

Abstract
Central to inflammatory bowel disease (IBD) pathogenesis is loss of mucosal barrier function. Emerging evidence implicates extracellular adenosine signaling in attenuating mucosal inflammation. We hypothesized that adenosine-mediated protection from intestinal barrier dysfunction involves tissue-specific signaling through the A2B adenosine receptor (Adora2b) at the intestinal mucosal surface. To address this hypothesis, we combined pharmacologic studies and studies in mice with global or tissue-specific deletion of the Adora2b receptor. Adora2b−/− mice experienced a significantly heightened severity of colitis, associated with a more acute onset of disease and loss of intestinal epithelial barrier function. Comparison of mice with Adora2b deletion on vascular endothelial cells (Adora2bfl/flVeCadCre+) or intestinal epithelia (Adora2bfl/flVillinCre+) revealed a selective role for epithelial Adora2b signaling in attenuating colonic inflammation. In vitro studies with Adora2b knockdown in intestinal epithelial cultures or pharmacologic studies highlighted Adora2b-driven phosphorylation of vasodilator-stimulated phosphoprotein (VASP) as a specific barrier repair response. Similarly, in vivo studies in genetic mouse models or treatment studies with an Adora2b agonist (BAY 60-6583) recapitulate these findings. Taken together, our results suggest that intestinal epithelial Adora2b signaling provides protection during intestinal inflammation via enhancing mucosal barrier responses.
INTRODUCTION
Inflammatory bowel disease (IBD) including Crohn’s disease (CD) and ulcerative colitis (UC) are relapsing-remitting conditions characterized by uncontrolled intestinal inflammation and tissue destruction.1 A recent study points to the increasing incidence and prevalence of both CD and UC in North America, highlighting the urgent need for effective therapeutic options.2 Genome-wide association studies have established a firm link between dysfunction in host–microbial responses and aberrant mucosal barrier protection with the development of IBD.1 As such, greater understanding of dysregulated mucosal homeostasis in IBD may provide novel therapeutic options.
Release of extracellular adenosine was previously implicated as an endogenous protective response during mucosal inflammation (reviewed in Aherne et al.3). Adenosine acts through a number of cell surface receptors to mediate its signaling responses.3 Studies to date have outlined a role for the A2A (A2A adenosine receptor (Adora2a))4–7 and A2B (Adora2b)8–13 adenosine receptors in acute inflammation. However, the functional consequences of adenosine release and signaling during mucosal inflammation observed in IBD remains to be defined. Previous observations indicate that generation of extracellular adenosine in models of IBD is profoundly protective.14–16 Adenosine signaling through Adora2a protects the intestinal mucosa in a chronic model of IBD;17 however, the role of the Adora2b receptor remains undefined. Recent findings highlight a tissue-protective role for Adora2b signaling during acute intestinal inflammation as occurs in IBD.12,18 These studies suggest that Adora2b expression at the intestinal epithelium is a key protective signaling pathway during acute inflammation. However, in separate studies, intestinal epithelial Adora2b signaling was observed to be deleterious in models of IBD.19–21 Reasons for these divergent findings have yet to elucidated, but may be due in part to genetic background differences in mice studied or in experimental design. We postulated that Adora2b functions in a tissue-specific manner at the mucosal surface to mediate protection in intestinal inflammation. Our aim was to investigate the function of Adora2b signaling at the mucosal surface during acute intestinal inflammation as experienced during IBD. To examine this, we generated novel mouse lines with tissue-specific deletion of the Adora2b receptor. Our studies demonstrated that mice with global knockout of Adora2b (Adora2b−/−) experience rapid, early onset of acute DSS (dextran sulfate sodium) colitis associated with premature loss of mucosal barrier function. Tissue-specific murine studies established a definitive role for intestinal epithelial Adora2b expression in driving mucosal protection during acute colitis. Supported by in vitro findings with Adora2b intestinal epithelial knockdown (KD) cells, we have outlined the Adora2b signaling pathway as a directly protective mechanism in repairing the damaged mucosal barrier through phosphorylation of vasodilator-stimulated phosphoprotein (VASP) in the intestinal epithelium.
Taken together, we observed that tissue-specific expression of the Adora2b receptor on the intestinal epithelium is an endogenous protective response to maintain mucosal barrier function during acute colitis. Pharmacological studies demonstrate that targeting the Adora2b receptor mediates mucosal protection from acute intestinal inflammation. This points to the exciting possibility of exploiting Adora2b signaling in a tissue-specific manner as a therapeutic approach in IBD.
RESULTS
Mice deficient in Adora2b demonstrate early onset and increased severity of DSS colitis
The exact role of the Adora2b receptor in IBD remains to be defined. In previous studies, Adora2b signaling on the intestinal epithelium was implicated as the relevant signaling pathway.12,19–21 However, these studies do not agree on whether Adora2b has a beneficial or deleterious role in models of IBD. We hypothesized that previous divergent findings may be due to distinct roles for Adora2b on different stromal tissue. To investigate this, we assessed mice with whole-body deletion of Adora2b (Adora2b−/−) over the course of DSS colitis. Decreased survival of Adora2b−/− mice compared with wild-type controls (Adora2b+/+) was noted when studies were performed using 4.5% DSS (Supplementary Figure S1 online). Owing to high mortality rates in Adora2b−/− mice at this percentage, we performed subsequent studies with 3–3.5% DSS. Monitoring mice during the course of acute colitis revealed increased weight loss in Adora2b−/− mice compared with wild-type controls (Figure 1a). Adora2b−/− mice exhibited significantly greater colonic shortening (Figure 1b) and loss of epithelial barrier function (Figure 1c), as measured by FITC (fluorescein isothiocyanate)-dextran permeability, compared with controls, from as early as day 3 following DSS. Upon termination of the study, Adora2b−/− mice demonstrated enhanced neutrophil numbers in the colon (Figure 1d) and dramatically increased severity of histological disease (Figure 1e, f). These findings indicate that deficiency of Adora2b results in earlier onset of tissue damage and loss of mucosal barrier function during DSS.
Figure 1.

Adora2b (A2B adenosine receptor)-deficient mice experience early onset of acute colitis. Gender-, age-, and weight-matched Adora2b-deficient mice (Adora2b−/−) or C57BL/6 wild-type controls (Adora2b+/+) were exposed to dextran sulfate sodium (DSS) (3–3.5%) over a time course of 7 days. (a) Daily weight measurements were obtained for each group of mice. (b) Following killing, colons were harvested and measured. Results are representative of one to two independent experiments with n 4–13 mice per group. (c) Mice were administered FITC (fluorescein isothiocyanate)-dextran by oral gavage (0.6 mg g−1 at 80 mg ml−1) 4 h before= killing. Serum was harvested at killing (days 1 and 3 after DSS) and fluoresence measurement was used to determine FITC levels; n = 2–9 mice per group from one independent experiment per time point. (d) Following colon harvest at day 7 after DSS, colonic lamina propria leukocytes were isolated and flow cytometry determined the frequency of GR-1+ (neutrophils), SiglecF+ (eosinophils), F4/80+ (macrophages), and MHCII+CD11cHi (dendritic cells) cells. Actual cell number for each cell type was calculated based on the frequency of cell type multiplied by cell counts following organ harvest. Results are representative of one independent experiment with n = 3–4 mice per group. (e) Representative histological sections from whole colon of Adora2b−/− or Adora2b+/+ mice harvested following 7 days DSS. Bar = 100 μm; images acquired at original magnification ×10. (f) Blinded histological analysis of whole colon from each group following DSS. Data represent 8–10 mice per group. Unless stated otherwise, results are representative of two independent experiments with 3–10 mice per group and are displayed as mean±s.e.m. Two-way analysis of variance (ANOVA) with post hoc Bonferroni t-test was used to determine statistical weight change, but in all other cases Student’s t-test was used. *P<0.5; **P<0.001; ***P<0.0001.
