

Abstract
Invasive M1T1 group A Streptococcus (GAS) can have a mutation in the regulatory system CovRS, and this mutation can render strains hypervirulent. Interestingly, via mechanisms that are not well understood, the host innate immune system's neutrophils select spontaneous M1T1 GAS CovRS hypervirulent mutants, thereby enhancing the pathogen's ability to evade immune killing. It has been reported that the DNase Sda1 is critical for the resistance of M1T1 strain 5448 to killing in human blood and provides pressure for in vivo selection of CovRS mutations. We reexamined the role of Sda1 in the selection of CovRS mutations and in GAS innate immune evasion. Deletion of sda1 or all DNase genes in M1T1 strain MGAS2221 did not alter emergence of CovRS mutants during murine infection. Deletion of sda1 in strain 5448 resulted in Δsda1 mutants with (5448 Δsda1M+ strain) and without (5448 Δsda1M− strain) M protein production. The 5448 Δsda1M+ strain accumulated CovRS mutations in vivo and resisted killing in the bloodstream, whereas the 5448 Δsda1M− strain lost in vivo selection of CovRS mutations and was sensitive to killing. The deletion of emm and a spontaneous Mga mutation in MGAS2221 reduced and prevented in vivo selection for CovRS mutants, respectively. Thus, in contrast to previous reports, Sda1 is not critical for in vivo selection of invasive M1T1 CovRS mutants and GAS resistance to innate immune killing mechanisms. In contrast, M protein and other Mga-regulated proteins contribute to the in vivo selection of M1T1 GAS CovRS mutants. These findings advance the understanding of the progression of invasive M1T1 GAS infections.
INTRODUCTION
The human pathogen group A Streptococcus (GAS) causes about 700 million cases of relatively mild, noninvasive pharyngitis and superficial skin infections annually. However, severe GAS infections, including severe invasive infections and acute rheumatic fever/rheumatic heart disease, can occur, causing approximately 517,000 deaths in the world each year (1). A globally disseminated M1T1 clone of serotype M1 GAS most frequently is associated with severe invasive infections in the United States (2–4). It is believed that the original M1T1 GAS clone evolved from the acquisitions of DNase Sda1- and superantigen SpeA-encoding prophages and an interserotype exchange of a 36-kb chromosomal region containing the toxin genes encoding NAD+-glycohydrolase (NADase) and streptolysin O (SLO) with serotype M12 GAS (5–7). More recently, hypervirulent M1T1 GAS strains have been isolated, usually having mutations in the two-component regulatory system CovRS (also known as CsrRS) (8–12).
CovRS is known to negatively regulate multiple virulence genes in GAS, including the capsule synthetase gene hasA, the interleukin-8 (IL-8)/CXC chemokine peptidase gene spyCEP, and the platelet-activating factor acetylhydrolase gene sse (13–18). Natural CovS mutations of invasive M1T1 isolates usually enhance the expression of these virulence genes and downregulate the production of the protease SpeB (SpeBA−, for the lack of the SpeB activity in culture supernatant), enhancing innate immune evasion, soft-tissue invasion, and systemic dissemination and resulting in hypervirulence. The lack of SpeB activity in culture supernatant is used to identify CovRS mutants of invasive M1T1 GAS (12).
While CovRS mutations of some M1T1 GAS strains can be rapidly selected in experimental soft-tissue infections of mice (8, 9, 12, 19–21), not all GAS strains are subject to the in vivo selection for CovRS mutations (20). It has been reported that the DNase Sda1 is required for the in vivo selection of SpeBA− variants of the M1T1 strain 5448, leading to a proposal that Sda1 helps GAS avoid killing by neutrophil extracellular traps in the absence of SpeB production and thereby provides pressure for selection of CovRS mutations with the SpeBA− phenotype (20). In addition, the hasA and M protein (emm) genes are required for selection of CovRS mutations of strain 5448 (21). The emm gene is positively regulated by Mga (for multiple gene regulator of GAS), which also directly activates several other virulence genes, including the C5a peptidase gene scpA (22).
Sda1 is encoded by a prophage which is present in invasive M1T1 GAS but is absent from the sequenced M1T1 strain SF370 (23). An sda1 deletion mutant of invasive M1T1 strain 5448 has been shown to have lost the resistance of 5448 to killing in human blood (24), and this same mutant also loses the capacity to switch to the SpeBA− phenotype in mouse infections (20). In the latter study, it was proposed that the acquisition of Sda1 by the invasive M1T1 clone provides pressure for the selection of SpeBA− variants by helping GAS escape killing in neutrophil extracellular traps. However, introduction of the sda1-encoding prophage into SF370 does not lead to in vivo selection of SpeBA− variants of the modified SF370 strain (25). Furthermore, an M98 GAS strain has a natural covS mutation even though the strain does not have the sda1 gene (26).
These findings raise several questions on the critical role of Sda1 in the selection of CovRS mutations and GAS resistance to killing by innate immune mechanisms. Is Sda1 required for resistance to killing and selection for CovRS mutations of invasive M1T1 other than strain 5448? If not, does Sda1 really provide selection pressure for SpeBA− variants of strain 5448 and confer its resistance to killing by innate immune cells? Can a secondary mutation acquired during construction of a deletion mutant or simple in vitro passage result in the loss of in vivo selection of CovRS mutations of M1T1 GAS? This study was designed to address these questions and, for the first time, accurately evaluate the role of Sda1 in the M1T1 GAS hyperinvasive phenotype. Our results indicate that Sda1 is not required for in vivo selection of CovRS mutations of invasive M1T1 GAS and GAS resistance to killing in human blood and that a secondary mutation that downregulates the production of the M protein posttranscriptionally or the transcription of the Mga regulon can lead to the loss of in vivo selection for M1T1 GAS CovRS mutants.
MATERIALS AND METHODS
Declaration of ethical approval.
All animal experimental procedures were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (27). The protocols for the experiments were approved by the Institutional Animal Care and Use Committee at MSU (permit numbers 2011-57 and 204-45). Blood was collected from healthy donors in accordance with a protocol approved by the Institutional Review Board at MSU (protocol no. BL120513). Written informed consent was provided by study participants and/or their legal guardians.
Bacterial strains and growth.
GAS strains MGAS2221 and 5448 are characterized isolates of invasive M1T1 GAS, which is associated with severe invasive infections (17, 20). A derivative of MGAS2221 with the loss of M protein production (the MGAS2221M− mutant) arising during laboratory passage has been described (28). These strains and their gene deletion mutants listed in Table 1 were statically grown at 37°C in 5% CO2 in Todd-Hewitt broth supplemented with 0.2% yeast extract (THY). Tryptose agar with 5% sheep blood and THY agar were used as solid media. pDCBB-covS- or pDCBB-covR-complemented SpeBA− variants of MGAS2221 were grown in THY broth or agar plates containing 10 μg/ml chloramphenicol. GAS bacteria used in animal infections were harvested at the exponential growth phase and washed three times with and resuspended in pyrogen-free Dulbecco's phosphate-buffered saline (DPBS) to the desired doses.
TABLE 1.
Group A Streptococcus strains and plasmids used in this study
| Strain or plasmid | Description | Reference |
|---|---|---|
| MGAS2221 | M1T1 GAS strain | 17 |
| MGAS2221 Δsda1 | sda1 deletion mutant of MGAS2221 | This study |
| MGAS2221 Δsda1 Δspd3 Δspd | Triple sda1 spd3 spd deletion mutant of MGAS2221 | This study |
| MGAS2221 Δemm | emm deletion mutant of MGAS2221 | This study |
| MGAS2221 ΔhasA | hasA deletion mutant of MGAS2221 | This study |
| MGAS2221M− | Laboratory-passaged derivative strain of MGAS2221 with diminished M protein production | 28 |
| MGAS2221M−/pDCBB-mga | MGAS2221M− carrying pDCBB-mga | This study |
| 5448 | M1T1 GAS strain | 20 |
| 5448 Δsda1M+ | sda1 deletion mutant of 5448 with normal M protein production | This study |
| 5448 Δsda1M− | sda1 deletion mutant of 5448 with diminished M protein production | This study |
| 5448 Δsda1M−/pDCBB-mga | 5448 Δsda1M− carrying pDCBB-mga | This study |
| p740-Δsda1 | Suicide plasmid for sda1 deletion | This study |
| p740-Δspd3 | Suicide plasmid for spd3 deletion | This study |
| p740-Δspd | Suicide plasmid for spd deletion | This study |
| p740-Δemm | Suicide plasmid for emm deletion | This study |
| p740-ΔhasA | Suicide plasmid for hasA deletion | This study |
| pDCBB-covS | covS of MGAS2221 cloned into pDCBB | 30 |
| pDCBB-covR | covR of MGAS2221 cloned into pDCBB | This study |
| pDCBB-mga | mga of MGAS2221 cloned into pDCBB | This study |
Construction of plasmids for gene deletions.
