

Background: NEIL2 (Nei-like 2) is a mammalian oxidized base-specific DNA glycosylase.
Results: Neil2-null mice accumulate oxidative damage in transcribed genes and are susceptible to inflammatory agents.
Conclusion: In long-lived species, NEIL2 plays a critical role in maintaining genomic integrity and tissue homeostasis.
Significance: We provide in vivo evidence for NEIL2's role in preferential repair of oxidized bases in active genes in mammals.
Keywords: DNA damage, DNA enzyme, gene knockout, inflammation, reactive oxygen species, DNA damage and repair, DNA glycosylase, NEIL2, knock-out animals, transcription-coupled repair
Abstract
Why mammalian cells possess multiple DNA glycosylases (DGs) with overlapping substrate ranges for repairing oxidatively damaged bases via the base excision repair (BER) pathway is a long-standing question. To determine the biological role of these DGs, null animal models have been generated. Here, we report the generation and characterization of mice lacking Neil2 (Nei-like 2). As in mice deficient in each of the other four oxidized base-specific DGs (OGG1, NTH1, NEIL1, and NEIL3), Neil2-null mice show no overt phenotype. However, middle-aged to old Neil2-null mice show the accumulation of oxidative genomic damage, mostly in the transcribed regions. Immuno-pulldown analysis from wild-type (WT) mouse tissue showed the association of NEIL2 with RNA polymerase II, along with Cockayne syndrome group B protein, TFIIH, and other BER proteins. Chromatin immunoprecipitation analysis from mouse tissue showed co-occupancy of NEIL2 and RNA polymerase II only on the transcribed genes, consistent with our earlier in vitro findings on NEIL2's role in transcription-coupled BER. This study provides the first in vivo evidence of genomic region-specific repair in mammals. Furthermore, telomere loss and genomic instability were observed at a higher frequency in embryonic fibroblasts from Neil2-null mice than from the WT. Moreover, Neil2-null mice are much more responsive to inflammatory agents than WT mice. Taken together, our results underscore the importance of NEIL2 in protecting mammals from the development of various pathologies that are linked to genomic instability and/or inflammation. NEIL2 is thus likely to play an important role in long term genomic maintenance, particularly in long-lived mammals such as humans.
Introduction
Endogenously generated reactive oxygen species in mammalian cells continuously target cellular macromolecules, including the genomic DNA (1–3). However, all cells are equipped with an arsenal of DNA repair proteins that continuously maintain the genome's integrity for proper cellular function and viability. Reactive oxygen species-induced oxidative DNA base modifications are primarily repaired via the base excision repair (BER)3 pathway, which is initiated with excision of the oxidized base by a DNA glycosylase/AP lyase, generating 3′-blocked ends and 5′-phosphate. The 3′ end is then processed to generate 3′-OH, which is necessary for DNA polymerase to incorporate the appropriate base using the nondamaged template base, and finally nick-sealing by a DNA ligase (4, 5). In human cells, five oxidized base-specific DNA glycosylases have been identified and characterized so far. Endonuclease III homolog 1 (NTH1) and 8-oxoguanine-DNA glycosylase (OGG1) were characterized initially by several groups; these enzymes preferentially excise oxidized pyrimidines and purines, respectively (6, 7). Several years later, we and others identified NEIL (Nei-like 1–3) DNA glycosylases, which are functionally similar to Escherichia coli MutM or Nei and excise both purine and pyrimidine oxidation products (8–12). All three NEILs excise base lesions from DNA bubble or single-stranded regions; in contrast, NTH1 and OGG1 are active only with duplex DNA (13–15). We have recently reported that NEIL1 is primarily involved in the repair of replicating genomes (16, 17), and our in vitro biochemical studies indicate that NEIL2 primarily removes the oxidized bases from transcribing genes via a transcription-coupled BER (TC-BER) pathway (18). We also identified a polymorphic variant of NEIL2 that occurs more frequently in human lung cancer patients than in normal individuals (19). Furthermore, depletion of NEIL2 caused a significant increase in the spontaneous mutation frequency in the HPRT gene of the V79 Chinese hamster lung cell line (20). All these studies collectively indicate that NEIL2 plays an important role in maintaining genomic integrity and preventing DNA mutagenesis in mammalian cells.
