

Introduction:
The goal of this study was to characterize the role of Endothelin (ET) type B receptors (ETB) on vascular function in healthy and diseased conditions and demonstrate how it affects the pharmacological activity of ET receptor antagonists (ERAs).
Methods:
The contribution of the ETB receptor to vascular relaxation or constriction was characterized in isolated arteries from healthy and diseased rats with systemic (Dahl-S) or pulmonary hypertension (monocrotaline). Because the role of ETB receptors is different in pathological vis-à-vis normal conditions, we compared the efficacy of ETA-selective and dual ETA/ETB ERAs on blood pressure in hypertensive rats equipped with telemetry.
Results:
In healthy vessels, ETB receptors stimulation with sarafotoxin S6c induced vasorelaxation and no vasoconstriction. In contrast, in arteries of rats with systemic or pulmonary hypertension, endothelial ETB-mediated relaxation was lost while vasoconstriction on stimulation by sarafotoxin S6c was observed. In hypertensive rats, administration of the dual ETA/ETB ERA macitentan on top of a maximal effective dose of the ETA-selective ERA ambrisentan further reduced blood pressure, indicating that ETB receptors blockade provides additional benefit.
Conclusions:
Taken together, these data suggest that in pathology, dual ETA/ETB receptor antagonism can provide superior vascular effects compared with ETA-selective receptor blockade.
Key Words: ETB receptor, endothelin, endothelium, macitentan, ambrisentan
INTRODUCTION
Endothelin (ET)-1, a 21-amino acid peptide produced by endothelial and other cells, is considered one of the most potent vasoconstrictors known. ET-1 binds to 2 G protein–coupled receptors, ETA and ETB. In physiological conditions, ETA receptors predominate on vascular smooth muscle cells and mediate vasoconstriction, whereas endothelial ETB receptors may mediate vasodilation through nitric oxide (NO) release and hemodilution.1,2 In certain pathological conditions, including systemic and pulmonary hypertension (PH), ETB receptors are upregulated in the media of blood vessels, and both ETA and ETB receptors contribute to the detrimental effects of ET-1.3–5
These observations suggest that the vascular function of the ETB receptor may vary, depending not only on cell type (eg, vasoconstricting ETB on smooth muscle cells vs. vasodilatory ETB on endothelial cells) but also on the status of the vessel (healthy vs. pathology). This change of function of the ETB receptor could have a differential impact on the response to ET receptor antagonists (ERAs) harboring different selectivity profiles toward this receptor. We hypothesized that in pathological conditions, endothelial ETB receptors cannot mediate NO-dependent vasorelaxation because of endothelial dysfunction, whereas smooth muscle ETB receptors could trigger vasoconstriction. This might suggest that ETA-selective ERAs may be less efficacious in antagonizing the deleterious effects of ET-1 in pathology than dual ERAs. To compare whether the dual ETA/B ERA macitentan may be more effective than the ETA-selective ERA ambrisentan, we designed a study in which conscious hypertensive rats were given a single maximal effective dose of ambrisentan corresponding to maximal blockade of ETA receptors, followed by a single maximal effective dose of macitentan, to observe the effect of additional blockade of ETB receptors.
The goal of this study was to investigate the contribution of ETB receptors to vascular reactivity in physiology and disease (PH and systemic hypertension) and demonstrate how it can affect the hemodynamic response to ERAs with different selectivity toward this receptor type.
METHODS
Animals
Two-month-old Wistar rats were ordered from Harlan (Venray, The Netherlands). All rats were maintained under identical conditions in accordance with the guidelines of the Swiss Animal Protection Law under licenses 164 and 185. The monocrotaline rat model was developed as follows: after an acclimatization period of 7 days, rats were allocated for monocrotaline injection. Monocrotaline (MCT; Sigma-Aldrich, St. Louis, MO) was dissolved in 1 N HCl at a concentration of 200 mg/mL, neutralized with 1 N NaOH and diluted with sterile 0.9% saline at 20 mg/mL. MCT was injected at a dose of 60 mg/kg subcutaneously 3 mL/kg. Rats were killed by pentobarbital administration (intraperitoneally) 4 weeks after monocrotaline injection.