Vascular endothelial expression of Adora2b does not have a role in acute colitis
Drawing from our findings in Adora2b−/− mice (Figure 1), we investigated a tissue-specific role for stromal expression of the Adora2b receptor using newly developed tissue-specific knockout mice. A previous study implicated vascular endothelial Adora2b as a key regulator of acute inflammation.8 Mice with deletion of Adora2b in vascular endothelial cells (Adora2bfl/flVeCadCre+) and their wild-type controls (VeCadCre+) were exposed to DSS. Genotyping PCR of tails (not shown) and whole colonic tissues (Supplementary Figure S2b) demonstrated Cre and Flox expression in desired tissue. Deletion of Adora2b in the vascular endothelium had no effect on acute inflammation experienced during DSS, as evidenced by no differences in weight loss, colon length, and histological damage between Adora2bfl/flVeCadCre+ and VeCadCre+ mice (Figure 2a–d). These findings indicate that Adora2b expression on the vascular endothelium has no significant role in acute intestinal inflammation as observed in IBD.
Figure 2.

Adora2b (A2B adenosine receptor) in the vascular endothelium does not contribute to DSS (dextran sulfate sodium) colitis disease activity. Mice with vascular endothelial specific deletion of Adora2b (Adora2bfl/flVeCadCre+) were generated and exposed to water or DSS (3%) along with their wild-type Cre controls (VeCadCre+). Following 7 days, mice were killed and the whole colon harvested by blunt dissection. (a) Daily weight measurements were taken for each group. (b) Upon harvest colon length was measured. (c) Representative whole colonic histological sections are displayed. Bar = 100 μm; images acquired at original magnification ×10. (d) Blinded histological analysis of whole colon from each group following DSS. Results are representative of 7–8 mice per DSS group and 2 mice per water group, and are displayed as mean±s.e.m.
Specific deletion of intestinal epithelial Adora2b results in increased severity of acute colitis
Having observed no significant impact of vascular endothelial-derived Adora2b on the outcome of DSS colitis, we investigated the function of intestinal epithelial-expressed Adora2b during acute colitis. Previous findings indicate that Adora2b is expressed to a high level on intestinal epithelial cells and its expression is induced during colonic inflammation.22 We generated mice with deletion of Adora2b in intestinal epithelial cells (Adora2bfl/flVillinCre+) and exposed them along with wild-type controls (VillinCre+) to DSS. Genotyping PCR of tails (not shown) and colonic intestinal epithelial cells (described in Flow Cytometry methods: Supplementary Figure S2c) demonstrated Cre and Flox expression in desired tissue. Adora2bfl/flVillinCre+ mice exhibited significantly increased weight loss during the course of DSS compared with VillinCre+ controls (Figure 3a). Mirroring our findings in whole-body knockout studies (Figure 1), Adora2bfl/flVillinCre+ mice experienced significantly greater colonic shortening and enhanced epithelial barrier permeability at early time points following DSS compared with VillinCre+ controls (Figure 3b, c, respectively). Tissue cytokine analysis revealed a trend towards an increase in cytokine levels in the tissue of Adora2bfl/flVillinCre+ mice compared with controls, particularly in interleukin-6 (IL-6) (Figure 3d). Finally, histological analysis of the distal colon (Figure 3e, f) demonstrated that Adora2bfl/flVillinCre+ mice experienced a significantly worse outcome of DSS colitis compared with wild-type controls. Our observations highlight a tissue-specific role for expression of Adora2b on intestinal epithelial cells as a protective response during the acute phase of intestinal inflammation.
Figure 3.

Epithelial-specific deletion of the Adora2b (A2B adenosine receptor) receptor results in significantly increased susceptibility to acute colitis. Mice with intestinal epithelial specific deletion of Adora2b (Adora2bfl/flVillinCre+) were generated and exposed to water or DSS (dextran sulfate sodium, 3–3.5%) along with their wild-type Cre controls (VillinCre+). Mice were killed at 3, 4, and 7 days after DSS, and colons harvested by blunt dissection. (a) Daily weight measurements were assessed for each group of mice. (b) Following harvest at day 3, 4, or 7 colon lengths were measured. Results are representative of one to two independent experiments per time point with 4–18 mice per group. (c) On day 4 after DSS, FITC (fluorescein isothiocyanate)-dextran was administered by oral gavage (0.6 mg g−1 at 80 mg ml−1) 4 h before killing and serum collection. Fluorescence measurement was used to determine FITC levels; n = 3–8 mice per group from one experiment. (d) Following harvest at day 7 after DSS, colon tissue was homogenized and tissue cytokines were measured by Meso Scale (Meso Scale Discovery, Rockville, MD). Results are displayed normalized to protein content and are representative of 4–6 mice per group. (e) Representative histological sections from distal colon of Adora2bfl/flVillinCre+ or VillinCre+ mice harvested following 7 days DSS. Bar = 100 μm; images are acquired at original magnification ×10. (f) Bar graph of blinded histological scoring of the distal colon following 7 days of DSS; n = 7–10 mice per group. Unless stated otherwise, results are representative of two to three independent experiments with 4–10 mice per DSS group and are displayed as mean±s.e.m. Two-way analysis of variance (ANOVA) with post hoc Bonferroni t-test was used to determine statistical weight change, but in all other cases Student’s t-test was used. *P<0.05.
An Adora2b-specific agonist is protective during acute colitis
Our studies reveal that endogenous expression of Adora2b on the intestinal epithelium is a protective mechanism during acute mucosal inflammation. However, the effect of an exogenous Adora2b agonist during acute colitis has not been investigated. We undertook studies using a specific Adora2b agonist (BAY 60-6583). This agonist has previously been demonstrated to have a high degree of specificity for the Adora2b receptor.23,24 We confirmed its ability to induce the second messenger cAMP, a known downstream signaling molecule of the Adora2b receptor in both CHO (Chinese hamster ovarian) cells expressing the human Adora2b receptor and T84 intestinal epithelial cells (Supplementary Figure S4). Continuous administration of the Adora2b agonist provided potent tissue protection as measured by weight loss, colonic shortening, tissue permeability, tissue cytokine levels, and histological damage of the distal colon in acute DSS colitis compared with vehicle-treated controls (Figure 4a–f, respectively). This provides the first evidence that an Adora2b agonist can provide mucosal protection in a model of IBD.
Figure 4.
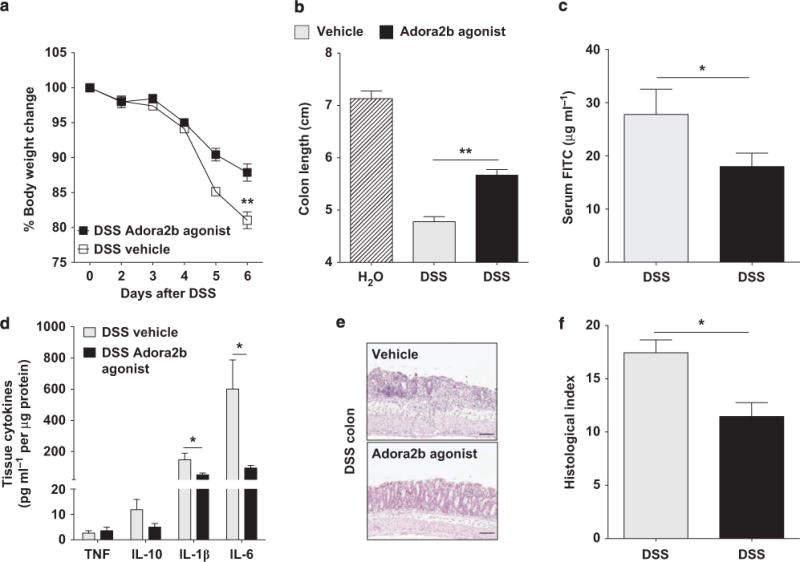
Treatment with an Adora2b (A2B adenosine receptor) agonist mediates mucosal protection observed in a model of acute colitis. Gender-, age-, and weight-matched C57BL/6 mice were treated with an Adora2b-specific agonist (BAY 60-6583; 1.2–1.25 mg kg−1 per day) or vehicle (30% Solutol HS15 in 0.9% saline) using a subcutaneous osmotic pump beginning 1 day before administration of DSS (dextran sulfate sodium, 3.5–4%) for 6 days. Mice exposed to DSS were orally gavaged with FITC (fluorescein isothiocyanate)-dextran (0.6 mg g−1 at 80 mg ml−1) 4 h before killing on day 6, serum collection, and blunt dissection of the colon. (a) Daily weight measurements were obtained for each group of mice. (b) Colon lengths were measured upon harvest. (c) Fluorescence measurement determined FITC levels in the serum on day 6 after DSS. (d) Following harvest, cytokines in colonic tissue were measured by Meso Scale. Results are displayed normalized to protein content and represent 4–5 mice per group. (e) Representative colonic histological images from vehicle and Adora2b agonist-treated mice exposed to DSS. Bar = 100 μm; images were acquired at original magnification ×10. (f) Bar graph of blinded histological scoring of the distal colon from vehicle and Adora2b agonist-treated mice exposed to DSS. Results are displayed as mean±s.e.m. and are representative of one to two independent experiments with 3–7 mice per group. Two-way analysis of variance (ANOVA) with post hoc Bonferroni t-test was used to determine statistical weight change, but in all other cases Student’s t-test was used. *P<0.5; **P<0.001.