To examine the biological significance of NEIL2, we generated Neil2-knock-out (KO) mice. The Neil2-KO mice were overtly normal and fertile; however, we found that they accumulated higher amounts of oxidized bases in the transcribed region of the genome as they aged. Moreover, Neil2-null MEFs showed a significantly higher frequency of telomere loss and genome instability, indicating a critical role of NEIL2 in long term genomic maintenance.
Experimental Procedures
Targeting Vector Construction and the Generation of Neil2-Knock-out (KO) Mice
To generate a gene-targeting vector, we screened a mouse genomic library, ES129SvJ, for clones containing Neil2. We then subcloned a DNA fragment containing Neil2 exons 1–3, and we introduced a loxP site 96 bp upstream from exon 2 (the first coding exon of Neil2) and a transcription unit containing the neomycin (neo) resistance gene under the transcriptional control of the phosphoglycerate kinase promoter into intron 2 in the opposite orientation relative to Neil2. A second loxP site was inserted immediately after the phosphoglycerate kinase-neo transcription element. The phosphoglycerate kinase-neo-loxP sequence was flanked by 2.9 kb of genomic DNA on the 5′ side and 3.2 kb on the 3′ side. We first generated mice that carried a Neil2 conditional allele (Neil2floxed), and we crossed them with C57BL/6-Tg (Zp3-Cre)93Knw (The Jackson Laboratory stock number 003651) to generate mice that carried the Neil2-null allele (Neil2KO). The mutants were subsequently backcrossed to C57BL/6J. All animal breeding and experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals. The protocol used was approved by the University of Texas Medical Branch Animal Care and Use Committee (Hazra, protocol number 0606029, and Boldogh, protocol number 0807044A).
Genotyping
To genotype mice, we used Southern blots and PCR. For Southern blots, genomic DNA was isolated from tails, digested with EcoRI, and hybridized with the 5′-flanking probe (a 446-bp EcoRI-BglII fragment, see Fig. 1). A pair of primers (Fig. 1A, blue arrowheads), N2loxPBglF (5′-CCTTATCCCTTTCCCAGCTC-3′) and N2loxPR (5′-TTAGCCCAGCATGTCTGATG-3′), was used for the genotyping of the Neil2floxed line. Another pair (Fig. 1A, green arrowheads), Neil2F (5′-TCAGCTGGTTTTTGGCATCT-3′) and Neil2R (5′-ATGTCACACCAAGAGCAAGG-3′), was used for the genotyping of the Neil2KO line.
FIGURE 1.
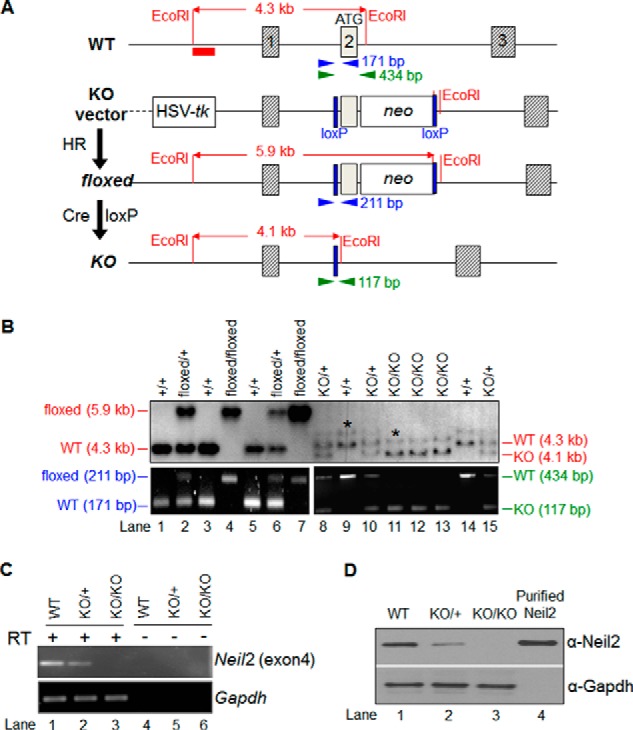
Neil2 gene targeting. A, Neil2 wild-type allele (WT), knock-out (KO) vector, conditional allele (floxed), and knock-out/null allele (KO) are shown. The red bar below the WT allele represents the 5′-flanking probe for a diagnostic Southern blot. EcoRI sites relevant to the Southern blot are shown. The arrowheads indicate PCR primers for genotyping. The size of each PCR amplicon is shown next to the reverse primer. Cre loxP, Cre-loxP recombination; neo, neomycin-resistance gene; HSV-tk, herpes simplex virus-thymidine kinase gene; HR, homologous recombination. B, genotyping of the mice. The upper panels show Southern blot data. The DNA was digested with EcoRI and separated in 1% agarose/TAE gels. The filters were hybridized with the 5′-flanking probe. The WT band (4.3 kb), floxed allele