In Vitro Vascular Reactivity
Rings of left pulmonary arteries or thoracic aortas (2 mm in length) were mounted in organ baths under isometric conditions and bubbled with the following respective gas mixtures: CO2 5%, O2 16%, N2 79% or CO2 5%, O2 95%. After a recovery period, vessels were stretched (2 × 0.5 g or 4 × 0.5 g for aortas and pulmonary arteries, respectively) and depolarized twice with KCl 60 mM. Endothelium-dependent relaxations were obtained with cumulative doses of acetylcholine (10−9 to 10−5 mol/L) after precontraction with norepinephrine corresponding to c.a. 80% of the contraction to KCl 60 mM. After a 45-minute recovery period, endothelium-independent relaxations were achieved with cumulative doses of sodium nitroprusside (Sigma-Aldrich, 10−10 to 10−6 mol/L). Endothelium-dependent relaxation to sarafotoxin S6c (Sigma-Aldrich), an ETB receptor agonist was assessed after in precontracted vessel by adding sarafotoxin S6c 10−9 mol/L in presence or absence of NO synthase inhibitor L-NAME (0.1 mM; Sigma-Aldrich; 30 minutes preincubation) or endothelium. Smooth muscle constriction to sarafotoxin S6c was assessed by performing a dose–response curve (10−9 to 3.10−7 mol/L) in nonprecontracted vessels without endothelium.
In Vivo Hemodynamics and Add-on Protocol
Two-month-old Dahl salt-sensitive (Dahl-S) rats were administered with 1% NaCl in drinking water. Five to 6 weeks after starting salt administration, a telemetry transmitter was implanted. Under sterile conditions, rats were instrumented microsurgically with a telemetry pressure transmitter implanted in the peritoneal cavity. The pressure catheter was inserted into the aorta, below the renal artery pointing upstream. The abdomen was closed and the transmitter sutured to the abdominal musculature. Blood pressure was collected continuously using the Dataquest ART Gold acquisition system (version 3.01). Systolic, mean, and diastolic arterial pressures and heart rate (HR) were collected at 5-minute intervals during the entire experiment, resulting in a series of 1152 data points per day for each animal. For both blood pressure and HR, each rat served as its own control, by using the data of the last 24 hours before drug or vehicle administration. An area between curves (ABC) was calculated for mean arterial pressure (MAP) versus time curves of the control and treatment periods (integrated differences between predose and postdose curves over time).
First, dose–response curves of each ERA were constructed in Dahl-S rats to determine the maximal effective dose and Tmax (time of observed maximal effect) of each ERA. Pharmacological effects on MAP and HR were measured up to 120 hours after a single gavage at doses ranging from 1 to 30 mg/kg for both compounds. To determine whether macitentan could provide superior pharmacological activity versus ambrisentan, we designed a study in which (1) macitentan was administered on top of the maximal effective dose of ambrisentan established by the dose–response curve (Table 1) and (2) the maximal effective dose of ambrisentan was administered on top of the maximal effective dose of macitentan. The maximal effective dose of the second compound was administered at Tmax of the first tested compound. As each compound had a relative rapid onset of action and the plateau effect lasted for around 6 hours, these pharmacodynamic characteristics enabled us to check whether an additive pharmacological activity of a first compound could be observed during the Tmax of a second compound.
TABLE 1.
Maximal MAP Decrease After Oral Administration of Vehicle, Macitentan, or Ambrisentan in Dahl-S Rats

Test Compounds
Macitentan and ambrisentan were supplied by Actelion Pharmaceuticals Ltd. Gelatin 7.5%, administered at 5 mL/kg, was used as vehicle for oral administration of the compounds by gavage.
Statistics
All results are presented as mean ± SEM. Area between the MAP versus time curves (ABC) is expressed in mm Hg·hour, MAP in mm Hg and HR in beats per minute. Differences between groups were analyzed using an unpaired Student's t test or one-way analysis of variance followed by Newman–Keuls for multiple comparisons test when required. Statistical significance was defined as P < 0.05.
RESULTS
ETB-mediated Vasodilation in Healthy Vessels Is NO and Endothelium Dependent
In left pulmonary arteries and aortas from healthy rats precontracted with phenylephrine, selective ETB receptor stimulation with sarafotoxin S6c 10−9 mol/L induced a relaxation that was inhibited by NO synthase blocker L-NAME or removal of the endothelium (Fig. 1).
