

Summary
Uniparental disomy is an alternative mechanism to achieve biallelic inactivation of important tumor suppressor genes in colorectal cancer, such as APC. Our analysis of recurrent UPDs/UPPs points to novel tumor suppressor genes in this disease.
Abstract
Colorectal cancer (CRC) is characterized by specific patterns of copy number alterations (CNAs), which helped with the identification of driver oncogenes and tumor suppressor genes (TSGs). More recently, the usage of single nucleotide polymorphism arrays provided information of copy number neutral loss of heterozygosity, thus suggesting the occurrence of somatic uniparental disomy (UPD) and uniparental polysomy (UPP) events. The aim of this study is to establish an integrative profiling of recurrent UPDs/UPPs and CNAs in sporadic CRC. Our results indicate that regions showing high frequencies of UPD/UPP mostly coincide with regions typically involved in genomic losses. Among them, chromosome arms 3p, 5q, 9q, 10q, 14q, 17p, 17q, 20p, 21q and 22q preferentially showed UPDs/UPPs over genomic losses suggesting that tumor cells must maintain the disomic state of certain genes to favor cellular fitness. A meta-analysis using over 300 samples from The Cancer Genome Atlas confirmed our findings. Several regions affected by recurrent UPDs/UPPs contain well-known TSGs, as well as novel candidates such as ARID1A, DLC1, TCF7L2 and DMBT1. In addition, VCAN, FLT4, SFRP1 and GAS7 were also frequently involved in regions of UPD/UPP and displayed high levels of methylation. Finally, sequencing and fluorescence in situ hybridization analysis of the gene APC underlined that a somatic UPD event might represent the second hit to achieve biallelic inactivation of this TSG in colorectal tumors. In summary, our data define a profile of somatic UPDs/UPPs in sporadic CRC and highlights the importance of these events as a mechanism to achieve the inactivation of TSGs.
Introduction
Copy number alterations (CNAs) are the defining feature of tumors of epithelial origin, including colorectal, providing a consistent landscape of genome-wide gains and losses in a tumor-type specific manner (1,2). These genomic imbalances might contain oncogenes in areas of amplification and tumor suppressor genes (TSGs) in regions commonly subjected to deletion. More recently, the usage of single nucleotide polymorphism (SNP) arrays allowed the identification of allele specific imbalances thus defining regions of copy number neutral loss of heterozygosity (cnLOH) or uniparental disomy (UPD) (3–5). UPD was firstly described by Engel (6), and arises when an individual inherits two copies of maternal or paternal chromosomes as a result of a meiotic error. While this gives rise to constitutional UPD associated with developmental disorders, this phenomenon has also been described in somatic cells, the so-called somatically acquired UPD (3–5). In fact, UPD has been recently described in several malignancies, including both solid tumors and hematological neoplasias (7–9), and it has been suggested as a mechanism to potentially alter the expression of driver genes involved in carcinogenesis (4). In addition, in many cancer cells of epithelial origin, whole genome duplications occur frequently, thus potentially resulting in trisomies and tetrasomies of chromosomes inherited from the same progenitor, a scenario designated as uniparental polysomy (UPP).
Colorectal cancer (CRC) is one of the most common cancers in Western Europe and North America (10). Continuous efforts are made to comprehensively characterize the genome of CRC cells in order to understand the genetic basis of this devastating disease and to identify biomarkers that can help with early detection and improve prognostication. In particular, genomic profiling of sporadic CRC has revealed consistent gains and losses during the emergence and evolution of these tumors (11). Based on the two-hit Knudson hypothesis, regions with LOH might contain known TSGs, which can be inactivated by either a genomic loss or a second inactivating mutation (12). In this context, UPD arises as an alternative mechanism to reach functional impairment of TSGs in cancer cells, in which one allele holds an inactivating mutation, and due to a duplication of the chromosome containing the mutated allele and a loss of the chromosome with the wild-type allele, the tumor cell becomes disomic with an inactivating mutation in homozygosis. Foremost intriguing is the question whether different profiles of UPD/UPP are observed depending on the tumor’s tissue of origin, and with what frequency UPD/UPP does occur in different cancer types. For example, deletion of chromosome 5q is very common on myelodysplastic syndrome and acute myeloid leukemia, but UPD at 5q is not extensively reported in these cancers types. On the other hand, UPD on chromosome 5q containing the TSG APC has been reported to be a common event in CRC (13–15) and in in vitro models (16,17). Moreover, it has been recently suggested that cnLOH affecting APC may play a role in earlier stages of tumorigenesis as this event was already found in adenomas (18).
In the present study, we aimed at establishing a map of UPD/UPP in sporadic CRC, and integrating these data with somatic CNAs and the methylation status of cancer-associated genes. Our results have been cross-compared to The Cancer Genome Atlas (TCGA) data to confirm which specific regions of the CRC genome are prone to develop UPD/UPP. In addition, combining mutation analysis and fluorescence in situ hybridization (FISH) of the gene APC provided insights into the mechanism by which UPDs result in biallelic inactivation of TSGs.
Materials and methods
Sample collection
Thirty colorectal adenocarcinomas provided by the Hospital Clínic of Barcelona/IDIBAPS Biobank were included in this study (Table 1). All patients signed the corresponding informed consent and the sample collection was approved by the local Ethics Committees. Fresh tumor and adjacent normal mucosa samples were collected immediately after surgical resection and preserved in DMEM culture medium supplemented with fetal bovine serum (Life Technologies, Carlsbad, CA), antibiotics and antifungals before storage at −80°C. An experienced pathologist macroscopically dissected the fresh tumor samples in order to minimize the inclusion of normal mucosa and necrotic tissue.
Table 1.