Suicide plasmid p740-Δsda1 (Table 1) for sda1 deletion was constructed as follows. The 5′ and 3′ flanking fragments, ∼1,000-bp long each, of an sda1 fragment (bases 421 to 849) to be deleted were amplified from MGAS2221 DNA using paired primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTAAGAGGAATGCAAGGATATG-3′ (primer 1)/5′-ATGTCTTGACATCAGTAGCAAGAAATAAGAATACCACCCACT-3′ (primer 2) and 5′-TTGCTACTGATGTCAAGACATATTGATTACCAAAACGGTGGA-3′ (primer 3)/5′-GGGGACCACTTTGTACAAGAAAGCTGGGTGGCATTGATTTATGGGGTAAT-3′ (primer 4), respectively. The two PCR fragments were fused together in subsequent overlay PCR via the 21-bp complementary sequences that are underlined in primers 2 and 3 using primers 1 and 4. The fused PCR product first was cloned into the donor vector pDONR221 in the BP Clonase reaction and then into pGRV-RFA (29) in the LR Clonase reaction using the Gateway cloning kit. pGRV-RFA was renamed p740-RFA. Other suicide plasmids, p740-Δspd3, p740-Δspd, p740-Δemm, and p740-ΔhasA, were constructed similarly. Paired PCR primers for flanking fragments of an spd3 fragment (bases 208 to 681) to be deleted were 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTACCTTTTGCTTTCAAAAATCC-3′/5′-ATGTCTTGACATCAGTAGCAAAAGATCGCCCAAAACTAATTG-3′ and 5′-TTGCTACTGATGTCAAGACATACATTAGATAACGTATCTCCT-3′/5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCTGACAAAGACGGTAACATTA-3′. Paired PCR primers for flanking fragments of an spd fragment (bases 187 to 648) to be deleted were 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTATGGCGCTAACAATATCATCT-3′/5′-ATGTCTTGACATCAGTAGCAAGAATGTCCAAGCTAATGCTTC-3′ and 5′-TTGCTACTGATGTCAAGACATCTTTATTATGAAGCTGCTCCA-3′/5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCATTATTTTGAATGCCGAAAC-3′. Paired PCR primers for flanking fragments of an emm fragment (bases 38 to 635) to be deleted were 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCTGATGCGTTCAGAGAAAGAAC-3′/5′-CAGATGAAAGTTGATCAAGTTCATAAGCGAATAGTGTCTATTCGTG-3′ and 5′-CACGAATAGACACTATTCGCTTATGAACTTGATCAACTTTCATCTG-3′/5′-GGGGACCACTTTGTACAAGAAAGCTGGGTGAGTTGTTTAGTTTGTGACCTCTC-3′. Paired PCR primers for flanking fragments of an hasA fragment (bases 326 to 767) to be deleted were 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTGATGTGAGAGTCACATGGCTAA-3′/5′-CTATGGTAGTCTCATCTAGGTATCTTCCAAAGAGTTGAAGTCTCTG-3′ and 5′-CAGAGACTTCAACTCTTTGGAAGATACCTAGATGAGACTACCATAG-3′/5′-GGGGACCACTTTGTACAAGAAAGCTGGGTGCAAGCTGTCTGCAAAGCTGATG-3′. The underlined sequences in the sets of the primers for each gene deletion were used for overlay PCRs to join the two flanking fragments together for each plasmid construct.
Generation of gene deletion mutant strains of MGAS2221.
For deletion of the sda1, emm, and hasA genes in MGAS2221, suicide plasmids for deletion of each gene were introduced into MGAS2221 via electroporation. Chloramphenicol-resistant merodiploid transconjugants were selected on THY agar with 10 μg/ml chloramphenicol and confirmed by PCR. One transconjugant was grown on THY agar without chloramphenicol selection (one passage), and the passage was repeated seven or more times. GAS colonies from the last passage were spotted in parallel on THY agar plates with and without chloramphenicol to identify chloramphenicol-sensitive colonies derived from a second homologous crossover. Chloramphenicol-sensitive strains then were analyzed by PCR to identify deletion mutants that had a shorter PCR product for the target gene than that of MGAS2221. Deletion mutants of MGAS2221 for these genes were confirmed by DNA sequencing. To generate a triple deletion mutant of MGAS2221 for the three DNase genes (MGAS2221 Δsda1 Δspd3 Δspd mutant), spd3 and spd in the MGAS2221 Δsda1 mutant were sequentially deleted using the suicide plasmids p740-Δspd3 and p740-Δspd in similar procedures.
Generation of sda1 deletion mutants of GAS 5448 with loss of M protein production (5448 Δsda1M− mutant).
5448 Δsda1 variants obtained as described above had normal M protein production and are referred to as 5448 Δsda1M+ mutants. For the generation of the 5448 Δsda1 mutant that had the loss of M protein production (5448 Δsda1M− strain), the second crossover was done by passaging a transconjugant of 5448 and p740-Δsda1 in THY broth, instead of THY agar plates, 10 times.
Complementation of 5448 Δsda1M− mutant.
In an attempt to construct pDCBB-sda1 for in trans complementation, sda1 was PCR amplified from MGAS2221 using primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTAGGAGGACATAAATATGTCTAAAC-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTGTGTGTTTCGTATG-3′. The PCR product was cloned into pDONR221 of the Gateway cloning kit using BP Clonase, yielding pDONR221-sda1. However, transfer of sda1 from pDONR221 to pDCBB-RFA (29) was not successful. Thus, the deleted sda1 gene in the 5440 Δsda1M− mutant was repaired by putting back the wild-type sda1 gene as follows. The sda1 gene and its flanking fragments were amplified by PCR using primers 1 and 4 that were used in the construction of p740-Δsda1. The PCR product first was cloned into pDONR221 and then transferred into p740-RFA using the Gateway cloning kit as described earlier. The resulting construct, p740-sda1, was introduced into the 5448 Δsda1M− mutant to obtain the 5448 Δsda1M−::p740-sda1 strain in the first crossover, and then the 5448 Δsda1M−-sda1 strain was obtained in the second crossover using the procedures that were used to generate the 5448 Δsda1M+ mutant.
In trans complementation of CovRS mutants.
Plasmids pDCBB-covS and pDCBB-covR were used for in trans complementation of MGAS2221 covRS mutants selected in vivo. pDCBB-covS has been described (30), and pDCBB-covR was constructed as follows. The wild-type covR gene from MGAS2221 was PCR amplified using Phusion DNA polymerase (New England BioLabs, MA) and paired primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTAGAGGATAAGGGTTGGTATAA-3′/5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTCCATATGACTTATTTCTCACGA-3′. The PCR products were cloned into pDONR221 in a BP Clonase reaction and then transferred into pDCBB-RFA in an LR Clonase reaction. pDCBB (vector control) (17), pDCBB-covS, or pDCBB-covR was introduced into test GAS strains via electroporation, and transformed GAS bacteria were selected with chloramphenicol.
Assay for DNase activity.
DNase activity in the culture supernatant of test strains was compared using calf thymus DNA (catalog no. D3664; Sigma) as the substrate. GAS was grown to an optical density at 600 nm (OD600) of 0.32, and cultures were centrifuged. One microgram of calf thymus DNA in 9.5 μl buffer (20 mM Tris-HCl, pH 7.5, 10 mM MgCl2, and 0.5 mM CaCl2) was mixed with 0.5 μl culture supernatant or THY control and incubated at 37°C for 5 min, and 2 μl 0.5 M EDTA was added to stop the reaction. Reaction samples were analyzed by agarose gel electrophoresis.
In vivo selection of SpeBA− variants.
C57BL/6J male mice were bred at the Animal Resource Center at Montana State University. Groups of 10 5-week-old male C57BL/6J mice were subcutaneously inoculated with 0.2 ml of GAS suspension at an OD600 of 0.9. Skin infection sites were collected at day 4 after inoculation, homogenized in DPBS by using a Kontes pestle, and plated on THY agar plates at appropriate dilutions. Forty-eight colonies from each mouse were randomly picked, inoculated in 200 μl THY in 96-well plates, and cultured overnight. Three microliters of 10% β-mercaptoethanol was added to each well, and the cultures in the plates were centrifuged at 3,500 rpm. The SpeB activity in 5 μl of the supernatant of the GAS cultures was detected by using a casein plate assay as described previously (12).