band (5.9 kb), and KO allele band (4.1 kb) are shown. The extra bands (highlighted by asterisks) likely occurred due to incomplete digestion of genomic DNA. The lower panels show PCR data. The mutant line with the floxed allele was genotyped by primers shown as blue arrowheads in A; the line with the KO allele was genotyped by primers shown as green arrowheads in A. C, expression of Neil2 transcript. Mock (−RT) and RT-PCR amplification profile of a segment of Neil2 (exon 4) mRNA from kidney tissue in WT, Neil2 heterozygous (KO/+), and Neil2-null mice (KO/KO) at 24 months. RT +/− indicates samples with or without treatment with RT. Mouse Gapdh was used as a loading control to confirm the integrity and equal loading of RNA in each lane. D, expression profile of NEIL2 protein as shown by Western blotting, using rabbit polyclonal anti-NEIL2 antibody. WT, KO/+, and KO/KO represent protein samples from kidney tissue of WT, Neil2 heterozygous, and Neil2-null mice, respectively. Purified NEIL2 (25 ng) was used as a positive control. GAPDH was used as a loading control to confirm equal loading of protein from each sample.
Isolating and Culturing Mouse Embryonic Fibroblast
Primary mouse embryonic fibroblast (MEF) cultures were established by standard procedures (21) from individual embryos of embryonic day 13.5 (E13.5) derived from heterozygous matings. Tissues were disaggregated with 0.25% trypsin containing 0.1 mm EDTA. The trypsin/EDTA was removed by centrifugation, and the cells were resuspended in the culture medium. The cells were then counted, plated at a density of 3 × 106 cells per plate, and cultured under low oxygen tension (3% O2, 92% N2, and 5% CO2) in DMEM Ham's F-12 (3:1) medium containing 10% FBS. The medium was changed on the 1st day, and cells were trypsinized and subcultured when they reached confluence (3–4 days). Genotyping was carried out by PCR. Primary (passage 2) cells were stored in liquid nitrogen for future studies.
RT-PCR and Western Analysis
Kidneys were harvested from WT, Neil2KO/+, and Neil2KO/KO mice at 24 months. RNA was then extracted from one part of the tissues (10 mg each) using RNeasy mini kits (Qiagen, catalog no. 74104) with on-column DNase digestion. cDNA was prepared from 1 μg of DNase-treated RNA using Superscript III First Strand Synthesis Super-Mix (Invitrogen, catalog no. 11752-250) and subsequently used for RT-PCR. RT-PCR was carried out with 2 μl of cDNA using Neil2 exon 4-specific oligos (Table 1) using Quick-load Taq 2× master mix (New England Biolabs, catalog no. M0271S) with the following thermal cycling conditions: 95 °C-3 min; (94 °C for 10 s, 55 °C for 15 s, and 68 °C for 30 s) for 30 cycles and 68 °C for 5 min. Mouse Gapdh-specific oligos (Table 1) were used as a control to confirm RNA integrity, as well as equal loading of RNA in each lane. Mock-treated cDNA (without RT enzyme mix) was used in each case to check for any possible amplification arising from DNA contamination of the isolated RNA. The expression of pol β, β-globin, NeuroD, and NanoG transcripts were examined in the kidney, liver, lung, and whole brain tissues of WT mice following a similar protocol to that described above with the oligos listed in Table 1.
