

Abstract
Clostridium difficile infection (CDI) is a major cause of health care-associated disease. CDI initiates with ingestion of C. difficile spores, germination in the gastrointestinal (GI) tract, and then colonization of the large intestine. The interactions between C. difficile cells and other bacteria and with host mucosa during CDI remain poorly understood. Here, we addressed the hypothesis that, in a mouse model of CDI, C. difficile resides in multicellular communities (biofilms) in association with host mucosa. To do this, we paraffin embedded and then sectioned the GI tracts of infected mice at various days postinfection (p.i.). We then used fluorescent in situ hybridization (FISH) with 16S rRNA probes targeting most bacteria as well as C. difficile specifically. The results revealed that C. difficile is present as a minority member of communities in the outer (loose) mucus layer, in the cecum and colon, starting at day 1 p.i. To generate FISH probes that identify bacteria within mucus-associated communities harboring C. difficile, we characterized bacterial populations in the infected mouse GI tract using 16S rRNA gene sequence analysis of bacterial DNA prepared from intestinal content. This analysis revealed the presence of genera of several families belonging to Bacteroidetes and Firmicutes. These data suggest that formation of multispecies communities associated with the mucus of the cecum and colon is an important early step in GI tract colonization. They raise the possibility that other bacterial species in these communities modulate the ability of C. difficile to successfully colonize and, thereby, cause disease.
INTRODUCTION
Every year, more than half a million people in the United States acquire Clostridium difficile infection (CDI). CDI initiates with ingestion of C. difficile spores, spore germination in the gastrointestinal (GI) tract, and then colonization of the large intestine (1). Symptoms can range from diarrhea to the severe inflammatory condition known as pseudomembranous colitis or, in the most severe cases, toxic megacolon (2). These pathologies likely involve multiple virulence factors, including toxins A and B.
Hosts employ a variety of mechanisms to resist infection of the GI tract by C. difficile. These include the colonization of the GI mucosa by microbial communities that interfere with C. difficile attachment and/or proliferation (3). Recent experiments suggest roles in resisting CDI for a number of bacterial species in the GI tract (4, 5). Resistance to C. difficile colonization can involve the inactivation of germinants (the molecules that cause the spore, the infectious form of C. difficile, to return to the vegetative state and produce toxins) (6) and production of inhibitory small molecules (7). Members of healthy human GI tract microbiota may also stimulate host immune responses that prevent C. difficile establishment (8). Competition for resources has also been suggested as a possible mechanism of C. difficile restriction (9).
Typically, the appearance of CDI follows treatment of the patient with antimicrobial therapy, which is likely to significantly alter or reduce the gastrointestinal microbiota and, as a result, allow C. difficile to successfully colonize (10). For example, an analysis of the microbiota in humans and mice with CDI suggested an association between the depletion of bacteria of the Ruminococcaceae, Lachnospiraceae, and butyric acid-producing Firmicutes species and active and severe C. difficile infection (4, 11). Possibly, this reduction of the obligate anaerobic bacterial population is accompanied by a metabolic shift that creates an environment permissive for C. difficile germination and growth (12–14). Antibiotic-induced dysbiosis also allows certain bacterial species to significantly increase in number (5, 10). For example, in several experimental models, the loss of Lachnospiraceae correlated with increased abundance of Enterobacteriaceae family members (5, 15).
The interactions between C. difficile and other bacteria, and with the host mucosa during CDI, especially at the onset of colonization, remain poorly understood. These interactions likely significantly influence the course of CDI and, potentially, relapsing disease. Nonetheless, recent evidence raised the possibility that C. difficile makes intimate contact with the mucosa and other members of the microbiota. For example, transmission electron microscopy revealed that during infection in the mouse, a mat of rod-shaped bacterial cells is found overlaying damaged microvilli (5). Scanning electron microscopic analysis of cecum and colon samples of infected hamsters showed C. difficile-like bacteria associated with mucus, over both the tissue surface and crypt crevasses, forming aggregates (16).
We hypothesize that, in the host, C. difficile resides in multicellular communities (biofilms) in association with host mucosa. Therefore, in this work, we sought to address two specific issues: (i) the locations of C. difficile communities in relation to the GI mucosa and (ii) the population structure of C. difficile-containing communities.
MATERIALS AND METHODS
Mouse model.
All animal procedures were performed in accordance with the NIH guidelines for housing and care of laboratory animals and were approved by the Loyola University Institutional Animal Care and Use Committee (approval no. LU108337 IACUC 10-007 P/D 012-10 Feb12/2010-2012 and LU 108337 IACUC10-043 P/D 061-10 Oct5/2010-2013).
C57BL/6 mice from Charles River Laboratories (Wilmington, MA) were housed and bred in the Comparative Medicine Facility at Loyola University Chicago. Mice (8 to 12 weeks old) were treated with antibiotics, including vancomycin (0.045 mg/ml), metronidazole (0.215 mg/ml), gentamicin (0.035 mg/ml), kanamycin (0.4 mg/ml), and colistin (850 U/ml), in sterile drinking water for 72 h and then were administered sterile drinking water without antibiotics for 48 h, followed by a single intraperitoneal injection of clindamycin (10 mg/kg of body weight). After 24 h, mice were challenged with 105 spores of epidemic C. difficile strain BI17/NAP1/027 by oral gavage (17). Mice were monitored for disease symptoms (diarrhea and weight loss) and fecal C. difficile CFU levels. Mice were euthanized by CO2 asphyxiation.
Two mice were sacrificed at each time point (1, 2, 4, 6, and 8 days postinfection [p.i.]). A noninfected C57BL/6 mouse that recovered after antibiotic treatment at day 12 and mice not given antibiotics were used as controls.
Spore preparation.
C. difficile strain BI17 was cultured anaerobically overnight in reduced-liquid brain heart infusion (BHI) medium supplemented with l-cysteine at 37°C. C. difficile was plated in a lawn on reduced blood agar plates and cultured anaerobically for 5 to 7 days at 37°C to induce sporulation. Vegetative cells were removed from sporulation cultures by pelleting cells in sterile phosphate-buffered saline (PBS) and then incubating them at 68°C for 2 h. Spores were washed 3 times with PBS, and CFU levels were determined by plating serial dilutions on brain heart infusion (BHI) agar containing taurocholate. Spore numbers were confirmed by phase-contrast microscopy.