To identify covRS mutations in some SpeBA− variants, a DNA fragment containing the covRS genes was amplified from test isolates using the primers 5′-TCGCTAGAAGACTATTTGAC-3′ and 5′-TTCATGTCATCCATCATTGC-3′ and the Phusion high-fidelity PCR kit. DNA sequencing of the amplified PCR products was performed using the BigDye Terminator v3.1 cycle sequencing kit and an Applied Biosystems 3130 genetic analyzer. Primers used for the sequencing were 5′-TCGCTAGAAGACTATTTGAC-3′, 5′-TTCATGTCATCCATCATTGC-3′, 5′-AACGGCTTCATCATATTTCC-3′, 5′-AAATCCACAAAACCGTTCAG-3′, 5′-TGATACACACGACCGATAG-3′, 5′-TTGATGACAGAAAGGGCAG-3′, 5′-TACGCGAACCATGTCTAAC-3′, and 5′-GTTGGGGTAAAGATGACAG-3′.
Bacterial growth in human blood.
A human blood growth assay was performed as previously described (31). GAS grown on a THY agar plate overnight was washed with DPBS twice and resuspended in DPBS to an OD600 of 0.02. Twenty microliters of GAS suspension (∼105 CFU) was inoculated into 0.5 ml of heparinized nonimmune human blood in triplicate. The samples were gently rotated for 4 h at 37°C. The numbers of viable GAS in the samples and inocula were determined by plating. The growth factor was defined as the ratio of CFU for each sample after 4 h of incubation to the CFU of the inoculum.
Other assays.
The production of M protein by GAS was assessed by Western blotting as described previously (28). Quantitative real-time reverse transcription-PCR (RT-PCR) analyses were performed using the All-in-One first-strand cDNA synthesis kit and the All-in-One SYBR quantitative PCR (qPCR) mix from GeneCopoeia (Rockville, MD), as described previously (30). Levels of mRNA for each target gene were normalized first to mRNA levels of the gyrA gene using the ΔΔCT method and then to those in MGAS2221. The platelet-activating factor (PAF) acetylhydrolase activity of Sse in culture supernatant of GAS was measured by a colorimetric assay using 2-thio-PAF as previously described (32). Statistical analysis of the percentages of SpeBA− variants among GAS recovered from mice (SpeBA−%) was performed using the two-tailed unpaired t test with Welch's correction of the Prism software program (GraphPad Software Inc.).
RESULTS
DNases are not required for in vivo selection of SpeBA− variants of MGAS2221.
To address whether Sda1 provides selection pressure for CovRS mutants of invasive M1T1 isolates other than strain 5448, we examined the role of Sda1 in the selection of SpeBA− variants of MGAS2221 in the subcutaneous infection of mice. A DNA fragment encoding amino acid residues 141 to 283 of Sda1 in MGAS2221 was deleted. An sda1 deletion mutant (MGAS2221 Δsda1 strain) was confirmed by diagnostic PCR (Fig. 1A) and DNA sequencing (data not shown). The MGAS2221 Δsda1 mutant had reduced DNase activity in its culture supernatant (Fig. 1B, lane 2). To rule out the possibility that the partial sda1 deletion did not completely abolish the Sda1 activity, the two other DNase genes in the MGAS2221 Δsda1 mutant, spd3 and spd (33), were deleted, resulting in an MGAS2221 Δsda1 Δspd3 Δspd triple deletion mutant. No DNase activity was detected in the culture supernatant of this triple deletion mutant (Fig. 1B, lane 3), indicating that sda1 was completely inactivated by the deletion of the internal sda1 fragment and that the remaining DNase activity of the MGAS2221 Δsda1 mutant was due to the production of Spd3 and/or Spd by the Δsda1 isolate.
FIG 1.

Deletion of sda1 gene in MGAS2221. (A) Picture of agarose gel electrophoresis for PCR analysis that confirms the deletion of a 429-bp fragment of the sda1 gene in MGAS2221. (B) Picture of agarose gel electrophoresis for comparing the DNase activity in the culture supernatant of various GAS strains. The DNase activity was assayed using calf thymus DNA as the substrate. Lanes: none, THY control; 1, MGAS2221; 2, MGAS2221 Δsda1 mutant; 3, MGAS2221 Δsda1 Δspd3 Δspd mutant; 4, an SpeBA− variant recovered from mice with MGAS221 Δsda1 mutant infection; 5, an SpeBA+ isolate from mice with MGAS2221 Δsda1 mutant infection; and 6, GAS strain SF370. (C) Western blot detecting the M protein produced by MGAS2221 (wt) and the MGAS2221 Δsda1 and MGAS2221 Δsda1 Δspd3 Δspd mutants.
The emm gene encoding the M protein is required for the selection of 5448 SpeBA− variants (21), and gene deletion mutants of MGAS2221 may have a secondary spontaneous mutation that abolishes M protein production (28), which in turn abolishes the selection of SpeBA− variants. Thus, it was crucial to make sure that derivative strains of MGAS2221 had normal M protein production in tests for selection of SpeBA− variants. As determined by Western blotting, MGAS2221 Δsda1, MGAS2221 Δsda1 Δspd3 Δspd, and MGAS2221 strains had similar levels of M protein (Fig. 1C), indicating that the two mutants did not have a secondary mutation that could downregulate the expression of the M protein.
To determine whether SpeBA− variants of the MGAS2221 Δsda1 mutant can be selected in vivo, groups of C57BL/6J mice were subcutaneously inoculated with MGAS2221 or the MGAS2221 Δsda1 mutant. On day 4 after inoculation, mice were sacrificed and viable GAS bacteria were recovered from skin lesions. SpeBA− variants among 48 randomly chosen colonies from each mouse were identified by checking the SpeB protease activity in supernatant of overnight cultures using the casein hydrolysis assay (12). On average, 38.8% (experiment 1) and 42.7% (experiment 2) (combined as 41.4%) of MGAS2221 Δsda1 mutant colonies recovered from mice in two independent experiments were SpeBA− (Fig. 2A and B). The sda1 deletion in MGAS2221 did not compromise the selection of MGAS2221 SpeBA− variants in skin infections. Actually, the average SpeBA−% of the MGAS2221 Δsda1 mutant recovered from mice was slightly higher than that of MGAS2221 recovered from mice (30.2%), although this difference was not significant (P = 0.1415). Thus, Sda1 does not serve as a selection force for SpeBA− variants of MGAS2221 during infection in mice.
FIG 2.
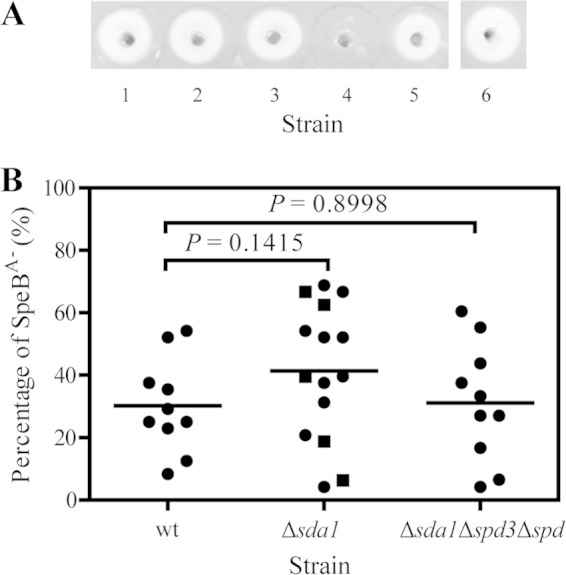
No detrimental effect of deletions of the DNase genes on in vivo selection of SpeBA− variants of MGAS2221 in subcutaneous mouse infection. (A) A casein gel picture showing the SpeB protease activity in the culture supernatant of test strains and representative SpeBA− and SpeBA+ isolates from mice with MGAS2221 Δsda1 mutant infection. Assay spots: 1, MGAS2221; 2, MGAS2221 Δsda1 mutant; 3, MGAS2221 Δsda1 Δspd3 Δspd mutant; 4, a representative SpeBA− isolate from a mouse with MGAS2221 Δsda1 mutant infection; 5, a representative SpeBA+ isolate from mouse with MGAS2221 Δsda1 mutant infection; and 6, strain SF370. (B) SpeBA−% of GAS isolates recovered from the skin infection sites of MGAS2221 (wt), MGAS2221 Δsda1 (Δsda1), and MGAS2221 Δsda1 Δspd3 Δspd (Δsda1Δspd3Δspd) strains in C57BL/6J mice. The MGAS2221 Δsda1 mutant data include those from an independent test, indicated by the square symbols, and the other data were from a parallel test.