FIGURE 1.
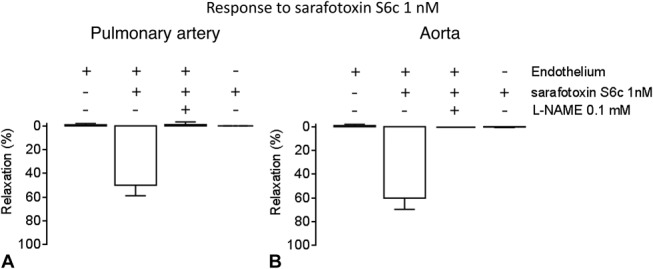
Response to ETB agonist sarafotoxin S6c 1 nM in (A) left pulmonary arteries (n = 8 per group) and (B) aortas from healthy Wistar rats (n = 4–10 per group) precontracted with phenylephrine (60%–80% of max KCl). The vasorelaxation observed in intact vessels was lost in the presence of nitric oxide synthase inhibitor L-NAME 0.1 mM or after removal of the endothelium.
ETB-mediated Vasodilation Is Lost in Rats With Pulmonary or Systemic Hypertension and Turns Into Vasoconstriction
Although precontracted aorta and left pulmonary arteries from Wistar rats showed ETB-mediated vasorelaxation to sarafotoxin S6c (10−9 mol/L), no vasoconstriction to this peptide was obtained in resting healthy vessels. In left pulmonary arteries from PH rats and aorta from Dahl-S rats, no relaxation to sarafotoxin S6c (10−9 mol/L) was observed (Fig. 2), whereas sarafotoxin S6c induced vasoconstriction in resting vessels (Fig. 3). In PH and Dahl-S rats, vascular dysfunction was observed: In PH rats, selective endothelial dysfunction was present as evidenced by impaired response to acetylcholine (Fig. 4A), whereas in Dahl-S rats impaired response to acetylcholine and NO donor sodium nitroprusside indicated both endothelial and smooth muscle cell dysfunction (Fig. 4B).
FIGURE 2.
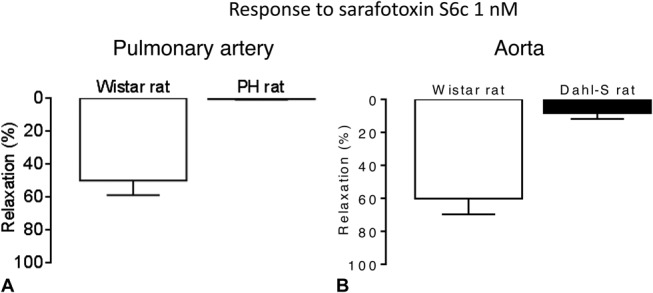
Vasorelaxation to ETB agonist sarafotoxin S6c 1 nM in (A) left pulmonary arteries from healthy Wistar rats (left) and diseased PH monocrotaline rats (right) precontracted with phenylephrine (n = 8 per group) and (B) aortas from healthy Wistar rats and diseased Dahl-S rats (n = 4–10 per group).
FIGURE 3.
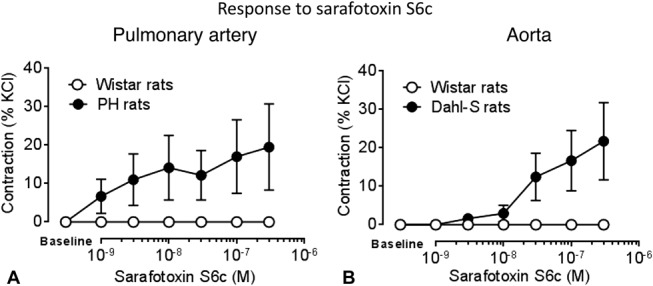
Dose–response curves to ETB agonist sarafotoxin S6c in resting (A) left pulmonary arteries from healthy Wistar rats and diseased PH monocrotaline rats (n = 8 per group) and (B) aortas from healthy Wistar rats and diseased Dahl-S rats (n = 8 per group).
FIGURE 4.