Clinical information and experimental setup
| Patient ID | Sex | Agea | Stage | Microsatellite instability status | aCGH | SNP array | Methylation array | APC sequencing | APC FISH |
|---|---|---|---|---|---|---|---|---|---|
| S1 | M | 51 | IIA | Negative | X | X | X | X | X |
| S2 | F | 59 | IIIB | Negative | X | X | X | X | X |
| S5 | M | 62 | I | n.a. | X | X | X | n.a. | X |
| S6 | M | 74 | IIIB | n.a. | X | X | X | n.a. | X |
| S7 | F | 62 | IV | n.a. | X | X | X | n.a. | X |
| S8 | F | 76 | IIA | Negative | X | X | X | X | X |
| S9 | F | 59 | I | n.a. | X | X | X | n.a. | X |
| S10 | F | 74 | IIIB | Negative | X | X | X | n.a. | X |
| S11 | M | 59 | IIIB | n.a. | X | X | X | n.a. | X |
| S12 | M | 85 | IIB | n.a. | X | X | X | n.a. | X |
| S13 | F | 65 | IIB | Negative | X | X | X | n.a. | X |
| S32 | M | 46 | IIIB | Negative | X | X | X | X | X |
| S33 | F | 80 | I | Negative | X | X | X | X | X |
| S34 | F | 80 | IIA | Negative | X | X | X | n.a. | n.d. |
| S35 | M | 46 | IIIB | Negative | X | X | X | n.a. | n.d. |
| S36 | M | 61 | IIB | Positive | X | X | X | n.a. | n.d. |
| S37 | M | 78 | IV | n.a. | X | X | X | X | n.d. |
| S38 | M | 84 | IIIB | n.a. | X | X | X | n.a. | n.d. |
| S39 | F | 44 | IIIB | Negative | X | X | X | n.a. | n.d. |
| S40 | M | 75 | IIA | n.a. | X | X | X | n.a. | n.d. |
| S41 | M | 68 | IV | n.a. | X | X | X | n.a. | n.d. |
| S42 | F | 42 | IV | n.a. | X | X | X | n.a. | n.d. |
| S43 | F | 78 | IIA | n.a. | X | X | X | X | n.d. |
| S44 | M | 68 | IIIB | Negative | X | X | X | n.a. | n.d. |
| S45 | F | 48 | I | Negative | X | X | X | X | n.d. |
| S46 | M | 58 | IV | Negative | X | X | X | X | n.d. |
| S47 | F | 38 | IIIB | Negative | X | X | n.d. | X | n.d. |
| S48 | M | 80 | I | n.a. | X | X | X | n.a. | n.d. |
| S49 | M | 62 | IIA | n.a. | X | X | X | n.a. | n.d. |
| S50 | M | 67 | IIA | n.a. | X | X | X | X | n.d. |
n.a., not available; n.d., not determined due to technical issues.
aAge of surgery.
DNA extraction
DNA from tumor and matched adjacent normal mucosa was extracted using phenol–chloroform as described previously (19). DNA concentration and purity was assessed using a NanoDrop ND-1000 (Thermo Scientific, Waltham, MA) spectrophotometer.
SNP array
Two hundred and fifty nanograms of DNA were processed for hybridization on the Genechip Human Mapping 250K Sty arrays (Affymetrix, Santa Clara, CA). The experimental procedure was performed according to recommendations of the manufacturer. After hybridization, the chips were processed using the GeneChip Fluidics Station 450, high-resolution microarray GeneChip Scanner 3000 and GCOS Instrument Control Workstation version 1.2 (Affymetrix). SNP calls were determined by GDAS version 3.0 with 25% level of confidence. Only samples with call rates >90% were included.
Array comparative genomic hybridization
Oligonucleotide-based array comparative genomic hybridization was performed according to the protocol provided by the manufacturer (Agilent Technologies, Santa Clara, CA), with minor modifications. Briefly, 3 μg of DNA from each sample were labeled with Cy3 and combined with DNA from matched normal colonic mucosa labeled with Cy5. Oligonucleotide-based Human Genome CGH Microarray (Agilent) with 244K features was hybridized, washed accordingly and scanned with an Agilent G2565BA scanner. Data were quality controlled and extracted using Agilent Technologies’ Feature Extraction (version 9.1). Visualization was performed using the software Nexus Copy Number version 7.5 (BioDiscovery, El Segundo, CA).
Data processing
Genome-wide LOH and CNA information was obtained using the Affymetrix array. The CRMA version 2.0 was used for normalizing allelic estimates of one tumor sample based on estimates from a single matched normal (20). Next, the Paired parent-specific circular binary segmentation algorithm, PSCBS version 0.4, was utilized (21). Different cutoffs, computed by the PSCBS algorithm, were used to consider allelic imbalances and CNAs for each pair of samples. When the distance between two adjacent segments was < 2.5Mb, a single region was considered. cnLOH was defined as a region without CNAs but with LOH, including UPD and UPP events. On the other hand, copy number gains with loss of heterozygosity were defined as gained regions with LOH, including solely UPP events. Both, cnLOH and copy number gains with loss of heterozygosity, were added up together when identifying regions of UPD or UPP. Finally, circos plots from different sets integrating both UPD/UPP and CNA regions were drawn using the Rcircos package (22). The Mann–Whitney U test was performed to assess the difference in the segment length and the Student’s t-test for the comparison between stages For the Agilent platform, the CBS algorithm was used to detect CNAs (23). For this purpose, R-functions available in the DNAcopy package were used with default parameter values. The segment list was then converted into a matrix format by aligning samples based on chromosome segments condensed by genes (human genome release: hg19). In order to measure the concordance of CNAs between Agilent and Affymetrix platforms, the Pearson correlation was computed using the CNAs condensed log2 ratio by gene in both platforms. Only CNAs computed from the Affymetrix platform were taken into consideration for further analysis. Finally, Gene Set Enrichment Analysis was performed to determine whether recurrent UPD/UPP contained a statistically significant number of TSGs. For this analysis, we considered the genes obtained from the TSGene database (http://bioinfo.mc.vanderbilt.edu/TSGene/) (24).