The MGAS2221 Δsda1 mutant still had detectable DNase activity in its culture supernatant (Fig. 1B, lane 2). It is possible that Spd and Spd3 in the MGAS2221 Δsda1 mutant provide pressure for the selection of its SpeBA− variants. To examine this possibility, we determined whether the MGAS2221 Δsda1 Δspd3 Δspd mutant can be subject to selection for SpeBA− variants in vivo. On average, 31.2% of MGAS2221 Δsda1 Δspd3 Δspd isolates recovered from mice on day 4 after inoculation were SpeBA−, which was not significantly different from the level for MGAS2221 (P = 0.8998) (Fig. 2B). Thus, Spd and Sdp3 do not serve as a selection force for the emergence of MGAS2221 SpeBA− variants. These results indicate that the DNases are not involved in selection for SpeBA− variants of MGAS2221.
Sda1 does not provide pressure for in vivo selection of SpeBA− variants of M1T1 GAS strain 5448.
Our MGAS2221 data are in contrast with the previous finding on the critical role of Sda1 in selection of 5448 SpeBA− variants. This discrepancy raises the question as to whether Sda1 is really a critical factor for the emergence of SpeBA− variants of strain 5448 during infection. To address this question, we deleted the sda1 gene in 5448 using the procedure of passaging on THY agar plates for the second crossover as described by Zhou et al. (28). As expected, a 5448 Δsda1 mutant had lost the majority of DNase activity in culture supernatant compared to that of the parent strain (Fig. 3A). This 5448 Δsda1 mutant also had levels of M protein similar to those of the parent strain (Fig. 3B, lanes Δsda1M+ and wt); therefore, it was referred to as the 5448 Δsda1M+ strain, for the 5448 Δsda1 mutant positive in M protein production. We then determined whether the 5448 Δsda1M+ mutant can switch to SpeBA− variants in mice. On average, 13.0% of 5448 Δsda1M+ colonies recovered from mice on day 4 after inoculation were SpeBA− (Fig. 3C). The average SpeBA−% of recovered 5448 Δsda1M+ colonies was slightly higher than, but not significantly different from, those of SpeBA− in 5448 controls (average, 11.5%; P = 0.7263). Apparently, Sda1 was not required for selection of 5448 SpeBA− variants in our tests.
FIG 3.

5448 Δsda1M+ strain, but not 5448 Δsda1M− strain, did not lose the capacity of strain 5448 to switch to the SpeBA− phenotype during mouse infection. (A) Picture of agarose gel electrophoresis for comparing the DNase activity in the culture supernatant of various GAS strains. Lanes: none, THY control; wt, strain 5448; Δsda1M+, 5448 Δsda1 mutant with normal M protein production; Δsda1M−, 5448 Δsda1 mutant negative in the production of the M protein; Δsda1M−-sda1, complement strain of Δsda1M− mutant. (B) Western blot showing that one 5448 Δsda1 strain (Δsda1M+) had levels of the M protein similar to those of the parent strain 5448, whereas another mutant (Δsda1M−) and its complement strain (Δsda1M−-sda1) were negative in the production of the M protein. (C) SpeBA−% of GAS isolates recovered from the skin infection sites of 5448 (wt), 5448 Δsda1M+, 5448 Δsda1M−, and 5448 Δsda1M−-sda1 strains in C57BL/6J mice.
Diminished in vivo selection of SpeBA− variants of 5448 Δsda1M− mutant.
MGAS2221 can acquire secondary mutations that downregulate M protein production during the construction of gene deletion mutants or simple passage in THY (28). Therefore, we investigated whether M protein production-negative mutants could be obtained during construction of 5448 Δsda1 mutants and lose selection pressure for 5448 SpeBA− variants. Instead of passaging a transconjugant on THY agar for the second crossover for construction of the 5448 Δsda1M+ mutant, the transconjugant was passed in THY broth for the second crossover to generate additional 5448 Δsda1 mutants for this investigation. Six 5448 Δsda1 mutants were obtained using this THY broth procedure, and four of them had undetectable M1 protein production in Western blotting, indicating that these four 5448 Δsda1 mutants had a secondary mutation that caused the loss of M protein production (Fig. 3B, lane Δsda1M−). One of these M protein production-negative mutants, referred to as the 5448 Δsda1M− mutant, was tested for selection of SpeBA− variants in mice. All 5448 Δsda1M− colonies recovered from six of 10 mice were SpeBA+, and just one of 48 colonies from each of three other mice was SpeBA−. The average SpeBA−% of the recovered 5448 Δsda1M− mutant, 0.8%, was significantly lower than that of recovered 5448 (11.5%) and 5448 Δsda1M+ (13.0%) strains (5448 versus 5448 Δsda1M− mutant, P = 0.0008; 5448 Δsda1M+ mutant versus 5448 Δsda1M− mutant, P = 0.022) (Fig. 3C).
To make sure that the loss of selection for SpeBA− variants of the 5448 Δsda1M− mutant was due not to the sda1 deletion but to a secondary mutation, we performed a complementation experiment. We first attempted the in trans expression of sda1 in the 5448 Δsda1M− mutant, but we could not clone the sda1 gene into pDCBB, even though it could be cloned into pDONR221 (see Materials and Methods for details). Thus, we repaired the sda1 deletion in the 5448 Δsda1M− mutant by putting back the wild-type sda1 gene. A resulting strain, the 5448 Δsda1M−-sda1 mutant, restored the normal levels of DNase activity in culture supernatant (Fig. 3A, lane Δsda1M−-sda1) but still lacked M protein production (Fig. 3B, lane Δsda1M−-sda1). This repaired strain still lost in vivo selection for SpeBA− variants (Fig. 3C). Therefore, a secondary mutation indeed can result in the loss of selection for GAS SpeBA− variants.
Deletion of sda1 does not alter the resistance of strains MGAS2221 and 5448 to killing in human blood.
It has been shown that Sda1 is critical for resistance of strain 5448 to innate immune cell killing in human blood (24). Since Sda1 is not critical for selection of SpeBA− variants of GAS 5448, it is important to reexamine the role of Sda1 in the survival of strain 5448 in human blood. We compared the growth and survival of 5448, 5448 Δsda1M+, and 5448 Δsda1M− strains in nonimmune human blood. After incubating in nonimmune human blood for 4 h, 5448 and 5448 Δsda1M+ strains could survive and grow, displaying growth factors of 6.2 and 5.8, respectively. In contrast, the surviving 5448 Δsda1M− mutant was just 8% of the inoculum (Fig. 4), which was expected, since the expression of the M protein was downregulated in this strain. The MGAS2221 Δsda1 mutant also was resistant to killing in human blood, whereas the MGAS2221M− spontaneous mutant of MGAS2221 with reduced M protein production was sensitive to immune killing (Fig. 4). These data indicate that Sda1 does not critically contribute to the resistance of invasive M1T1 GAS to killing by innate immune cells in human blood, whereas the strains without normal M protein production are sensitive to killing.
FIG 4.
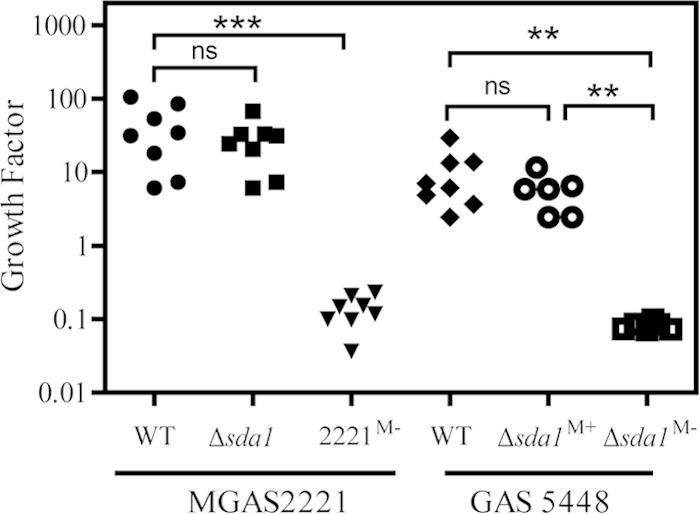
MGAS2221 Δsda1 and 5448 Δsda1M+ strains, but not 5448 Δsda1M− and MGAS2221M− strains, resist killing in blood. Shown are growth factors of the test strains in nonimmune human blood after 4 h of incubation. The growth factor was defined as the ratio of CFU for each sample after 4 h of incubation to the CFU of inoculum. Significance was determined by one-way analysis of variance (ANOVA) with Newman-Keuls tests: ns, not significant; ***, P < 0.0001; **, P < 0.001.