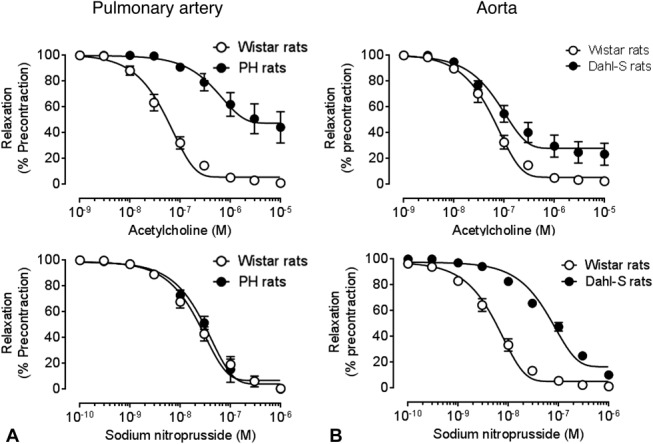
Dose–response curves to acetylcholine (top panel) and nitric oxide donor sodium nitroprusside (bottom panel) in (A) left pulmonary arteries from healthy Wistar rats and diseased monocrotaline (PH) rats (n = 4 per group) and (B) aortas from healthy Wistar rats and diseased Dahl-S rats (n = 6 per group).
Blockade of ETA and ETB Receptors Is More Effective Than ETA-selective Blockade in Hypertensive Rats
Dahl-S rats developed an increase of MAP of approximately 180 mm Hg. Acute oral administration of macitentan and ambrisentan dose-dependently decreased MAP without affecting HR (Table 1). At a dose of 30 mg/kg macitentan, the maximal decrease in MAP was 35 ± 4 mm Hg, about 24 hours after administration (Tmax). This effect was superior (P < 0.01) to that observed with the maximal effective dose of 30 mg/kg ambrisentan that decreased MAP by 17 ± 2 mm Hg (Table 1), with a Tmax around 6 hours.
Based on these data, the maximal effective dose of 30 mg/kg of macitentan and ambrisentan was selected for the add-on study. These maximal effective doses were confirmed using the add-on protocol: ambrisentan 30 mg/kg administered orally when the maximal effect of a previous dose of ambrisentan 30 mg/kg was reached, did not show an additional MAP decrease compared with vehicle on top of ambrisentan, and the maximal MAP decrease were 30 ± 3 and 25 ± 5 mm Hg, respectively (Fig. 5A). Oral administration of macitentan 30 mg/kg to Dahl-S rats, when the maximal effect of ambrisentan 30 mg/kg had been reached, induced an additional MAP decrease by 17 mm Hg compared with vehicle administered on top of ambrisentan 30 mg/kg. The maximal decrease induced by macitentan on top of ambrisentan was −42 ± 3 mm Hg (vs. −25 ± 5 mm Hg for vehicle P < 0.05) (Fig. 5B). Conversely, ambrisentan, administered orally at 30 mg/kg, when the maximal effect of macitentan 30 mg/kg had been reached, did not induce an additional MAP decrease compared with vehicle administered on top of macitentan 30 mg/kg (Fig. 5C).
FIGURE 5.
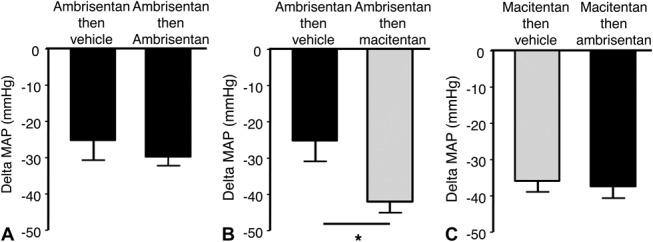
Superior pharmacological effect of macitentan on top of ambrisentan in conscious Dahl-S rats. Although addition of ambrisentan 30 mg/kg on top of ambrisentan 30 mg/kg did not induce further MAP reduction, confirming that a plateau of effect had been reached with ambrisentan (A), addition of macitentan 30 mg/kg on top of ambrisentan 30 mg/kg further decreased blood pressure by 17 mm Hg, *P < 0.05 versus vehicle (B). Conversely, addition of ambrisentan 30 mg/kg on top of macitentan 30 mg/kg did not induce further blood pressure reduction (C) (n = 6 per group).
