

Abstract
Dendrimer-based gene delivery has been constrained by intrinsic toxicity and suboptimal nanostructure. Conjugation of neutral morpholino oligonucleotides (ONs) with PAMAM dendrimers resulted in neutral, uniform, and ultra-small (~10nm) nanoconjugates. The nanoconjugates dramatically enhanced cellular delivery of the ONs in cancer cells. After release from the dendrimer in the cytosol, the ONs produced potent functional activity without causing significant cytotoxicity. When carrying an apoptosis-promoting ON, the nanoconjugates produced cancer cell killing directly. Thus, the dendritic nanoconjugates may provide an effective tool for delivering ONs to tumors and other diseased tissues.
Oligonucleotides (ONs) provide an opportunity for treating serious, life-threatening diseases that have limited therapeutic options using traditional small-molecule and antibody drugs. Antisense and siRNA ONs can modulate the expression of any gene and thus can target any protein by inducing enzyme-dependent degradation of target mRNAs.1 Further, steric-blocking ONs, including splice switching ONs (SSOs), and antagomers of microRNA and long non-coding RNAs, block the access of cellular machinery to pre-mRNA or mRNA without causing enzymatic degradation of the RNA.2 For example, a morpholino antisense ON, capable of inducing exon skipping in dystrophin pre-mRNA, has shown to restore dystrophin function in patients with Duchenne muscular dystrophy in a phase II clinical trial.3 Despite the enormous therapeutic potential, the development of ONs as therapeutic agents has been constrained by the inability of these hydrophilic and often charged macromolecules to reach their intracellular sites of action.4
Utilization of nanoparticles as delivery vehicle holds promise for unleashing the tremendous therapeutic potential of ONs. In this context, cationic dendrimers such as poly(amidoamine) (PAMAM) dendrimers have been widely used in ON delivery by condensing anionic ONs into nanoparticles.5 However, the use of dendrimers in biological systems is constrained by their inherent toxicity, which is attributed to the interaction of surface cationic residues of dendrimers with negatively charged biological membranes.5c Further, the method of complexation of cationic dendrimers with negatively charged ONs often leads to large (typically >100nm in diameter), heterogeneous and polydisperse structures, causing the problems such as limited biodistribution and low reproducibility. In this study, we use chemical conjugation methods to construct ultra-small neutral dendritic nanoconjugates that combine superior ON delivery and reduced cytotoxicity.
The overall strategy of this study is to link multiple neutrally charged ONs6 to a single molecule of PAMAM dendrimer via a reductively responsive linkage (Scheme 1).
Scheme 1.
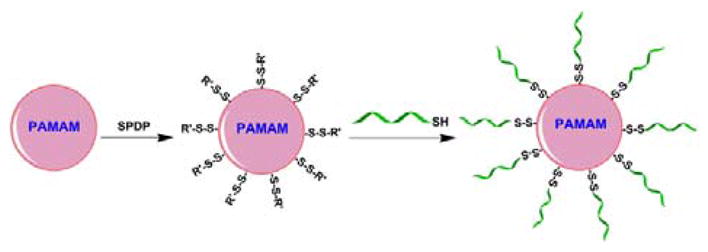
Preparation of dendritic nanoconjugates.
The SSO623 (5′-GTTATTCTTTAGAATGGTGC-3′)7 and Mcl-1 SSO (5′-CGAAGCATGCCTGAGAAAGAAAAGC-3′)8 were custom synthesized by Gene Tools, LLC (Philomath, OR). These ONs were phosphorodiamidate morpholino oligomers (PMOs) functionalized with a disulfide amide for sulfhydryl linkage at the 3′ position. PAMAM dendrimers G5 (Sigma-Aldrich) were reacted with a bifunctional crosslinker N-succinimidyl 3-(2-pyridyldithio) propionate (SPDP, Thermo Fisher Scientific) at a 1:15 molar ratio of dendrimer to linker in PBS (pH7.5) for 1h at room temperature. The excess amount of SPDP was removed by gel filtration using a PD-10 Column (GE Healthcare). The average number of 2-pyridyldithio group (the sulfhydryl-reactive portion of SPDP) linked to dendrimer was determined as 12 by observing the release of pyridine-2-thione (λmax of 343nm) from the intermediate PAMAM-SPDP conjugates after being treated with an excess amount of DTT (Sigma-Aldrich). The thiol group on the PMO needed for conjugation with the 2-pyridyldithio group on the dendrimer was freshly generated by treatment with 10mM DTT for 1h and any residual DTT was removed by gel filtration using a PD-10 Column. The 2-pyridyldithio groups on the dendrimer were then reacted with the thiol group of PMO at a 1:15 molar ratio of dendrimer to PMO in PBS with 1mM EDTA (pH7.0) overnight, and the final product was purified by gel filtration using a Sephadex G-100 gel column (GE Healthcare) to remove unreacted free PMOs. The number of PMOs linked to the dendrimer was estimated by measurement of pyridine-2-thione formation after the 3′-thiol PMO reacted with the SPDP-conjugated dendrimer as well as quantification of the PMO contents in the final product using OD260. These measurements led to close agreement with 9–11 oligonucleotides linked per dendrimer in various preparations. The nanoconjugates were then termed PAMAM-PMO10. Preparation of fluorescent Dylight650-labelled nanoconjugates is described in the supporting information.
