

Abstract
Malignant brain tumors are characterized by destructive growth and neuronal cell death making them one of the most devastating diseases. Neurodegenerative actions of malignant gliomas resemble mechanisms also found in many neurodegenerative diseases such as Alzheimer's and Parkinson's diseases and amyotrophic lateral sclerosis. Recent data demonstrate that gliomas seize neuronal glutamate signaling for their own growth advantage. Excessive glutamate release via the glutamate/cystine antiporter xCT (system xc-, SLC7a11) renders cancer cells resistant to chemotherapeutics and create the tumor microenvironment toxic for neurons. In particular the glutamate/cystine antiporter xCT takes center stage in neurodegenerative processes and sets this transporter a potential prime target for cancer therapy. Noteworthy is the finding, that reactive oxygen species (ROS) activate transient receptor potential (TRP) channels and thereby TRP channels can potentiate glutamate release. Yet another important biological feature of the xCT/glutamate system is its modulatory effect on the tumor microenvironment with impact on host cells and the cancer stem cell niche. The EMA and FDA-approved drug sulfasalazine (SAS) presents a lead compound for xCT inhibition, although so far clinical trials on glioblastomas with SAS were ambiguous. Here, we critically analyze the mechanisms of action of xCT antiporter on malignant gliomas and in the tumor microenvironment. Deciphering the impact of xCT and glutamate and its relation to TRP channels in brain tumors pave the way for developing important cancer microenvironmental modulators and drugable lead targets.
Keywords: GBM, glutamate, neuron, NMDA, ROS, systemX, TRP, xCT.
INTRODUCTION
Primary brain tumors are derived from glial cells or glial progenitors or stem cells. Malignant or high grade gliomas such as the glioblastoma are the most malignant human entities [1, 2]. Despite multimodal therapy regimens including cytoreduction, radiotherapy and cytotoxic chemotherapy (i.e. temozolomide), the prognosis of glioma patients remains poor, i.e. less than 3% of patients survive more than five years [3, 4]. Rapid proliferation, diffuse brain invasion as well as tumor-induced brain edema and neurodegeneration are pathological hallmarks of these tumors and determine the unfavorable prognosis [5]. A common concept concerning the development of cancer considers deregulation of specifically metabolic-associated genes with link to the redox system [6, 7]. Generally, metabolic processes are associated with the formation of reactive oxygen species (ROS) such as hydroxyl radical (-OH), hydrogen peroxide (H2O2), superoxide radical (O2-) and oxygen (O2) which contribute to TRP channel activation, Ca2+ shifts and oncogenic challenges. Oncogenic transformation via oncogenic activation and loss of tumor suppressor genes conversely drives metabolism and thereby initiate a vicious circle with fostered aerobic glycolysis at the expense of the host organism. The price cancer cells have to pay for increased cell growth and survival is their thirst for glucose due to high consumption of carbon skeletons, NADPH, and ATP. To a great extent nutrient transporters meet this task and in addition are also involved in regulating redox homeostasis and cell metabolism [8, 9]. Current knowledge on nutrient transporters in cancer points to the fact that cancer cells adjust their increased demand for nutrients and excessive glucose metabolism via these transporters [10-13]. To keep up with the increased energy demand, cancer cells express nutrient transporters for lactate and amino acids which in particular are primarily related to augmentation of ROS and redox cycle regulation [14, 15]. Another clinical feature of increased transporter action is the influence on the host tissue. Nutrient transporters such as the monocarboxylate/lactate transporters have direct impact on the tumor microenvironment influencing various processes such as cell survival, immune response and angiogenesis [13, 16].
BRAIN TUMORS HIJACK THE GLUTAMATE SIGNALING SYSTEM FOR THEIR GROWTH ADVANTAGE
There exist now ample evidence that the cystine/glutamate antiporter xCT is expressed in various malignant tumors such as leukemias [17, 18], lymphomas [19-21], Karposi´s sarcoma [21, 22], pancreatic cancer [23], breast cancer [24, 25], squamous cell carcinoma/epithelial carcinomas [26, 27] and brain tumors [5, 28, 29]. There, the cystine/glutamate antiporter system xc- is composed of the catalytic domain xCT (solute carrier family 7 member 11, SLC7a11) and its heavy chain chaperone CD98/4F2hc (SLC3A2) (Fig. 1). In this complex, xCT functions as a Na-independent electro-neutral exchange system for cystine/glutamate with cystine entry and glutamate efflux in a 1:1 molar ratio [30, 31]. Intracellular cystine becomes reduced to cysteine required for the tripeptide glutathione (GSH) and protein biosynthetic pathways (Fig. 1). Importantly, cysteine is the rate-limiting substrate for GSH synthesis and GSH is required for proliferation, redox cycling and antioxidative defense [32, 33]. This dual function of system xc- (xCT) and its interrelation to cell proliferation led to the concept of targeting xCT in cancer. The idea on which inhibition of xCT is based on was to reduce intracellular glutathione levels and thereby subsequently disturbing cellular redox balance leading eventually to cancer cell death. Such cytotoxic strategy is especially appealing when the drug target acts specifically on cancer cells and target inhibition does not awry normal cell functioning within the therapeutic window.
Fig. (1).