TABLE 1.
List of oligonucleotides used in the study
| Primers | 5′ to 3′ Nucleotide sequence | Tm | Purpose |
|---|---|---|---|
| pol β LA FP | TAT CTC TCT TCC TCT TCA CTT CTC CCC TGG | 61.5 °C | LA-qPCR forward primer for mouse pol β |
| pol β LA RP | CGT GAT GCC GCC GTT GAG GGT CTC CTG | 68 °C | LA-qPCR reverse primer for mouse pol β |
| β-Globin LA FP | TTG AGA CTG TGA TTG GCA ATG CCT | 59.2 °C | LA-qPCR forward primer for mouse β-globin |
| β-Globin LA RP | CCT TTA ATG CCC ATC CCG GAC T | 59.4 °C | LA-qPCR reverse primer for mouse β-globin |
| NeuroD LA FP | CTCGCAGGTGCAATATGAATC | 57 °C | LA-qPCR forward primer for mouse NeuroD |
| NeuroD LA RP | GCAACTGCATGGGAGTTTTCT | 59 °C | LA-qPCR reverse primer for mouse NeuroD |
| NanoG LA FP | GGACACAGGACGGAGCAATATAAG | 57.1 °C | LA-qPCR forward primer for mouse NanoG |
| NanoG LA RP | CCTGTGAGTGGTCAGGAGTTAAAG | 57 °C | LA-qPCR reverse primer for mouse NanoG |
| pol β SA FP | TATGGACCCCCATGAGGAACA | 57.8 °C | Forward primer for amplification of short fragment from mouse pol β |
| pol β SA RP | AACCGTCGGCTAAAGACGTG | 57.6 °C | Reverse primer for amplification of short fragment from mouse pol β |
| β-Globin SA FP | ACACTACTCAGAGTGAGACCCA | 56.8 °C | Forward primer for amplification of short fragment from mouse β-globin |
| β-Globin SA RP | ATACCCAATGCTGGCTCCTG | 57.4 °C | Reverse primer for amplification of short fragment from mouse β-globin |
| NeuroD SA FP | CTGCAAAGGTTTGTCCCAGC | 59.9 °C | Forward primer for amplification of short fragment from mouse NeuroD, RT-PCR and qChIP |
| NeuroD SA RP | CTGGTGCAGTCAGTTAGGGG | 60 °C | Reverse primer for amplification of short fragment from mouse NeuroD, RT-PCR and qChIP |
| NanoG SA FP | GTCCATTAGTCCTGTCCCTTTC | 54.9 °C | Forward primer for amplification of short fragment from mouse NanoG |
| NanoG SA RP | GCAATGGATGCTGGGATACT | 55 °C | Reverse primer for amplification of short fragment from mouse NanoG |
| Mouse pol β FP | CATGGGTGTTTGCCAGCTTC | 60.0 °C | Forward primer for RT-PCR and qChIP of mouse pol β |
| Mouse pol β FP | CTCACTGTCCACAGGAAGGG | 59.6 °C | Reverse primer for RT-PCR and qChIP of mouse pol β |
| Mouse β-globin FP | N/A RealTimePrimers | Forward primer for RT-PCR of mouse β-globin | |
| Mouse β-globin RP | N/A RealTimePrimers | Reverse primer for RT-PCR of mouse β-globin | |
| Mouse NanoG FP | N/A RealTimePrimers | Forward primer for RT-PCR and qChIP of mouse NanoG | |
| Mouse NanoG RP | N/A RealTimePrimers | Reverse primer for RT-PCR and qChIP of mouse NanoG | |
| Mouse HPRT FP | N/A RealTimePrimers | Forward primer for RT-PCR and qChIP of mouse Hprt | |
| Mouse HPRT RP | N/A RealTimePrimers | Reverse primer for RT-PCR and qChIP of mouse Hprt | |
| Mouse Neil-2 exon4 FP | TTTAGTGGTGGTGGCTTCCT | 56.3 °C | Forward primer for RT-PCR of mouse Neil-2 |
| Mouse Neil-2 exon4 RP | AGTGTGTAGCACACAGGCTA | 58.6 °C | Reverse primer for RT-PCR of mouse Neil-2 |
| Mouse GAPDH FP | TCTCTGCTCCTCCCTGTTCC | 60 °C | Forward primer for RT-PCR of mouse Gapdh |
| Mouse GAPDH RP | CAATCTCCACTTTGCCACTGC | 60 °C | Reverse primer for RT-PCR of mouse Gapdh |
Total protein was extracted from another part of the kidney tissue from WT, Neil2KO/+, and Neil2KO/KO mice at 24 months, according to a reported protocol (22) for Western analysis. Twenty five μg of total protein was loaded onto a 4–12% Bis-Tris gel (Invitrogen). After electrophoretic transfer of proteins to nitrocellulose membranes, the membranes were probed with rabbit polyclonal anti-NEIL2 antibody (Ab, developed in-house, dilution 1:500). The anti-rabbit GAPDH antibody (Genetex Inc., catalog no. GTX100118) was used for examining the level of GAPDH as loading control in each lane. In each case, the blot was stripped using Restore Plus stripping buffer (Thermo Scientific) and reprobed with a second Ab.