The final product of the nanoconjugates was analysed with size-exclusion chromatography using a Varian HPLC system (ProStar/Dynamax, Walnut Creek, CA) equipped with a Yarra SEC-3000 column (Phenomenex, Torrance, CA). The PMO-containing samples were detected by OD260. As shown in Fig S1, the nanoconjugates eluted earlier than free PMOs in the column with the retention time of 8.2 and 10.1 min, respectively, indicating that the PMOs were successfully linked to the PAMAM dendrimers and the following purification step using the Sephadex G-100 gel column removed the remaining free PMOs from the nanoconjugates.
The average particle sizes and zeta potentials of the nanoconjugates were estimated using Zetasizer Nano (Malvern Instruments, Malvern, UK). We used polyplexes of the dendrimer with negatively charged ONs9 as a control. Average particle sizes for the starting material G5 PAMAM dendrimers, the nanoconjugates, and the polyplexes are summarized in Table 1 and a representative size distribution graph is shown in Fig. 1A. The diameters for dendrimers, nanoconjugates, and polyplexes were 6.0, 9.6, and 500.9 nm, respectively. The nanoconjugates were visualized by transmission electron microscopy (LEO Electron Microscopy, Oberkochen, Germany), which revealed a uniform size distribution with a diameter averaging 10 nm (Fig. 1B).
Table 1.
Particle sizes, polydispersity index (P.I.) and zeta potentials of the nanoconjugates and polyplexes. Results are expressed as mean ± SD (n = 3 or 5)
| Particle Size (nm) | P.I. | Zeta Potential (mV) | |
|---|---|---|---|
| PAMAM | 6.0 ± 0.1 | 0.36 ± 0.04 | 16.2 ± 0.7 |
| Nanoconjugates | 9.6 ± 0.2 | 0.41 ± 0.03 | 1.9 ± 0.5 |
| Polyplexes | 500.9 ± 60.2 | 0.164 ± 0.12 | 10.1 ± 0.4 |
Fig. 1.
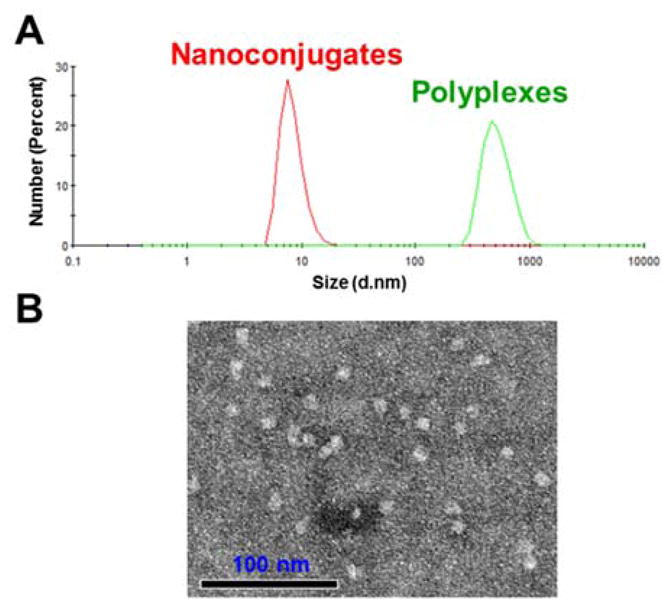
Characterization of Nanoconjugates. A. Overlay of DLC histograms of the nanoconjugates (red) and PAMAM polyplexes (green). B. TEM image of the nanoconjugates.