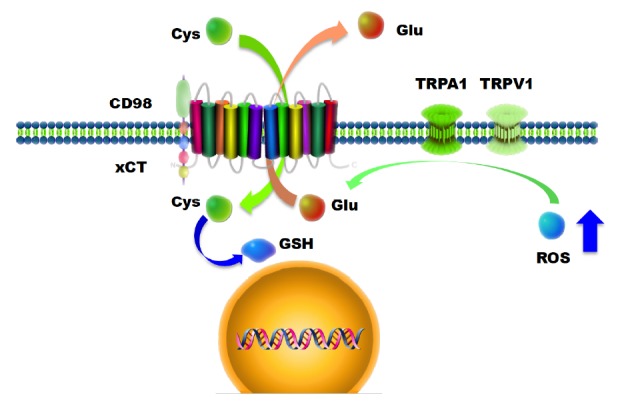
The heterodimeric glutamate-cystine antiporter system Xc-/xCT (SCL7a11) and its relation to TRP channels. xCT is an integral membrane protein with 12 transmembrane domains and forms a complex with CD98. xCT transports Na+-independent glutamate out in exchange for cystine. Cystine is further processed to its reduced form cysteine which is further acquired for protein and glutathione (GSH) biosynthesis. The ‘glutathione-only’ hypothesis defines this particular function essential for cancer cells. A competing concept sets glutamate in the focus of the biology of brain tumors (glutamate-microenvironment hypothesis). The glutamate-microenvironment hypothesis is supported by data demonstrating that xCT knock down or genetic xCT deficiency is not essential for cell growth. Further, TRP channels are ROS-sensitive and can mediate glutamate release. In this scheme it is speculated that TRP channels may challenge xCT transporter activity. Abbreviations: Cys, cysteine; Glu, glutamate; GSH, glutathione; ROS, reactive oxygen species.
Indeed, many cancer cells express elevated levels of xCT and one explanation for this phenomenon is that cancer cells facilitate xCT for their glutathione (GSH) demands required for recovering the increased glutathione consumptions [28, 34, 35]. However, recent data indicate an alternative scenario which opens another biological function apart from the ‘glutathione-only’ concept (Fig. 1). Independent experimental evidences show that increased xCT expression in tumor cells does not necessarily recover intracellular glutathione levels even though xCT overexpression gains cell survival under glutathione and glutamine depletion [19, 24, 36]. This discloses another biological role of the antiporter xCT with respect to the extracellular space. In addition to the intracellular cis-action xCT modulates also the extracellular space by releasing high amounts of glutamate (glutamate-microenvironment hypothesis) [6, 36].
This overlooked aspect of the xCT antiporter biology comes from the perception that the metabolite glutamate which is exported in exchange to cystine has been viewed solely as a byproduct of the GSH cycle. In fact the amino acid glutamate represents a potent signaling molecule and neurotransmitter acting on ionotropic and metabotropic glutamate receptors [37-39]. While in the brain glutamate signaling evokes Ca2+-dependent depolarization of the membrane, excessive glutamate release and hence glutamate receptor activation can lead to excitatory neuronal cell death [40, 41]. In particular in primary brain tumors elevated xCT expression is interconnected with increased extracellular glutamate levels in the peritumoral zone [5, 42] resulting in neuronal damage, brain swelling and tumor-associated seizures [5, 43]. Numerous studies demonstrated in experimental and clinical settings that glioma cells secrete high levels of the neurotransmitter glutamate, resulting in neuronal damage [5, 44-46]. Conversely, antagonizing ionotropic glutamate receptors alleviate neuronal degeneration in the tumor vicinity and lessen glioma growth in vivo, suggesting that neurodegeneration is a prerequisite for rapid glioma progression [42]. Excessive glutamate release by glioma cells has also been linked to decreased activities of glial glutamate transporters such as EAAT1 (GLAST/SLC1A3) and EAAT2 (GLT-1/SLC1A2). This can in addition result in decreased glutamate incorporation and consequently reduced extracellular glutamate clearance.
In consequence, reduced EAAT1 and EAAT2 expression and concurrently abundant xCT (system Xc-) activity lead to a net balance shifted towards extracellular glutamate release in favor of glioma progression. In line with these findings, enhancing EAAT2 expression in C6 rat glioma cell grafts reduced tumor size and elevated the lifespan of tumor-bearing animals [47]. Moreover, inhibition of glutamate release via system Xc- (old term for the xCT (SLC7A11) and CD98 (SLC3A2, 4F2HC) heterodimer), profoundly decelerates the malignant phenotype of gliomas in vivo [5, 48, 49] and in addition mitigates tumor-induced brain swelling and microenvironmental disturbances [5]. Even though the glioma-promoting properties of glutamate release by glioma cells appear to be without doubt, the underlying mechanisms are still under investigation. Besides neurotoxic effects, there is good evidence for extracellular glutamate as a promoter for glioma and cancer growth [50, 51]. The mode of action of the growth promoting effects of glutamate could possibly be in an autocrine or paracrine fashion via activation of the fast transmitting ionotropic glutamate receptors on gliomas. Here, especially the NMDA and AMPA receptor subtypes are in focus of current research activities [52].
CROSSTALK BETWEEN TRP CHANNELS AND GLUTAMATE RELEASE
Recent data indicate that TRP channels can modulate and potentiate glutamate release in a ROS dependent manner [53-55]. In particular this has been demonstrated for TRPA1, TRPM3 and TRPV1 in spinal cord and brain. Although this connection between TRP channels and glutamate release has been shown for pre-synaptic terminals, it is likely that such regulatory mechanisms exist also in glial-derived primary brain tumors (Fig. 1). These data point to an orphan role of TRP channels in glutamate signaling with wide ranging implications. Since xCT and subsequently glutamate signaling in gliomas is associated with resistance towards chemotherapeutic agents (such as temozolomide and carmustine), tumor invasion and neuronal cell death, TRP channels may provide a new starting point for targeting this system (Table 1). This is in particular relevant since TRP channels have been considered as targets of chemotherapeutics and drug carriers enabling the efficacy of tamoxifen or all-trans retinoic acid on cancer cells.
Table 1.
TRP channel tissue distribution and expression in tumors.
| TRP Subfamily | Organ Distribution | Cancer Types | Expression in Tumors | Relevation to Glutamate |
|---|---|---|---|---|
| TRPV1 | Brain, Skin,Prostate | Glioma | ↑↑ | ++ |
| TRPV2 | Brain, Prostate, | Glioma | ↓↓ | ? |
| TRPA1 | Skin, Brain | ? | ? | ++ |
| TRPC5 | Breast, Brain | ? | ? | ? |
| TRPC6 | Breast, Brain, Prostate | Glioma | ↑ | ? |
| TRPM2 | Prostate | Prostate cancer | ↑↑ | ? |
| TRPM3 | Brain | Ependymoma | ↑↑ | ++ |
| TRPM7 | Breast | Breast cancer | ↑↑ | ? |
Overview of the expression pattern of TRP channels in relation to brain and glutamate signalling. Boxes with question marks indicate missing published data.