Data were deposited in the NCBI Gene Expression Omnibus (GSE64114).
The Cancer Genome Atlas data analysis
With the purpose to validate our results, level 1 SNP6 array data from the colon adenocarcinoma dataset and pair-matched normal DNA obtained from the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/) were systematically curated. Only those samples showing less than 500 segments after segmentation were included in the analysis. Finally, PSCBS algorithm was carried out, and 303 samples were considered for analysis.
Methylation analysis
One microgram of purified DNA from each paired tumor-normal sample was bisulfite converted using the EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA). Subsequently, methylation status of 1505 CpG sites representing 807 cancer-related genes were evaluated by using the Illumina GoldenGate Methylation Cancer Panel I Array (Illumina, San Diego, CA) as described previously (25). Methylation probes representing a change of beta values >50% and a false discovery rate ≤ 0.001 using the Benjamini–Hochberg procedure were considered differentially expressed (26).
Mutational screening
A subset of tumors and their matched normal mucosa DNA (n = 10) underwent PCR amplification and were screened for sequence variations at the APC mutational cluster region (exon 16: one fragment from nucleotide 3081–3210, and another fragment from nucleotide 3529–4767, NCBI hg19) by Sanger sequencing. All samples were sequenced in either forward or reversed orientation to ensure the highest amount of sequence overlapping among PCR products (Supplementary Table 1, available at Carcinogenesis Online). Only PCR products with a single amplicon went forward for sequencing. Sequences were visualized and analyzed using the dedicated software 4Peaks (Nucleobytes, Aalsmeer, The Netherlands). Any identified mutation was verified against the Catalogue of Somatic Mutations in Cancer database (COSMIC, http://cancer.sanger.ac.uk/cosmic).
FISH
FISH analysis was performed on tissue microarrays containing two replicates of the tumor and two replicates of the normal adjacent mucosa per sample. Two BAC clones were used: one covering the APC gene (RP11-3B10), kindly provided by Dr B. Espinet (IMIM-Hospital del Mar, Barcelona, Spain), and a second one located at 5q31.2 (RP11-461O14), kindly provided by Dr X. Estivill (Center for Genomic Regulation, Barcelona, Spain), that was used as a control. They were labeled with Spectrum Orange and Spectrum Green (Abbott Molecular, Des Plaines, IL), respectively. DNA extraction, nick translation and hybridization were performed as described previously (19). Signal quantification was carried out with a Nikon Eclipse 50i fluorescence microscope in 100 nuclei per sample, and image acquisition was done by using the Isis Fluorescence Imaging System (MetaSystems, Altlussheim, Germany).
Results
Minimal regions of CNAs
Thirty primary colorectal adenocarcinomas were analyzed with the GeneChip Human Mapping 250K Sty SNP arrays and oligonucleotide-based Human Genome CGH microarrays. The mean concordance of CNAs between both platforms was 92%. CNAs were found in 28 out of 30 samples (93.3%), with a mean of 22 gains and 7 losses per sample. One of the two samples without CNAs showed microsatellite instability, while the other sample belonged to a patient with familial CRC Type X (27). Genomic profiling of frequent gains and losses based on SNP-arrays and PSCBS analysis is displayed in a circos plot (Figure 1A). Chromosomes 7, 8q, 13 and 20q were gained in more than 40% of the samples. Chromosomes 1p, 4, 5q, 8p, 14, 15, 17p, 18 and 20p were lost in more than 20% of the cases. The most frequently gained region was 20q11.21 (70%), and the most frequently lost region was 18q21.2-q22.3 (40%). Detailed frequencies of gains and losses in a gene-centered manner are presented in Supplementary Table 2, available at Carcinogenesis Online. Furthermore, a comparison between samples with stages I and II, defined as low stage tumors, and samples with stages III and IV, defined as high stage tumors, was performed. Only genomic regions 6p21.31, 6p24.1-p25.3 and 19q13.32-q13.43 were found significantly lost in high stage compared to low stage colorectal adenocarcinomas (false discovery rate < 0.25).
Figure 1.
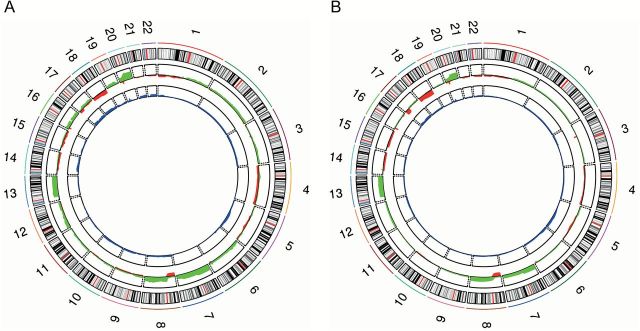
Profile of CNAs and UPDs/UPPs from our study (A) and from the TCGA cohort (B). Chromosomes are represented in the outer ring of the circos plot. In the middle, gained regions are in green and lost regions in red. The inner ring shows frequency of UPDs/UPPs in blue. To note, regions with high frequency of UPD/UPP matched with regions with high frequency of copy number loss.
UPD/UPP profiling
Genome-wide UPD/UPP profiling was performed on 30 adenocarcinomas by applying the PSCBS algorithm to the SNP array dataset. Overall, 21 out of 30 samples (70%) showed UPD/UPP segments, ranging from 1 to 17. A total of 163 genomic segments corresponded to cnLOH, including both events UPD and UPP. In addition, 22 segments corresponded to copy number gains with loss of heterozygosity, including only UPP.