Histology.
Mouse GI tissue were removed and placed into Carnoy solution (18) with retention of feces. Three colon segments from the proximal colon (about 2 cm down from the cecum), the distal colon (about 1 to 2 cm up from the rectum), and the region between the proximal colon and the distal colon as well as cecum segments of approximately 1 to 2 mm were removed and placed into histology tissue cassettes which were processed according to a standard protocol, paraffin embedded (without formalin), and then sectioned longitudinally. Histology was examined by hematoxylin and eosin (H&E) and alcian blue staining of 3.5-μm-thick tissue sections.
FISH.
Probes used in fluorescent in situ hybridization (FISH) experiments were synthesized and purified by high-performance liquid chromatography (HPLC; Integrated DNA Technologies, Inc.) and are listed in Table 1 (19–25). After paraffin removal, 3.5-μm-thick sections were treated with 100 μg/ml lysozyme and 20 U of mutanolysin (Sigma) in STE buffer (10 mM Tris [pH 7.4], 1 mM EDTA, 100 mM NaCl) for 1 h at 37°C and then rinsed three times in PBS buffer and once in 100% ethanol for 10 min. Sections were covered with 80 μl of hybridization solution (30 mM Tris [pH 7.2], 0.9 M NaCl, 0.1% SDS) containing 100 ng of each probe and incubated overnight in the dark at 50°C. After hybridization, slides were washed three times for 10 min each time with prewarmed washing solution (100 mM Tris [pH 7.2], 0.9 M NaCl) and were then stained with Hoechst 33342 solution (Thermo Scientific). Finally, slides were rinsed with distilled water, allowed to dry at 40°C, and mounted in PermaFluor (Thermo Scientific).
TABLE 1.
FISH probes
| Name of the probe—fluorescent label | Sequence | Specificity | Reference |
|---|---|---|---|
| Cd198—Cy5 | 5′-CATCCTGTACTGGCTCAC-3′ | Clostridium difficile | 19 |
| Eub338—Cy3 | 5′-GCTGCCTCCCGTAGGAGT-3′ | Most bacteria | 20 |
| NonEub—Cy3 | 5′-CGACGGAGGGCATCCTCA-3′ | Nonsense probe | 21 |
| Erec482—Cy3 | 5′-GCTTCTTAGTCARGTACCG-3′ | Most of the Clostridium coccoides-Eubacterium rectale group (Clostridium clusters XIVa and XIVb) | 22 |
| Lab158—Cy3 | 5′-GGTATTAGCAYCTGTTTCCA-3′ | Lactobacilli, enterococci | 23 |
| Ent183—Cy3 | 5′-CTCTTTGGTCTTGCGACG-3′ | Enterobacteriaceae | 24 |
| Bac303—Cy3 | 5′-CCAATGTGGGGGACCTT-3′ | Most Bacteroidaceae and Prevotellaceae, some Porphyromonadaceae | 25 |
Samples were imaged with a LSM 510 microscope (Carl Zeiss Inc.). Cy3- and Cy5-labeled probes were detected with a 561-nm-wavelength laser and a 633-nm-wavelength laser, respectively. Hoechst 33342 staining was detected with a 405-nm-wavelength laser. Images were processed using Zeiss LSM image examiner software.
Immunodetection of mucin.
After the FISH procedure was performed, slides were blocked with 2% bovine serum albumin (BSA; Fisher Scientific)–PBS for 1 h at room temperature (RT). Rabbit antibodies against mucin 2 (H300; sc15334, Santa Cruz Biotechnology, Inc.) were added at a 1:50 dilution in PBS and incubated for 1 h at 37°C. Slides were washed 3 times for 10 min each time with PBS containing 0.1% Tween 20, and then goat anti-rabbit immunoglobulin conjugated with Alexa 488 (Molecular Probes) was added at a 1:100 dilution in PBS for 1 h at RT. Slides were washed two times with PBS containing 0.1% Tween 20, stained with Hoechst 33342 solution, rinsed with distilled water, and then mounted and imaged as described above. Alexa 488-conjugated antibody was imaged using an LSM 510 microscope and an Argon laser.
DNA extraction, 16S rRNA gene amplicon library preparation, and sequencing.
After removing fragments of Carnoy-fixed tissue for preparation of paraffin sections, luminal contents from cecum and colon were combined for DNA extraction using a PowerSoil Mo Bio kit (Mo Bio Laboratories, Inc.). The protocol used for DNA extraction, with implemented modifications, is described in the guidelines of the Human Microbiome Initiative (26). The DNA sample was stored at −20°C. PCR was performed using a TaKaRa Ex Taq enzyme mixture (Clontech) as described in reference 27. Specimens were multiplexed using barcoded primers 806rcbs1 through 806rcbs20 (27). PCR products containing 16S RNA gene regions V4 and V5 were purified and pooled in equal amounts. Each pool of PCR products was subjected to gel purification and used for sequencing in an Illumina MiSeq machine according to the recommended protocol (27). Reads were demultiplexed using Illumina software preinstalled on the MiSeq sequencer. Separate fastq files were generated for each specimen.
Microbial taxonomy analysis.
Analysis of the microbiota was conducted according to an established standard procedure (28). We utilized Mothur software (http://www.mothur.org/) (29) to generate a set of unique, filtered, high-quality trimmed reads for each sample while retaining data on the frequency of each unique read. After chimera trimming, we constructed both an abundance matrix of reference operational taxonomic units (OTUs) and a phylogenetic tree derived from the Silva reference set. We also generated rarefaction curves and calculated Simpson diversity indices (29). The Good's coverage index was calculated as G = 1 − n/N, where n is the number of singleton phylotypes and N is the total number of sequences in the sample. Principal coordinate analysis (PCoA) based on the Bray-Curtis similarity matrix was used to visualize sample distributions. Metastats (open-source software [http://metastats.cbcb.umd.edu/]) (30) was used to perform statistical analysis of differentially abundant community members. Multivariable statistical analyses (PCoA and analysis of similarities [ANOSIM]) were applied using Primer v6 software.