Both PAMAM and the polyplexes showed strong positive zeta potential of over 10mV, while the PMO modified PAMAM showed low zeta potential of 1.9mV (Table 1), indicating that conjugation of neutral PMOs shielded the surface charge of PAMAM dendrimers. Similar charge shielding was observed when cationic nanoparticles were modified by neutral polymer polyethylene glycol (PEG).10 Since high positive charge is the main cause of cytotoxicity of PAMAM, conjugation with neutral ONs may decrease the toxicity of the dendrimer.
We then tested the serum stability of the disulfide linkage in the nanoconjugates by incubation in PBS with 20% serum at 37 °C for 16h. The incubated samples were eluted using a Sephadex G-100 gel, which separates the nanoconjugates from the PMOs. For the control and serum treated samples, the PMO contents (measured by OD260) eluted within the first 8 fractions (Fig. 2), indicating the PMOs were not cleaved by serum treatment and thus the nanoconjugates were stable in serum for at least 16h. Then, we tested whether intracellular sulfhydryls might release the PMOs from the nanoconjugates using PBS containing 10mM L-glutathione and 100μM cysteine, the typical free thiol concentrations in the cytosol.11 In gel filtration of the treated sample, over 84% of PMO contents eluted slower and overlapped the peak of free PMOs (Fig. 2), indicating that the majority of the PMOs were released from the nanoconjugates by the sulfhydryls. Thus, the ONs can be released from the nanoconjugates by cytosolic glutathione when delivered into the target cells.
Fig. 2.
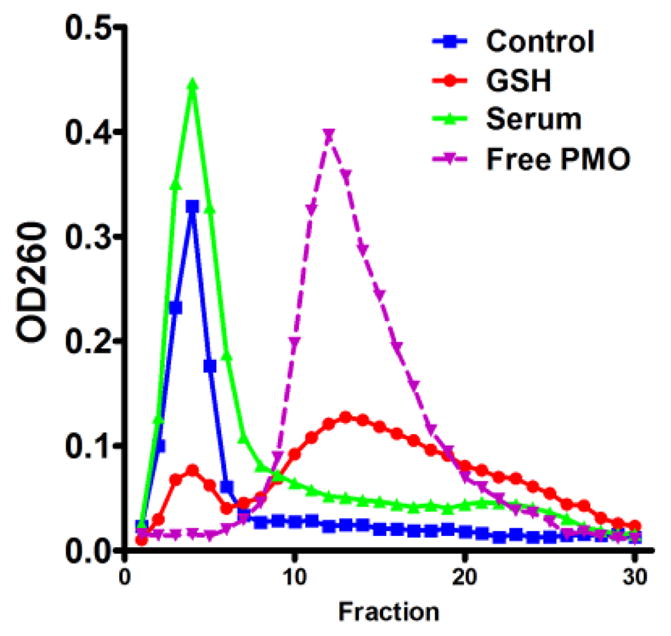
Stability of disulfide linkage in serum and sulfhydryls. The nanoconjugates were incubated in serum-containing PBS for 16h or in PBS containing 10mM glutathione (GSH) and 100μM cysteine for 4h, using PBS as a control. After incubation, samples were eluted using a Sephadex G-100 column. The ON content in the fractions was then detected by OD260.
Total cellular uptake of the nanoconjugates and free PMOs was evaluated by incubating A375 cells with these molecules for 4h and then measuring total cell-associated fluorescence by flow cytometry. As seen in Fig. 3A, there was a 273-fold greater uptake of the nanoconjugate as compared to free PMOs. In a previous study, antisense ONs were covalently conjugated to an anionic dendrimer and cellular uptake of the conjugates was 4-fold greater than naked ON.12 Thus, the dendritic conjugates in this study may provide more potent cellular delivery of ONs. Pharmacological inhibitors were used to identify possible endocytotic pathways. A375 cells were pre-treated with the inhibitors for 30 min and then treated with the nanoconjugates for 4h in the presence of the inhibitors followed by flow cytometry analysis. Four pharmacological inhibitors were used in the study to examine the possible endocytosis pathways: chlorpromazine (12.5μM), an inhibitor of the clathrin pathway; genistein, an inhibitor of caveolae pathway; amiloride (100μM), an inhibitor of macropinocytosis; and dynasore (30μM), a dynamin inhibitor.13 Treatment with dynasore abolished the uptake of the nanoconjugates, while genistein decreased the uptake in dose-dependent manner (Fig. 3B). Thus, the nanoconjugates may undergo cell entry by the caveolae pathway.