Downregulation of the Mga regulon prevents emergence of SpeBA− variants.
To better understand the nature of the secondary mutation for the downregulation of M protein production in the 5448 Δsda1M− mutant, pDCBB-mga, which has the mga gene encoding the activator of the Mga regulon, including the M protein-encoding emm gene (34), was introduced into the 5448 Δsda1M− mutant, and M protein production and emm transcripts were measured. In trans expression of mga did not restore M protein production in the 5448 Δsda1M− strain (Fig. 5A). Consistent with this result, both 5448 Δsda1M− and 5448 Δsda1M−/pDCBB-mga strains had normal levels of emm and scpA transcripts (Fig. 5B). These results indicate that an unknown secondary mutation in the 5448 Δsda1M− strain caused the loss of M protein production posttranscriptionally.
FIG 5.

Respective transcriptional and posttranscriptional downregulation of emm expression in MGAS221M− and 5448 Δsda1M− strains and the contribution of the M protein, Mga regulon, and capsule to in vivo selection of MGAS2221 SpeBA− variants. (A and B) Western blot detecting M protein production (A) and relative levels of hasA, scpA, and emm transcripts (B) in MGAS2221, MGAS2221 Δemm, MGAS2221M−, MGAS2221M−/pDCBB-mga, 5448 Δsda1M−, and 5448 Δsda1M−/pDCBB-mga strains. (C) SpeBA−% of GAS isolates recovered from the skin infection sites of MGAS2221, MGAS2221 Δemm, MGAS2221M−, MGAS2221M−/pDCBB-mga, and MGAS2221 ΔhasA strains in C57BL/6J mice.
To examine the role of the M protein in selection of SpeBA− variants of MGAS2221 during infection, an MGAS2221 Δemm mutant and an MGAS2221M− variant were tested for selection of SpeBA− variants during infection in mice. The SpeBA−% of isolates of the MGAS2221 Δemm mutant recovered from mice at day 4 after inoculation was 9.6% ± 8.9%, which was significantly lower than that (43.1% ± 23.6%) of MGAS2221 (P = 0.0021) (Fig. 5C). Thus, the M protein significantly contributes to in vivo selection for SpeBA− variants of MGAS2221. This result is similar to but not the same as the previous finding that the emm gene is required for the selection of 5448 SpeBA− variants (21). In contrast, the MGAS2221M− strain almost completely lost in vivo selection for SpeBA− variants (P = 0.0023 for the MGAS2221 Δemm mutant versus MGAS2221M−) (Fig. 5C), indicating that additional factors other than the diminished M protein production in MGAS2221M− further reduced in vivo selection for SpeBA− variants of MGAS2221M−. MGAS2221M− was a derivative of MGAS2221 with reduced M protein production that was obtained during in vitro passage (28). In contrast with the 5448 Δsda1M− strain, MGAS2221M− had 328- and 33-fold downregulation in emm and scpA transcription, respectively, compared with MGAS2221 (Fig. 5B), and there was a mutation in the mga gene, which will be reported elsewhere. In trans expression of mga in MGAS2221M− restored emm and scpA transcription (Fig. 5B), M protein production (Fig. 5A), and, more importantly, selection for SpeBA− variants (P values for SpeBA− selection were the following: MGAS2221M− versus MGAS2221M−/pDCBB-mga, 0.0008; MGAS2221M−/pDCBB-mga versus MGAS2221, 0.9698) (Fig. 5C). These data indicate that the downregulation of the M protein and another Mga-regulated protein(s) in MGAS2221 prevents the emergence of SpeBA− variants.
The hasA gene is required for in vivo selection for MGAS2221 SpeBA− variants.
Since the hyaluronic acid capsule has been shown to be essential for the selection of 5448 SpeBA− variants (21), we also evaluated the contribution of the hasA gene to in vivo selection for MGAS2221 SpeBA− variants using an hasA deletion mutant of MGAS2221. No SpeBA− variants of the MGAS2221 ΔhasA strain were detected (Fig. 5C), indicating that the capsule also is essential for the in vivo selection of MGAS2221 SpeBA− variants.
Novel CovR mutations that confer the SpeBA−/SseA+ phenotype.
The SpeBA− phenotype has been shown to be a valid marker for CovRS mutations of invasive M1T1 GAS (12). However, mutations in ropB, the regulator of the speB gene, also can lead to the SpeBA− phenotype and commonly occur in clinical GAS isolates (35–38). SpeBA−-causing mutations of CovRS, but not RopB, also enhance the expression of several virulence factors of M1T1 GAS, including the platelet-activating factor acetylhydrolase Sse (12–17). To make sure that SpeBA− variants of the MGAS2221 Δsda1 mutant indeed were caused by CovRS mutations, we first sequenced the covRS genes of 10 randomly chosen SpeBA− variants of the MGAS2221 Δsda1 mutant from mice. Three SpeBA− isolates from a mouse have either a base insertion or deletion in the covS gene, resulting in truncated CovS mutants, and three other SpeBA− isolates from the same mouse had a missense mutation in the covR gene that caused the CovRG61S point mutation (Table 2). The CovRG61S mutation also was found in three SpeBA− isolates from three other mice. In addition, there was one adenine base insertion at position 505 of the covR gene in one SpeBA− variant, resulting in a truncated CovR mutant. Thus, all analyzed SpeBA− variants of the MGAS2221 Δsda1 strain had covS or covR mutation.
TABLE 2.
CovRS mutations and in vitro Sse expression of SpeBA− variants of MGAS2221 Δsda1 mutant isolated from subcutaneous mouse infection
| Isolatea | covRS mutation | Mutated CovS or CovRc | Production of: |
Mouse no. | Complementation to SpeBA+/SseA− | |
|---|---|---|---|---|---|---|
| SpeB | Sseb | |||||
| 1 | 381TT382 deletion in covS | Truncated CovS | − | + | 1 | Yes |
| 2 | 929A insertion in covS | Truncated CovS | − | + | 1 | Yes |
| 3 | 1048A insertion in covS | Truncated CovS | − | + | 1 | Yes |
| 4 | 181G to A in covR | CovRG61S | − | + | 1 | Yes |
| 5 | 181G to A in covR | CovRG61S | − | + | 1 | Yes |
| 6 | 181G to A in covR | CovRG61S | − | + | 1 | Yes |
| 7 | 181G to A in covR | CovRG61S | − | + | 2 | Yes |
| 8 | 505A insertion in covR | Truncated CovR | − | + | 3 | Yes |
| 9 | 181G to A in covR | CovRG61S | − | + | 4 | Yes |
| 10 | 181G to A in covR | CovRG61S | − | + | 5 | Yes |
| 11 | Wild type | Wild type | + | − | ||
Isolates 1 through 10 all were SpeBA− variants detected among GAS isolates from mice infected with the MGAS2221 Δsda1 mutant. Isolate 11 was the MGAS2221 Δsda1 mutant.
The Sse production status was assigned according to data like those depicted in Fig. 5B.
CovRG61S has been detected in a clinical isolate in reference 44.
We next checked the expression of virulence genes of a randomly chosen SpeBA− variant, which had a covS mutation causing CovS truncation. As expected, this MGAS2221 Δsda1 SpeBA− variant had higher levels of hasA, spyCEP, and sse transcripts than the MGAS2221 Δsda1 strain (Fig. 6A), and the platelet-activating factor acetylhydrolase activity of Sse was detected (SseA+, for Sse activity-positive phenotype) in its culture supernatant but not in those of the MGAS2221 Δsda1 strain and an MGAS2221 SpeBA+ isolate from mice (Fig. 6B). The SpeBA− variants with the CovRG61S mutation and CovR truncation also had the SpeBA−/SseA+ phenotype (Fig. 6C and D). Third, we performed complementation. These CovS and CovR mutants restored the wild-type SpeBA+/SseA− phenotype when they were complemented in trans by the wild-type covS and covR genes, respectively, indicating that the SpeBA−/SseA+ phenotype of these MGAS2221 Δsda1 variants was caused by the detected CovS and CovR mutations (Fig. 5D and 6C). It is known that covR deletion increases SpeB production (14, 17). It was unexpected that the variant with the CovR truncation also had the SpeBA− phenotype.