RESULTS
Visualizing C. difficile communities in the GI tract.
To study the association of C. difficile cells with host mucosa and other bacteria in an infected animal, we employed the mouse model of CDI established by Chen at al. (31). To visualize C. difficile in the GI tract of infected mice, we harvested tissue from the cecum and colon of mice at 1, 2, 4, 6, and 8 days postinfection (p.i.) and generated thick sections for analysis by microscopy. First, we visualized mucus in the sections using staining with alcian blue; second, we visualized the pathology with hematoxylin and eosin (Fig. 1A to G). As expected in this infection model, we detected inflammation, edema, epithelial damage, and immune cell infiltration (Fig. 1A, C, H, and I).
FIG 1.
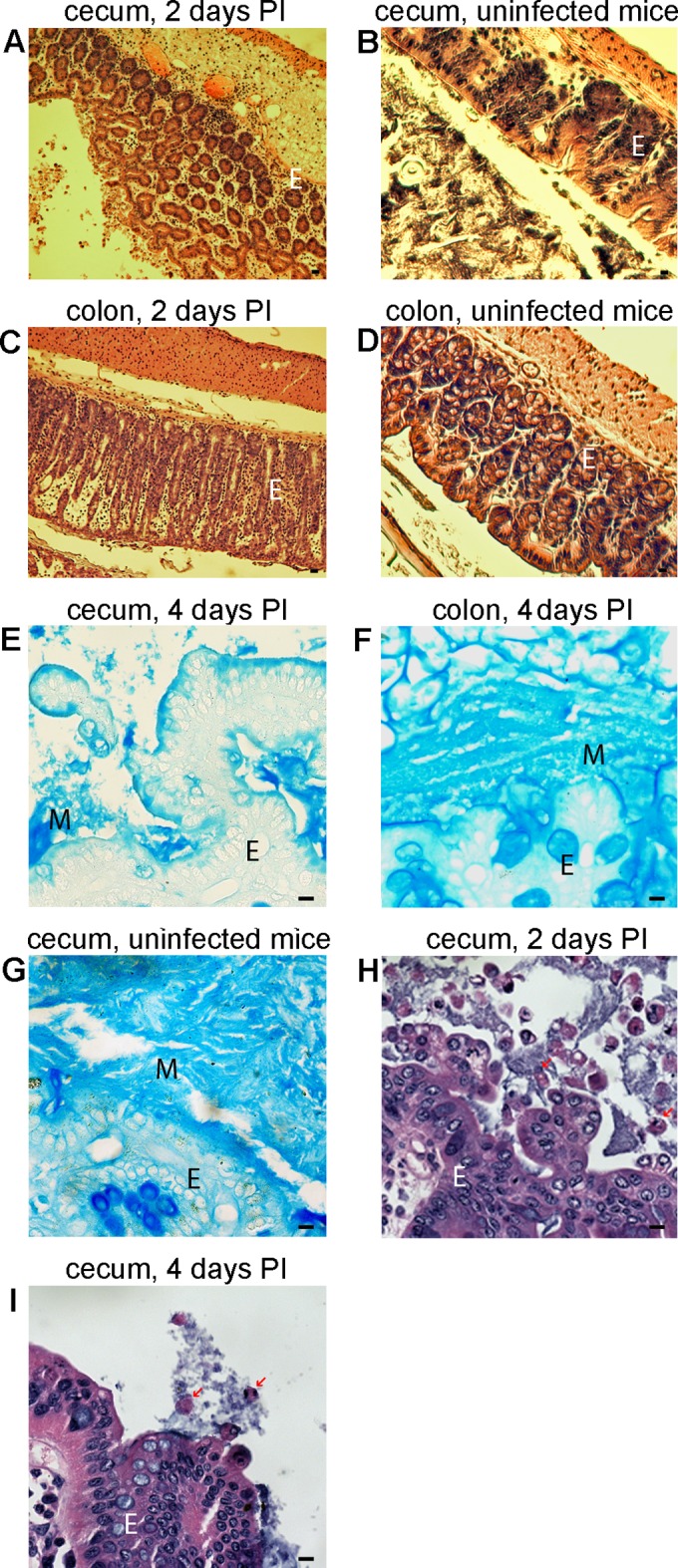
Light-microscopic analysis of stained sections of cecum and colon from mice infected with C. difficile. Cecum sections (A, B, E, and G to I) or colon sections (C, D, and F) were prepared from infected (A, C, E, F, H, and I) or uninfected (B, D, and G) mice. Sections from infected tissues were harvested after either 2 (A, C, and H) or 4 (E, F, and I) days and are representative of early infection and late infection, respectively. Sections were stained with hematoxylin and eosin (A to D, H, and I) or alcian blue (E to G); blue staining indicates the presence of mucus (M). E, epithelial cells; PI, postinfection. Arrows show bacteria interacting with neutrophils. Bars, 10 μm.
Next, using 16S rRNA probes, we localized bacteria in these sections by FISH. To visualize C. difficile, we employed a previously characterized C. difficile-specific probe (Cd198 [19]). To identify most other bacteria, we used a well-characterized domain-specific probe (Eub338 [20]) (Table 1). We readily detected bacteria, and C. difficile in particular, in association with the mucus of the cecum and the large intestine (Fig. 2). We found that in mucus-associated bacterial communities harboring C. difficile, C. difficile was a minority member of the population (as judged using the Eub338 probe) (Fig. 2 and 3) (see also Fig. 5). The C. difficile cells had the morphology expected of vegetative cells (rods of about 4 μm in length) and were labeled positively with both Cd198 and Eub338 probes. We think it is unlikely that our methodology would detect C. difficile spores, given that we were unable to visualize spores produced in a laboratory culture using our FISH protocol (data not shown). Nonetheless, we consider it plausible that spores are present in these communities, as discussed later.