In order to establish an overview of regions simultaneously involved in UPD/UPP and CNAs, data have been displayed in an integrative circos plot (Figure 1A). Interestingly, most of the regions showing high frequency of UPD/UPP matched with those regions of the genome showing high frequency of losses. However, the median length of the segments involved in UPD/UPP was higher than those affected by genomic losses (41.7 versus 34.8Mb) (Figure 2). While copy number losses are more frequently affecting interstitial chromosome segments (28.8 versus 14.8%; P < 0.05), UPD/UPP regions displayed a tendency for whole chromosome events (35.2 versus 23.7%). On the other hand, UPD/UPP and copy number losses shared a similar frequency of telomeric events (34.3 versus 33.1%) (Table 2).
Figure 2.

Distribution of the length of UPD/UPP events and genomic losses. The interquartile range is represented by the black box inside the violin plots. The median is represented by the white dot. Events smaller than 2.5Mb have been discarded.
Table 2.
Distribution of UPDs/UPPs and genomic losses
| Type of event | UPDs/UPPs | Genomic losses | P valuee |
|---|---|---|---|
| Whole chromosomea | 38 (35.2%) | 28 (23.7%) | 0.0785 |
| Span centromereb | 17 (15.7%) | 17 (14.4%) | 0.8531 |
| Telomeric pc | 18 (16.7%) | 20 (16.9%) | 1 |
| Telomeric qc | 19 (17.6%) | 19 (16.1%) | 0.8591 |
| Interstitiald | 16 (14.8%) | 34 (28.8%) | 0.0156 |
aEvents including the entire chromosome.
bEvents including the centromere but not the entire chromosome.
cEvents including one of the two telomeres but not the centromere.
dEvents comprised between two location within the same chromosome arm not involving the centromere nor the telomeres.
eFisher’s exact test was performed to assess statistical significance.
Genomic regions showing a frequency of UPD/UPP greater than 10% were located at chromosomes 1, 3, 5, 8, 9, 10, 14, 17, 18, 19, 20, 21 and 22 (Table 3). Remarkably, there are several genomic regions whose frequency of UPD/UPP events is equal or higher than the frequency of copy number losses. This is the case for genomic regions located at chromosome arms 3p, 5q, 9q, 10q, 14q, 17p, 17q, 20p, 21q and 22q, indicating a prevalence of UPD/UPP over genomic loss. Among these regions, TSGs such as APC and TP53 map to 5q (20% of UPD/UPP and 20% of losses) and 17p (23.3% of UPD/UPP and 23.3% of losses), respectively. Here, we report a novel region of UPD/UPP in CRC at 10q11.23-q26.3 containing the TSG PTEN (13.3% of UPD/UPP and 6.7% of copy number losses) as well as other tumor suppressor candidates, such as TCF7L2 and DMBT1 (16.7% of UPD/UPP and 6.7% of copy number losses). On the other hand, genomic regions with a higher prevalence of copy number losses than UPDs/UPPs are located on chromosomes 1p, 8p and 18. The analysis of the minimal regions of interest led to the identification of several candidate TSGs reported in the COSMIC database, such as ARID1A at 1p35.3 (13.3% of UPD/UPP, 20% of copy number losses), DLC1 at 8p22 (16.7% of UPD/UPP, and 33.3% of copy number losses), and SMAD4 at 18q21.1 (20% of UPD/UPP and 36.7% of losses). Furthermore, the Gene Set Enrichment Analysis showed a statistically significant presence of TSGs in the recurrent regions of UPD/UPP (P < 0.05) (Table 3).
Table 3.
Minimal regions of frequent UPD/UPPa
| Region | Size (bp) | Loss (%) | UPD/UPP (%) | LOH (%)b | Number of genesc | Genes of interestd |
|---|---|---|---|---|---|---|
| 1p35.1-p36.33 | 15209290 | 16.7–20 | 13.3 | 30–33.3 | 243 | ARID1A |
| 1q44 | 372585 | 3.3 | 13.3 | 16.7 | 9 | |
| 3p21.31-p26.3 | 46624171 | 3.3–6.7 | 13.3 | 16.7–20 | 314 | VHL; MLH1 |
| 3q25.1 | 442601 | 0 | 13.3 | 13.3 | 7 | |
| 5q14.1-q31.2 | 101633161 | 6.7–20 | 13.3–20 | 20–40 | 743 | APC |
| 8p12-p23.3 | 33954420 | 26.7–33.3 | 13.3–16.7 | 40–50 | 277 | DLC1 |
| 8p11.21-p11.23 | 2364608 | 10–16.7 | 13.3 | 23.3–30 | 36 | |
| 8p11.21-p11.21 | 9093559 | 0–3.3 | 13.3 | 13.3–16.7 | 41 | |
| 8q21.13 | 333470 | 0 | 13.3 | 13.3 | 1 | |
| 8q24.3 | 286530 | 0 | 13.3 | 13.3 | 1 | |
| 9q21.13-q34.3 | 64365620 | 10–13.3 | 13.3 | 23.3–26.7 | 599 | PTCH1; DAPK1 |
| 10q11.23-q26.3 | 83250609 | 3.3–10 | 13.3–16.7 | 16.7–26.7 | 624 | PTEN; TCF7L2; DMBT1; PLCE1 |
| 14q11.2-q24.2 | 52186574 | 10–16.7 | 13.3–20 | 30 | 425 | |
| 14q31.3-q32.33 | 21331905 | 16.7–20 | 13.3 | 30–33.3 | 286 | |
| 17p13.3-q25.3 | 80999143 | 6.7–26.7 | 13.3–23.3 | 20–46.7 | 1434 | TP53; NF1 |
| 18p11.32-q23 | 77842670 | 30–40 | 16.7–26.7 | 53.3–60 | 363 | SMAD4 |
| 19p13.2-p13.3 | 9862965 | 0 | 13.3 | 13.3 | 307 | |
| 19q13.32-q13.43 | 12657480 | 0–3.3 | 13.3 | 13.3 | 569 | PEG3 |
| 20p12.1 | 780860 | 10–23.3 | 13.3–20 | 23.3–43.3 | 1 | |
| 20p12.1 | 96087 | 10 | 13.3 | 23.3 | 1 | |
| 21p11.21-q22.3 | 37157795 | 0–10 | 16.7–23.3 | 20–30 | 307 | |
| 22q11.1-q13.33 | 34352671 | 10 | 20 | 30 | 558 | MYO18B |
aOnly regions showing UPDs/UPPs in more than 10% of the cases are listed.