Adora2b signaling does not effect epithelial barrier breakdown
To date, we observed premature onset of acute colonic inflammation in mice deficient in the Adora2b receptor during DSS colitis (Figure 1). Tissue-specific deletion identified Adora2b expression on the intestinal epithelium as the key protective signaling pathway in this model (Figure 3). Considering that intestinal barrier permeability was significantly enhanced early in the course of DSS in both Adora2b−/− (Figure 1c) and Adora2bfl/flVillinCre+ mice (Figure 3c), we sought to determine how intestinal epithelial Adora2b might modulate epithelial barrier breakdown.
A number of intrinsic and extrinsic mechanisms have been identified that regulate epithelial barrier breakdown during acute colitis. These include cytokine-induced epithelial barrier dysfunction,25 alterations in the phosphorylation status of myosin light chain (MLC),26 nuclear factor-κB (NF-κB) activity,27 intestinal epithelial cell apoptosis,28 and mucin-2 expression.29 First, we analyzed the effect of Adora2b signaling on cytokine-induced epithelial barrier breakdown (Figure 5a). Barrier studies were performed in T84 intestinal epithelial cells as they express high levels of the Adora2b receptor22 in comparison with other epithelial cell lines that have been used to study Adora2b receptor function (Supplementary Figure S5a).30 T84 intestinal epithelial monolayers were cotreated with the Adora2b agonist or vehicle and a mixture of tumor necrosis factor-α (TNFα), IL-1β, and interferon γ (IFNγ) (“cytomix”). Following 72 h of treatment, a FITC-flux assay was performed to determine the flux rate across the barrier and thereby the permeability of the monolayer. Adora2b agonist treatment did not suppress the cytokine-induced increase in barrier permeability (Figure 5a).
Figure 5.
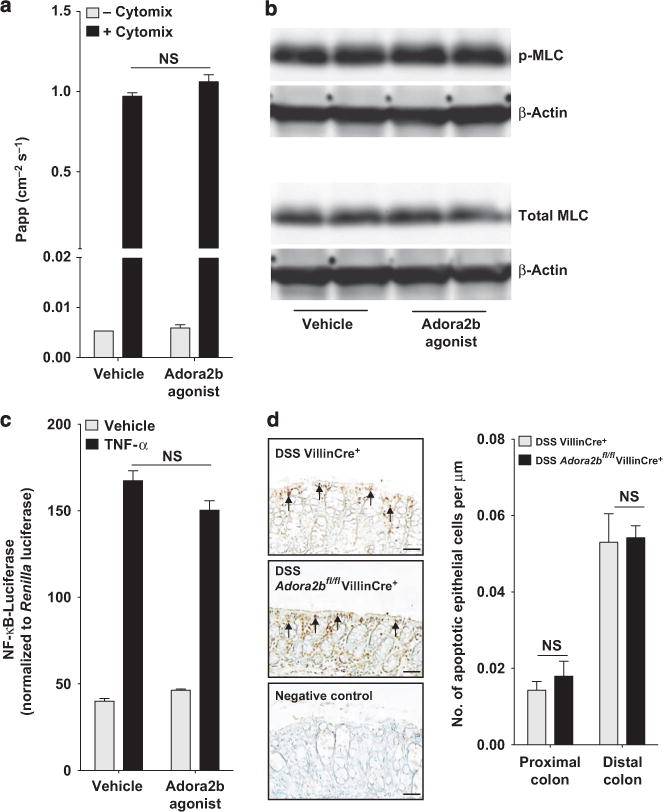
Epithelial Adora2b (A2B adenosine receptor) signaling does not affect epithelial barrier breakdown. (a) A combination of tumor necrosis factor-α (TNFα), interleukin-1β (IL-1β), and interferon γ (IFNγ) (all 10 ng ml−1) or media alone was added to the basolateral aspect of polarized T84 intestinal epithelial cells. Vehicle or the Adora2b agonist (BAY 60-6583; 10 μM) was added to both chambers. Following 72 h, FITC (fluorescein isothiocyanate)-labeled dextran (3 kDa) was added to the apical chamber and a flux assay was performed. The permeability of the monolayer was assessed by measuring the concentration of FITC in the basolateral chamber over time, calculated as the apparent permeability (Papp: cm−2 s−1). Results are representative of three independent experiments with 2–4 wells per group. (b) Confluent T84 intestinal epithelial cells were treated on the basolateral and apical aspect with vehicle or Adora2b-specific agonist (BAY 60-6583; 10 μM) for 30 min before cell harvest and total protein extraction. p-MLC (myosin light chain) (Ser19), total MLC, and β-actin levels were determined by Western blot analysis. Results are representative of three independent experiments with 2–3 wells per experiment. (c) Caco-2 intestinal epithelial cells were transfected with an equal amount of a nuclear factor-κB (NF-κB)-responsive promoter attached to a firefly luciferase reporter (NF-κB-luciferase) and a control reporter vector (Renilla luciferase). Cells were treated in triplicate for 6 h with tumor necrosis factor-α (TNF-α) (10 ng ml−1) and the Adora2b agonist (BAY 60-6583; 10 μM). Equal volumes of cell lysate were assayed for luciferase activity. Data were normalized to Renilla luciferase and are representative of three independent experiments. (d) Mice with intestinal epithelial specific deletion of Adora2b (Adora2bfl/flVillinCre+) were exposed to water or DSS (dextran sulfate sodium, 3%) along with their wild-type Cre controls (VillinCre+) for 4 days and colons harvested by blunt dissection. Representative images are displayed of apoptotic colonic epithelial cells identified by TUNEL (terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling) staining of paraffin-embedded colonic sections. Arrows point to positively stained cells. Bar = 50 μm; images were acquired at original magnification ×20. Terminal deoxynucleotidyl transferase was omitted as a negative control. Bar graph of the number of apoptotic colonic epithelial cells in the proximal or distal colon of each group of mice. The number is displayed relative to the length of basement membrane. Scoring was carried out in a blinded manner in three randomly selected mice per group of 6, with a total of six to eight sections per mouse. All results are displayed as the mean±s.e.m. Statistical analysis was performed using the Student’s t-test.
The phosphorylation status of MLC has been demonstrated to be intimately involved in epithelial tight junction paracellular permeability,31 with a recent study highlighting a key role for MLC phosphorylation in intestinal permeability in IBD.26 Treatment of polarized T84 intestinal epithelial monolayers for 30 min (Figure 5b) or 180 min (not shown) did not alter MLC phosphorylation. Of note, an experiment performed with polarized Caco-2 cells yielded similar findings (not shown).
The master transcription factor NF-κB has a controversial role in regulating intestinal epithelial barrier function in acute colitis.27 Studies suggest that adenosine signaling through the Adora2b receptor inhibits NF-κB transcriptional activity.24,30,32 In line with previous findings,33 we observed no effect of cotreatment with the Adora2b agonist on TNFα- (Figure 5c) or IL-1β- (not shown) induced NF-κB transcriptional activity in intestinal epithelial cells using an NF-κB-driven luciferase reporter.
Increased epithelial cell apoptosis may result in loss of epithelial barrier function during DSS colitis.28,34 TUNEL (terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling) staining revealed no differences in the number of apoptotic epithelial cells in the colon of Adora2bfl/flVillinCre+ mice compared with VillinCre+ controls at the time point when enhanced barrier dysfunction was observed during DSS (Figure 5d).