FIG 2.

Fluorescent in situ hybridization analysis of sections of cecum and colon from mice infected with C. difficile. Cecum sections (A, C, E, G, I, and K) or colon sections (B, D, F, H, J, and L) were prepared from uninfected tissues (A and B) or from infected tissues harvested at 1 (C and D), 2 (E and F), 4 (G and H), 6 (I and J), or 8 (K and L) days postinfection (using the same intestinal samples as those presented in Fig. 1). Images were taken from a representative section. Hoechst 33342 was used to stain DNA (blue). Bacteria were visualized with the domain-specific Eub338 probe (red), and C. difficile cells were visualized with the C. difficile-specific Cd198 probe (green). E, epithelial cells; im, the inner mucin layer in colon. Bars, 10 μm. Brackets indicate locations of bacterial communities on the mucin layer.
FIG 3.
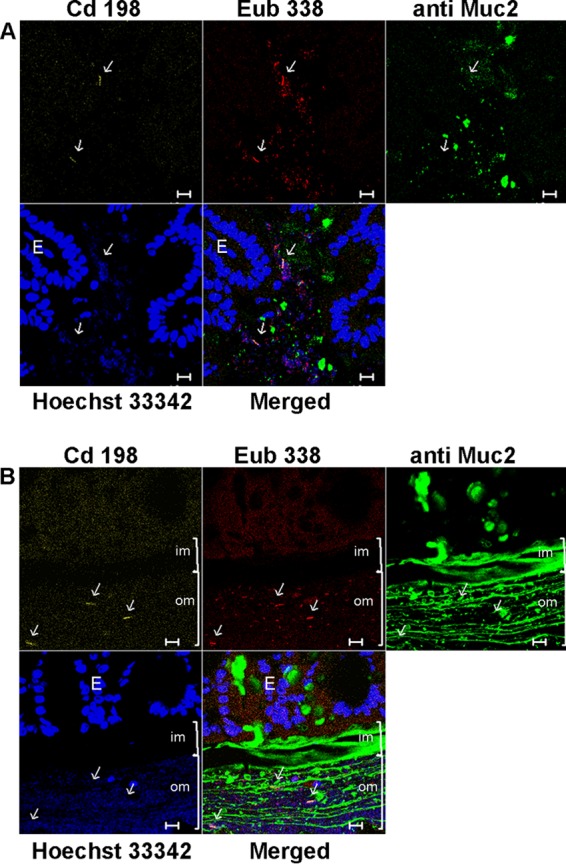
Fluorescence in situ hybridization analysis of sections of cecum and colon from mice infected with C. difficile followed by immunodetection of mucin. Sections were prepared from mouse cecum at 4 days p.i. (A) and from mouse colon at 6 days p.i. (B). Hoechst 33342 was used to stain DNA (blue). Bacteria were visualized with the domain-specific Eub338 probe (red), C. difficile cells were visualized with the C. difficile-specific Cd198 probe (yellow), and mucus was visualized with Muc2-specific antibody (green). E, epithelial cells. Brackets indicate the position of mucus: im, inner mucus; om, outer mucus. Arrows indicate positions of C. difficile cells. Bars, 10 μm.
FIG 5.
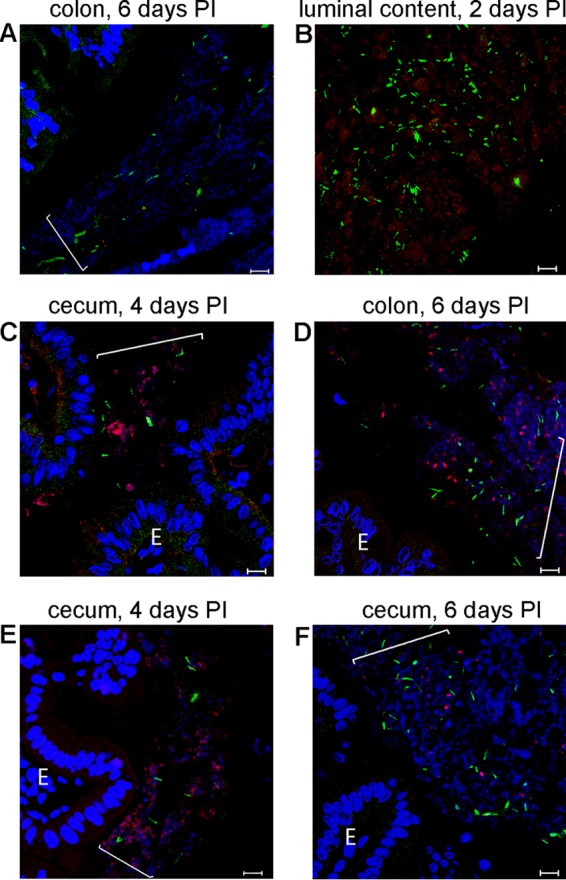
Fluorescent in situ hybridization analysis of groups of bacteria in sections of cecum and colon from mice infected with C. difficile. Cecum (C, E, and F) or colon (A, B, and D) sections were collected from animals at 2 (B), 4 (C and E), or 6 (A, D, and F) days postinfection. Hoechst 33342 was used to stain DNA (blue). C. difficile cells were visualized with the C. difficile-specific probe Cd198 (green). Red staining indicates binding of taxon-specific probe Bac303 (C), Lab158 (B and F), Erec482 (E), or Ent183 (D). In panel A, a nonsense probe (red; not detected because there was no binding) was applied as a negative control. E, epithelial cell. Brackets indicate locations of bacterial communities. Bars, 10 μm.
We observed bacterial communities in cecum and colon sections beginning on day 1 p.i. The frequencies with which we observed these communities did not detectably differ between days 1 and 8 (Fig. 2E to L). We also did not see any differences in binding between animals by the use of the Cd198 and Eub 338 probes, at any single time point. We did not detect any binding either of Cd198 applied to sections from uninfected mice or of a nonsense probe (non-Eub) applied to sections from infected mice (Fig. 2A and B) (see also Fig. 5A). We quantified the number of C. difficile cells in mucus-associated bacterial communities by counting mucin-associated C. difficile cells in close proximity to the epithelium (i.e., separated from the epithelium by 150 μm or less). As shown in Fig. S1 in the supplemental material, the numbers of cells per site differed significantly in both colon and cecum at every time point. On days 4 and 6 p.i., the numbers of cells slightly increased.