LA-qPCR for DNA Damage Analysis
Long amplicon quantitative-PCR (LA-qPCR) assays were carried out essentially following the reported protocols (19, 23–25) with some minor modifications. Briefly, livers (15 mg), kidneys (20 mg), lungs (20 mg), and whole brain (15 mg) were harvested from WT and Neil2KO/KO mice (2, 8, or 24 months old, as has been indicated in the figure legends) immediately after the sacrifice of the animals, and tissues were stored in 20% DMSO in liquid nitrogen for future use. Genomic DNA extraction was performed using the genomic-tip 20/G kit (Qiagen, catalog no. 10223, with corresponding buffer sets) per the manufacturer's directions. This kit has the advantage of minimizing DNA oxidation during the isolation steps, and thus it can be used reliably for isolation of high molecular weight DNA with excellent template integrity to detect endogenous DNA damage using LA-qPCR. After precise quantitation of the DNA by Pico Green (Molecular Probes) in a 96-well black-bottomed plate, the genomic DNA (300 ng) was digested with the E. coli enzymes Fpg and Nei to induce strand breaks at the sites of the unrepaired oxidized base lesion. Gene-specific LA-qPCR analyses for measuring DNA damage were performed using Long Amp TaqDNA polymerase (New England Biolabs). LA-qPCR was carried out to amplify a 6.5-kb region of pol β, 8.7 kb of β-globin, 7.2 kb of NeuroD, and 7.4 kb of NanoG in mouse genomic DNA using the primer sets described in Table 1. The numbers of cycles and DNA concentrations were standardized in each case before the actual reaction, so that the PCR remains within the linear range of amplification (23–25). The final PCR condition was optimized at 94 °C for 30 s (94 °C for 30 s, 55–60 °C for 30 s depending on the oligo annealing temperature, 65 °C for 10 min) for 25 cycles and 65 °C for 10 min. Fifteen ng of DNA template was used in each case, and the LA-qPCR was set for all the genes under study from the same stock of Fpg/Nei-treated diluted genomic DNA samples, to avoid variations in PCR amplification due to sample preparation. Because amplification of a small region would be independent of DNA damage, a small DNA fragment for each gene (Table 1) was also amplified to normalize the amplification of large fragments. The PCR conditions were 94 °C for 30 s (94 °C for 30 s, 54 °C for 20 s, and 68 °C for 30 s) for 25 cycles and 68 °C for 5 min. Fifteen ng of template from the same Fpg/Nei-digested DNA aliquot was used for short PCR. The amplified products were then visualized on gels and quantitated with an ImageJ automated digitizing system (National Institutes of Health) based on three independent replicate PCRs. The extent of damage was calculated in terms of lesions/10 kb of genome following Poisson's distribution according to reported methods (26).