Apart from glutamate release, the xCT antiporter is involved in cysteine uptake, which is further metabolized into glutathione (GSH). GSH belongs to the main cellular reactive oxygen scavenging systems balancing cytosolic reactive oxygen species (ROS, H2O2) and gaseous messenger molecules (O2, H2C, CO2). Noteworthy is the fact that TRP channels such as TRPA1, TRPV1, TRPV4, TRPM2, TRPM7 and TRPC5 can act as sensors for ROS and molecular oxygens and subsequently can be activated upon GSH and ROS shifts [56]. Thus, the glutamate-cystine antiporter xCT could potentially regulate TRP channel activation by its ROS regulating impact in the cytosol. Vice versa, TRP channels may impact on xCT and drive glutamate release thereby modulating glutamate signaling. The basis for such ROS sensitivity lays in the redox sensitive cysteine residues present in TRP channels. It has been shown that ROS can modify covalently cysteine residues and thereby alters the responsiveness to stimuli [55]. Alternatively, ROS challenges in the extracellular space can affect agonists of TRP channels and therefore influence the TRP activation in a paracrine mode.
In conclusion, there is now increasing evidence for a role of TRP channels in malignant brain tumors (Table 1). First, TRPV1, TRPV2, and TRPC6 have been found to be expressed and up-regulated in gliomas [53, 57, 58]. Second, TRP channels can transit directly glioma cell cycle progression and thereby contribute to glioma development as shown for TRPC6 [58].
The relation of TRP and brain tumors becomes less linear when considering TRP vanilloid type channels. Noteworthy, it has been shown that TRPV1 and TRPV2 stimulation can cause cell death in malignant glioma [59, 60]. Moreover, there is a role for TRPV channels also in neuro-inflammation (Table 1). Thus, future studies will unravel whether TRP channels are involved in tumor-immune responses and tumor-associated microglial modulation.
GLUTAMATE MODULATES THE TUMOR MICRO-ENVIRONMENT
Malignant gliomas can disturb the extracellular glutamate homeostasis in the brain via enhanced xCT antiporter activity. Interestingly, in contrast to glial cells or neurons, gliomas can cope with toxic glutamate levels and utilize the amino acid for their growth advantage [6]. Conversely, brain cells are sensitive even to subtle glutamate challenges and are prone to excessive glutamate levels. Neurons, microglial cells and macroglial cells (i.e. astrocytes and oligodendrocytes) have been shown to express ionotropic and metabotropic glutamate receptors in a temporal and spatial manner [61-63]. Neurons and oligodendrocytes are in particular sensitive to micro-molar elevations of glutamate and enduring stimulation leads to neuronal cell death around the initial ischemic area in stroke, a process also termed as penumbra. A recent investigation revealed that peri-infarct depolarization in ischemia is driven by elevated xCT expression [64]. This study by Soria and colleagues uncovered for the first time a role of xCT in stroke contributing to neurodegeneration as has been shown in brain tumors [5]. Although the penumbra in stroke and peritumoral zone in gliomas has different underlying mechanisms the events of neurodegeneration are in both cases glutamate-driven. Astrocytes can buffer neurotoxic glutamate concentrations to a certain extent and provide protection for neurons. However, exposure of excessive glutamate levels to astrocytes eventually leads to gliotoxicty as well [65]. Microglial cells express ionotropic and metabotropic glutamate receptors which are implicated in microglial activation and motility [66]. Up to now the precise microglial activation status (M1 or M2) upon glutamate needs yet to be defined. Following brain damage glutamate levels burst potentially from dying neurons and astrocytes and activated microglial cells migrate to the lesion site and subsequently proliferate and phagocytose damaged cells and debris. There, the spatial and temporal activation of microglial cells is decisive whether microglia contributes in a beneficial or detrimental manner. Under chronic conditions microglial function is less unambiguous indicating a correlation of microglial activation and ongoing neurodegenerative diseases. There, microglial cells have the capacity to release large amount of detrimental factors and ROS thereby poisoning their microenvironment. Interestingly, microglial redox balance has been indicated to be controlled by the induction of xCT [67].
What is the biological advantage for malignant gliomas to release high amounts of glutamate?
First, malignant gliomas show destructive characteristics to their environment and are hallmarked by the induction of cell death and neurodegeneration. Second, this tumor entity is surrounded by a unique environment which is restricted in space due to the bony scull. This makes the environment of primary brain tumors so different to those of non-CNS tumors.
Thus, the physical constrains in space and the hallmark of massive neuronal destruction made the hypothesis appealing that glioma-derived glutamate release is implicated in the mechanisms for creating space for tumor growth (Fig. 2). The level of tumor-derived extracellular glutamate has been determined in vitro and in vivo and glutamate levels above 250 µM have been monitored [44, 45, 68], a concentration known to be neurotoxic. Also, there are independent evidences that malignant gliomas destroy the peritumoral area which is also reflected by the development of cytotoxic edema. Moreover, these data are confirmed by studies utilizing the competitive NMDA receptor antagonist MK801 or uncompetitive NMDA receptor antagonist memantine in gliomas where neuronal cell death could be inhibited. However, pharmacological or genetic inhibition of xCT revealed that glioma cells grow unlimited albeit their neurodegenerative potential is restricted. Thus, the concept that the neurodegenerative potential of gliomas creates space for unrestricted tumor growth in the brain is thus not likely to be the main biological role of xCT in brain tumors. In favor for alternative explanation is the finding that tumor-derived glutamate induces a plethora of signaling events beside neuronal damage (Fig. 2).