DISCUSSION
The vascular ET system plays a modest role in the maintenance of vascular tone in physiological conditions as demonstrated by the moderate or absence effect of ERAs on blood pressure in healthy animals6–8 and human volunteers.9 Conversely, in pathological conditions associated with a vascular activation of the ET system, ERAs are expected to exert a stronger blood pressure lowering effects than normotensive conditions. Indeed, dysregulation of the vascular ET system leads to endothelial dysfunction and vascular remodeling.10,11 This feature of the system is of interest when considering the blockade of the ET receptor as a therapeutic strategy because the pharmacological activity of the antagonists will be driven by the local dysregulation of the ET system. This is especially the case for PH where the pulmonary vascular ET system is dysregulated and patients present increased mean pulmonary arterial hypertension without systemic hypertension. In the monocrotaline rat model of PH, administration of an ERA to conscious animals equipped with a double telemetry system has been shown to selectively decrease mean pulmonary arterial pressure without affecting mean arterial blood pressure and HR.12 In patients with PH, the incidence of hypotension observed with ERAs seems to be lower than with nonselective vasodilators such as guanylate cyclase activators or calcium channel blockers.13,14 Therefore, vascular response to ERAs depends on the level of vascular ET system activation.
The choice of the selectivity profile of an ERA is a strategic decision that must be taken based on understanding of the ET system not only in physiological but also in pathological situations. It is commonly assumed that the endothelial ETB should not be blocked as it plays a role in vasodilation through production of NO. Our findings regarding the vasorelaxing role of ETB receptors in left pulmonary arteries and aortas from healthy animals are in line with previous publications demonstrating their endothelial localization and involvement of NO.15,16 Other groups reported a loss of endothelial ETB relaxation17 or development of ETB vasoconstriction18 in disease conditions (eg, salt-sensitive hypertension and diabetes, respectively). Several studies have shown that in pathological situations such as hypertension, diabetes mellitus, and hypercholesterolemia, endothelial ETB expression is downregulated, whereas ETB expression is upregulated in smooth muscle cells where it mediates vasoconstriction.3 In addition to a decrease in endothelial ETB receptor expression, impairment of endothelial function can be a cause of the loss of ETB-mediated relaxation: increased generation of reactive oxygen species in pathological conditions such as obesity lead to a decrease of NO bioavailability and endothelial dysfunction and hence, together with our findings, could contribute to a loss of function of the endothelial ETB/NO pathway.19 Therefore, for pathological vessels, a selective ETA blockade strategy might not benefit from unblocked endothelial ETB receptors and may miss to block the upregulated smooth muscle ETB receptors. Using isolated pulmonary arteries, we showed that ETB-mediated relaxation is dependent on endothelial function as it can be abolished either by removal of the endothelium or NO synthase inhibition. In pulmonary vessels from PH rats or aorta from hypertensive rats both associated with severe endothelial dysfunction, no ETB-dependent relaxation was found. In patients with cardiovascular pathologies such as atherosclerosis20 or type II diabetes mellitus,21 ETB-mediated vasodilation is lost as well. These data suggest that preservation of endothelial vasodilatory ETB function by use of ETA-selective ERAs is not relevant in pathology, including PH, where decreased NO production, a marker of endothelial cells integrity, was observed.22 For instance, a study by Liang et al23 showed that only combination of an ETA-selective ERA but not of a dual ETA/ETB ERA with a phosphodiesterase 5 inhibitor allowed a synergism between endothelial ETB-derived NO and inhibition of NO second messenger cyclic guanosine monophosphate. Interestingly, this effect was abolished in absence of endothelium confirming that beneficial endothelial ETB-dependent effects are unlikely to occur in pathology. Besides endothelium, smooth muscle ETB receptors contribute to vasoconstriction in pathology as evidenced here in monocrotaline and Dahl-S rats. We confirmed in vivo that dual blockade of ET receptors was able to achieve a superior effect on blood pressure reduction. We also showed that dual blockade of ET receptors was able to further decrease blood pressure when administered on top of the maximal ETA-selective receptor blockade. This was demonstrated by comparing the effect of the maximal effective doses of each compound administered on top of the maximal effective dose of the other one as previously described.24 In pathologies like essential hypertension or insulin resistance, ETB receptors have indeed been demonstrated to contribute to vasoconstriction as forearm vasodilation achieved by dual ETA/ETB ERAs was superior to ETA-selective agents.25,26 In PH, smooth muscle ETB receptors are upregulated27 suggesting a contribution to deleterious vascular effects as evidenced in our PH rats that developed ETB-dependent vasoconstriction.