FIG 6.

SpeBA−/SseA+ phenotype of SpeBA− variants of MGAS2221 Δsda1 mutant was caused by CovR or CovS mutations. (A) Relative expression levels of hasA, spyCEP, and sse transcripts as measured by real-time RT-PCR in the MGAS2221 Δsda1 mutant and a randomly chosen SpeBA− or SpeBA+ colony recovered from a mouse with MGAS2221 Δsda1 mutant infection. (B) Enhanced PAF acetylhydrolase activity of Sse in the culture supernatant of the SpeBA− isolate shown in panel A compared to that of MGAS2221, MGAS2221 Δsda1, and SpeBA+ isolates from mice infected with the MGAS2221 Δsda1 strain. (C) Restoration in SpeB production of SpeBA− variants with distinct CovS (spots 1 to 3) and CovR (spots 4 and 5) mutations listed in Table 2 by in trans complementation with pDCBB-covS and pDCBB-covR, respectively. pDCBB was used for a vector control. (D) Enhanced levels of the Sse PAF acetylhydrolase activity in culture supernatants of SpeBA− variants with CovS and CovR mutations were reduced to undetectable levels as in that of MGAS2221 by in trans complementation with pDCBB-covS and pDCBB-covR, respectively, but not by control vector pDCBB.
DISCUSSION
In this report, we present findings from an examination of the role of DNases and the Mga regulon in in vivo selection of CovRS mutations of invasive M1T1 GAS and GAS resistance to killing by innate immune cells. First, Sda1 and the two other DNases are not required for in vivo selection of CovRS mutations of strain MGAS2221. Second, the deletion of Sda1 in GAS strain 5448 does not affect the in vivo selection of CovRS mutations. Third, Sda1 does not contribute to the resistance of M1T1 strains MGAS2221 and 5448 to killing in human blood. Fourth, loss of M protein and other Mga-controlled proteins due to secondary mutations prevents the emergence of CovRS mutants by reducing the fitness of the population for survival in the infected host and leads to the loss of GAS resistance to killing in human blood. The first three findings are contrary to the previous reports that Sda1 is required for in vivo selection of CovRS mutants of invasive M1T1 GAS (20) and the resistance of GAS to blood killing (24). In addition to the M protein, the other proteins regulated by Mga also contribute to the emergence of CovRS mutants. Our results also highlight the critical importance of ensuring the normal expression of the Mga regulon in targeted gene deletion mutants to avoid false conclusions on functions of targeted genes. In addition, we detected two CovR mutations that lead to the SpeBA−/SseA+ phenotype. These findings contribute to the understanding of the progression of invasive M1T1 GAS infections.
The main finding of this study is the demonstration of the insignificant role of Sda1 in the selection of SpeBA− variants or CovRS mutations of invasive M1T1 GAS. Kazmi et al. first reported the in vivo SpeBA+-to-SpeBA− phase shift of M1T1 strain 5448 (39), and Aziz et al. subsequently analyzed the SpeB phase-shift phenotype (19). Walker et al. later reported that Sda1 provides the pressure for selection for 5448 SpeBA− variants (20). Our results establish that Sda1 is not required for the selection of SpeBA− variants of invasive M1T1 GAS. This finding is consistent with the recent report that introduction of the Sda1-encoding prophage into GAS strain SF370 does not lead to in vivo selection of SpeBA− variants of modified SF370 (25). Engleberg et al. first reported the spontaneous mutations of CovRS in experimental GAS infection of mice with invasive M1T1 GAS (8). Characterized SpeBA− variants of GAS strain 5448 all have CovRS mutations (9–12, 20, 21). The SpeBA− phenotype is a valid marker for in vivo CovRS mutations of invasive M1T1 GAS (12). All analyzed SpeBA− variants of the MGAS2221 Δsda1 strain carry CovS or CovR mutations. Thus, in contrast to the previous report (20), Sda1 is not required for in vivo selection of CovRS mutations of invasive M1T1 GAS.
Targeted deletion of sda1 of M1T1 GAS could generate Δsda1 mutants that are negative in M protein production due to secondary mutations. Deletion of the sagA gene and simple in vitro passage of MGAS2221 also yield ΔsagA mutants and MGAS2221 derivatives that are negative in M protein production due to undesired secondary mutations (28). These findings indicate that in vitro manipulation of GAS frequently results in spontaneous mutations that reduce M protein production. The results in this study indicate that such mutations either affect transcription of the genes of the Mga regulon or affect M protein production posttranscriptionally. Failing to rule out a spontaneous mutation that affects the expression of emm and other genes at transcriptional or posttranscriptional levels in targeted gene deletion mutants may lead to false conclusions on the function of target genes. The incorrect assignment of the role of the sagA gene in the regulation of the M protein gene (40) appears to be such an example (28).
The contradictory results on the role of Sda1 in the selection for SpeBA− variants of M1T1 GAS and GAS resistance to killing in blood between this study and the studies of Walker et al. (20) and Buchanan et al. (24) might be due to the status of the production of the M protein and other Mga-regulated proteins. It is likely that the 5448 Δsda1 mutant in the studies of Walker et al. (20) and Buchanan et al. (24) were negative in M protein production as a result of a secondary mutation. First, this could explain why the mutant lost the capacity to switch to the SpeBA− phenotype, since the M protein is required for the selection of 5448 SpeBA− variants (21). Our results indicate that in vitro manipulation of strain 5448 indeed can lead to frequent spontaneous mutations that cause the downregulation of the M protein and that our M protein production-negative, but not M protein production-positive, 5448 Δsda1 mutant and MGAS2221M− mutants lose the capacity to switch to the SpeBA− phenotype. Further, the possibility that the 5448 Δsda1 mutant of Walker et al. was negative in M protein production was consistent with the finding that the same 5448 Δsda1 mutant lost the resistance of 5448 to killing in blood (24). We could not repeat this detrimental effect of the sda1 deletion on the resistance of 5448 to killing in blood using the 5448 Δsda1M+ mutant but showed that the 5448 Δsda1M− and MGAS2221M− strains were sensitive to killing in nonimmune human blood. The M protein confers resistance to phagocytic killing by neutrophils. The downregulation of M protein expression is expected to compromise GAS resistance to neutrophil killing. In addition, the M protein production-negative phenotype and loss of in vivo selection for SpeBA− variants of our 5448 Δsda1M− mutant cannot be complemented. However, the loss of selection for SpeBA− variants of the 5448 Δsda1 mutant of Walker et al. can be complemented by sda1 (20). We cannot think of a reason for this discrepancy.
The lost capacity of the 5448 Δsda1 mutant to switch to the SpeBA− phenotype and its loss in resistance to killing in human blood in the studies of Walker et al. (20) and Buchanan et al. (24) have been interpreted as the consequence of the loss of the function of Sda1 to help GAS escape killing in neutrophil extracellular traps (20, 24). Since we have shown that sda1 deletion is not directly linked to these phenotypes, this interpretation most likely is incorrect, and evading the neutrophil extracellular trap-mediated GAS killing by DNases should not be as critical as concluded in these previous studies. The phenotype of the 5448 Δsda1 mutant in the study of Walker et al. is similar to that of our 5448 Δsda1M− mutant, suggesting that M protein production, but not Sda1, plays the critical role in the evasion of neutrophil-mediated killing.
Although we cannot duplicate the previous findings that Sda1 plays a critical role in the selection for CovRS mutations of M1T1 GAS during infection and in M1T1 GAS resistance to killing in blood, we can at least partially confirm the previous findings that hasA and emm genes are critical for in vivo selection for SpeBA− variants of M1T1 GAS. It has been shown that the M protein and hyaluronic acid capsule are essential for in vivo selection of SpeBA− variants of GAS strain 5448 (20). We found that the hasA deletion abolishes the selection for SpeBA− variants of MGAS2221 in vivo, indicating that the capsule is a common essential factor for in vivo selection of CovRS mutations of M1T1 GAS. The deletion of the emm gene significantly reduces, but does not completely abolish, in vivo selection for SpeBA− variants of MGAS2221. Furthermore, other proteins in the Mga regulon contribute to in vivo selection of MGAS2221 SpeBA− variants. Apparently, the capsule and the Mga-regulated protein(s) contribute to the in vivo selection of M1T1 GAS CovRS mutants.