bLOH represents frequencies of losses and UPDs/UPPs altogether.
cOnly RefSeq genes have been taken into account.
dGenes of recurrent somatic mutation (>8%) based on COSMIC database.
In order to further narrow down novel TSGs potentially driving the selection of UPD/UPP events, we performed a cancer-gene oriented CpG methylation analysis. Our data show that the genes VCAN, FLT4, SFRP1 and GAS7 were located in regions of recurrent UPD/UPP (>10%) and showed high levels of hypermethylation in CpG islands (Δβ > 0.5). A total of 23.3% of the samples showed UPD/UPP events affecting the hypermethylated gene GAS7. In addition, 46.7% of the remaining samples showed genomic losses in 17p involving this gene. Moreover, 13.3% of the samples showed that the highly hypermethylated genes VCAN and FLT4 were included in regions with UPD/UPP. Finally, the gene SFRP1, which is located in the commonly gained region 8p11.21 in colorectal (and other) cancers, showed 13.3% of UPD/UPP and high levels of hypermethylation of the promoter or exon of the gene in all samples displaying a genomic gain.
Overall, a total of 14873 genes mapped to regions of UPD/UPP present in at least 10% of the samples, whereas only 9092 genes were included in genomic losses. A detailed table showing gene-centralized frequencies for each event (i.e. gains, losses, UPDs/UPPs and LOH) is presented in Supplementary Material (Supplementary Table 2, available at Carcinogenesis Online).
Meta-analysis using TCGA dataset
Next, we cross-compared the results obtained in our analysis using an independent sample set. Genome-wide CNAs and UPD/UPP events were analyzed from 303 normal-matched colon adenocarcinomas from the TCGA Level 1 dataset. An integrative circos plot with CNAs and UPD/UPP data is presented in Figure 1B. A very similar profile of gains and losses was observed when comparing CNAs from both cohorts. Despite the high concordance of the regions affected by UPD/UPP, we detected a decreased frequency of these events in the TCGA cohort. Regions showing a frequency of UPD/UPP higher than 10% in both datasets were located at 3p, 5q, 17, 18, 20, and 22. Of note, the frequency of UPD/UPP at 5q involving the TSG APC was very similar in both cohorts, showing a higher frequency of UPD/UPP (10.2–19.8% and 13.3–20% in the TCGA and our cohort, respectively) than copy number losses (6.3–17.2% in TCGA and 6.7–20% in our cohort). Regions with focal UPDs/UPPs were identified at 3p14.2, 16p13.3 and 20p12.1, which also matched with a high incidence of focal deletions. Interestingly, the gene C20orf133 located at 20p12.1 is the most commonly affected gene by UPD/UPP in both cohorts (26.7% in our cohort and 22.1% in TCGA). A gene-centered table indicating all genomic events and their associated frequencies from the TCGA dataset is presented in Supplementary Table 3, available at Carcinogenesis Online.
Mutational status and FISH analysis of APC
We then explored whether UPDs/UPPs affecting the TSG APC act as a “second hit” by analyzing if a mutation, when present, could be homozygous. Sequence variations in the APC mutational cluster region were found in three out of six samples with UPD/UPP events at the chromosome arm 5q. All mutations identified were nonsense. In sample 1, the mutation c.3871C>T was found to be heterozygous in the tumor, showing a lower frequency of the inactivating allele. On the other hand, in samples 33 and 50 nonsense mutations c.4360A>T and c.3856G>T, respectively, were also found in heterozygosity in the tumor; however, the frequency of the mutated allele was much higher than the normal one, suggesting that the inactivating allele is present in the majority of the tumor cells (Figure 3A and B). In fact, we cannot discard that partial heterozygosity is due to the presence of non-tumoral infiltrating cells. In addition, fluorescent-labeled DNA probes covering APC and a control region at 5q31.2 were used to perform FISH analysis onto two tissue microarrays containing all samples included in our dataset. Interestingly, cases 33 and 1 displayed two signals of APC in 84% and 86% of all the cells, respectively, confirming that these samples displayed UPD/UPP in 5q (Figure 3C).
Figure 3.