The differences of hemodynamic changes observed after acute administration of ERAs in this study could also translate on sustained effects on blood pressure reduction and tissue remodeling. Several chronic studies in models of hypertension demonstrate that ERAs can contribute to a sustained decrease in blood pressure by improvement of renal28 and endothelial function,29 decrease of sympathetic activity,30 and vascular remodeling.28,29 Beyond control of vascular tone, one should keep in mind that in addition of ETA, ETB receptors are also involved in cell proliferation.31 As a consequence, chronic blockade of both receptors was shown to lead to a superior effect on vascular remodeling versus selective blockade of ETA receptors.29 More generally, chronic dual ET receptor blockade could confer an advantage over ETA-selective blockade on remodeling processes and hence on long-term benefit as suggested by improved survival in preclinical models of PH and chronic heart failure.32,33 Safety might possibly also be improved with dual ERAs, as chronic stimulation of unantagonized ETB receptors may explain some secondary effects of ETA-selective antagonists, such as aggravation of cell proliferation34 or more fluid retention compared with dual ERAs.35,36 Clinical trials in idiopathic pulmonary fibrosis showed increased clinical worsening in patients treated with an ETA-selective antagonist but not in patients treated with dual ERAs.37–39 In addition, less side effects related to fluid retention (eg, edema) are observed in PH patients treated with dual40 than with ETA-selective ERAs.41 Although no head-to-head clinical trial is available, these observations may suggest that dual ETA and ETB receptor blockade favors a better efficacy and safety profile.
In conclusion, our data demonstrate that vascular ETB receptors have different activities in physiological versus pathological situations. In healthy vessels, endothelial ETB receptors can contribute to vasorelaxation, whereas in pathology, endothelial ETB-mediated relaxation is lost and smooth muscle ETB-mediated constriction appears. This functional shift has important consequences regarding the vascular response to ERAs with different selectivity profile toward the ETB receptor.
Footnotes
Supported by Actelion Pharmaceuticals Ltd.
The authors are employees of Actelion Pharmaceuticals Ltd.
REFERENCES
- 1.Takayanagi R, Kitazumi K, Takasaki C, et al. Presence of non-selective type of endothelin receptor on vascular endothelium and its linkage to vasodilation. FEBS Lett. 1991;282:103–106. [DOI] [PubMed] [Google Scholar]
- 2.Filep JG, Fournier A, Foldes-Filep E. Effects of the ETA/ETB receptor antagonist, bosentan on endothelin-1-induced myocardial ischaemia and oedema in the rat. Br J Pharmacol. 1995;116:1745–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kakoki M, Hirata Y, Hayakawa H, et al. Effects of hypertension, diabetes mellitus, and hypercholesterolemia on endothelin type B receptor-mediated nitric oxide release from rat kidney. Circulation. 1999;99:1242–1248. [DOI] [PubMed] [Google Scholar]
- 4.Clozel M, Hess P, Rey M, et al. Bosentan, sildenafil, and their combination in the monocrotaline model of pulmonary hypertension in rats. Exp Bio Med (Maywood). 2006;231:967–973. [PubMed] [Google Scholar]
- 5.Iglarz M, Clozel M. At the heart of tissue: endothelin system and end-organ damage. Clin Sci (Lond). 2010;119:453–463. [DOI] [PubMed] [Google Scholar]
- 6.Fryer RM, Rakestraw PA, Banfor PN, et al. Blood pressure regulation by ETA and ETB receptors in conscious, telemetry-instrumented mice and role of ETA in hypertension produced by selective ETB blockade. Am J Physiol Heart Circ Physiol. 2006;290:H2554–H2559. [DOI] [PubMed] [Google Scholar]