Bacterial communities harboring C. difficile were associated with regions of tissue showing pathology as determined by H&E staining (Fig. 1A and C). Often, these communities were closely associated with massive neutrophil infiltrates (Fig. 1H and I and 2E to G and I). However, C. difficile was also present where tissue appeared healthy. Mucin-associated C. difficile cells were present at a lower density than cells in the lumen, defined operationally as those cells that were separated from the epithelium by at least 150 μm (32) (see Fig. 5B). We did not detect binding of the Cd198 probe in sections of the ileum.
Recent observations showed that mucosally associated bacteria can be present in an outer, loosely attached mucin layer in colon (32). To determine whether communities harboring C. difficile occupy the mucin layer, we performed FISH using, in addition, Muc2 antibodies as a counterstain for mucin. As expected from our other results and previous observations, the inner (firm) mucin was largely free of bacteria (Fig. 3) (32). We detected single C. difficile cells in the inner mucus layer very rarely (data not shown).
In the distal colon, all the bacterial communities that we observed were present at high density in the outer mucus, which was clearly demarcated from the rest of the luminal content (referred to as the “interlaced” layer by Swidsinski and colleagues [33]) (Fig. 1F, 2F, H, J, and L, and 3B; see also Fig. S2 in the supplemental material). C. difficile-bearing communities in the proximal colon and in the region between the proximal colon and the distal colon were present but at a lower frequency than in the distal colon. In the cecum, we detected C. difficile cells and other bacteria attached to mucus which appeared to be only partly attached to the epithelium (Fig. 2G, I, and K; see also Fig. S2). Most likely, the partial attachment was due to the diarrhea that these animals experience. We rarely detected C. difficile cells attached to the surface of the epithelium in the cecum (Fig. 2).
Microbial community profiling.
Our results using FISH identified the presence of a complex bacterial community in animals infected with C. difficile. To begin to characterize this population by using more-specific FISH probes, we identified bacterial taxa in the GI tract, using 16S rRNA gene sequencing of bacterial DNA prepared from intestinal cecum and colon content.
A total 968,300 sequencing reads were generated from these samples, with an average of 74,484 ± 11,474 reads per sample. In order to facilitate subsequent analysis, samples were normalized to equal numbers of reads (n = 10,000). From these sequences, 6,870 operational taxonomic units (OTUs) were identified based on 97% DNA similarity. An average of 592 OTUs were identified per sample; 60 OTUs had a relative abundance of more than 1% in at least one sample. Good's coverage level results, achieved by sampling of 10,000 reads per sample, indicated that 94% of OTUs in our study were sampled more than once (34). We assessed the bacterial community diversity in mice at various days p.i. using the Simpson diversity index (SDI), which measures the probability that two bacteria randomly selected from a population would belong to the same species (35). The intestinal microbiota of uninfected mice had a diverse community (SDI < 0.1), while antibiotic treatment followed by C. difficile infection drastically reduced the community diversity (Fig. 4A). We observed the lowest level of diversity at days 1 and 2 p.i. Days 4 to 8 p.i. were characterized by a slow increase in community diversity, consistent with the process of partial recovery from infection, but the SDI determined for the infected mice still differed from the SDI for the uninfected mice as well as from the SDI for a mouse that recovered after antibiotic treatment (Fig. 4A).
FIG 4.

(A) Simpson diversity index data. Communities for analyses were harvested from uninfected non-antibiotic-treated mice (lanes UI), from an antibiotic-treated, uninfected mouse (12 days after treatment; lane atbt), or from infected mice at day 8 (lanes 8d), 6 (lanes 6d), 4 (lanes 4d), 2 (lanes 2d), or 1 (lanes 1d) postinfection. Two mice were analyzed in each case (except for lane atbt). (B) Principal coordinate (PCO) analysis of mouse microbiota. Luminal contents were harvested from infected animals on days 1, 2, 4, 6, and 8 p.i. or from animals that were left uninfected (UI) or were treated with an antibiotic (AtbT). A Bray-Curtis similarity matrix was constructed from samples standardized using total and square root-transformed data. The encircled clusters represent groups of related samples. (C) Distributions of bacterial families representing at least 10% of the sequences. DNA for analyses was harvested from mouse GI tracts at 1, 2, 4, 6, or 8 days p.i. Two mice were analyzed at each day as indicated (1d-1 represents one of the two mice analyzed on day 1, 1d-2 represents the other mice analyzed on day 1, etc.). Controls: DNA from an antibiotic-treated but uninfected mouse, 12 days after cessation of antibiotics, and DNA from GI tract and feces from non-antibiotic-treated, uninfected mice.
Since the gut bacterial community was clearly affected by antibiotic treatment, we used a phylotyping approach to study the temporal dynamics of the GI tract microbial community. Sequences were assembled into phylotypes according to their genus-level classifications.
A Bray-Curtis similarity matrix was constructed using the square root-transformed standardized values of genera abundances and was used to visualize the spatial distribution of samples in a principal coordinate analysis (PCoA). PCoA (Fig. 4B) identified 3 groups defined by a 60% similarity between samples. Each community in each group was at least 60% similar to the other communities in its group based on the Bray-Curtis similarity index. The fact that these groups were significantly different was confirmed by analysis of similarities (ANOSIM) (P = 0.001). Group 1 included samples collected at days 1 and 2 p.i. Group 2 contained samples collected at days 4, 6, and 8 p.i. Group 3 contained samples collected on the 8th day p.i. from uninfected mice, from antibiotic-treated but uninfected mice, and from one fully recovered mouse. Principal coordinate 1 (PC1) accounts for 61.6% of the total variation and separates the infected and control samples. PC2 (capturing 18.6% of the total variation) separates the samples taken at days 1 and 2 after infection from the later samples (days 4, 6, and 8 p.i.), indicating the percentage of total variation attributable to partial recovery of the bacterial community composition.