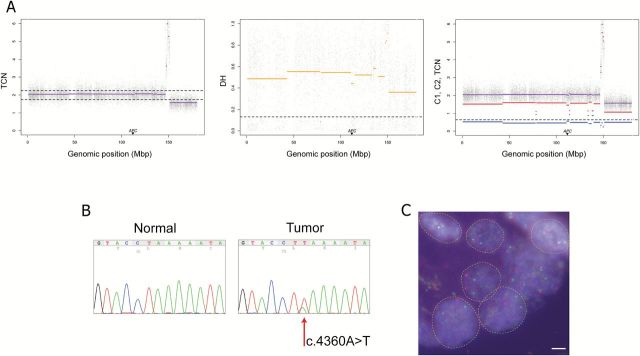
UPD of chromosome 5 accompanied by a homozygous nonsense mutation of sample 33. (A) Tumor-matched normal PSCBS profiling of SNP array data showing total copy number (panel 1), decrease of heterozygosity (panel 2) and allele-specific copy number (panel 3). Dotted horizontal lines represent specific thresholds for each parameter. As illustrated, APC is located in a region with cnLOH. (B) Sanger sequencing of the gene APC showing the existence of a nonsense mutation (c.4360A>T) in the tumor. The minority allele could represent intratumoral heterogeneity and/or infiltration of normal cells. (C) Microscopic assessment of FISH signals using fluorescent probes covering the gene APC (red) and a control probe at 5q31.2 (green). Single isolated nuclei displaying two copies of APC and the control probe confirmed that most tumor population showed a disomic chromosome 5q within this sample. Scale bar = 5 µm.
Discussion
Our study generated a profile of recurrent UPD/UPP regions in sporadic CRC to identify novel TSGs important for this disease. By utilizing the PSCBS algorithm in two different sample cohorts, we confirmed that chromosomal regions with the highest frequency of UPD/UPP mapped to chromosome arms 5q, 17p and 18q, containing the TSGs APC, TP53, and SMAD4, respectively (13,14,16,18). In addition, we have also identified regions of UPD/UPP affecting chromosomes 1p, 3p, 8p, 9q, 10q, 14q, 19, 20p, 21q and 22q, several of which have been previously reported as genomic losses in CRC (1). Although UPD/UPP profiles are very similar between our cohort and the TCGA sample set, some regions showed differences, most probably due to the sample size and heterogeneity. Of note is the high frequency of UPD/UPP events affecting chromosomes 6p and 12 in the TCGA cohort. Frequent allelic losses at these chromosomes have been described previously in mismatch repair deficient carcinomas (28). While only one sample in our cohort showed microsatellite instability, many more microsatellite instability positive samples are included in the TCGA cohort, thus possibly explaining the frequency of such alterations.
The identification of LOH and high-level amplifications has led to the discovery of driver TSGs and oncogenes, respectively (29,30). However, fewer studies have focused on regions with recurrent UPD/UPP. Our data show that these regions frequently coincide with genomic losses, suggesting that these events might also be guiding the inactivation of TSGs. In fact, several efforts have attempted to identify TSGs on chromosome arm 8p due to its recurrent loss in CRC (31). Integrative analysis of our data unveiled that the frequency of LOH in this genomic location is between 40 and 50%, including UPD/UPP events (13.3–16.7%) and copy number losses (26.7–33.3%). Among all genes encompassed within this area, DLC1, which encodes a RhoGAP protein that catalyzes the conversion of GTP-bound Rho to the inactive GDP-bound form, falls within the minimal region of UPD/UPP. DLC1, has been suggested to function as a TSG in several common cancers including hepatocellular carcinoma and CRC (32–34). Furthermore, mutations in this gene have been identified in nearly 10% of all CRC reported in the COSMIC database, thus representing a highly mutated gene and a good candidate for biallelic inactivation via UPD/UPP or genomic loss. Furthermore, here we also report novel regions of UPD/UPP that have not previously been associated with CRC. Among the 624 genes encompassing the 10q11.23-q26.3 region, PTEN stands out having already been considered a TSG in CRC (35). Likewise, TCF7L2, encoding a transcription factor that plays a key role in the Wnt/β-catenin pathway, is also located within this region and has been found to be mutated in 9% of CRC (1,36). Moreover, a candidate TSG located at 1p35.3, ARID1A, a subunit of the SWI/SNF chromatin remodeler and transcription regulator of MYC, showed a high frequency of UPD/UPP (16.7%) and has been also found recurrently mutated (5%) in CRC (1,37).
The genome-wide integration of regions with UPD/UPP and methylation patterns has provided insights into the identification of novel putative TSGs. Of note, the genes VCAN, FLT4, SFRP1 and GAS7 were among the highest hypermethylated genes and were consistently involved in UPDs/UPPs. Foremost interesting is the gene SFRP1, which is a soluble modulator of the Wnt/β-catenin signaling pathway and is located at 8p11.21, a genomic region with a high incidence of copy number gains in CRC (38). Our results revealed that in all cases with copy number gains of SFRP1, simultaneous hypermethylation of the promoter or the exon close to the promoter occurred. SFRP1 has been already identified as being hypermethylated in CRC (39); however, our data suggest the mechanism by which biallelic inactivation of this candidate TSG might have arisen in the presence of copy number gains. Importantly, gene expression data extracted from the Oncomine Portal (https://www.oncomine.org) and the Cancer Genomics Browser (https://genome-cancer.ucsc.edu) supported the hypothesis that the aforementioned genes might be considered TSGs as three out of four candidates (SFRP1, GAS7 and FLT4) showed downregulation in colorectal tumors compared to normal mucosa (data not shown).
Besides whole chromosome or chromosomal arm UPD/UPP events, we have also detected focal regions of UPD/UPP at 3p14.2, 16p13.3 and 20p12.1, containing the genes FHIT, RBFOX1 and C20orf133, respectively. Interestingly, these regions have been also described as recurrent focal deletions in several cancer types, and the genes located at these genomic regions are among the largest genes in the genome (2). Whether deletions of these genes have an impact on tumorigenesis or whether they occur as a consequence of the genome plasticity in these regions (e.g. the presence of fragile sites or the fact that they encode structural proteins which carry tandem repeats prone to recombination) remains unknown (40,41). Further analysis of genes with high incidence of UPD/UPP is required in order to understand their functional role as TSGs.