Finally, expression of the mucin, Muc-2, has been demonstrated to be essential in maintaining intestinal epithelial barrier function.29 Previous findings suggest that adenosine signaling can upregulate Muc-2 in epithelial cells.35 We observed no appreciable difference in Muc-2 expression in Adora2b−/− mice or Adora2bfl/flVillinCre+ mice compared with their controls at the time point when barrier dysfunction occurred (Supplementary Figure S3). Taken together, these findings suggest that the intestinal epithelial Adora2b receptor does not protect the epithelial barrier by suppressing cytokine-induced epithelial barrier permeability, by altering MLC phosphorylation status, by altering NF-κB transcriptional activity, or by affecting either intestinal epithelial cell apoptosis or Muc-2 expression.
Adora2b regulates epithelial barrier resealing
Having observed that Adora2b signaling was not effective in altering mechanisms associated with enhanced epithelial barrier breakdown, we directed our studies to look at alternate mechanisms by which Adora2b receptor signaling could improve epithelial barrier function. To mimic our in vivo model, we genetically deleted Adora2b in T84 intestinal epithelial cells in vitro (Supplementary Figure S5b). We used a calcium switch assay involving chelation of calcium with EDTA, which has been demonstrated to cause junction disassembly and a rapid increase in epithelial barrier permeability that is recoverable by the reintroduction of calcium.36,37 This allows for the assessment of epithelial junction regulation during acute disruption of the barrier such as that observed in DSS. Importantly, this assay allows us to assess the ability of the epithelial barrier to recover. Barrier recovery was monitored by measuring the rate at which a FITC-labeled dextran moved from the apical to the basolateral aspect of epithelial monolayers once calcium was reintroduced (Figure 6). The method by which this rate was calculated is discussed in Supplementary Materials. The rate at which control knockdown (KD) cells or vehicle-treated cells repair is defined as 1 and data are displayed as a fold change relative to this rate to account for inter-experiment variability. Adora2b KD cells demonstrated a significantly slower recovery of barrier function compared with their control KD cells (Figure 6a). A previous study implicated the phosphorylation of the focal adhesion protein VASP at Ser157 in cAMP-induced epithelial barrier repair following acute disruption.37 One of the major intracellular pathways downstream of the Adora2b receptor is the cAMP-protein kinase A (PKA) pathway.3 We have demonstrated that Adora2b signaling rapidly enhances cAMP levels in T84 intestinal epithelial cells (within 30 min: Supplementary Figure S4c). Within 60 min of calcium reintroduction the epithelial barrier rapidly recovers.37 In this instance, a marked increase in VASP phosphorylation at Ser157 was observed by 60 min, which diminished thereafter.37 We monitored protein levels of p-VASP (Ser157) and total VASP in our cells at the indicated time points of recovery (Figure 6b). Of note, the total VASP antibody also recognizes the phosphorylated form of VASP, confirming our results with p-VASP (Ser157) antibody. Western blot confirmed that VASP is phosphorylated at Ser157 during the first 60 min of barrier recovery, as observed previously (Figure 6b).37 Importantly, Adora2b KD cells were unable to phosphorylate VASP (Ser157) to the same level as their control cells at time points analyzed and did not exhibit any alteration in the timecourse of VASP (Ser157) phosphorylation. In contrast, T84 wild-type cells treated from the onset of recovery with an Adora2b-specific agonist (BAY 60-6583; 10 μM) exhibited an almost 40% increase in the rate of barrier recovery compared with vehicle-treated controls (Figure 6c). We noted a very rapid and sustained elevation of p-VASP (Ser157) in agonist-treated cells over that in vehicle-treated controls during barrier recovery (Figure 6d). The position at which VASP is detected by Western blot makes loading quantification hard to determine. Therefore, our Western blot data demonstrate a qualitative trend in alteration of VASP phosphorylation as a consequence of Adora2b signaling. Agonist specificity for Adora2b-induced VASP (Ser157) phosphorylation was confirmed by pretreatment of T84 cells with a specific Adora2b antagonist before agonist cotreatment. Under these conditions, the Adora2b agonist failed to phosphorylate VASP (Ser157) (Supplementary Figure S6). Finally, we confirmed our Western blot findings by immunofluorescence staining (Figure 6e). We observed a substantial portion of p-VASP (Ser157) localizing to E-cadherin-positive T84 epithelial cell junctions within 30 min of Adora2b agonist treatment during barrier recovery compared with vehicle controls (Figure 6e). This is consistent with a more rapid rate of barrier recovery in agonist-treated cells (Figure 6c). These results outline a direct effect of Adora2b signaling on epithelial barrier function, which our results suggest is mediated through the phosphorylation of VASP (Ser157).
Figure 6.
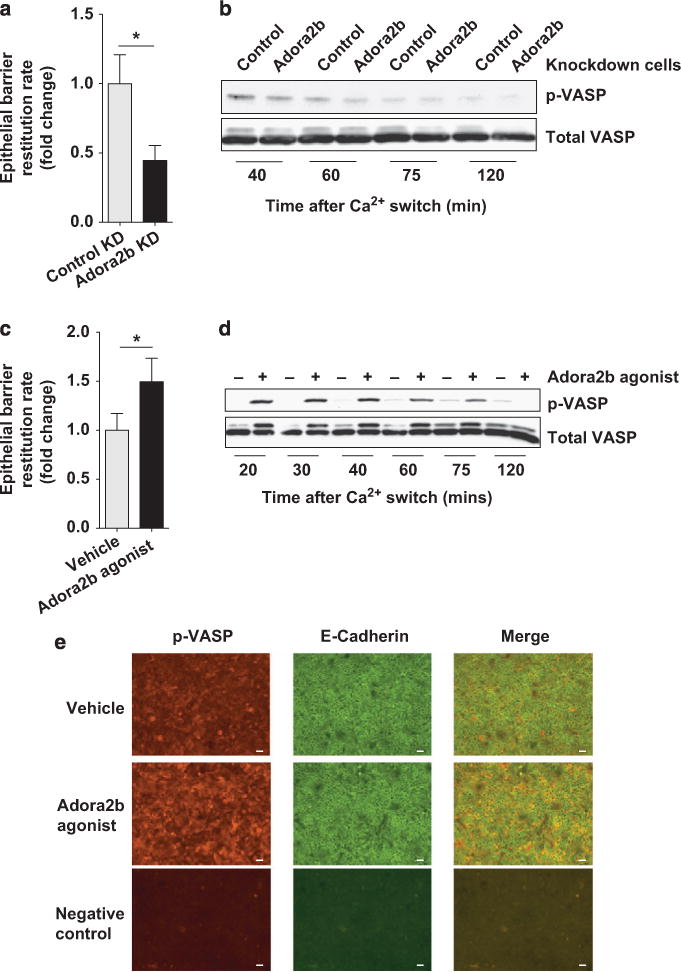
Epithelial Adora2b (A2B adenosine receptor) promotes barrier restitution in intestinal epithelial cells. Calcium switch assays were performed using intestinal epithelial cells with a fully competent barrier. Cells were placed in calcium-depleting conditions until barrier function was fully compromised and subsequently allowed to recover in calcium complete media for the time points outlined. (a) Barrier recovery studies were performed with T84 intestinal epithelial cells with constitutive Adora2b knockdown (Adora2b KD) or nonspecific gene KD (control KD) as mentioned. FITC (fluorescein isothiocyanate)-labeled dextran (10 kDa) was added to the apical aspect of the cells at the start of the recovery period. Media from the basolateral aspect were sampled every 15 min for 105 min and fluorescence in the media at each point was measured to analyze rate of barrier restitution. Results are displayed as fold change in the rate of restitution and represent two independent experiments with 4–6 wells per experiment. (b) Constitutive Adora2b or control KD cells were used in calcium switch assays as described above and allowed to recover for the indicated times. Total protein was extracted and phosphorylated vasodilator-stimulated phosphoprotein (p-VASP) (Ser157) and total VASP levels were determined by Western blot analysis. Results are representative of three independent experiments with 2 wells per time point per experiment. (c) T84 intestinal epithelial cells were used in calcium switch assays. Vehicle or the Adora2b-specific agonist (BAY 60-6583; 10 μM) was added to both the basolateral and apical aspects of the cells at the beginning of the recovery period. Barrier restitution rate was determined as described in a. Results are representative of three independent experiments with 4–6 wells per experiment. (d) T84 intestinal epithelial cells were treated as described in c and cells were harvested at different time points during the recovery period as indicated. Total protein was extracted and p-VASP (Ser157) and total VASP levels were determined by Western blot analysis. Results are representative of three independent experiments with 2 wells per time point per experiment. (e) Calcium switch assays were performed with T84 cells as described in c. Cells were allowed to recover for 30 min in the presence of vehicle or Adora2b agonist before fixation with 2% paraformaldehyde. Cells were costained with a p-VASP (Ser157) and an E-cadherin antibody followed by fluorescent secondary antibody labeling. Primary antibodies were omitted as negative control. Representative images were acquired at original magnification ×20. Bar = 50 μm. Results are representative of three independent experiments with at least 2 wells per group. Results are displayed as mean±s.e.m. Statistical analysis was performed using the Student’s t-test. *P<0.05.