Fig. 3.
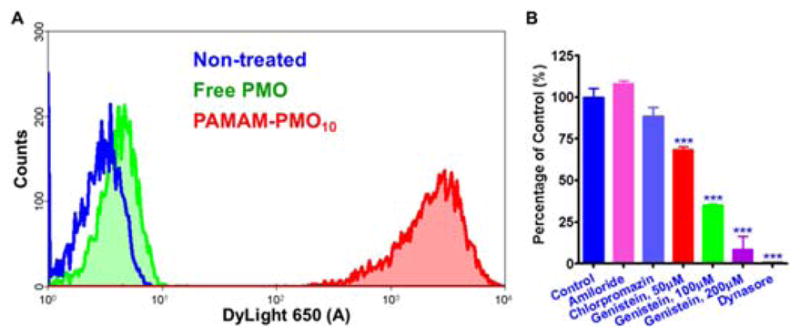
Intracellular uptake of nanoconjugates and effects of endocytosis inhibitors. Total cellular uptake of the Dylight650 labelled nanoconjugates and free PMOs (100nM) was evaluated by incubating cells with these molecules for 4h followed by flow cytometry. In an uptake inhibition experiment, the cells were pre-treated with the inhibitors and then treated with the nanoconjugates for 4 h in the presence of the inhibitors followed by flow cytometry analysis. ***P < 0.001.
To further understand the intracellular trafficking of the nanoconjugates after cellular entry, we utilized chimeras of GFP with marker proteins for specific endomembrane compartments to visualize the subcellular distribution of the targeted nanoconjugates in live cells. As seen in Fig. 4, there was considerable co-localization of the fluorescent nanoconjugates with Rab7 and Lamp1, markers for late endosome14 and lysosome15, respectively, indicating that the nanoconjugates were transported to late endosomes and lysosomes. This was confirmed by the substantial co-localization of the nanoconjugates with the lysosomal probe LysoTracker Green (Life technologies) (Fig. 4). In contrast, there was little co-localization of the nanoconjugate with Rab5, the early endosome marker (Fig. 4) and with the markers of mitochondria, Golgi network, and ER (Fig. S2). After trafficking to the late endosomes and lysosomes, the SSOs may undergo endosomal release and then transport to the nucleus to exert their pharmacological action.
Fig. 4.
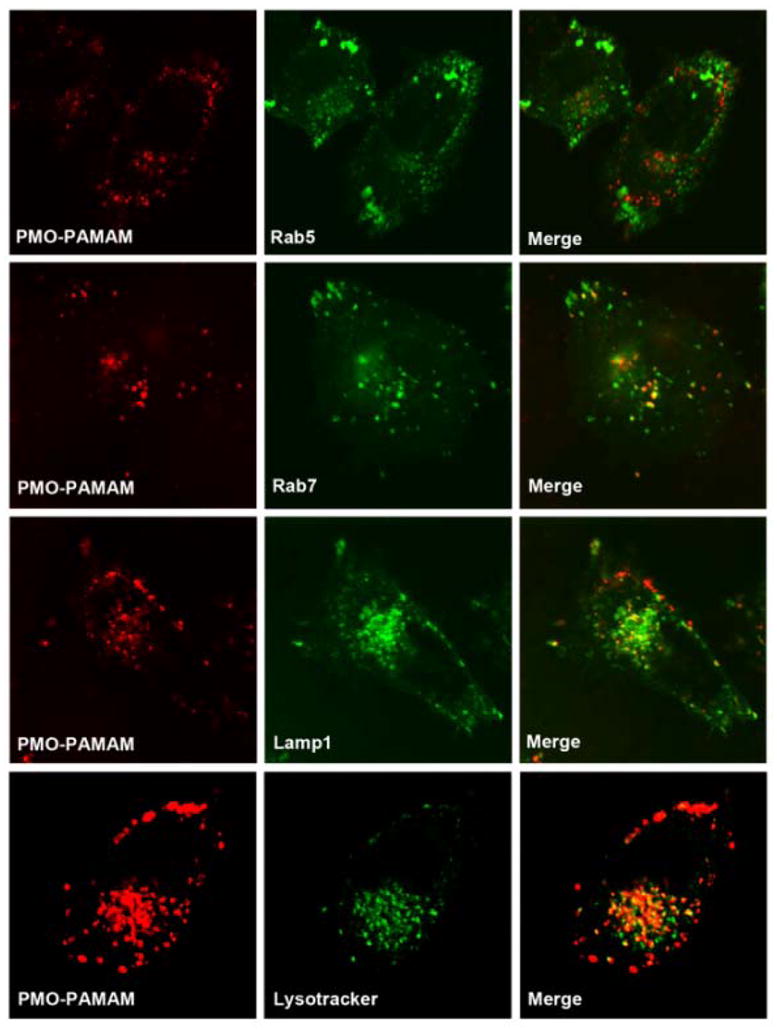
Subcellular localization of the nanoconjugates. A375 cells were transfected with expression vectors for GFP chimeras that serve as markers for several endomembrane compartments (Rab5, early endosomes; Rab7, late endosomes; Lamp1, lysosome). Thereafter, cells were incubated with the fluorescent nanoconjugates (100nM) for 4h. Live cells were observed by confocal microscopy. In co-localization with LysoTracker, the cells were treated with LysoTracker and the nanoconjugates for 4h followed by imaging.