Fig. (2).

The tumor microenvironment in brain tumors. Scheme of the brain tumor microenvironment and its various compartments. To cover the heterogeneity of malignant gliomas the tumor zone model classifies gliomas into three distinct tumor zones [3]. Left, given is the tumor core or tumor zone I (TZI, black spot), peritumoral zone (yellow encircled), and the tumor zone III (margenda encircled). Right, higher magnification of the boxed area. The glioma microenvironment impacts on host cells such as neurons (orange), vessels (pink), astrocytes (blue) and microglia (purple). Glioma cells are given in green. The glutamate gradient is shown in red dots. Abbreviations: TZ, tumor zones (numbered I to III).
These may have not yet been recognized such as effects on the blood-brain barrier, blood flow and metabolism. The not well perceived events are focus of ongoing research. Up to now there is solid evidence that xCT is the essential glutamate transporter in malignant gliomas related to excessive glutamate release.
XCT AND GLUTAMATE: DRUGABLE TARGETS FOR THERAPY
Now, there exist robust data on the consequences of xCT expression in gliomas which call for clinical interventions. So how can we block xCT in tumors and what is the expected outcome for patients?
Up to now two compounds are commonly in use for the pharmacological disruption of xCT. First, the phenylglycine derivate (S)-4-carboxyphenylglycine (S-4CPG) has been described as competitive antagonist for Group I metabotropic glutamate receptors with an approx. IC50 of 4x10-5 M [69-71]. Later, S4CPG was discovered to competitively inhibit system xc- in a non-substrate dependent manner with a Ki of 5x10-6 M and IC80 of 5x10-5 M [31]. In primary brain tumor cells xCT-drug targeting with S-4CPG in micro molar range can potently inhibit glutamate secretion [49]. In vivo, S-4CPG reduces tumor-induced neuronal cell death [5]. In addition, S-4CPG can exert antiproliferative and cytotoxic effects at higher concentrations in some glioma cell lines, pointing to the fact that xCT inhibition does not act generally cytotoxic for cancer. Indeed, genetic deletion of xCT by homologous recombination (xCT-/-) or by naturally occurring mutations (in subtle grey [Sut] mice) display no malformation or alterations in cell proliferation in vivo [72-74]. This indicates that xCT is dispensable for normal cell proliferation and stem cell growth. Notably, the xCT knockout mouse and the spontaneous occurring xCT mutant sut differ in particular behavioral and chemical tests and thus further investigations are required for valid xCT mouse models [75].
Second, Sulfasalazine (brand name Azulfidine in the U.S., Salazopyrin in Europe) is a FDA approved drug for the treatment of inflammatory bowel disease and rheumatoid arthritis. In addition Sulfasalazine (SAS) has been described for the inhibition of xCT/system xc- in normal and tumor cells [30, 76]. However, antiproliferative effects of SAS on tumor cells are not likely to be exclusively attributable to xCT inhibition since SAS actions are generally pleiotropic. It has been reported that SAS can inhibit NFκB [77, 78] and glutathione transferase [79], and also shows a wealth of immunomodulatory actions [80]. Thus, so far with the two available drugs in hand information on xCT in tumor biology is limited since off-target effects and xCT-independent effects cannot clearly be distinguished from sole xCT inhibitory effects.
This may be able to be overcome by more recently developed small molecule inhibitors. Currently, one study reported on erastin and sorafenib (Nexavar) as potential xCT inhibitors in the micromolar range [81]. This is in particular interesting since erastin is known as an antitumor agent selective for tumor cells bearing oncogenic RAS. Formerly erastin was supposed to be acting on mitochondrial voltage-dependent anion channel (VDAC) gating.
Recently, histone deacetylase inhibitors (HDACi) as an entirely new drug family have been shown to specifically target the glutamate transporter xCT [82]. The HDACi vorinostat (suberoylanilide hydroxamic acid or SAHA) is a clinically established drug especially used for chemotherapeutically resistant tumors. High xCT expression in tumors is associated with chemoresistancy [36]. The finding that epigenetic modulation by histone deacetylase inhibitors impacts xCT expression opens new avenues for modulating the brain tumor microenvironment and treating neurodegeneration.
Thus, it can be expected in the future that better drugs and eventually small molecule inhibitors for xCT will expanding the experimental tools to further decipher xCT function in physiology and in cancer. Moreover, the relation of TRP channel activation and glutamate release opens a new path to control glioma-derived glutamate signaling. Future investigations will decipher whether TRP channels and their pharmacological targets offer an option for regulating tumor-induced glutamate-dependent excitotoxicity.
CONCLUSION AND FUTURE SUBJECT
What can we expect as an outcome when blocking the xCT transporter in cancer?
The initial ‘glutathione-only’ hypothesis that xCT biology is primarily based on the role of glutathione in cell proliferation is probably only one side of the coin. Evidences exist that equally important for the clinical settings are the effects of xCT on the tumor microenvironment. There, xCT regulates extracellular glutamate levels as in the case of brain tumors gives cancer cells a survival advantage over neurons. Another important aspect is the regulation of the extracellular cysteine concentration by xCT. Here, xCT directly operates on the net lipid peroxidation level at the plasma membrane and thus can lead to resistance towards glutathione depleting anticancer drugs [23]. Thus, inhibition of xCT transporter in cancer extents the cytotoxic approach as xCT inhibition can reduce intracellular glutathione levels and at the same time modulates the tumor microenvironment. Dependent on the type of cancer one or the other effect may dominate the outcome of treatment and hence determines its efficacy in cancer therapy. Another rising topic is the modulation of xCT which could be in principle achieved by TRP channels. Whether this is a valid path also in brain tumors will be deciphered in future investigations.