Natural CovRS mutations are frequently associated with invasive GAS isolates (36, 41), and invasive isolates with CovRS mutations usually have high virulence (8–11, 26, 30, 42). It is well established that null covS mutations enhance the expression of multiple virulence factors, including hasA, spyCEP, and sse, and, at the same time, reduce the expression of SpeB (9, 11, 13–18); this is referred as the SpeBA−/SseA+ phenotype in this study. In contrast, deletion of the covR gene increases the expression of speB and many CovRS-controlled virulence factors (14, 17). Various single-amino-acid replacements of CovR have been detected in clinical isolates (43), including the CovRG61S mutation (44), and most of them have not been characterized for their effects on the expression of virulence factors and speB. This CovRG61S mutation causes the SpeBA−/SseA+ phenotype. CovRR119H (17) and CovRI205F (12) have been shown to cause the SpeBA−/SseA+ phenotype as well. The SpeBA−/SseA+ phenotype of CovR G61S and CovR R119H mutants is not surprising, because they are in the receptor domain of CovR for CovS, may interrupt interaction between CovR and CovS, and likely are equivalent to null CovS mutations. However, the 505A insertion in the covR gene, which also confers the SpeBA−/SseA+ phenotype, is surprising. This insertion caused a reading frameshift, resulting in a truncated CovR fragment containing its first 168 amino acid residues and two additional amino acids. This truncated CovR mutant lacks this part of the DNA-binding domain, which is located in the CovR/DNA interface (43). The SpeBA− phenotype of this covR insertion mutant is in contrast to the known finding that covR deletion enhances the expression of both SpeB and other virulence factors (14, 17). Regardless of the basis for the SpeBA−/SseA+ phenotype of these mutations, our results indicate that covR mutations can be selected in vivo to confer the SpeBA−/SseA+ phenotype as covS mutations do.
CovRS mutations of invasive M1T1 GAS are selected mainly by neutrophils for better survival against the innate immune system (12). It is well documented that the enhanced expression of CovRS-controlled virulence factors contributes to the hypervirulence of M1T1 GAS CovRS mutants (8–12, 30, 42). The downregulation of SpeB production has been proposed to contribute to the hypervirulence of the mutant by preserving virulence factor proteins (19, 20); however, the SpeBA− phenotype of CovRS mutants recently was proposed to be a by-product of CovRS mutations (45). It appears to be difficult to verify these proposals experimentally. The requirement of the capsule for in vivo selection of CovRS mutations apparently has nothing to do with whether SpeB is produced. However, the contribution of the Mga regulon to in vivo selection of MGAS2221 SpeBA− variants supports the proposed preservation model; that is, the downregulation of SpeB production in natural CovRS mutants of M1T1 GAS is not a by-product of CovRS mutation but is a selection force for preservation of the virulence proteins for GAS survival against innate immune responses. This model concisely explains the emergence of hypervirulent strains and provides an opportunity to study effective mechanisms that abrogate this selection process and thereby protect the host against the potentially fatal consequences of invasive GAS infections.
ACKNOWLEDGMENTS
This work was supported in part by grants AI095704, AI097703, and GM110732 from the National Institutes of Health, USDA Animal Formula Fund, and the Montana State Agricultural Experimental Station.
We thank Debra Weinstein at the University of Maryland for editorial assistance.
REFERENCES
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 2.Cleary PP, Kaplan EL, Handley JP, Wlazlo A, Kim MH, Hauser AR, Schlievert PM. 1992. Clonal basis for resurgence of serious Streptococcus pyogenes disease in the 1980s. Lancet 339:518–521. doi: 10.1016/0140-6736(92)90339-5. [DOI] [PubMed] [Google Scholar]
- 3.Musser JM, Kapur V, Szeto J, Pan X, Swanson DS, Martin DR. 1995. Genetic diversity and relationships among Streptococcus pyogenes strains expressing serotype M1 protein: recent intercontinental spread of a subclone causing episodes of invasive disease. Infect Immun 63:994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Loughlin RE, Roberson A, Cieslak PR, Lynfield R, Gershman K, Craig A, Albanese BA, Farley MM, Barrett NL, Spina NL, Beall B, Harrison LH, Reingold A, Van Beneden C. 2007. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis 45:853–862. doi: 10.1086/521264. [DOI] [PubMed] [Google Scholar]
- 5.Sumby P, Porcella SF, Madrigal AG, Barbian KD, Virtaneva K, Ricklefs SM, Sturdevant DE, Graham MR, Vuopio-Varkila J, Hoe NP, Musser JM. 2005. Evolutionary origin and emergence of a highly successful clone of serotype M1 group a Streptococcus involved multiple horizontal gene transfer events. J Infect Dis 192:771–782. doi: 10.1086/432514. [DOI] [PubMed] [Google Scholar]
- 6.Aziz RK, Kotb MZ. 2008. Rise and persistence of global M1T1 clone of Streptococcus pyogenes. Emerg Infect Dis 14:1511–1517. doi: 10.3201/eid1410.071660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nasser W, Beres SB, Olsen RJ, Dean MA, Rice KA, Long SW, Kristinsson KG, Gottfredsson M, Vuopio J, Raisanen K, Caugant DA, Steinbakk M, Low DE, McGeer A, Darenberg J, Henriques-Normark B, Van Beneden CA, Hoffmann S, Musser JM. 2014. Evolutionary pathway to increased virulence and epidemic group A Streptococcus disease derived from 3,615 genome sequences. Proc Natl Acad Sci U S A 111:E1768–E1776. doi: 10.1073/pnas.1403138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engleberg NC, Heath A, Miller A, Rivera C, DiRita VJ. 2001. Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J Infect Dis 183:1043–1054. doi: 10.1086/319291. [DOI] [PubMed] [Google Scholar]
- 9.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kansal RG, Datta V, Aziz RK, Abdeltawab NF, Rowe S, Kotb M. 2010. Dissection of the molecular basis for hypervirulence of an in vivo-selected phenotype of the widely disseminated M1T1 strain of group A Streptococcus bacteria. J Infect Dis 201:855–865. doi: 10.1086/651019. [DOI] [PubMed] [Google Scholar]
- 11.Li J, Zhu H, Feng W, Liu M, Song Y, Zhang X, Zhou Y, Bei W, Lei B. 2013. Regulation of inhibition of neutrophil infiltration by the two-component regulatory system CovRS in subcutaneous murine infection with group A Streptococcus. Infect Immun 81:974–983. doi: 10.1128/IAI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Liu G, Feng W, Zhou Y, Liu M, Wiley JA, Lei B. 2014. Neutrophils select hypervirulent CovRS mutants of M1T1 group A Streptococcus during subcutaneous infection of mice. Infect Immun 82:1579–1590. doi: 10.1128/IAI.01458-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levin JC, Wessels MR. 1998. Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol Microbiol 30:209–219. doi: 10.1046/j.1365-2958.1998.01057.x. [DOI] [PubMed] [Google Scholar]
- 14.Heath A, DiRita VJ, Barg NL, Engleberg NC. 1999. A two-component regulatory system, CsrR-CsrS, represses expression of three Streptococcus pyogenes virulence factors, hyaluronic acid capsule, streptolysin S, and pyrogenic exotoxin B. Infect Immun 67:5298–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Federle MJ, McIver KS, Scott JR. 1999. A response regulator that represses transcription of several virulence operons in the group A Streptococcus. J Bacteriol 181:3649–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edwards RJ, Taylor GW, Ferguson M, Murray S, Rendell N, Wrigley A, Bai Z, Boyle J, Finney SJ, Jones A, Russell HH, Turner C, Cohen J, Faulkner L, Sriskandan S. 2005. Specific C-terminal cleavage and inactivation of interleukin-8 by invasive disease isolates of Streptococcus pyogenes. J Infect Dis 192:783–790. doi: 10.1086/432485. [DOI] [PubMed] [Google Scholar]
- 17.Treviño J, Perez N, Ramirez-Peña E, Liu Z, Shelburne SA III, Musser JM, Sumby P. 2009. CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect Immun 77:3141–3149. doi: 10.1128/IAI.01560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu H, Liu M, Sumby P, Lei B. 2009. The secreted esterase of group A Streptococcus is important for invasive skin infection and dissemination in mice. Infect Immun 77:5225–5232. doi: 10.1128/IAI.00636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aziz RK, Pabst MJ, Jeng A, Kansal R, Low DE, Nizet V, Kotb M. 2004. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol Microbiol 51:123–134. [DOI] [PubMed] [Google Scholar]
