therein. [Google Scholar]
- 7.Ogata M, Ueda J-y, Hoshi M, Hashimoto J, Nakashima T, Anzai K, Takagi M, Shin-ya K. J Antibiot. 2007;60:645. doi: 10.1038/ja.2007.83. [DOI] [PubMed] [Google Scholar]
- 8.(a) Cole RJ, Kirksey JW, Wells JM. Can J Microbiol. 1974;20:1159. doi: 10.1139/m74-179. [DOI] [PubMed] [Google Scholar]; (b) Springer JP, Clardy J, Wells JM, Cole RJ, Kirksey JW. Tetrahedron Lett. 1975;16:2531. [Google Scholar]
- 9.(a) Brnardic E, Doherty JB, Dorsey J, Ellwood C, Fillmore M, Malaska M, Nelson K, Soukri M. Preparation of Indole Diterpene Alkaloids as Maxi-K Channel Blockers for the Treatment of Glaucoma. 2009/048559. Patent WIPO. 2009; (b) Garcia ML, Goetz MA, Kaczorowski GJ, McManus OB, Monaghan RL, Strohl WR, Tkacz JS. Novel Maxi-K Channel Blockers, Methods of Use and Process for Making the Same. 0239863. US Patent. 2005; (c) Brnardic E, Doherty JB, Ellwood C, Fillmore M, Malaska M. Maxi-K Channel Blockers and Methods of Use. 048558. US Patent. 2009
- 10.Sheehan JJ, Benedetti BL, Barth AL. Epilepsia. 2009;50:711. doi: 10.1111/j.1528-1167.2008.01888.x. [DOI] [PubMed] [Google Scholar]
- 11.(a) Springer JP, Clardy J. Tetrahedron Lett. 1980;21:231. [Google Scholar]; (b) Dorner JP, Cole RJ, Cox RH, Cunfer BM. J Agric Food Chem. 1984;32:1069. [Google Scholar]
- 12.(a) Smith AB, III, Mewshaw R. J Am Chem Soc. 1985;107:1769. [Google Scholar]; (b) Smith AB, III, Leenay TL. Tetrahedron Lett. 1988;29:2787. [Google Scholar]; (c) Smith AB, III, Leenay TL. Tetrahedron Lett. 1988;29:2791. [Google Scholar]; (d) Mewshaw RE, Taylor MD, Smith AB., III J Org Chem. 1989;54:3449. [Google Scholar]; (e) Smith AB, III, Leenay TL. J Am Chem Soc. 1989;111:5761. [Google Scholar]; (f) Smith AB, III, Sunazuka T, Leenay TL, Kingery-Wood J. J Am Chem Soc. 1990;112:8197. [Google Scholar]; (g) Smith AB, III, Kingery-Wood J, Leenay TL, Nolen EG, Sunazuka T. J Am Chem Soc. 1992;114:1438. [Google Scholar]; (h) Zou Y, Melvin JE, Gonzales SS, Spafford MJ, Smith AB., III J Am Chem Soc. 2015;137:7095. doi: 10.1021/jacs.5b04728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Guile SD, Saxton JE, Thornton-Pett MJ. Chem Soc, Perkin Trans. 1992;1:1763. [Google Scholar]; (b) Clark JS, Myatt J, Roberts L, Walshe N. Synlett. 2005:697–699. [Google Scholar]; (c) Enomoto M, Kuwahara S. J Org Chem. 2010;75:6286. doi: 10.1021/jo101258f. [DOI] [PubMed] [Google Scholar]; (d) Oikawa M, Hashimoto R, Sasaki M. Eur J Org Chem. 2011;2011:538. [Google Scholar]; (e) Isaka T, Hasegawa M, Toshima H. Biosci, Biotechnol, Biochem. 2011;75:2213. doi: 10.1271/bbb.110511. [DOI] [PubMed] [Google Scholar]; (f) Adachi M, Higuchi K, Thasana N, Yamada H, Nishikawa T. Org Lett. 2012;14:114. doi: 10.1021/ol202895u. [DOI] [PubMed] [Google Scholar]; (g) Okano K, Yoshii Y, Tokuyama H. Heterocycles. 2012;84:1325. [Google Scholar]
- 14.(a) Churruca F, Fousteris M, Ishikawa Y, Rekowski MW, Hounsou C, Surrey T, Giannis A. Org Lett. 2010;12:2096. doi: 10.1021/ol100579w. [DOI] [PubMed] [Google Scholar]; (b) Bradshaw B, Etxebarria-Jardi G, Bonjoch J. J Am Chem Soc. 2010;132:5966. doi: 10.1021/ja101994q. [DOI] [PubMed] [Google Scholar]; (c) Enomoto S, Morita A, Kuwahara S. Angew Chem, Int Ed. 2012;51:12833. doi: 10.1002/anie.201206299. [DOI] [PubMed] [Google Scholar]; (d) Bian M, Wang Z, Xiong X, Sun Y, Matera C, Nicolaou KC, Li A. J Am Chem Soc. 2012;134:8078. doi: 10.1021/ja302765m. [DOI] [PubMed] [Google Scholar]; (e) Goetz AE, Silberstein AL, Corsello MA, Garg NK. J Am Chem Soc. 2014;136:3036. doi: 10.1021/ja501142e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Nicewicz DA, Satterfield DA, Schmitt DS, Johnson JS. J Am Chem Soc. 2008;130:17281. doi: 10.1021/ja808347q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Campbell MJ, Johnson JS. J Am Chem Soc. 2009;131:10370. doi: 10.1021/ja904136q. [DOI] [PubMed] [Google Scholar]; (c) Sanders S, Ruiz-Olalla A, Johnson JS. Chem Commun. 