Whole-chromosome UPD/UPP can occur in cancer cells as a consequence of segregation errors in mitosis through anaphase lagging and/or non-disjunction events (4). On the other hand, homologous recombination, with the purpose of repairing a double-strand break, has been proposed as a possible mechanism of segmental UPD/UPP formation in cancer (42). In fact, mitotic recombination as a result of double strand breaks on the long arm of chromosome 5 has been suggested to be the most frequent mechanism to achieve cnLOH affecting APC in patients with familial adenomatous polyposis (43). Moreover, a high frequency of segmental UPD has been described in base excision repair deficient MUTYH-associated polyposis carcinomas (44). In our sample set, the frequency of UPD in the genomic region 5q21 is as high as the frequency of genomic loss, suggesting that UPD is indeed an alternative mechanism to acquire biallelic inactivation of APC. The higher prevalence of UPD/UPP in chromosome 5q, as well as in other chromosomes, compared to the frequency of genomic loss, suggests that tumor cells need to preserve such chromosomes in two or more copies in order to maintain cellular viability and fitness. Despite the fact that most of the cases showed segmental UPD for 5q, one specific sample displayed cnLOH involving the centromere region of chromosome 5, thus indicating that one possibility for the origin of such rearrangement was through a chromosome segregation error. As FISH analysis proved that 16% of cells in this tumor showed monosomy or trisomy for the chromosome arm 5q, it is plausible to hypothesize that a non-disjunction event occurred early in the tumor development and originated the UPD, which resulted in biallelic representation of a nonsense mutation required for tumor progression. Furthermore, the TCGA dataset showed several samples with whole chromosome 5 UPD events. Altogether, this would indicate that mitotic recombination is not the only mechanism to generate cnLOH affecting APC, and a missegregation event resulting in a whole chromosome UPD/UPP might as well represent the second hit to achieve homozygosity for an inactivating mutation.
In summary, sporadic CRC shows a specific pattern of UPD/UPP, which greatly coincides with regions of recurrent genomic losses. Further research efforts are needed to determine whether other cancer types present unique landscapes of UPD/UPP as well. The integrative analysis of regions with UPDs/UPPs, CNAs, methylation patterns, and mutational gene status provided further evidence of novel putative TSGs. In fact, we propose that UPD/UPP events act as the second hit suggested by Knudson. Finally, our data show that cnLOH affecting APC can occur by either chromosome segregation errors or mitotic recombination events, thus generating whole chromosome or segmental UPD, respectively.
Supplementary material
Supplementary Tables 1–3 can be found at http://carcin.oxfordjournals.org/
Funding
Intramural program of National Institutes of Health; Ministerio de Educación y Ciencia (SAF2007-64167); Ministerio de Economía y Competitividad (SAF2012-40017-C02-02); RTICC and FEDER (RD06/0020/1020); Generalitat de Catalunya (2009 SGR 1107, 2014 SGR 0135, 2014 SGR 0903); Catalonian Biobank Network; the European Comission (COLONGEVA); the Universitat Autònoma de Barcelona (PIF fellowship to K.T.); Asociación Española Contra el Cáncer (AIO2011 to J.C.); Instituto de Salud Carlos III (CP13/00160 to J.C.); CIBERehd program (J.J.L); the International Agency for Research on Cancer (Fellowship program to N.F.J) and Spanish Ministry (FPU program to I.Q.).
Supplementary Material
Acknowledgements
We thank Drs Jean-Claude Zenklusen, Margaret C. Cam and Yuri Kotliarov from the Neuro-Oncology Branch, NCI/NIH for their technical and bioinformatics support, and Drs Xavier Estivill and Blanca Espinet for supplying BAC clones.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- CNA
copy number alteration
- cnLOH
copy number neutral loss of heterozygosity
- CRC
colorectal cancer
- FISH
fluorescence in situ hybridization
- PSCBS
parent-specific circular binary segmentation algorithm
- LOH
loss of heterozygosity
- SNP
single nucleotide polymorphism
- TSG
tumor suppressor gene
- UPD
uniparental disomy
- UPP
uniparental polysomy
References
- 1. The Cancer Genome Atlas Network. (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature, 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beroukhim R., et al. (2010) The landscape of somatic copy-number alteration across human cancers. Nature, 463, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lapunzina P., et al. (2011) The consequences of uniparental disomy and copy number neutral loss-of-heterozygosity during human development and cancer. Biol. Cell, 103, 303–317. [DOI] [PubMed] [Google Scholar]
- 4. Tuna M., et al. (2009) Uniparental disomy in cancer. Trends Mol. Med., 15, 120–128. [DOI] [PubMed] [Google Scholar]
- 5. Makishima H., et al. (2011) Pathogenesis and consequences of uniparental disomy in cancer. Clin. Cancer Res., 17, 3913–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Engel E. (1980) A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am. J. Med. Genet., 6, 137–143. [DOI] [PubMed] [Google Scholar]
- 7. Frankel A., et al. (2014) Genome-wide analysis of esophageal adenocarcinoma yields specific copy number aberrations that correlate with prognosis. Genes Chromosomes Cancer, 53, 324–338. [DOI] [PubMed] [Google Scholar]
- 8. Beà S., et al. (2009) Uniparental disomies, homozygous deletions, amplifications, and target genes in mantle cell lymphoma revealed by integrative high-resolution whole-genome profiling. Blood, 113, 3059–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. O'Keefe C., et al. (2010) Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood, 115, 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Siegel R., et al. (2012) Cancer statistics. CA Cancer J. Clin., 62, 10–29. [DOI] [PubMed] [Google Scholar]
- 11. Ried T., et al. (1996) Comparative genomic hybridization reveals a specific pattern of chromosomal gains and losses during the genesis of colorectal tumors. Genes Chromosomes Cancer, 15, 234–245. [DOI] [PubMed] [Google Scholar]