Functional delivery by the nanoconjugates was tested in A375/eGFP654 cells that had been stably transfected with the eGFP gene interrupted by an abnormally spliced intron.16 Successful delivery of SSO623, a model ON, to the cell nucleus leads to upregulation of eGFP expression, providing a positive read-out. A375/eGFP654 cells were treated with the nanoconjugates carrying SSO623 or with controls for 4h. After another 24h-culutre, eGFP induction in A375/eGFP654 cells was measured using flow cytometry. For comparison, we included the gold standard transfection reagent Lipofectamine 2000 and prepared its complexes with negatively charged phosphorothioate (PS) SSO623 as described previously.17 As indicated in Fig. 5A, treatment with the nanoconjugates produced a dose-dependent increase in eGFP expression compared to little expression with free PMO. Compared to Lipofectamine 2000 complexes, the nanoconjugates demonstrated lower cytotoxicity and more uniform transfection (Fig. 5B). The dose of the SSO623 in the Lipofectamine 2000 complexes could only reach 200nM to avoid severe cytotoxicity. At this concentration, only 46% of A375/eGFP654 cells showed increased eGFP expression (Fig. 5B). The nanoconjugates produced homogenous eGFP induction at all doses, and when the SSO concentration increased to 800nM, over 95% of the cells showed eGFP induction (Fig. 5B) but no cytotoxicity was observed (Fig. S3). Thus, dendritic nanoconjugates may provide superior therapy in treating diseases that require uniform effects in all diseased cells, such as cancer.
Fig. 5.
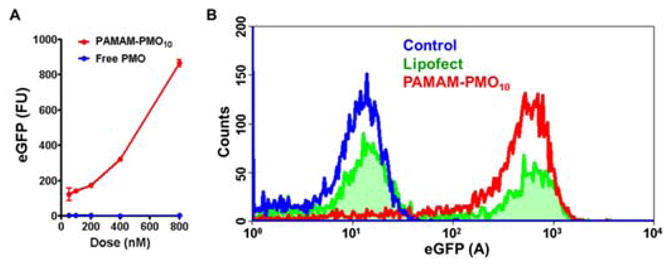
Functional delivery of SSOs by the nanoconjugates. A. Comparison of dose-dependent eGFP induction by treatments with PMO and PAMAM-PMO10. B. Comparison of eGFP induction by the treatments of PAMAM-PMO10 and Lipoplexes of PS SSO.
The nanoconjugates achieve excellent delivery and low cytotoxicity. The nanoconjugates with neutral PMOs on the surface showed a moderate positive charge of 1.9mV, and thus cytotoxicity was likely reduced. As we designed a reductively responsive linkage between the PMOs and PAMAM, the PMOs are probably released inside the endosomes of the cells. This may expose the strong positive charge of the dendrimers, causing endosomal membrane disruption and release of the PMOs.