2009:5135. doi: 10.1039/b911765b. [DOI] [PubMed] [Google Scholar]; (d) Campbell MJ, Johnson JS. Synthesis. 2010:2841. [Google Scholar]; (e) Greszler SN, Malinowski JT, Johnson JS. Org Lett. 2011;13:3206. doi: 10.1021/ol2011192. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Malinowski JT, McCarver SJ, Johnson JS. Org Lett. 2012;14:2878. doi: 10.1021/ol301140c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Malinowski JT, Sharpe RJ, Johnson JS. Science. 2013;340:180. doi: 10.1126/science.1234756. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Sharpe RJ, Malinowski JT, Johnson JS. J Am Chem Soc. 2013;135:17990. doi: 10.1021/ja409944u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Sharpe RJ, Malinowski JT, Sorana F, Luft JC, Bowerman CJ, DeSimone JM, Johnson JS. Bioorg Med Chem. 2015;23:1849. doi: 10.1016/j.bmc.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharpe RJ, Johnson JS. J Am Chem Soc. 2015;137:4968. doi: 10.1021/jacs.5b02631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Chu Y, White JB, Duclos BA. Tetrahedron Lett. 2001;42:3815. [Google Scholar]; (b) Iguchi M, Niwa M, Yamamura S. J Chem Soc D. 1971:974. [Google Scholar]
- 18.(a) Stewart IC, Ung T, Pletnev AA, Berlin JM, Grubbs RH, Schrodi Y. Org Lett. 2007;9:1589. doi: 10.1021/ol0705144. [DOI] [PubMed] [Google Scholar]; (b) Vorfalt T, Leuhausser S, Plenio H. Angew Chem, Int Ed. 2009;48:5191. doi: 10.1002/anie.200900935. [DOI] [PubMed] [Google Scholar]
- 19.McMurry JE, Fleming MP, Kees KL, Krepski LR. J Org Chem. 1978;43:3255. [Google Scholar]
- 20.Gharpure SJ, Reddy SRB. Org Lett. 2009;11:2519. doi: 10.1021/ol900721q. [DOI] [PubMed] [Google Scholar]
- 21.Breit B, Zahn SK. J Org Chem. 2001;66:4870. doi: 10.1021/jo015634i. [DOI] [PubMed] [Google Scholar]
- 22.The selectivity was determined via the collective NOESY analyses of 19, 20, and the ethyl ketone precursor of 21. The same selectivity was observed by Deslongchamps and Germain in their synthesis of momilactone A. See: Germain J, Deslongchamps P. J Org Chem. 2002;67:5269. doi: 10.1021/jo025873l.
- 23.(a) Gianturco MA, Friedel P. Tetrahedron. 1963;19:2039. doi: 10.1016/0040-4020(63)85019-x. [DOI] [PubMed] [Google Scholar]; (b) House HO. Modern Synthetic Reactions. Benjamin; Menlo Park: 1972. pp. 520–532. [Google Scholar]; c Stowell JC. Carbanions in Organic Synthesis. Wiley; New York: 1979. pp. 205–210. [Google Scholar]
- 24.Demir AS, Enders DJ. J Prakt Chem/Chem-Ztg. 1997;339:553. [Google Scholar]
- 25.The current method for preparing these compounds involves bisalkylation of 1,5-dimethoxy-1,4-cyclohexadiene. See: Piers E, Grierson JR. J Org Chem. 1977;42:3755.
- 26.Watanabe H, Iwamoto M, Nakada M. J Org Chem. 2005;70:4652. doi: 10.1021/jo050349a. [DOI] [PubMed] [Google Scholar]
- 27.(a) Katoh T, Mizumoto S, Fudesaka M, Nakashima Y, Kajimoto T, Node M. Synlett. 2006;37:2076. [Google Scholar]; (b) Katoh T, Mizumoto S, Fudesaka M, Takeo M, Kajimoto T, Node M. Tetrahedron: Asymmetry. 2006;17:1655. [Google Scholar]
- 28.Becke AD. J Chem Phys. 1993;98:5648.Lee CT, Yang WT, Parr RG. Phys Rev B: Condens Matter Mater Phys. 1988;37:785. doi: 10.1103/physrevb.37.785.Hariharan PC, Pople JA. Theor Chim Acta. 1973;28:213.Francl MM, Pietro WJ, Henre WJ, Binley JS, Gordon MS, Frees DJ, Pople JA. J Chem Phys. 1982;77:3654.See Supporting Information for additional computational details..