To identify genera that differed in abundance among groups 1, 2, and 3, we used the Metastats methodology (30). After correction for false discovery, we defined the P value cutoff for differentially abundant genera as <0.01. Comparison of groups 1 and 2 revealed 7 differentially abundant genera. Members of the Clostridium XVIII cluster and the genus Parabacteroides were decreased in abundance in group 2 compared to group 1, while the abundances of the genera Blautia, Anaerostipes, and Lactobacillus increased in group 2 compared to group 1. We detected 14 genera that were differentially abundant between groups 2 and 3. Members of the genus Bacteroides, along with members of the genera Blautia, Anaerostipes, Proteus, and Parabacteroides, were significantly decreased in abundance in group 3 compared with group 2. We found only a small increase in the abundance of the minor genera Oscillibacter and Dorea in group 3 compared with group 2 (see Table SA1 in the supplemental material). Antibiotic treatment, followed by C. difficile infection, significantly affected bacterial community composition. We observed the highest level of Bacteroidaceae (70%) at day 1 p.i. (Fig. 4C). Moreover, Bacteroidaceae (including the genera Alistipes and Bacteroides) remained the most abundant group throughout the course of disease (day 1 to 8 median abundance, 50%). At days 1 and 2 p.i., members of the Enterobacteriaceae (day 1 to 2 median abundance, 6%) and Enterococcaceae (day 1 to 2 median abundance, 1% to 5%) (genus Enterococcus) families, the unclassified Betaproteobacteria (median abundance, 17%), and the Clostridium XVII cluster (median abundance, 11%) (Erysipelotrichaceae family) were also abundant. At days 4 to 8, in addition to Bacteroidaceae, the families Lactobacillaceae (day 4 to 8 median abundance, 7%) (genus Lactobacillus), Porphyromonadaceae (median abundance, 9%) (genus Parabacteroides), and Lachnospiraceae (median abundance, 12%) (genera Blautia and Clostridium XIV cluster) represented more than 10% of the total number of sequences (Fig. 4C; see also Table SA2).
FISH with group-specific probes.
The results of microbial community profiling allowed us to choose 16S rRNA gene-specific fluorescent probes to identify bacteria within mucus-associated communities harboring C. difficile (Table 1). We selected probes specific to already known mucus-associated bacterial genera. Each of these genera represented more than 10% of the community at several stages of disease. Colabeling of mouse intestinal samples with the Cd198 probe and one of the group-specific probes allowed us to visualize bacteria present in C. difficile communities (Fig. 5).
Using the Bac303 probe to localize the most abundant bacteria belonging to the family Bacteroidaceae, we detected small rod-shaped bacteria in the mucus, in samples from both the distal colon and cecum (Fig. 5C). The densities of the Bac303-positive communities varied depending on location. The highest density of Bac303-positive bacteria was detected in the lumen at 1 to 2 days p.i. We found that Enterobacteriaceae-containing communities (in experiments using the Ent183 probe) were mostly in the interlaced layer of the distal colon (Fig. 5D). These communities were associated with loose mucus and contained rod-shaped bacteria. At 4 to 6 days p.i., we found that the luminal content stained intensely with the Ent183 probe. We detected members of the Lactobacillaceae and Enterococcaceae at low density (using the Lab158 probe) on loose mucus in the cecum as well as in the distal colon, rarely in association with sites of pathology (Fig. 5F). Bacteria that stained positively with the Lab158 probe formed small aggregates and were abundant in the lumen at day 4 p.i. (Fig. 5B). Using the Erec482 probe, we detected members of Clostridium coccoides-Eubacterium rectale group often in communities associated with pathology. These bacteria appeared to be significantly smaller in size than other bacteria in these communities and were present at low density in any given community (Fig. 5E). We also detected bacteria with this probe in the lumen at day 6 to day 8 p.i. Application of group-specific probes to samples from uninfected mice as well as to samples from antibiotic-treated but uninfected mice allowed detection of the groups of bacteria which were most abundant on the basis of the results of microbiota profiling (Fig. 4C; see also Fig. S3 in the supplemental material).
DISCUSSION
In this study, we used FISH and 16S rRNA gene sequence analysis to draw four major conclusions about CDI in the mouse: (i) during infection, C. difficile is found in communities in the cecum and colon, starting at day 1 p.i.; (ii) these communities are associated with the loose, outer layer of the mucus; (iii) C. difficile is a minority member of these communities; and (iv) the communities contain bacteria of several families of Bacteroidetes and Firmicutes.
Our observation of the locations of C. difficile during infection is consistent with the findings of Neumann et al., who identified C. difficile associated with mucosa at low density in vivo, using confocal laser endomicroscopy and FISH with a C. difficile-specific probe, in colonic biopsy samples (36). In contrast to the results reported by Lawley and colleagues (5), we did not detect dense mats of cells covering damaged enterocytes. Rather, we found that C. difficile forms mucin-associated communities in the cecum and colon up to at least 8 days p.i. We did not detect any C. difficile cells in direct association with the epithelium or within crypts, as was shown previously (16). This difference could be explained by differences in our infection models and/or properties of the strain we used for infection. Differences in the distributions of different strains have been documented in the hamster model previously, in that C. difficile strain 630 was associated predominantly with the deep crypts and strain B1 was found primarily within the mucosal tissue associated with inflammatory cells (37).