- 7.Teerlink JR, Loffler BM, Hess P, et al. Role of endothelin in the maintenance of blood pressure in conscious rats with chronic heart failure. Acute effects of the endothelin receptor antagonist Ro 47-0203 (bosentan). Circulation. 1994;90:2510–2518. [DOI] [PubMed] [Google Scholar]
- 8.Munter K, Ehmke H, Kirchengast M. Maintenance of blood pressure in normotensive dogs by endothelin. Am J Physiol. 1999;276:H1022–H1027. [DOI] [PubMed] [Google Scholar]
- 9.Taddei S, Virdis A, Ghiadoni L, et al. Vasoconstriction to endogenous endothelin-1 is increased in the peripheral circulation of patients with essential hypertension. Circulation. 1999;100:1680–1683. [DOI] [PubMed] [Google Scholar]
- 10.Amiri F, Paradis P, Reudelhuber TL, et al. Vascular inflammation in absence of blood pressure elevation in transgenic murine model overexpressing endothelin-1 in endothelial cells. J Hypertens. 2008;26:1102–1109. [DOI] [PubMed] [Google Scholar]
- 11.Amiri F, Virdis A, Neves MF, et al. Endothelium-restricted overexpression of human endothelin-1 causes vascular remodeling and endothelial dysfunction. Circulation. 2004;110:2233–2240. [DOI] [PubMed] [Google Scholar]
- 12.Rey M, Weber EW, Hess PD. Simultaneous pulmonary and systemic blood pressure and ECG Interval measurement in conscious, freely moving rats. J Am Assoc Lab Anim Sci. 2012;51:231–238. [PMC free article] [PubMed] [Google Scholar]
- 13.Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369:330–340. [DOI] [PubMed] [Google Scholar]
- 14.Medarov BI, Judson MA. The role of calcium channel blockers for the treatment of pulmonary arterial hypertension: how much do we actually know and how could they be positioned today? Respir Med. 2015;109:557–564. [DOI] [PubMed] [Google Scholar]
- 15.Higashi T, Ishizaki T, Shigemori K, et al. Pharmacological characterization of endothelin-induced rat pulmonary arterial dilatation. Br J Pharmacol. 1997;121:782–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tykocki NR, Gariepy CE, Watts SW. Endothelin ET(B) receptors in arteries and veins: multiple actions in the vein. J Pharmacol Exp Ther. 2009;329:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Llorens S, Miranda FJ, Alabadi JA, et al. Different role of endothelin ETA and ETB receptors and endothelial modulators in diabetes-induced hyperreactivity of the rabbit carotid artery to endothelin-1. Eur J Pharmacol. 2004;486:43–51. [DOI] [PubMed] [Google Scholar]
- 18.Hirata Y, Hayakawa H, Suzuki E, et al. Direct measurements of endothelium-derived nitric oxide release by stimulation of endothelin receptors in rat kidney and its alteration in salt-induced hypertension. Circulation. 1995;91:1229–1235. [DOI] [PubMed] [Google Scholar]
- 19.Virdis A, Duranti E, Rossi C, et al. Tumour necrosis factor-alpha participates on the endothelin-1/nitric oxide imbalance in small arteries from obese patients: role of perivascular adipose tissue. Eur Heart J. 2015;36:784–794. [DOI] [PubMed] [Google Scholar]
- 20.Bohm F, Ahlborg G, Johansson BL, et al. Combined endothelin receptor blockade evokes enhanced vasodilatation in patients with atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22:674–679. [DOI] [PubMed] [Google Scholar]
- 21.Cardillo C, Campia U, Bryant MB, et al. Increased activity of endogenous endothelin in patients with type II diabetes mellitus. Circulation. 2002;106:1783–1787. [DOI] [PubMed] [Google Scholar]
- 22.Girgis RE, Champion HC, Diette GB, et al. Decreased exhaled nitric oxide in pulmonary arterial hypertension: response to bosentan therapy. Am J Respir Crit Care Med. 2005;172:352–357. [DOI] [PubMed] [Google Scholar]
- 23.Liang F, Yang S, Yao L, et al. Ambrisentan and tadalafil synergistically relax endothelin-induced contraction of rat pulmonary arteries. Hypertension. 2012;59:705–711. [DOI] [PubMed] [Google Scholar]