PKA activity mediates Adora2b-driven VASP (Ser157) phosphorylation
PKA has been directly implicated in epithelial barrier recovery and VASP (Ser157) phosphorylation.37 PKA is a known downstream mediator of Adora2b signaling.3 Treating T84 cells with the PKA inhibitor (H89; 30 μM) before administration of the Adora2b-specific agonist (BAY 60-6583; 1 and 10 μM) revealed a concentration-dependent increase in p-VASP (Ser157) that was significantly attenuated by the inhibition of PKA (Figure 7a). Analyzing the Adora2b signaling pathway during barrier recovery revealed a profound effect of PKA inhibition on the ability of the Adora2b agonist to phosphorylate VASP (Ser157) (Figure 7b). We conclude that Adora2b signaling through PKA plays a significant role in VASP (Ser157) phosphorylation during barrier recovery.
Figure 7.
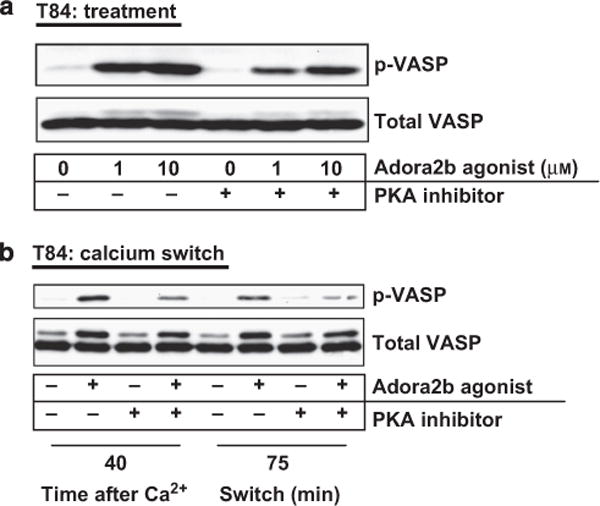
Epithelial vasodilator-stimulated phosphoprotein (VASP) (Ser157) phosphorylation is induced by Adora2b (A2B adenosine receptor) signaling through protein kinase A (PKA). (a) T84 intestinal epithelial cells were pretreated on both the basolateral and apical aspect of the cell monolayer with the PKA inhibitor (H89, 30 μM) for 45 min before 5 min treatment with increasing concentrations of the Adora2b-specific agonist (BAY 60-6583; 1 and 10 μM) in the presence or absence of the PKA inhibitor. Cells were harvested, total protein extracted, and p-VASP (Ser157) and total VASP levels were determined by Western blot analysis. (b) Calcium switch assays were performed using T84 intestinal epithelial cells with a fully competent barrier. Cells were placed in calcium-depleting conditions until barrier function was fully compromised and subsequently allowed to recover in calcium complete media for the time points outlined. Cells were treated on both the basolateral and apical aspect with the Adora2b-specific agonist (BAY 60-6583; 10 μM) in the presence or absence of the PKA inhibitor (H89, 30 μM) from the beginning of the recovery period for the time points indicated. Cells were harvested, total protein extracted, and p-VASP (Ser157) and total VASP levels were determined by Western blot analysis. Results are representative of three independent experiments with 2–3 wells per time point per experiment.
Epithelial VASP (Ser157) phosphorylation is significantly diminished in mice deficient in the epithelial Adora2b receptor during DSS
To assess the relevance of p-VASP (Ser157) in our in vivo model, colonic tissue from Adora2bfl/flVillinCre+ and VillinCre+ mice was stained with p-VASP (Ser157; red) and an epithelial-specific antibody (E-cadherin; green) at 4 and 7 days after DSS (Figure 8a, b). 4′,6-diamidino-2-phenylindole (DAPI) (blue) served as a nuclear counterstain. Of note, the predominant source of p-VASP (Ser157) staining in the colon was in E-cadherin-positive cells, indicating that p-VASP (Ser157) is enriched in the colonic epithelium. In particular, p-VASP (Ser157) expression was largely observed at the E-cadherin-positive epithelial cell junctions during DSS (Figure 8a, b). A profound loss of p-VASP (Ser157) in E-cadherin-positive epithelial cell junctions was noted in Adora2bfl/flVillinCre+ compared with VillinCre+ mice at both 4 and 7 days after DSS (Figure 8a, b, respectively). Differences were quantified by relating the percentage of p-VASP (Ser157) staining in E-cadherin-positive areas of the tissue (Figure 8c). Our data reveal a profound reduction in VASP (Ser157) phosphorylation in epithelial cells in Adora2bfl/flVillinCre+ mice compared with VillinCre+ mice in DSS.
Figure 8.

Loss of epithelial Adora2b (A2B adenosine receptor) expression results in significantly reduced localization of p-VASP (phosphorylated vasodilator-stimulated phosphoprotein) (Ser157) to the intestinal epithelial barrier during acute colitis. Mice with intestinal epithelial-specific deletion of Adora2b− (Adora2bfl/flVillinCre+) were exposed to DSS (dextran sulfate sodium) (3%) along with their wild-type Cre controls (VillinCre+). Following 4 and 7 days DSS, colons were harvested by blunt dissection and formalin fixed. (a and b) Formalin-fixed colons were deparaffinized and rehydrated. Slides were costained with p-VASP (Ser157) and E-cadherin antibodies and appropriate fluorescent secondary antibodies. DAPI (4′,6-diamidino-2-phenylindole) served as a nuclear counterstain. Omission of primary antibodies was used as a negative control. Representative images were acquired at original magnification ×20. Bar = 50 μm. (c) p-VASP (Ser157) costaining of E-cadherin-positive cells was calculated using Olympus software (Shinjuku, Tokyo, Japan). Image analysis was performed in a blinded manner using images acquired at the same settings, with a defined threshold of intensity applied to all images. Results represent the percentage staining of three identically sized randomly selected E-cadherin-positive regions of interest from each image. Day 4 results represent one image per mouse with four mice in each group. Day 7 results represent 4–5 images per mouse with 5–7 mice per group. Data are displayed as the mean±s.e.m. Statistical analysis was performed using the Student’s t-test. *P<0.05 and ***P<0.0001.
An Adora2b agonist induces epithelial VASP (Ser157) phosphorylation during DSS colitis
We demonstrated that continuous administration of an Adora2b-specific agonist protects the intestinal epithelial barrier during acute DSS colitis (Figure 4). In vitro analysis indicates that Adora2b agonist-mediated induction of VASP (Ser157) phosphorylation assists in rapid repair of the intestinal epithelial barrier (Figure 6c, d). We therefore analyzed p-VASP (Ser157) expression in vivo following Adora2b agonist treatment during DSS (Figure 9). Immunofluorescence analysis was performed in colonic tissue following vehicle or Adora2b agonist administration for 6 days during DSS, as discussed above (Figure 4). Adora2b agonist-treated mice exhibited a significantly higher level of p-VASP (Ser157) expression in E-cadherin-positive cells in comparison with vehicle-treated mice (Figure 9a). A large portion of epithelial p-VASP (Ser157) was observed to localize to E-cadherin-positive epithelial cell junctions following Adora2b agonist treatment (Figure 9a). p-VASP (Ser157) levels in the epithelium were quantified by measuring the percentage of staining in standardized sections of E-cadherin-positive tissue (Figure 9b). These findings indicate that an Adora2b agonist can induce epithelial VASP (Ser157) phosphorylation to facilitate a barrier-protective response in DSS colitis.