- 29.Other examples of remote direction in alkene functionalization: Zhang W, Yamamoto H. J Am Chem Soc. 2007;129:286. doi: 10.1021/ja067495y.Wan KK, Iwasaki K, Umotoy JC, Wolan DW, Shenvi RA. Angew Chem, Int Ed. 2015;54:2410. doi: 10.1002/anie.201411493.
- 30.(a) Shapiro RH, Heath MJ. J Am Chem Soc. 1967;89:5734. [Google Scholar]; (b) Shapiro RH. Org React (NY) 1975;23:405. [Google Scholar]
- 31.(a) Stork G, Darling SD. J Am Chem Soc. 1960;82:1512. [Google Scholar]; (b) Stork G, Rosen P, Goldman N, Coombs RV, Tsuji J. J Am Chem Soc. 1965;87:275. [Google Scholar]
- 32.Vilkas M. Bull Soc Chem Fr. 1959:1401. [Google Scholar]
- 33.(a) Stork G, Kahn M. J Am Chem Soc. 1985;107:500. [Google Scholar]; (b) Stork G, Sher PM. J Am Chem Soc. 1986;108:303. [Google Scholar]
- 34.(a) Ireland RE, Mueller RH. J Am Chem Soc. 1972;94:5897. doi: 10.1021/ja00765a079. [DOI] [PubMed] [Google Scholar]; (b) Ireland RE, Mueller RH, Willard AK. J Am Chem Soc. 1976;98:2868. [Google Scholar]
- 35.(a) Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]; (b) Neufeldt SR, Sanford MS. Org Lett. 2010;12:532. doi: 10.1021/ol902720d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu C, Tang P, Yu B. J Am Chem Soc. 2008;130:5872. doi: 10.1021/ja801669r. [DOI] [PubMed] [Google Scholar]
- 37.Siler DA, Mighion JD, Sorensen EJ. Angew Chem, Int Ed. 2014;53:5332. doi: 10.1002/anie.201402335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krasovskiy A, Kopp F, Knochel P. Angew Chem, Int Ed. 2006;45:497. doi: 10.1002/anie.200502485. [DOI] [PubMed] [Google Scholar]
- 39.Abad A, Agullo C, Arno M, Domingo LR, Zaragoza RJ. J Org Chem. 1990;55:2369. and references therein. [Google Scholar]
- 40.(a) Evans DA, Morrissey M. J Am Chem Soc. 1984;106:3866. [Google Scholar]; (b) Evans DA, Morrissey M. Tetrahedron Lett. 1984;25:4637. [Google Scholar]
- 41.(a) St André AF, MacPhillamy HB, Nelson JA, Shabica AC, Scholz CR. J Am Chem Soc. 1952;74:5506. [Google Scholar]; (b) Gabbard RB, Segaloff A. Steroids. 1983;41:791. doi: 10.1016/0039-128x(83)90054-5. [DOI] [PubMed] [Google Scholar]; (c) Haddad M, Blazejewski JC, Wakselman C, Dorai V, Duc I. Eur J Med Chem. 1994;29:627. [Google Scholar]; (d) Khripach VA, Zhabinskii VN, Fando GP, Kuchto AI, Lyakhov AS, Govorova AA, Groen MB, Van Der Louw J, De Groot A. Steroids. 2004;69:495. doi: 10.1016/j.steroids.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 42.Gassman PG, van Bergen TJ, Gilbert DP, Cue BW., Jr J Am Chem Soc. 1974;96:5495. [Google Scholar]
- 43.Breit B, Zahn SK. J Org Chem. 2001;66:4870. doi: 10.1021/jo015634i. [DOI] [PubMed] [Google Scholar]
- 44.Abecassis K, Gibson SE. Eur J Org Chem. 2010;2010:2938. [Google Scholar]
- 45.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, revision D.01. Gaussian, Inc; Wallingford, CT: 2004. [Google Scholar]
- 46.Xiang S-K, Zhang Bo, Zhang L-H, Cui Y, Jiao N. Chem Commun. 2011;47:8097. doi: 10.1039/c1cc12220g. [DOI] [PubMed] [Google Scholar]
- 47.Zhang H-B, Liu L, Liu Y-L, Chen Y-J, Wang J, Wang D. Synth Commun. 2007;37:173. [Google Scholar]
- 48.Westermaier M, Mayr H. Org Lett. 2006;8:4791. doi: 10.1021/ol0618555. [DOI] [PubMed] [Google Scholar]
- 49.Zimmerman HE, Wang P. J Org Chem. 2002;67:9216. doi: 10.1021/jo026187p. [DOI] [PubMed] [Google Scholar]
- 50.Van Goor FF, Burton WL. Compositions for Treatment of Cystic Fibrosis and Other Chronic Diseases. 61254180. US Patent. 2011
- 51.Hettche F, Hoffmann RW. New J Chem. 2003;27:172. [Google Scholar]
- 52.Archer CH, Thomas NR, Gani D. Tetrahedron: Asymmetry. 1993;4:1141. [Google Scholar]
- 53.Nyfeler E, Renaud P. Org Lett. 2008;10:985. doi: 10.1021/ol702832x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.