ACKNOWLEDGEMENTS
We thank all members of the Cell Biology & Neuro-oncology lab for continuous support and critical discussions during the course of preparation of the manuscript. Tina Sehm and Ali Ghoochani are gratefully acknowledged for their support in xCT and glutamate analysis. Our group is supported by the German Research Council (Deutsche Forschungsgemeinschaft, DFG), the China Scholarship Council (CSC: 2011627126 to Z.F.) and the Verein zur Förderung des Tumorzentrums der Universität Erlangen-Nürnberg.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Dolecek T.A., Propp J.M., Stroup N.E., Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-oncol. 2012;14(Suppl. 5):v1–v49. doi: 10.1093/neuonc/nos218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Furnari F.B., Fenton T., Bachoo R.M., Mukasa A., Stommel J.M., Stegh A., Hahn W.C., Ligon K.L., Louis D.N., Brennan C., Chin L., DePinho R.A., Cavenee W.K. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21(21):2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 3.Eyupoglu I.Y., Buchfelder M., Savaskan N.E. Surgical resection of malignant gliomas-role in optimizing patient outcome. 2013. [DOI] [PubMed]
- 4.Wen P.Y., Kesari S. Malignant gliomas in adults. N. Engl. J. Med. 2008;359(5):492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 5.Savaskan N.E., Heckel A., Hahnen E., Engelhorn T., Doerfler A., Ganslandt O., Nimsky C., Buchfelder M., Eyüpoglu I.Y. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat. Med. 2008;14(6):629–632. doi: 10.1038/nm1772. [DOI] [PubMed] [Google Scholar]
- 6.Savaskan N.E., Eyüpoglu I.Y. xCT modulation in gliomas: relevance to energy metabolism and tumor microenvironment normalization. Ann. Anat. 2010;192(5):309–313. doi: 10.1016/j.aanat.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Flavahan W.A., Wu Q., Hitomi M., Rahim N., Kim Y., Sloan A.E., Weil R.J., Nakano I., Sarkaria J.N., Stringer B.W., Day B.W., Li M., Lathia J.D., Rich J.N., Hjelmeland A.B. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013;16(10):1373–1382. doi: 10.1038/nn.3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geier E.G., Chen E.C., Webb A., Papp A.C., Yee S.W., Sadee W., Giacomini K.M. Profiling solute carrier transporters in the human blood-brain barrier. Clin. Pharmacol. Ther. 2013;94(6):636–639. doi: 10.1038/clpt.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269–270. [PubMed] [Google Scholar]
- 11.Ganapathy V., Thangaraju M., Prasad P.D. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. 2009. [DOI] [PubMed]
- 12.Dang C.V. Links between metabolism and cancer. Genes Dev. 2012;26(9):877–890. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Outschoorn U., Sotgia F., Lisanti M.P. Tumor microenvironment and metabolic synergy in breast cancers: critical importance of mitochondrial fuels and function. 2014. [DOI] [PubMed]
- 14.Kroemer G., Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 2008 doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Semenza G.L. Angiogenesis in ischemic and neoplastic disorders. Annu. Rev. Med. 2003;54:17–28. doi: 10.1146/annurev.med.54.101601.152418. [DOI] [PubMed] [Google Scholar]
- 16.Kato Y., Ozawa S., Miyamoto C., Maehata Y., Suzuki A., Maeda T., Baba Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013;13(1):89. doi: 10.1186/1475-2867-13-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang W., Trachootham D., Liu J., Chen G., Pelicano H., Garcia-Prieto C., Lu W., Burger J.A., Croce C.M., Plunkett W., Keating M.J., Huang P. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012;14(3):276–286. doi: 10.1038/ncb2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garnier N., Redstone G.G., Dahabieh M.S., Nichol J.N., del Rincon S.V., Gu Y., Bohle D.S., Sun Y., Conklin D.S., Mann K.K., Miller W.H., Jr The novel arsenical darinaparsin is transported by cystine importing systems. Mol. Pharmacol. 2014;85(4):576–585. doi: 10.1124/mol.113.089433. [DOI] [PubMed] [Google Scholar]
- 19.Banjac A., Perisic T., Sato H., Seiler A., Bannai S., Weiss N., Kölle P., Tschoep K., Issels R.D., Daniel P.T., Conrad M., Bornkamm G.W. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene. 2008;27(11):1618–1628. doi: 10.1038/sj.onc.1210796. [DOI] [PubMed] [Google Scholar]
- 20.Chen R.S., Song Y.M., Zhou Z.Y., Tong T., Li Y., Fu M., Guo X.L., Dong L.J., He X., Qiao H.X., Zhan Q.M., Li W. Disruption of xCT inhibits cancer cell metastasis via the caveolin-1/beta-catenin pathway. Oncogene. 2009;28(4):599–609. doi: 10.1038/onc.2008.414. [DOI] [PubMed] [Google Scholar]
- 21.Dai L., Cao Y., Chen Y., Parsons C., Qin Z. Targeting xCT, a cystine-glutamate transporter induces apoptosis and tumor regression for KSHV/HIV-associated lymphoma. J. Hematol. Oncol. 2014;7:30. doi: 10.1186/1756-8722-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qin Z., Freitas E., Sullivan R., Mohan S., Bacelieri R., Branch D., Romano M., Kearney P., Oates J., Plaisance K., Renne R., Kaleeba J., Parsons C. Upregulation of xCT by KSHV-encoded microRNAs facilitates KSHV dissemination and persistence in an environment of oxidative stress. PLoS Pathog. 2010;6(1):e1000742. doi: 10.1371/journal.ppat.1000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Y., Dai Z., Barbacioru C., Sadée W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005;65(16):7446–7454. doi: 10.1158/0008-5472.CAN-04-4267. [DOI] [PubMed] [Google Scholar]