Figure 9.

Adora2b (A2B adenosine receptor) agonist treatment results in significantly increased localization of p-VASP (phosphorylated vasodilator-stimulated phosphoprotein) (Ser157) to the intestinal epithelial barrier in vivo during DSS (dextran sulfate sodium) colitis. Gender-, age-, and weight-matched C57BL/6 mice were treated with an Adora2b-specific agonist (BAY 60-6583; 1.2–1.25 mg/kg−1 per day) or vehicle for 6 days during DSS as described in Figure 4. Colons were harvested by blunt dissection and formalin fixed. (a) Formalin-fixed colons were deparaffinized and rehydrated. Sections were costained with p-VASP (Ser157) and E-cadherin antibodies as described in Figure 7a and b. Representative images were acquired at original magnification ×20. Bar = 50 μm. (b) p-VASP (Ser157) staining of E-cadherin-positive cells was calculated as described in Figure 7c. Results are representative of 1–3 images per mouse with 5–7 mice per group. Statistical analysis was performed using the Student’s t-test. Data are displayed as the mean±s.e.m. ***P<0.0001.
DISCUSSION
Current studies point to the key role played by dysregulated mucosal homeostasis in the development of IBD.1 As such, better understanding of mucosal responses in the context of IBD is of great relevance to the identification of novel therapeutic options.
The release of adenosine from inflamed tissues and its receptor signaling pathways have been highlighted as an endogenous mechanism to dampen inflammation.3 In the context of IBD, maintaining high tissue levels of adenosine has been demonstrated to protect the intestinal mucosa.14–16 However, the predominant adenosine receptor signaling pathway that mediates protection during IBD remains to be elucidated. There is evidence that adenosine signaling through the Adora2a receptor is protective in a T-cell-mediated murine model of IBD.17,38 However, an Adora2a agonist was ineffective in an acute colitis model, indicating a potential tissue- and context-specific role for adenosine signaling in the inflamed intestine.39 This is further highlighted when considering the potential role of the Adora2b adenosine receptor in IBD. To date, studies have indicated both a protective12,18 and deleterious effect19–21 of Adora2b receptor signaling at the level of the intestinal epithelium in murine models of colitis. Divergent results observed may be because of specific effects of Adora2b signaling at alternative stromal tissue sites. Indeed vascular endothelial expression of the Adora2b receptor has been demonstrated to be an important protective pathway in another model of acute mucosal inflammation.8 Therefore, we were interested in defining the Adora2b signaling events occurring on stromal tissue during acute intestinal inflammation. We postulated that the Adora2b receptor had a tissue-specific role to play during the development of IBD.
To dissect the role of Adora2b on stromal tissue during acute colitis, we examined mice lacking the Adora2b receptor (Adora2b−/−) over a time course of DSS (Figure 1). We observed significant increases in disease activity parameters early in the time course of disease in Adora2b−/− mice compared with wild-type controls (Figure 1). As early as day 3, Adora2b−/− mice exhibited pronounced colonic shortening and loss of epithelial barrier function (Figure 1b, c, respectively). This culminated in considerably greater histological disease (Figure 1e–f) and robust recruitment of neutrophils to the colonic lamina propria of Adora2b−/− mice in comparison with wild-type controls (Figure 1d). Taken together, our findings suggest a protective role for Adora2b signaling during acute intestinal inflammation.
Previous findings in a model of hypoxia-driven inflammation indicate that Adora2b receptors on the vascular endothelium mediate a protective response in acute inflammation.8 Using a novel Adora2b-flox mouse line, we generated mice with tissue-specific deletion of Adora2b on the vascular endothelium. These mice did not exhibit any alteration in disease outcome compared with their genetic controls during DSS colitis (Figure 2), prompting us to consider an alternative stromal source of Adora2b-mediated mucosal protection.
Intestinal epithelial cells express a high level of the Adora2b receptor, which can be upregulated by mediators associated with colitis.22 Using a tissue-specific gene approach, we demonstrated that genetic loss of Adora2b in intestinal epithelial cells led to significantly greater disease activity during DSS (Figure 3). As in whole-body Adora2b deletion (Figure 1), we observed pronounced early onset of disease in mice lacking epithelial Adora2b (Adora2bfl/flVillinCre) culminating in worsened DSS colitis outcome in Adora2bfl/flVillinCre mice in all parameters measured (Figure 3).
Given our current findings, we hypothesized that pharmacologic targeting of the Adora2b receptor may be of therapeutic benefit in acute colitis. Mice administered an Adora2b-specific agonist were profoundly protected from development of DSS colitis (Figure 4). Importantly, Adora2b agonist treatment improved intestinal epithelial barrier function (Figure 4c), suggesting agonist-directed mucosal protection in acute colitis. While previous studies illustrated that an Adora2b antagonist is deleterious in colitis,12 this is the first study demonstrating that Adora2b signaling can be therapeutically targeted for tissue protection in a model of IBD.
Our observations to date suggest that endogenous epithelial Adora2b is a barrier-protective signaling pathway in acute colitis. We therefore concentrated our efforts on elucidating the functional role of the Adora2b receptor on the intestinal epithelium during the acute response in DSS. Generally, DSS colitis is characterized by elevated tissue cytokine levels, enhanced epithelial cell apoptosis, and loss of intestinal barrier function.28,40,41 We pursued studies to interrogate the role of the Adora2b receptor in processes known to be involved in these features of early mucosal barrier dysfunction. Previous study suggests that Adora2b signaling induces the release of the proinflammatory cytokine, IL-6,42 and the anti-inflammatory cytokine, IL-10,12 from the intestinal epithelium. Adora2bfl/flVillinCre mice were observed to have a slightly elevated tissue content of IL-6 but no significant alteration in IL-10 compared with control mice (Figure 3d). In contrast, Adora2b agonist-treated mice exhibited a significant decrease in IL-6 tissue levels and a reduction in IL-10 compared with vehicle controls (Figure 4d). Taking these findings in the context of the previous studies mentioned, our results suggest that direct regulation of cytokine release at the epithelium was not the predominant function of the epithelial Adora2b receptor in our model. We next investigated the potential role of Adora2b receptor signaling in mechanisms that promote epithelial barrier breakdown during DSS colitis. However, we failed to observe an effect of Adora2b signaling on cytokine-induced epithelial barrier permeability, MLC phosphorylation, NF-κB transcriptional activation, epithelial cell apoptosis (Figure 5a–d, respectively), or mucin-2 expression (Supplementary Figure S3). These studies were predominantly performed in vitro and were used to direct additional in vitro and in vivo studies. We cannot abandon the possibility that one of the mechanisms excluded by our in vitro studies may contribute to the phenotype we observe in our animal model.
Having ruled out direct effects of epithelial Adora2b receptor signaling on epithelial cytokine release and processes implicated in epithelial barrier breakdown, we considered a potential role for the Adora2b in mechanisms involved in epithelial barrier repair. To do this, we used an in vitro model with polarized intestinal epithelial cells. First, we confirmed the capability of the Adora2b-specific agonist to induce known receptor second messenger pathways and demonstrated efficient increase in intracellular cAMP following agonist treatment (Supplementary Figure S4). We subsequently generated genetic knockout of Adora2b in intestinal epithelial cells to mirror our in vivo model (Supplementary Figure S5b). To model epithelial tight junction rearrangement and barrier dysfunction observed in IBD, we used a calcium switch assay in vitro.36,37 Importantly, in this model epithelial junction reassembly and repair can be studied. Adora2b KD cells exhibited impaired recovery of barrier function following junction disassembly (Figure 6a), whereas agonist treatment during recovery significantly enhanced the rate of barrier restitution when compared with vehicle treatment (Figure 6c). Considering our observations that Adora2b signaling was capable of enhancing barrier recovery (Figure 6) and also elevated intracellular cAMP (Supplementary Figure S4), we noted that cAMP-mediated phosphorylation of VASP had previously been implicated in recovery of epithelial barrier function.37 In this study, expression of VASP in the intestinal epithelium was demonstrated to be necessary for the recovery of epithelial barrier function, with phosphorylated VASP (p-VASP Ser157) localizing to the junction during barrier recovery. Using these previous findings, we examined our Adora2b-deficient cells and demonstrated an appreciable decrease in the level of p-VASP (Ser157) during barrier recovery compared with control cells (Figure 6b). In addition, Adora2b agonist treatment of wild-type cells enhanced p-VASP (Ser157) expression during barrier recovery over that in vehicle-treated cells (Figure 6d). Importantly, an increased level of p-VASP (Ser157) was observed to colocalize with E-cadherin within 30 min of recovery following agonist treatment compared with vehicle, suggesting an accumulation of this protein at the repairing junction (Figure 6e). Using a PKA antagonist, we demonstrated that induction of PKA was a major pathway by which the Adora2b receptor induced VASP (Ser157) phosphorylation either in intact or repairing epithelial cells (Figure 7).