To test therapeutic activity of the nanoconjugates, we prepared nanoconjugates carrying Mcl-1 SSO, which can redirect Mcl-1 splicing from anti-apoptotic Mcl-1L to pro-apoptotic Mcl-1S and thereby induce cancer cell apoptosis.8 By eliminating a cancer-permissive splice variant and inducing an apoptotic splice variant simultaneously, Mcl-1 SSO can potentially achieve greater cancer suppression than a Mcl-1L inhibitor that only acts on a single function. A375 cells were treated with nanoconjugates carrying Mcl-1 SSO (800nM) and other controls for 4h. Total RNA was isolated from the treated cells after 24-h culture and RT-PCR of Mcl-1 was performed using a method described in the Supporting Information. As shown Fig. 6A, treatment with the nanoconjugates carrying Mcl-1 SSO induced a substantial shift in Mcl-1 splicing from anti-apoptotic Mcl-1L to pro-apoptotic Mcl-1S, while treatment with the nanoconjugates carrying a mismatch SSO had no effect on splicing. We tested the cytotoxic activity of the nanoconjugates carrying Mcl-1 SSO. A375 cells were treated with the nanoconjugates for 4h. Cell viability was measured in the treated cells after 72-h culture using the Alamar Blue assay. The nanoconjugates carrying Mcl-1 SSO caused death of 52% of A375 cells, which is comparable to that caused by Lipofectamine transfection of negatively charge Mcl-1 SSO (Fig. 6B). Again, treatment with the nanoconjugates carrying the mismatch SSO was not toxic to A375 cells, indicating that the toxicity was by functional delivery of Mcl-1 SSO by the nanoconjugates.
Fig. 6.
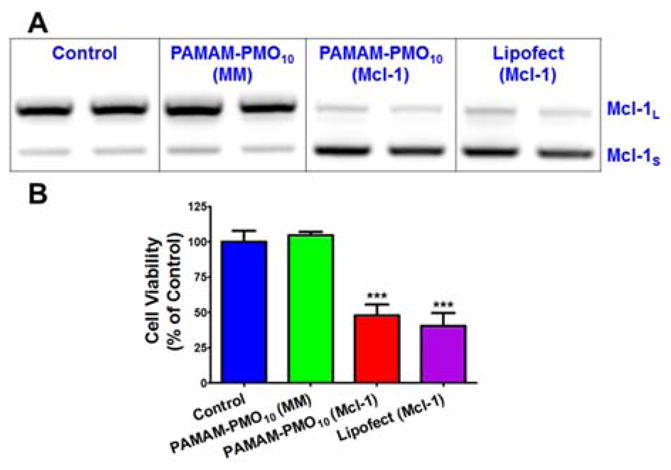
Functional delivery of Mcl-1 SSO using nanoconjugates. (a) RT-PCR of total RNA from A375 cells after treatments. (b) In vitro cytotoxicity caused by functional delivery of Mcl-1 SSO. *** p < 0.001.
The results shown in Fig. 6 indicated therapeutic potential of neutral ON-dendrimer conjugates. Other SSOs that cause cancer cell killing include Bcl-x SSO18 and STAT3 SSO19, and the nanoconjugates could also use them for cancer cell killing. Neutral ONs can be also used as antagomers of microRNA and long non-coding RNAs.2b Thus, this may provide a platform technology for using ONs in tumors and other diseased tissues.
Conclusions
Severe cytotoxicity is the main hurdle for in vivo application of dendrimers as drug carriers.5 Interaction of dendrimers with biological membranes results in membrane disruption via nanohole formation, membrane thinning and erosion.5c Thus, PEGylation is the main approach to decrease the toxicity associated with dendrimers.5 Morpholino antisense oligonucleotides resemble PEG in many aspects including good water solubility, neutral charge, high stability, and nontoxicity.6 Further, the morpholino oligo itself is a therapeutic agent. Thus, conjugation of dendrimers with PMOs achieves dual purposes of enhancing cellular delivery and decreasing cytotoxicity. Even with reduced direct interaction of nanoconjugates with cell membranes, the dendritic nanoconjugates can still use the caveolae pathway to enter the cells. After reaching the endosomes in the cells, cytosolic glutathione cleaves disulphide bonds and releases PMOs from the dendrimers. The unconjugated dendrimers might disrupt the endosomal membrane to release the ONs to their sites of action in the nucleus, and thereby produce therapeutic actions.
In conclusion, we have a utilized simple conjugation approach to construct neutrally charged and ultra-small nanoconjugates for ON delivery. The dendritic nanoconjugates showed excellent cellular uptake in cancer cells, and produced a dramatic increase in functional ON delivery.