- 24.Timmerman L.A., Holton T., Yuneva M., Louie R.J., Padró M., Daemen A., Hu M., Chan D.A., Ethier S.P., van ’t Veer L.J., Polyak K., McCormick F., Gray J.W. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24(4):450–465. doi: 10.1016/j.ccr.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang F., Yang Y. Suppression of the xCT-CD44v antiporter system sensitizes triple-negative breast cancer cells to doxorubicin. Breast Cancer Res Treat. 2014 doi: 10.1007/s10549-014-3068-6. [DOI] [PubMed] [Google Scholar]
- 26.Yoshikawa M., Tsuchihashi K., Ishimoto T., Yae T., Motohara T., Sugihara E., Onishi N., Masuko T., Yoshizawa K., Kawashiri S., Mukai M., Asoda S., Kawana H., Nakagawa T., Saya H., Nagano O. xCT inhibition depletes CD44v-expressing tumor cells that are resistant to EGFR-targeted therapy in head and neck squamous cell carcinoma. Cancer Res. 2013;73(6):1855–1866. doi: 10.1158/0008-5472.CAN-12-3609-T. [DOI] [PubMed] [Google Scholar]
- 27.Toyoda M., Kaira K., Ohshima Y., Ishioka N.S., Shino M., Sakakura K., Takayasu Y., Takahashi K., Tominaga H., Oriuchi N., Nagamori S., Kanai Y., Oyama T., Chikamatsu K. Prognostic significance of amino-acid transporter expression (LAT1, ASCT2, and xCT) in surgically resected tongue cancer. Br. J. Cancer. 2014;110(10):2506–2513. doi: 10.1038/bjc.2014.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J.Y., Kanai Y., Chairoungdua A., Cha S.H., Matsuo H., Kim D.K., Inatomi J., Sawa H., Ida Y., Endou H. Human cystine/glutamate transporter: cDNA cloning and upregulation by oxidative stress in glioma cells. Biochim. Biophys. Acta. 2001;1512(2):335–344. doi: 10.1016/S0005-2736(01)00338-8. [DOI] [PubMed] [Google Scholar]
- 29.Chairoungdua A., Kanai Y., Matsuo H., Inatomi J., Kim D.K., Endou H. Identification and characterization of a novel member of the heterodimeric amino acid transporter family presumed to be associated with an unknown heavy chain. J. Biol. Chem. 2001;276(52):49390–49399. doi: 10.1074/jbc.M107517200. [DOI] [PubMed] [Google Scholar]
- 30.Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 1986;261(5):2256–2263. [PubMed] [Google Scholar]
- 31.Patel S.A., Warren B.A., Rhoderick J.F., Bridges R.J. Differentiation of substrate and non-substrate inhibitors of transport system xc(-): an obligate exchanger of L-glutamate and L-cystine. Neuropharmacology. 2004;46(2):273–284. doi: 10.1016/j.neuropharm.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 32.Reddy N.M., Kleeberger S.R., Bream J.H., Fallon P.G., Kensler T.W., Yamamoto M., Reddy S.P. Genetic disruption of the Nrf2 compromises cell-cycle progression by impairing GSH-induced redox signaling. Oncogene. 2008;27(44):5821–5832. doi: 10.1038/onc.2008.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seiler A., Schneider M., Förster H., Roth S., Wirth E.K., Culmsee C., Plesnila N., Kremmer E., Rådmark O., Wurst W., Bornkamm G.W., Schweizer U., Conrad M. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8(3):237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Okuno S., Sato H., Kuriyama-Matsumura K., Tamba M., Wang H., Sohda S., Hamada H., Yoshikawa H., Kondo T., Bannai S. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br. J. Cancer. 2003;88(6):951–956. doi: 10.1038/sj.bjc.6600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robert S.M., Ogunrinu-Babarinde T., Holt K.T., Sontheimer H. Role of glutamate transporters in redox homeostasis of the brain. Neurochem. Int. 2014;73:181–191. doi: 10.1016/j.neuint.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savaskan N.E., Seufert S., Hauke J., Tränkle C., Eyüpoglu I.Y., Hahnen E. Dissection of mitogenic and neurodegenerative actions of cystine and glutamate in malignant gliomas. Oncogene. 2011;30(1):43–53. doi: 10.1038/onc.2010.391. [DOI] [PubMed] [Google Scholar]
- 37.Uchihori Y., Puro D.G. Glutamate as a neuron-to-glial signal for mitogenesis: role of glial N-methyl-D-aspartate receptors. 1993. [DOI] [PubMed]
- 38.Nedergaard M., Takano T., Hansen A.J. Beyond the role of glutamate as a neurotransmitter. Nat. Rev. Neurosci. 2002;3(9):748–755. doi: 10.1038/nrn916. [DOI] [PubMed] [Google Scholar]
- 39.Martel M.A., Ryan T.J., Bell K.F., Fowler J.H., McMahon A., Al-Mubarak B., Komiyama N.H., Horshburgh K., Kind P.C., Grant S.G., Wyllie D.J., Hardingham G.E. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. 2012. [DOI] [PMC free article] [PubMed]
- 40.Beal M.F. Mechanisms of excitotoxicity in neurologic diseases. FASEB J. 1992;6(15):3338–3344. [PubMed] [Google Scholar]
- 41.Gupta K., Hardingham G.E., Chandran S. NMDA receptor-dependent glutamate excitotoxicity in human embryonic stem cell-derived neurons. Neurosci. Lett. 2013;543:95–100. doi: 10.1016/j.neulet.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takano T., Lin J.H., Arcuino G., Gao Q., Yang J., Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001;7(9):1010–1015. doi: 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- 43.Buckingham S.C., Campbell S.L., Haas B.R., Montana V., Robel S., Ogunrinu T., Sontheimer H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011;17(10):1269–1274. doi: 10.1038/nm.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Behrens P.F., Langemann H., Strohschein R., Draeger J., Hennig J. Extracellular glutamate and other metabolites in and around RG2 rat glioma: an intracerebral microdialysis study. 2000. [DOI] [PubMed]