Having identified the phosphorylation of VASP (Ser157) as a likely mechanism by which the Adora2b receptor repairs the intestinal epithelial barrier in vitro, we wanted to investigate the potential relevance of this in vivo. Adora2bfl/flVillinCre mice demonstrated a significantly diminished expression of p-VASP (Ser157), particularly at epithelial cell junctions during the early onset of DSS (day 4; Figure 8a). This was also the case following the complete time course of DSS (day 7; Figure 8b). Early loss of p-VASP (Ser157) in the epithelial junctions coincided with our observation of enhanced epithelial barrier permeability in Adora2bfl/flVillinCre mice compared with control mice (Figure 3c). In contrast, mice treated with the Adora2b agonist during DSS were observed to have a pronounced increase in p-VASP (Ser157) at epithelial cell junctions compared with vehicle-treated controls (Figure 9). This matched with our finding of significantly reduced epithelial barrier permeability in agonist-treated mice compared with vehicle controls at the same time point (Figure 4c). These observations suggest that Adora2b signaling in the intestinal epithelium in vivo regulates VASP (Ser157) phosphorylation to enhance epithelial barrier function during acute inflammation. Of note, we did not analyze the expression of other junctional proteins in these mice and therefore cannot exclude the possibility that in vivo Adora2b signaling may have additional effects on epithelial junction proteins.
In this study, we used a whole-body and tissue-specific genetic approach to define the contribution of Adora2b to mucosal inflammation. Initial studies in mice with whole-body deletion of Adora2b (Adora2b−/−) demonstrated rapid, early onset of mucosal inflammation in Adora2b−/− mice during experimental colitis. Excitingly, we determined that intestinal epithelial-specific Adora2b signaling is a key pathway in mediating mucosal barrier protection during acute colitis. In vitro and in vivo analysis points to Adora2b phosphorylation of VASP (Ser157) and its localization to epithelial cell junctions as a potential mechanism by which endogenous Adora2b mediates a barrier protective effect. Herein, we present novel studies demonstrating for the first time that Adora2b signaling can be exploited as a therapeutic pathway in a model of IBD. Taken together, we have defined that specific signaling through intestinal–epithelial Adora2b receptors provides potent protection during acute intestinal inflammation, an important finding that may be exploited as a novel therapeutic strategy in IBD.
METHODS
DSS colitis
Adora2b-deficient mice,8 mice with tissue-specific Adora2b deletion (for more details see Supplementary Materials), matched genetic controls, or C57BL/6 mice were used in DSS studies (3–4.5%), as described previously.13,44,45 Adora2b agonist (BAY 60-6583; Tocris Bioscience, Bristol, UK) or vehicle (30% SolutolHS15 in 0.9% saline; BASF, Florham Park, NJ) was administered by subcutaneous osmotic pump (Alzet; DURECT Corporation, Cupertino, CA) at 1.2–1.25 mg kg−1 mouse per day.
Flow cytometric analysis
Flow cytometric analysis of lamina propria leukocytes was performed with antibodies against GR-1 (RB6-8C5), SiglecF (E50-2440), F4/80 (BM8), CD11c (N418), MHCII (M5/114.15.2), and CD45 (30-F11), as outlined previously.13,43
In vivo permeability
Tissue permeability was measured using FITC-labeled dextran as described previously.13
Cell culture and generation of short hairpin RNA KD
Cells were cultured as outlined in Supplementary Materials.30,46–48 Constitutive KD of Adora2b was generated using MISSION shRNA technology (as described in Supplementary Materials).
In vitro FITC-dextran flux assay
FITC-dextran (3 kDa) flux assays were performed in T84 intestinal epithelial cells following cytomix treatment (TNFa, IL-1b, and IFNg; 10ng/ml) in the presence of vehicle or Adora2b agonist (BAY 60-6583, 10 mM) as described13,49 (for more details see Supplementary Materials).
Luciferase reporter assay
Caco-2 intestinal epithelial cells were transfected with equal amounts of NF-κB luciferase and Renilla luciferase plasmids. Following 24 h, cells were treated with vehicle, TNFα (10 ng/ml), the Adora2b agonist alone (BAY 60-6583, 10 μM), or in combination with TNFα. Luminescence was detected as described in Supplementary Materials.
TUNEL staining
Apoptotic epithelial cells were detected by TUNEL staining of formalin–fixed, paraffin-embedded tissue (for details see Supplementary Materials).
Calcium switch assay
Calcium switch assays in T84 intestinal epithelial cells were performed as outlined previously.36 Barrier restitution rate was determined by the addition of FITC-dextran (10 kDa) to the apical aspect at the time of recovery and sampling of FITC in the basolateral compartment (for further details see Supplementary Methods).
Western blot analysis
Equal amounts of whole-cell lysates were separated by gel electrophoresis, transferred, and membranes incubated with either anti-human p-VASP (Ser157) (Cell Signaling, Danvers, MA), anti-human anti-VASP (BD Transduction Laboratories, San Jose, CA), anti-human p-MLC-2 (Ser19) (Cell Signaling), anti-human MLC-2 (Cell signaling), or anti-actin (Calbiochem).
Immunofluorescence
Paraffin-embedded sections or paraformaldehyde fixed cell membranes were incubated with anti-human p-VASP (Ser157) or anti-human E-cadherin followed by fluorescent detection.
Statistical analysis
GraphPad Prism Analysis software (GraphPad Software, La Jolla, CA) was used to perform statistical analysis. Analysis of variance (ANOVA) followed by post-test or Student’s t-test was used where appropriate and results are expressed as mean±s.e.m.
For further details see Supplementary Materials.
Supplementary Material
Acknowledgments
We acknowledge technical assistance from Maria I. Wong, Kristann Magee, Melissa Ledezma, Chelsea Ruller, Caleb Kelly, Aneta Gandjeva, and Dan Koyanagi. Consultation from Simon J. Kelley was greatly appreciated. Human T84 intestinal epithelial cells were a kind gift from Stephen J. Keely, PhD, Royal College of Surgeons Dublin. CHO wild-type cells were a gift from Twila Jackson, PhD, University of Colorado Denver. CHO-Adora2b-overexpresssing cells were a gift from Prof. Bertil Fredholm, Karolinska Institute. The NF-κB-luciferase reporter construct was kindly provided by Prof. Sean Colgan, University of Colorado Denver. The present study was supported by National Institute of Health grants R01 DK097075, R01-HL092188, R01-HL098294, POI-HL114457, and R01-HL119837 and a grant from the Crohn’s and Colitis Foundation (CCFA) to HKE. This work was also supported by a National Institute of Health grant K01 DK099485, the President’s Research Scholar Award from the American Gastroenterological Association (AGA), and grants from the Crohn’s and Colitis Foundation of America (CCFA no.s 2865 and 276536) to CMA.
Footnotes
DISCLOSURE
The authors declare no conflict of interest.