Supplementary Material
Footnotes
The authors gratefully acknowledge Prof. Rudy L. Juliano (University of North Carolina, Chapel Hill, NC, USA) for invaluable discussion on the study and proof-reading of the manuscript. This work was supported by NIH grants 5U54CA151652 and 5R01CA151964 and an Innovation Award from the University Cancer Research Fund (UNC Lineberger Comprehensive Cancer Center). Ahu Yuan was sponsored by the China Scholarship Council.
Electronic Supplementary Information (ESI) available: See DOI: 10.1039/c000000x/
Notes and references
- 1.Bennett CF, Swayze EE. Annu Rev Pharmacol Toxicol. 2010;50:259. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 2.(a) Wahlestedt C. Nature reviews. 2013;12:433. doi: 10.1038/nrd4018. [DOI] [PubMed] [Google Scholar]; (b) Kole R, Krainer AR, Altman S. Nature reviews. 2012;11:125. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F. Lancet. 2011;378:595. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ming X. Expert Opin Drug Deliv. 2011;8:435. doi: 10.1517/17425247.2011.561313. [DOI] [PubMed] [Google Scholar]
- 5.(a) Shcharbin D, Shakhbazau A, Bryszewska M. Expert Opin Drug Deliv. 2013;10:1687. doi: 10.1517/17425247.2013.853661. [DOI] [PubMed] [Google Scholar]; (b) Dutta T, Jain NK, McMillan NA, Parekh HS. Nanomedicine. 2010;6:25. doi: 10.1016/j.nano.2009.05.005. [DOI] [PubMed] [Google Scholar]; (c) Jain K, Kesharwani P, Gupta U, Jain NK. Int J Pharm. 2010;394:122. doi: 10.1016/j.ijpharm.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 6.(a) Summerton J, Weller D. Antisense Nucleic Acid Drug Dev. 1997;7:187. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]; (b) Amantana A, Iversen PL. Current opinion in pharmacology. 2005;5:550. doi: 10.1016/j.coph.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Ming X, Carver K, Wu L. Biomaterials. 2013;34:7939. doi: 10.1016/j.biomaterials.2013.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shieh JJ, Liu KT, Huang SW, Chen YJ, Hsieh TY. J Invest Dermatol. 2009;129:2497. doi: 10.1038/jid.2009.83. [DOI] [PubMed] [Google Scholar]
- 9.(a) Yoo H, Juliano RL. Nucleic Acids Res. 2000;28:4225. doi: 10.1093/nar/28.21.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yoo H, Sazani P, Juliano RL. Pharm Res. 1999;16:1799. doi: 10.1023/a:1018926605871. [DOI] [PubMed] [Google Scholar]
- 10.Kumar V, Qin J, Jiang Y, Duncan RG, Brigham B, Fishman S, Nair JK, Akinc A, Barros SA, Kasperkovitz PV. Molecular therapy. Nucleic acids. 2014;3:e210. doi: 10.1038/mtna.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deneke SM. Current topics in cellular regulation. 2000;36:151. doi: 10.1016/s0070-2137(01)80007-8. [DOI] [PubMed] [Google Scholar]
- 12.Hussain M, Shchepinov M, Sohail M, Benter IF, Hollins AJ, Southern EM, Akhtar S. J Control Release. 2004;99:139. doi: 10.1016/j.jconrel.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Ivanov AI. Methods Mol Biol. 2014;1174:3. doi: 10.1007/978-1-4939-0944-5_1. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz SL, Cao C, Pylypenko O, Rak A, Wandinger-Ness A. J Cell Sci. 2008;121:246. doi: 10.1242/jcs.015909. [DOI] [PubMed] [Google Scholar]
- 15.Howe CL, Granger BL, Hull M, Green SA, Gabel CA, Helenius A, Mellman I. Proc Natl Acad Sci U S A. 1988;85:7577. doi: 10.1073/pnas.85.20.7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ming X, Feng L. Mol Pharm. 2012;9:1502. doi: 10.1021/mp300113c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ming X, Sato K, Juliano RL. J Control Release. 2011;153:83. doi: 10.1016/j.jconrel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bauman JA, Li SD, Yang A, Huang L, Kole R. Nucleic acids research. 2010;38:8348. doi: 10.1093/nar/gkq731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zammarchi F, de Stanchina E, Bournazou E, Supakorndej T, Martires K, Riedel E, Corben AD, Bromberg JF, Cartegni L. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17779. doi: 10.1073/pnas.1108482108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.