- 24.Iglarz M, Bossu A, Wanner D, et al. Comparison of pharmacological activity of macitentan and bosentan in preclinical models of systemic and pulmonary hypertension. Life Sci. 2014;118:333–339. [DOI] [PubMed] [Google Scholar]
- 25.Shemyakin A, Bohm F, Wagner H, et al. Enhanced endothelium-dependent vasodilatation by dual endothelin receptor blockade in individuals with insulin resistance. J Cardiovasc Pharmacol. 2006;47:385–390. [DOI] [PubMed] [Google Scholar]
- 26.Cardillo C, Kilcoyne CM, Waclawiw M, et al. Role of endothelin in the increased vascular tone of patients with essential hypertension. Hypertension. 1999;33:753–758. [DOI] [PubMed] [Google Scholar]
- 27.Bauer M, Wilkens H, Langer F, et al. Selective upregulation of endothelin B receptor gene expression in severe pulmonary hypertension. Circulation. 2002;105:1034–1036. [DOI] [PubMed] [Google Scholar]
- 28.Iglarz M, Binkert C, Morrison K, et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327:736–745. [DOI] [PubMed] [Google Scholar]
- 29.Amiri F, Ko EA, Javeshghani D, et al. Deleterious combined effects of salt-loading and endothelial cell restricted endothelin-1 overexpression on blood pressure and vascular function in mice. J Hypertension. 2010;28:1243–1251. [DOI] [PubMed] [Google Scholar]
- 30.Bruno RM, Sudano I, Ghiadoni L, et al. Interactions between sympathetic nervous system and endogenous endothelin in patients with essential hypertension. Hypertension. 2011;57:79–84. [DOI] [PubMed] [Google Scholar]
- 31.Yang Z, Krasnici N, Luscher TF. Endothelin-1 potentiates human smooth muscle cell growth to PDGF: effects of ETA and ETB receptor blockade. Circulation. 1999;100:5–8. [DOI] [PubMed] [Google Scholar]
- 32.Jasmin JF, Lucas M, Cernacek P, et al. Effectiveness of a nonselective ET(A/B) and a selective ET(A) antagonist in rats with monocrotaline-induced pulmonary hypertension. Circulation. 2001;103:314–318. [DOI] [PubMed] [Google Scholar]
- 33.Mulder P, Richard V, Derumeaux G, et al. Role of endogenous endothelin in chronic heart failure: effect of long-term treatment with an endothelin antagonist on survival, hemodynamics, and cardiac remodeling. Circulation. 1997;96:1976–1982. [DOI] [PubMed] [Google Scholar]
- 34.Hocher B, Kalk P, Slowinski T, et al. ETA receptor blockade induces tubular cell proliferation and cyst growth in rats with polycystic kidney disease. J Am Soc Nephrol. 2003;14:367–376. [DOI] [PubMed] [Google Scholar]
- 35.Stuart D, Chapman M, Rees S, et al. Myocardial, smooth muscle, nephron, and collecting duct gene targeting reveals the organ sites of endothelin A receptor antagonist fluid retention. J Pharmacol Exp Ther. 2013;346:182–189. [DOI] [PubMed] [Google Scholar]
- 36.Vercauteren M, Strasser D, Vezzali E, et al. Vasopressin is involved in endothelin receptor antagonist-induced fluid retention in rat. Differential effect of selective ETA and dual ETA/ETB receptor antagonists. Eur Respir J. 2012;40:716S. [Google Scholar]
- 37.King TE, Jr, Brown KK, Raghu G, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184:92–99. [DOI] [PubMed] [Google Scholar]
- 38.Raghu G, Behr J, Brown KK, et al. ; ARTEMIS-IPF Investigators. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158:641–649. [DOI] [PubMed] [Google Scholar]
- 39.Raghu G, Million-Rousseau R, Morganti A, et al. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomised controlled MUSIC trial. Eur Respir J. 2013;42:1622–1632. [DOI] [PubMed] [Google Scholar]
- 40.Pulido T, Adzerikho I, Channick RN, et al. ; SERAPHIN Investigators. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–818. [DOI] [PubMed] [Google Scholar]
- 41.Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117:3010–3019. [DOI] [PubMed] [Google Scholar]