- 45.Marcus H.J., Carpenter K.L., Price S.J., Hutchinson P.J. In vivo assessment of high-grade glioma biochemistry using microdialysis: a study of energy-related molecules, growth factors and cytokines. J. Neurooncol. 2010;97(1):11–23. doi: 10.1007/s11060-009-9990-5. [DOI] [PubMed] [Google Scholar]
- 46.Ye Z.C., Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59(17):4383–4391. [PubMed] [Google Scholar]
- 47.Vanhoutte N., Abarca-Quinones J., Jordan B.F., Gallez B., Maloteaux J.M., Hermans E. Enhanced expression of the high affinity glutamate transporter GLT-1 in C6 glioma cells delays tumour progression in rat. Exp. Neurol. 2009;218(1):56–63. doi: 10.1016/j.expneurol.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 48.Lyons S.A., Chung W.J., Weaver A.K., Ogunrinu T., Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007 doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chung W.J., Lyons S.A., Nelson G.M., Hamza H., Gladson C.L., Gillespie G.Y., Sontheimer H., Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J. Neurosci. 2005;25(31):7101–7110. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshida Y., Tsuzuki K., Ishiuchi S., Ozawa S. Serum-dependence of AMPA receptor-mediated proliferation in glioma cells. Pathol. Int. 2006;56(5):262–271. doi: 10.1111/j.1440-1827.2006.01954.x. [DOI] [PubMed] [Google Scholar]
- 51.Ishiuchi S., Yoshida Y., Sugawara K., Aihara M., Ohtani T., Watanabe T., Saito N., Tsuzuki K., Okado H., Miwa A., Nakazato Y., Ozawa S., Oszawa S. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J. Neurosci. 2007;27(30):7987–8001. doi: 10.1523/JNEUROSCI.2180-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ishiuchi S., Tsuzuki K., Yoshida Y., Yamada N., Hagimura N., Okado H., Miwa A., Kurihara H., Nakazato Y., Tamura M., Sasaki T., Ozawa S. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002;8(9):971–978. doi: 10.1038/nm746. [DOI] [PubMed] [Google Scholar]
- 53.Nabissi M., Morelli M.B., Amantini C., Farfariello V., Ricci-Vitiani L., Caprodossi S., Arcella A., Santoni M., Giangaspero F., De Maria R., Santoni G. TRPV2 channel negatively controls glioma cell proliferation and resistance to Fas-induced apoptosis in ERK-dependent manner. Carcinogenesis. 2010;31(5):794–803. doi: 10.1093/carcin/bgq019. [DOI] [PubMed] [Google Scholar]
- 54.Yue H.Y., Jiang C.Y., Fujita T., Kumamoto E. Zingerone enhances glutamatergic spontaneous excitatory transmission by activating TRPA1 but not TRPV1 channels in the adult rat substantia gelatinosa. J. Neurophysiol. 2013;110(3):658–671. doi: 10.1152/jn.00754.2012. [DOI] [PubMed] [Google Scholar]
- 55.Nishio N., Taniguchi W., Sugimura Y.K., Takiguchi N., Yamanaka M., Kiyoyuki Y., Yamada H., Miyazaki N., Yoshida M., Nakatsuka T. Reactive oxygen species enhance excitatory synaptic transmission in rat spinal dorsal horn neurons by activating TRPA1 and TRPV1 channels. Neuroscience. 2013;247:201–212. doi: 10.1016/j.neuroscience.2013.05.023. [DOI] [PubMed] [Google Scholar]
- 56.Shimizu S., Takahashi N., Mori Y. TRPs as chemosensors (ROS, RNS, RCS, gasotransmitters). Handbook Exp. Pharmacol. 2014;223:767–794. doi: 10.1007/978-3-319-05161-1_3. [DOI] [PubMed] [Google Scholar]
- 57.Amantini C., Mosca M., Nabissi M., Lucciarini R., Caprodossi S., Arcella A., Giangaspero F., Santoni G. Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J. Neurochem. 2007;102(3):977–990. doi: 10.1111/j.1471-4159.2007.04582.x. [DOI] [PubMed] [Google Scholar]
- 58.Ding X., He Z., Zhou K., Cheng J., Yao H., Lu D., Cai R., Jin Y., Dong B., Xu Y., Wang Y. Essential role of TRPC6 channels in G2/M phase transition and development of human glioma. J. Natl. Cancer Inst. 2010;102(14):1052–1068. doi: 10.1093/jnci/djq217. [DOI] [PubMed] [Google Scholar]
- 59.Stock K., Kumar J., Synowitz M., Petrosino S., Imperatore R., Smith E.S., Wend P., Purfürst B., Nuber U.A., Gurok U., Matyash V., Wälzlein J.H., Chirasani S.R., Dittmar G., Cravatt B.F., Momma S., Lewin G.R., Ligresti A., De Petrocellis L., Cristino L., Di Marzo V., Kettenmann H., Glass R. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat. Med. 2012;18(8):1232–1238. doi: 10.1038/nm.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nabissi M., Morelli M.B., Santoni M., Santoni G. Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis. 2013;34(1):48–57. doi: 10.1093/carcin/bgs328. [DOI] [PubMed] [Google Scholar]
- 61.Gottlieb M., Matute C. Expression of ionotropic glutamate receptor subunits in glial cells of the hippocampal CA1 area following transient forebrain ischemia. J. Cereb. Blood Flow Metab. 1997;17(3):290–300. doi: 10.1097/00004647-199703000-00006. [DOI] [PubMed] [Google Scholar]
- 62.Dzamba D., Honsa P., Anderova M. NMDA Receptors in Glial Cells: Pending Questions. Curr. Neuropharmacol. 2013;11(3):250–262. doi: 10.2174/1570159X11311030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Butt A.M., Fern R.F., Matute C. Neurotransmitter signaling in white matter. Glia. 2014;62(11):1762–1779. doi: 10.1002/glia.22674. [DOI] [PubMed] [Google Scholar]