- 12. Knudson A.G., Jr (1971) Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA, 68, 820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Andersen C.L., et al. (2007) Frequent occurrence of uniparental disomy in colorectal cancer. Carcinogenesis, 28, 38–48. [DOI] [PubMed] [Google Scholar]
- 14. Melcher R., et al. (2011) LOH and copy neutral LOH (cnLOH) act as alternative mechanism in sporadic colorectal cancers with chromosomal and microsatellite instability. Carcinogenesis, 32, 636–642. [DOI] [PubMed] [Google Scholar]
- 15. Kurashina K., et al. (2008) Chromosome copy number analysis in screening for prognosis-related genomic regions in colorectal carcinoma. Cancer Sci., 99, 1835–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gaasenbeek M., et al. (2006) Combined array-comparative genomic hybridization and single-nucleotide polymorphism-loss of heterozygosity analysis reveals complex changes and multiple forms of chromosomal instability in colorectal cancers. Cancer Res., 66, 3471–3479. [DOI] [PubMed] [Google Scholar]
- 17. Melcher R., et al. (2007) SNP-Array genotyping and spectral karyotyping reveal uniparental disomy as early mutational event in MSS- and MSI-colorectal cancer cell lines. Cytogenet. Genome Res., 118, 214–221. [DOI] [PubMed] [Google Scholar]
- 18. Zarzour P., et al. (2015) Single nucleotide polymorphism array profiling identifies distinct chromosomal aberration patterns across colorectal adenomas and carcinomas. Genes Chromosome Cancer, 54, 303–314. [DOI] [PubMed] [Google Scholar]
- 19. Prat E., et al. (2008) Genomic imbalances in urothelial cancer: intratumor heterogeneity versus multifocality. Diagn. Mol. Pathol., 17, 134–140. [DOI] [PubMed] [Google Scholar]
- 20. Bengtsson H., et al. (2009) A single-array preprocessing method for estimating full-resolution raw copy numbers from all Affymetrix genotyping arrays including GenomeWideSNP 5 & 6. Bioinformatics, 25, 2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Olshen A.B., et al. (2011) Parent-specific copy number in paired tumor-normal studies using circular binary segmentation. Bioinformatics, 27, 2038–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang H., et al. (2013) RCircos: an R package for Circos 2D track plots. BMC Bioinformatics, 14, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olshen A.B., et al. (2004) Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics, 5, 557–572. [DOI] [PubMed] [Google Scholar]
- 24. Zhao M., et al. (2013) TSGene: a web resource for tumor suppressor genes. Nucleic Acids Res., 41, D970–D976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bibikova M., et al. (2009) GoldenGate assay for DNA methylation profiling. Methods Mol. Biol., 507, 149–163. [DOI] [PubMed] [Google Scholar]
- 26. Benjamini Y., et al. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B, 57, 289–300. [Google Scholar]
- 27. Goel A., et al. (2010) Aberrant DNA methylation in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology, 138, 1854–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van Puijenbroek M., et al. (2008) Genome-wide copy neutral LOH is infrequent in familial and sporadic microsatellite unstable carcinomas. Fam. Cancer, 7, 319–330. [DOI] [PubMed] [Google Scholar]
- 29. Beroukhim R., et al. (2007) Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc. Natl. Acad. Sci. USA, 104, 20007–20012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dulak A.M., et al. (2012) Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res., 72, 4383–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zimonjic D.B., et al. (2012) Role of DLC1 tumor suppressor gene and MYC oncogene in pathogenesis of human hepatocellular carcinoma: potential prospects for combined targeted therapeutics (review). Int. J. Oncol., 41, 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xue W., et al. (2008) DLC1 is a chromosome 8p tumor suppressor whose loss promotes hepatocellular carcinoma. Genes Dev., 22, 1439–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Low J.S., et al. (2011) A novel isoform of the 8p22 tumor suppressor gene DLC1 suppresses tumor growth and is frequently silenced in multiple common tumors. Oncogene, 30, 1923–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peng H., et al. (2013) Downregulation of DLC-1 gene by promoter methylation during primary colorectal cancer progression. Biomed. Res. Int., 2013, 181384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nassif N.T., et al. (2004) PTEN mutations are common in sporadic microsatellite stable colorectal cancer. Oncogene, 23, 617–628. [DOI] [PubMed] [Google Scholar]
- 36. Angus-Hill M.L., et al. (2011) T-cell factor 4 functions as a tumor suppressor whose disruption modulates colon cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA, 108, 4914–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu R.C., et al. (2014) The emerging roles of ARID1A in tumor suppression. Cancer Biol. Ther., 15, 655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Camps J., et al. (2008) Chromosomal breakpoints in primary colon cancer cluster at sites of structural variants in the genome. Cancer Res., 68, 1284–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rawson J.B., et al. (2011) Promoter methylation of Wnt antagonists DKK1 and SFRP1 is associated with opposing tumor subtypes in two large populations of colorectal cancer patients. Carcinogenesis, 32, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bignell G.R., et al. (2010) Signatures of mutation and selection in the cancer genome. Nature, 463, 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rajaram M., et al. (2013) Two distinct categories of focal deletions in cancer genomes. PLoS One, 8, e66264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moynahan M.E., et al. (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol., 11, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Howarth K., et al. (2009) A mitotic recombination map proximal to the APC locus on chromosome 5q and assessment of influences on colorectal cancer risk. BMC Med. Genet., 10, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Middeldorp A., et al. (2008) High frequency of copy-neutral LOH in MUTYH-associated polyposis carcinomas. J. Pathol., 216, 25–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.