Our finding that C. difficile is associated with other species is an important outcome of this study. We found that bacterial communities harboring C. difficile contain bacterial species belonging to multiple Bacteroidetes and Firmicutes families. The potential for significant impact of specific components of the microbiota on C. difficile pathogenesis, in terms of both the course of disease and overall severity, is an important emerging concept. Several studies found overall changes in the composition of the microbiota during CDI that resembled those found here, although the specific bacteria were not always identified (4, 15, 38, 39). Lawley and colleagues documented that antibiotic treatment and C. difficile infection affect bacterial community structure, causing a depletion of numbers of obligate anaerobic bacteria and a bloom of facultative anaerobes, including Enterobacteriaceae (5). As another example of this effect, clindamycin treatment in mice was found to cause an increase in the abundance of members of the Enterobacteriaceae family and of enterococci (40). A similar result was also demonstrated in a chemostat model, where C. difficile germination and proliferation after ceftriaxone treatment were associated with a reduction of Bifidobacterium species population levels and an increase in Enterococcus species population levels (41).
We interpret our results as suggesting that the presence of C. difficile communities in mucus is a key step in the pathogenesis of CDI. In this regard, it is interesting that about 1% of the microbiota resident in humans can use mucin as a carbon source (42). An interesting speculative possibility is that the species composition of the communities that we detect at the mucus is, at least in part, an adaptation that allows community members to metabolize mucin. Members of Bacteroides, Bifidobacteria, Enterobacteria, and Clostridia can participate in mucin degradation (42–44). The possibility that mucin degradation is specifically involved in pathogenesis is supported by data from studies of both Salmonella and C. difficile bacteria, which can use mucin-derived monosaccharides, available due to dysbiosis, to invade the host GI tract (12). Mucin degradation requires multiple enzymatic activities which, plausibly, could come from contributions by several community members.
This study raised an important question: are C. difficile spores present in the communities that we identified in the mucosa? Our inability to detect spores with our current methodology is a limitation of the present study and is under investigation in our laboratory. Nonetheless, results of studies of C. difficile communities formed in laboratory cultures (biofilms) suggest that, at least under those conditions, spores are readily formed (45, 46). Therefore, it is plausible that spores are present in C. difficile-containing communities in the mucosa during infection. This could have important implications for recurrent disease, as spores can persist for very long periods of time and are insensitive to antibiotics. Therefore, they are potential causes of recurrent disease (1).
Our data show that C. difficile is present with other bacteria in discrete communities within the outer mucin layer of the GI tract during CDI. The identities of these bacteria, and their specific interactions with C. difficile, are likely to be important factors with respect to pathogenesis and disease outcomes. Microbial community profiling reveals a rich population structure in the GI tract and raises the possibility that many more species remain to be identified within C. difficile-containing communities. We expect that the application of additional 16S rRNA gene probes targeting high-level taxa to better characterize the composition of these communities will reveal important information regarding the local environment of C. difficile in the host during infection and will provide useful insights into improved treatment of CDI.
Supplementary Material
ACKNOWLEDGMENTS
We thank Karen Dempsey and Lourdcymole Pazhampally for mouse tissue processing, Mathew Perisin for technical assistance with DNA sequencing, and Linda Fox for advice on confocal microscopy.
This work was funded by grants from the Falk Foundation and the National Institutes of Health (R21AI097934) to A.D. and was partly funded by a National Institutes of Health grant (NIH 1R21AI099713-01) to V.A.P.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00145-15.
REFERENCES
- 1.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 2.Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas V, Rochet V, Boureau H, Ekstrand C, Bulteau S, Dore J, Bourlioux P. 2002. Molecular characterization and spatial analysis of a simplified gut microbiota displaying colonization resistance against Clostridium difficile. Microb Ecol Health Dis 14:203–210. doi: 10.1080/08910600310002082. [DOI] [Google Scholar]
- 4.Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, Rand KH, Wang GP. 2013. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol 51:2884–2892. doi: 10.1128/JCM.00845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C, Deakin LJ, Pickard DJ, Duncan SH, Flint HJ, Clark TG, Parkhill J, Dougan G. 2012. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog 8:e1002995. doi: 10.1371/journal.ppat.1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sorg JA, Sonenshein AL. 2008. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol 190:2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rea MC, Alemayehu D, Casey PG, O'Connor PM, Lawlor PG, Walsh M, Shanahan F, Kiely B, Ross RP, Hill C. 2014. Bioavailability of the anti-clostridial bacteriocin thuricin CD in gastrointestinal tract. Microbiology 160:439–445. doi: 10.1099/mic.0.068767-0. [DOI] [PubMed] [Google Scholar]
- 8.Kamada N, Seo SU, Chen GY, Nunez G. 2013. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 13:321–335. doi: 10.1038/nri3430. [DOI] [PubMed] [Google Scholar]
- 9.Wilson KH, Perini F. 1988. Role of competition for nutrients in suppression of Clostridium difficile by the colonic microflora. Infect Immun 56:2610–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pham TA, Lawley TD. 2014. Emerging insights on intestinal dysbiosis during bacterial infections. Curr Opin Microbiol 17:67–74. doi: 10.1016/j.mib.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Britton RA, Young VB. 2012. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol 20:313–319. doi: 10.1016/j.tim.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, Sonnenburg JL. 2013. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502:96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Theriot CM, Koenigsknecht MJ, Carlson PE Jr, Hatton GE, Nelson AM, Li B, Huffnagle GB, Li JZ, Young VB. 2014. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 5:3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferreyra JA, Wu KJ, Hryckowian AJ, Bouley DM, Weimer BC, Sonnenburg JL. 2014. Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe 16:770–777. doi: 10.1016/j.chom.