- 20.Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG, Simpson AJ, Buchanan JT, Chhatwal GS, Kotb M, Nizet V. 2007. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med 13:981–985. doi: 10.1038/nm1612. [DOI] [PubMed] [Google Scholar]
- 21.Cole JN, Pence MA, von Köckritz-Blickwede M, Hollands A, Gallo RL, Walker MJ, Nizet V. 2010. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. mBio 1:e00191-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ribardo DA, McIver KS. 2006. Defining the Mga regulon: comparative transcriptome analysis reveals both direct and indirect regulation by Mga in the group A streptococcus. Mol Microbiol 62:491–508. doi: 10.1111/j.1365-2958.2006.05381.x. [DOI] [PubMed] [Google Scholar]
- 23.Aziz RK, Ismail SA, Park HW, Kotb M. 2004. Post-proteomic identification of a novel phage-encoded streptodornase, Sda1, in invasive M1T1 Streptococcus pyogenes. Mol Microbiol 54:184–197. doi: 10.1111/j.1365-2958.2004.04255.x. [DOI] [PubMed] [Google Scholar]
- 24.Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, Feramisco J, Nizet V. 2006. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol 16:396–400. doi: 10.1016/j.cub.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 25.Venturini C, Ong CL, Gillen CM, Ben-Zakour NL, Maamary PG, Nizet V, Beatson SA, Walker MJ. 2013. Acquisition of the Sda1-encoding bacteriophage does not enhance virulence of the M1 Streptococcus pyogenes strain SF370. Infect Immun 81:2062–2069. doi: 10.1128/IAI.00192-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsatsaronis JA, Hollands A, Cole JN, Maamary PG, Gillen CM, Zakour NL, Kotb M, Nizet V, Beatson SA, Walker MJ, Sanderson-Smith ML. 2013. Streptococcal collagen-like protein A and general stress protein 24 are immunomodulating virulence factors of group A Streptococcus. FASEB J 27:2633–2643. doi: 10.1096/fj.12-226662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 28.Zhou Y, Hanks TS, Feng W, Li J, Liu G, Liu M, Lei B. 2013. The sagA/pel locus does not regulate the expression of the M protein of the M1T1 lineage of group A Streptococcus. Virulence 4:698–706. doi: 10.4161/viru.26413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu G, Liu M, Xie G, Lei B. 2013. Characterization of streptococcal platelet-activating factor acetylhydrolase variants that are involved in innate immune evasion. Infect Immun 81:3128–3138. doi: 10.1128/IAI.00398-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stetzner ZW, Li D, Feng W, Liu M, Liu G, Wiley J, Lei B. 2015. Serotype M3 and M28 group A streptococci have distinct capacities to evade neutrophil and TNF-α responses and to invade soft tissues. PLoS One 10:e0129417. doi: 10.1371/journal.pone.0129417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu M, Hanks TS, Zhang J, McClure MJ, Siemsen DW, Elser JL, Quinn MT, Lei B. 2006. Defects in ex vivo and in vivo growth and sensitivity to osmotic stress of group A Streptococcus caused by interruption of response regulator gene vicR. Microbiology 152:967–978. doi: 10.1099/mic.0.28706-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu M, Zhu H, Li J, Garcia CC, Feng W, Kirpotina LN, Hilmer J, Tavares LP, Layton AW, Quinn MT, Bothner B, Teixeira MM, Lei B. 2012. Group A Streptococcus secreted esterase hydrolyzes platelet-activating factor to impede neutrophil recruitment and facilitate innate immune evasion. PLoS Pathog 8:e1002624. doi: 10.1371/journal.ppat.1002624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sumby P, Barbian KD, Gardner DJ, Whitney AR, Welty DM, Long RD, Bailey JR, Parnell MJ, Hoe NP, Adams GG, DeLeo FR, Musser JM. 2005. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc Natl Acad Sci U S A 102:1679–1684. doi: 10.1073/pnas.0406641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hondorp ER, McIver KS. 2007. The Mga virulence regulon: infection where the grass is greener. Mol Microbiol 66:1056–1065. doi: 10.1111/j.1365-2958.2007.06006.x. [DOI] [PubMed] [Google Scholar]
- 35.Kappeler KV, Anbalagan S, Dmitriev AV, McDowell EJ, Neely MN, Chaussee MS. 2009. A naturally occurring Rgg variant in serotype M3 Streptococcus pyogenes does not activate speB expression due to altered specificity of DNA binding. Infect Immun 77:5411–5417. doi: 10.1128/IAI.00373-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikebe T, Ato M, Matsumura T, Hasegawa H, Sata T, Kobayashi K, Watanabe H. 2010. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog 6:e1000832. doi: 10.1371/journal.ppat.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carroll RK, Shelburne SA III, Olsen RJ, Suber B, Sahasrabhojane P, Kumaraswami M, Beres SB, Shea PR, Flores AR, Musser JM. 2011. Naturally occurring single amino acid replacements in a regulatory protein alter streptococcal gene expression and virulence in mice. J Clin Investig 121:1956–1968. doi: 10.1172/JCI45169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olsen RJ, Laucirica DR, Watkins ME, Feske ML, Garcia-Bustillos JR, Vu C, Cantu C, Shelburne SA III, Fittipaldi N, Kumaraswami M, Shea PR, Flores AR, Beres SB, Lovgren M, Tyrrell GJ, Efstratiou A, Low DE, Van Beneden CA, Musser JM. 2012. Polymorphisms in regulator of protease B (RopB) alter disease phenotype and strain virulence of serotype M3 group A Streptococcus. J Infect Dis 205:1719–1729. doi: 10.1093/infdis/jir825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kazmi SU, Kansal R, Aziz RK, Hooshdaran M, Norrby-Teglund A, Low DE, Halim AB, Kotb M. 2001. Reciprocal, temporal expression of SpeA and SpeB by invasive M1T1 group a streptococcal isolates in vivo. Infect Immun 69:4988–4995. doi: 10.1128/IAI.69.8.4988-4995.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mangold M, Siller M, Roppenser B, Vlaminckx BJ, Penfound TA, Klein R, Novak R, Novick RP, Charpentier E. 2004. Synthesis of group A streptococcal virulence factors is controlled by a regulatory RNA molecule. Mol Microbiol 53:1515–1527. doi: 10.1111/j.1365-2958.2004.04222.x. [DOI] [PubMed] [Google Scholar]
- 41.Shea PR, Beres SB, Flores AR, Ewbank AL, Gonzalez-Lugo JH, Martagon-Rosado AJ, Martinez-Gutierrez JC, Rehman HA, Serrano-Gonzalez M, Fittipaldi N, Ayers SD, Webb P, Willey BM, Low DE, Musser JM. 2011. Distinct signatures of diversifying selection revealed by genome analysis of respiratory tract and invasive bacterial populations. Proc Natl Acad Sci U S A 108:5039–5044. doi: 10.1073/pnas.1016282108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miyoshi-Akiyama T, Ikebe T, Watanabe H, Uchiyama T, Kirikae T, Kawamura Y. 2006. Use of DNA arrays to identify a mutation in the negative regulator, csrR, responsible for the high virulence of a naturally occurring type M3 group A streptococcus clinical isolate. J Infect Dis 193:1677–1684. doi: 10.1086/504263. [DOI] [PubMed] [Google Scholar]
- 43.Horstmann N, Sahasrabhojane P, Suber B, Kumaraswami M, Olsen RJ, Flores A, Musser JM, Brennan RG, Shelburne SA III. 2011. Distinct single amino acid replacements in the control of virulence regulator protein differentially impact streptococcal pathogenesis. PLoS Pathog 7:e1002311. doi: 10.1371/journal.ppat.1002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoe NP, Vuopio-Varkila J, Vaara M, Grigsby D, De Lorenzo D, Fu YX, Dou SJ, Pan X, Nakashima K, Musser JM. 2001. Distribution of streptococcal inhibitor of complement variants in pharyngitis and invasive isolates in an epidemic of serotype M1 group A Streptococcus infection. J Infect Dis 183:633–639. doi: 10.1086/318543. [DOI] [PubMed] [Google Scholar]
- 45.Flores AR, Sahasrabhojane P, Saldaña M, Galloway-Peña J, Olsen RJ, Musser JM, Shelburne SA. 2014. Molecular characterization of an invasive phenotype of group A Streptococcus arising during human infection using whole genome sequencing of multiple isolates from the same patient. J Infect Dis 209:1520–1523. doi: 10.1093/infdis/jit674. [DOI] [PMC free article] [PubMed] [Google Scholar]