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/mi
References
- 1.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molodecky NA, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54 e42. doi: 10.1053/j.gastro.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Aherne CM, Kewley EM, Eltzschig HK. The resurgence of A2B adenosine receptor signaling. Biochim Biophys Acta. 2011;1808:1329–1339. doi: 10.1016/j.bbamem.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lukashev D, Ohta A, Apasov S, Chen JF, Sitkovsky M. Cutting edge: physiologic attenuation of proinflammatory transcription by the Gs protein-coupled A2A adenosine receptor in vivo. J Immunol. 2004;173:21–24. doi: 10.4049/jimmunol.173.1.21. [DOI] [PubMed] [Google Scholar]
- 5.Impellizzeri D, et al. CGS 21680, an agonist of the adenosine (A2A) receptor, decreases acute lung inflammation. Eur J Pharmacol. 2011;668:305–316. doi: 10.1016/j.ejphar.2011.06.049. [DOI] [PubMed] [Google Scholar]
- 6.Chouker A, et al. Critical role of hypoxia and A2A adenosine receptors in liver tissue-protecting physiological anti-inflammatory pathway. Mol Med. 2008;14:116–123. doi: 10.2119/2007-00075.Chouker. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 8.Eckle T, et al. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111:2024–2035. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Csoka B, et al. A2B adenosine receptors protect against sepsis-induced mortality by dampening excessive inflammation. J Immunol. 2010;185:542–550. doi: 10.4049/jimmunol.0901295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koeppen M, et al. Adora2b signaling on bone marrow derived cells dampens myocardial ischemia-reperfusion injury. Anesthesiology. 2012;116:1245–1257. doi: 10.1097/ALN.0b013e318255793c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schingnitz U, et al. Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol. 2010;184:5271–5279. doi: 10.4049/jimmunol.0903035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frick JS, et al. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J Immunol. 2009;182:4957–4964. doi: 10.4049/jimmunol.0801324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aherne CM, et al. Neuronal guidance molecule netrin-1 attenuates inflammatory cell trafficking during acute experimental colitis. Gut. 2012;61:695–705. doi: 10.1136/gutjnl-2011-300012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Louis NA, et al. Control of IFN-alphaA by CD73: implications for mucosal inflammation. J Immunol. 2008;180:4246–4255. doi: 10.4049/jimmunol.180.6.4246. [DOI] [PubMed] [Google Scholar]
- 15.Friedman DJ, et al. From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci USA. 2009;106:16788–16793. doi: 10.1073/pnas.0902869106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antonioli L, et al. Inhibition of adenosine deaminase attenuates inflammation in experimental colitis. J Pharmacol Exp Ther. 2007;322:435–442. doi: 10.1124/jpet.107.122762. [DOI] [PubMed] [Google Scholar]
- 17.Naganuma M, et al. Cutting edge: critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol. 2006;177:2765–2769. doi: 10.4049/jimmunol.177.5.2765. [DOI] [PubMed] [Google Scholar]
- 18.Alam M, Wilson J, Ernst P. Role of adenosine A2B receptors in regulating inflammatory responses of TNBS-induced experimental colitis. J Immunol. 2010;184(87):2. [Google Scholar]
- 19.Kolachala VL, et al. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology. 2008;135:861–870. doi: 10.1053/j.gastro.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolachala V, et al. Blockade of adenosine A2B receptors ameliorates murine colitis. Br J Pharmacol. 2008;155:127–137. doi: 10.1038/bjp.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingersoll SA, et al. A((2)B)AR expression in non-immune cells plays an important role in the development of murine colitis. Dig Liver Dis. 2012;44:819–826. doi: 10.1016/j.dld.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolachala V, et al. TNF-alpha upregulates adenosine 2b (A2b) receptor expression and signaling in intestinal epithelial cells: a basis for A2bR overexpression in colitis. Cell Mol Life Sci. 2005;62:2647–2657. doi: 10.1007/s00018-005-5328-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckle T, et al. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- 24.Baraldi PG, Tabrizi MA, Fruttarolo F, Romagnoli R, Preti D. Recent improvements in the development of A(2B) adenosine receptor agonists. Purinergic Signal. 2008;4:287–303. doi: 10.1007/s11302-008-9097-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. 2010;177:512–524. doi: 10.2353/ajpath.2010.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su L, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. 2013;145:407–415. doi: 10.1053/j.gastro.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 28.Araki Y, Mukaisyo K, Sugihara H, Fujiyama Y, Hattori T. Increased apoptosis and decreased proliferation of colonic epithelium in dextran sulfate sodium-induced colitis in mice. Oncol Rep. 2010;24:869–874. doi: 10.3892/or.2010.869. [DOI] [PubMed] [Google Scholar]
- 29.Van der Sluis M, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131:117–129. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 30.Khoury J, Ibla JC, Neish AS, Colgan SP. Antiinflammatory adaptation to hypoxia through adenosine-mediated cullin-1 deneddylation. J Clin Invest. 2007;117:703–711. doi: 10.1172/JCI30049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turner JR, et al. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273:C1378–C1385. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- 32.Majumdar S, Aggarwal BB. Adenosine suppresses activation of nuclear factor-kappaB selectively induced by tumor necrosis factor in different cell types. Oncogene. 2003;22:1206–1218. doi: 10.1038/sj.onc.1206184. [DOI] [PubMed] [Google Scholar]
- 33.Bergmann S, et al. Adenosine and homocysteine together enhance TNF-mediated cytotoxicity but do not alter activation of nuclear factor-kappa B in L929 cells. J Immunol. 1994;153:1736–1743. [PubMed] [Google Scholar]
- 34.Tambuwala MM, et al. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology. 2010;139:2093–2101. doi: 10.1053/j.gastro.2010.06.068. [DOI] [PubMed] [Google Scholar]
- 35.McNamara N, et al. Adenosine up-regulation of the mucin gene, MUC2, in asthma. FASEB J. 2004;18:1770–1772. doi: 10.1096/fj.04-1964fje. [DOI] [PubMed] [Google Scholar]
- 36.Parkos CA, et al. Intestinal epithelia (T84) possess basolateral ligands for CD11b/CD18-mediated neutrophil adherence. Am J Physiol. 1995;268:C472–C479. doi: 10.1152/ajpcell.1995.268.2.C472. [DOI] [PubMed] [Google Scholar]
- 37.Lawrence DW, Comerford KM, Colgan SP. Role of VASP in reestablishment of epithelial tight junction assembly after Ca2+ switch. Am J Physiol Cell Physiol. 2002;282:C1235–C1245. doi: 10.1152/ajpcell.00288.2001. [DOI] [PubMed] [Google Scholar]
- 38.Odashima M, et al. Activation of A2A adenosine receptor attenuates intestinal inflammation in animal models of inflammatory bowel disease. Gastroenterology. 2005;129:26–33. doi: 10.1053/j.gastro.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 39.Selmeczy Z, Csoka B, Pacher P, Vizi ES, Hasko G. The adenosine A2A receptor agonist CGS 21680 fails to ameliorate the course of dextran sulphate-induced colitis in mice. Inflamm Res. 2007;56:204–209. doi: 10.1007/s00011-006-6150-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan Y, et al. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS One. 2009;4:e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alex P, et al. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sitaraman SV, et al. Neutrophil-epithelial crosstalk at the intestinal lumenal surface mediated by reciprocal secretion of adenosine and IL-6. J Clin Invest. 2001;107:861–869. doi: 10.1172/JCI11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collins CB, et al. Inhibition of N-terminal ATPase on HSP90 attenuates colitis through enhanced Treg function. Mucosal Immunol. 2013;6:960–971. doi: 10.1038/mi.2012.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siegmund B, et al. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1264–R1273. doi: 10.1152/ajpregu.2001.281.4.R1264. [DOI] [PubMed] [Google Scholar]
- 45.Burns RC, et al. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn’s disease in mice. Gastroenterology. 2001;121:1428–1436. doi: 10.1053/gast.2001.29568. [DOI] [PubMed] [Google Scholar]
- 46.Schulte G, Fredholm BB. Human adenosine A(1), A(2A), A(2B), and A(3) receptors expressed in Chinese hamster ovary cells all mediate the phosphorylation of extracellular-regulated kinase 1/2. Mol Pharmacol. 2000;58:477–482. [PubMed] [Google Scholar]
- 47.Haeberle HA, et al. Oxygen-independent stabilization of hypoxia inducible factor (HIF)-1 during RSV infection. PLoS One. 2008;3:e3352. doi: 10.1371/journal.pone.0003352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gessi S, et al. Adenosine receptors in colon carcinoma tissues and colon tumoral cell lines: focus on the A(3) adenosine subtype. J Cell Physiol. 2007;211:826–836. doi: 10.1002/jcp.20994. [DOI] [PubMed] [Google Scholar]
- 49.Keely S, et al. Dexamethasone-pDMAEMA polymeric conjugates reduce inflammatory biomarkers in human intestinal epithelial monolayers. J Control Rel. 2009;135:35–43. doi: 10.1016/j.jconrel.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.