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reeves AE, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. 2011. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile Infection. Gut Microbes 2:145–158. doi: 10.4161/gmic.2.3.16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buckley AM, Spencer J, Candlish D, Irvine JJ, Douce GR. 2011. Infection of hamsters with the UK Clostridium difficile ribotype 027 outbreak strain R20291. J Med Microbiol 60:1174–1180. doi: 10.1099/jmm.0.028514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnston PF, Gerding DN, Knight KL. 2014. Protection from Clostridium difficile infection in CD4 T cell- and polymeric immunoglobulin receptor-deficient mice. Infect Immun 82:522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuo K, Ota H, Akamatsu T, Sugiyama A, Katsuyama T. 1997. Histochemistry of the surface mucous gel layer of the human colon. Gut 40:782–789. doi: 10.1136/gut.40.6.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bloedt K, Riecker M, Poppert S, Wellinghausen N. 2009. Evaluation of new selective culture media and a rapid fluorescence in situ hybridization assay for identification of Clostridium difficile from stool samples. J Med Microbiol 58:874–877. doi: 10.1099/jmm.0.009811-0. [DOI] [PubMed] [Google Scholar]
- 20.Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA. 1990. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol 56:1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daims H, Bruhl A, Amann R, Schleifer KH, Wagner M. 1999. The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst Appl Microbiol 22:434–444. doi: 10.1016/S0723-2020(99)80053-8. [DOI] [PubMed] [Google Scholar]
- 22.Swidsinski A, Loening-Baucke V, Verstraelen H, Osowska S, Doerffel Y. 2008. Biostructure of fecal microbiota in healthy subjects and patients with chronic idiopathic diarrhea. Gastroenterology 135:568–579. doi: 10.1053/j.gastro.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 23.Salzman NH, de Jong H, Paterson Y, Harmsen HJM, Welling GW, Bos NA. 2002. Analysis of 16S libraries of mouse gastrointestinal microflora reveals a large new group of mouse intestinal bacteria. Microbiology 148:3651–3660. doi: 10.1099/00221287-148-11-3651. [DOI] [PubMed] [Google Scholar]
- 24.Friedrich U, Van Langenhove H, Altendorf K, Lipski A. 2003. Microbial community and physicochemical analysis of an industrial waste gas biofilter and design of 16S rRNA-targeting oligonucleotide probes. Environ Microbiol 5:183–201. doi: 10.1046/j.1462-2920.2003.00397.x. [DOI] [PubMed] [Google Scholar]
- 25.Manz W, Amann R, Ludwig W, Vancanneyt M, Schleifer KH. 1996. Application of a suite of 16S rRNA-specific oligonucleotide probes designed to investigate bacteria of the phylum cytophaga-flavobacter-bacteroides in the natural environment. Microbiology 142(Pt 5):1097–1106. doi: 10.1099/13500872-142-5-1097. [DOI] [PubMed] [Google Scholar]
- 26.McInnes P, Cutting M. 2010. Manual of procedures for Human Microbiome Project core microbiome sampling protocol A: HMP protocol no. 07-001, version no. 12.0. NIH, Bethesda, MD: http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/GetPdf.cgi?id=phd003190.2. [Google Scholar]
- 27.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White JR, Nagarajan N, Pop M. 2009. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol 5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, Kelly CP. 2008. A mouse model of Clostridium difficile-associated disease. Gastroenterology 135:1984–1992. doi: 10.1053/j.gastro.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Johansson ME, Larsson JM, Hansson GC. 2011. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A 108:4659–4665. doi: 10.1073/pnas.1006451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swidsinski A, Loening-Baucke V, Lochs H, Hale LP. 2005. Spatial organization of bacterial flora in normal and inflamed intestine: a fluorescence in situ hybridization study in mice. World J Gastroenterol 11:1131–1140. doi: 10.3748/wjg.v11.i8.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Good IJ. 1953. The Population frequencies of species and the estimation of population parameters. Biometrika 40:237–264. [Google Scholar]
- 35.Simpson EH. 1949. Measurement of diversity. Nature 163:688. doi: 10.1038/163688a0. [DOI] [Google Scholar]
- 36.Neumann H, Vieth M, Raithel M, Mudter J, Kiesslich R, Neurath MF. 2010. Confocal laser endomicroscopy for the in vivo detection of intraepithelial neoplasia in Peutz-Jeghers polyps. Endoscopy 42(Suppl 2):E139–E140. doi: 10.1055/s-0029-1244052. [DOI] [PubMed] [Google Scholar]
- 37.Goulding D, Thompson H, Emerson J, Fairweather NF, Dougan G, Douce GR. 2009. Distinctive profiles of infection and pathology in hamsters infected with Clostridium difficile strains 630 and B1. Infect Immun 77:5478–5485. doi: 10.1128/IAI.00551-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, Young VB. 2008. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis 197:435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- 39.Koenigsknecht MJ, Theriot CM, Bergin IL, Schumacher CA, Schloss PD, Young VB. 2015. Dynamics and establishment of Clostridium difficile infection in the murine gastrointestinal tract. Infect Immun 83:934–941. doi: 10.1128/IAI.02768-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, Ubeda C, Xavier J, Pamer EG. 2012. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun 80:62–73. doi: 10.1128/IAI.05496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baines SD, Crowther GS, Todhunter SL, Freeman J, Chilton CH, Fawley WN, Wilcox MH. 2013. Mixed infection by Clostridium difficile in an in vitro model of the human gut. J Antimicrob Chemother 68:1139–1143. doi: 10.1093/jac/dks529. [DOI] [PubMed] [Google Scholar]
- 42.Derrien M, van Passel MW, van de Bovenkamp JH, Schipper RG, de Vos WM, Dekker J. 2010. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes 1:254–268. doi: 10.4161/gmic.1.4.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Probert HM, Gibson GR. 2002. Bacterial biofilms in the human gastrointestinal tract. Curr Issues Intest Microbiol 3:23–27. [PubMed] [Google Scholar]
- 44.Macfarlane S, Woodmansey EJ, Macfarlane GT. 2005. Colonization of mucin by human intestinal bacteria and establishment of biofilm communities in a two-stage continuous culture system. Appl Environ Microbiol 71:7483–7492. doi: 10.1128/AEM.71.11.7483-7492.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Semenyuk EG, Laning ML, Foley J, Johnston PF, Knight KL, Gerding D, Driks A. 2014. Spore formation and toxin production in Clostridium difficile biofilms. PLoS One 9:e87757. doi: 10.1371/journal.pone.0087757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ðapa T, Leuzzi R, Ng YK, Baban ST, Adamo R, Kuehne SA, Scarselli M, Minton NP, Serruto D, Unnikrishnan M. 2013. Multiple factors modulate biofilm formation by the anaerobic pathogen Clostridium difficile. J Bacteriol 195:545–555. doi: 10.1128/JB.01980-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.