- 64.Soria F.N., Pérez-Samartín A., Martin A., Gona K.B., Llop J., Szczupak B., Chara J.C., Matute C., Domercq M. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J. Clin. Invest. 2014;124(8):3645–3655. doi: 10.1172/JCI71886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rosenberg P.A. Accumulation of extracellular glutamate and neuronal death in astrocyte-poor cortical cultures exposed to glutamine. Glia. 1991;4(1):91–100. doi: 10.1002/glia.440040111. [DOI] [PubMed] [Google Scholar]
- 66.Domercq M., Vázquez-Villoldo N., Matute C. Neurotransmitter signaling in the pathophysiology of microglia. Front. Cell. Neurosci. 2013;7:49. doi: 10.3389/fncel.2013.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kigerl K.A., Ankeny D.P., Garg S.K., Wei P., Guan Z., Lai W., McTigue D. System x(c)(-) regulates microglia and macrophage glutamate excitotoxicity in vivo. Exp. Neurol., 2012, 233, 333-41. 2012. [DOI] [PMC free article] [PubMed]
- 68.Klegeris A., Walker D.G., McGeer P.L. Regulation of glutamate in cultures of human monocytic THP-1 and astrocytoma U-373 MG cells. J. Neuroimmunol. 1997;78(1-2):152–161. doi: 10.1016/S0165-5728(97)00094-5. [DOI] [PubMed] [Google Scholar]
- 69.Eaton S.A., Jane D.E., Jones P.L., Porter R.H., Pook P.C., Sunter D.C., Udvarhelyi P.M., Roberts P.J., Salt T.E., Watkins J.C. Competitive antagonism at metabotropic glutamate receptors by (S)-4-carboxyphenylglycine and (RS)-alpha-methyl-4-carboxyphenylglycine. Eur. J. Pharmacol. 1993;244(2):195–197. doi: 10.1016/0922-4106(93)90028-8. [DOI] [PubMed] [Google Scholar]
- 70.Hayashi Y., Sekiyama N., Nakanishi S., Jane D.E., Sunter D.C., Birse E.F., Udvarhelyi P.M., Watkins J.C. Analysis of agonist and antagonist activities of phenylglycine derivatives for different cloned metabotropic glutamate receptor subtypes. J. Neurosci. 1994;14(5 Pt 2):3370–3377. doi: 10.1523/JNEUROSCI.14-05-03370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bedingfield J.S., Kemp M.C., Jane D.E., Tse H.W., Roberts P.J., Watkins J.C. Structure-activity relationships for a series of phenylglycine derivatives acting at metabotropic glutamate receptors (mGluRs). Br. J. Pharmacol. 1995;116(8):3323–3329. doi: 10.1111/j.1476-5381.1995.tb15142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sato H., Shiiya A., Kimata M., Maebara K., Tamba M., Sakakura Y., Makino N., Sugiyama F., Yagami K., Moriguchi T., Takahashi S., Bannai S. Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 2005;280(45):37423–37429. doi: 10.1074/jbc.M506439200. [DOI] [PubMed] [Google Scholar]
- 73.Liu R.R., Brown C.E., Murphy T.H. Differential regulation of cell proliferation in neurogenic zones in mice lacking cystine transport by xCT. Biochem. Biophys. Res. Commun. 2007;364(3):528–533. doi: 10.1016/j.bbrc.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 74.Iuchi Y., Kibe N., Tsunoda S., Okada F., Bannai S., Sato H., Fujii J. Deficiency of the cystine-transporter gene, xCT, does not exacerbate the deleterious phenotypic consequences of SOD1 knockout in mice. Mol. Cell. Biochem. 2008;319(1-2):125–132. doi: 10.1007/s11010-008-9885-3. [DOI] [PubMed] [Google Scholar]
- 75.McCullagh E.A., Featherstone D.E. Behavioral characterization of system xc- mutant mice. Behav. Brain Res. 2014;265:1–11. doi: 10.1016/j.bbr.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 76.Gout P.W., Buckley A.R., Simms C.R., Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15(10):1633–1640. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- 77.Robe P.A., Bentires-Alj M., Bonif M., Rogister B., Deprez M., Haddada H., Khac M.T., Jolois O., Erkmen K., Merville M.P., Black P.M., Bours V. In vitro and in vivo activity of the nuclear factor-kappaB inhibitor sulfasalazine in human glioblastomas. Clin. Cancer Res. 2004;10(16):5595–5603. doi: 10.1158/1078-0432.CCR-03-0392. [DOI] [PubMed] [Google Scholar]
- 78.Pentikäinen V., Suomalainen L., Erkkilä K., Martelin E., Parvinen M., Pentikäinen M.O., Dunkel L. Nuclear factor-kappa B activation in human testicular apoptosis. Am. J. Pathol. 2002;160(1):205–218. doi: 10.1016/S0002-9440(10)64364-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hayeshi R., Mutingwende I., Mavengere W., Masiyanise V., Mukanganyama S. The inhibition of human glutathione S-transferases activity by plant polyphenolic compounds ellagic acid and curcumin. Food Chem. Toxicol. 2007;45(2):286–295. doi: 10.1016/j.fct.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 80.Haskó G., Szabó C., Németh Z.H., Deitch E.A. Sulphasalazine inhibits macrophage activation: inhibitory effects on inducible nitric oxide synthase expression, interleukin-12 production and major histocompatibility complex II expression. Immunology. 2001;103(4):473–478. doi: 10.1046/j.1365-2567.2001.01272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dixon S.J., Patel D.N., Welsch M., Skouta R., Lee E.D., Hayano M., Thomas A.G., Gleason C.E., Tatonetti N.P., Slusher B.S., Stockwell B.R. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wolf I.M., Fan Z., Rauh M., Seufert S., Hore N., Buchfelder M., Savaskan N.E., Eyüpoglu I.Y. Histone deacetylases inhibition by SAHA/Vorinostat normalizes the glioma microenvironment via xCT equilibration. Sci. Reports. 2014 doi: 10.1038/srep06226. [DOI] [PMC free article] [PubMed] [Google Scholar]