Analysis of NEIL2-associated Proteins by Co-immunoprecipitation (Co-IP) from WT and Neil2 KO Mice Tissue
Approximately 250 mg of intact liver tissue from freshly sacrificed WT and Neil2-null mice were sliced into small pieces. The tissue pieces were collected in a pre-chilled, sterile all-glass homogenizer (Thomas, PHILA USA C55506) with a large clearance pestle and hand-homogenized with 4 volumes of ice-cold homogenization buffer (0.25 m sucrose, 15 mm Tris-HCl, pH 7.9, 60 mm KCl, 15 mm NaCl, 5 mm EDTA, 1 mm EGTA, 0.15 mm spermine, 0.5 mm spermidine, 1 mm dithiothreitol (DTT), 0.1 mm phenylmethylsulfonyl fluoride (PMSF), and protease inhibitors (Roche Applied Science)) with ∼20 strokes to disrupt tissues (27–29). Homogenization was continued until single-cell slurry was obtained (monitored under a microscope to ensure that the cells were dissociated), incubated on ice for 15 min, and centrifuged at 1,000 × g to obtain the cell pellet. Nuclear extracts were then prepared from the cells for subsequent co-IP analysis according to an established protocol by Aygun et al. (30), with modifications as applicable. The immunoprecipitates were examined for the presence of various interacting proteins using the appropriate Abs (NEIL2, ligase III, APE1 (all anti-rabbit in-house Abs), polynucleotide kinase 3′-phosphatase (BioBharati Life Sciences Pvt. Ltd., Kolkata, India), polymerase β (gift from Dr. Sam Wilson), CSB (Santa Cruz Biotechnology, sc-25370), TFIIH (Santa Cruz Biotechnology, Sc-293), XRCC4 (Genetex, GTX109632), and H-5 RNAP II (Covance, MMS-129R)).
Chromatin Immunoprecipitation (ChIP) and Re-ChIP Assay from WT and NEIL2 KO Mice Tissue
ChIP and ChIP/re-ChIP assays were performed from fresh liver tissue of WT and Neil2 KO mice as described (18, 31) with minor modifications. Briefly, 80 mg of fresh tissue were chopped into small pieces (between 1 and 3 mm3) using a sterile scalpel razor and fixed in 1% formaldehyde for 15 min with gentle agitation at room temperature to cross-link DNA to bound proteins. The sample was centrifuged at 440 × g for 5 min at room temperature followed by the addition of 0.125 m glycine to terminate the cross-linking reaction. The sample was washed 2–3 times with ice-cold PBS (containing protease inhibitor mixture) and centrifuged each time at 440 × g for 4 min at 4 °C. The pellet was resuspended in 1 ml of ice-cold lysis buffer (10 mm EDTA, 1% (w/v) SDS, 50 mm Tris-HCl, pH 7.5) with freshly added protease inhibitors and PMSF for 15 min on ice and homogenized slowly (∼20 strokes on ice) with a hand homogenizer and tight pestle to make a single-cell suspension. After homogenization, the sample was transferred to pre-cooled 1.5-ml microcentrifuge tubes and centrifuged at 2260 × g for 5 min. The pellet was further resuspended in ice-cold lysis buffer (500 μl) and subjected to sonication to generate ∼400 bp of DNA fragments (sonicated 7× for 3 min for each pulse (21 min total)). The sample was centrifuged at 20,780 × g for 30 min at 4 °C, and the supernatant (the sheared chromatin with bound protein) was collected for subsequent ChIP as described previously (18). The sheared chromatin was immunoprecipitated for 6 h at 4 °C with 10 μg of isotype control IgG (Santa Cruz Biotechnology, Sc-2027) or the following antibodies: anti-NEIL2 (32) and anti-RNAP II (N20, Santa Cruz Biotechnology, Sc-899).
For re-ChIP assays, the eluant of the primary immunocomplex, obtained with the anti-RNAP II Ab, was diluted 10-fold with dilution buffer (20 mm Tris-HCl, pH 8.0, 1 mm EDTA, 150 mm NaCl, 1% Triton X-100, and protease inhibitors) and then subjected to further immunoprecipitation with the anti-NEIL2 Ab or control IgG (18). After the recovery of DNA with proteinase K treatment followed by phenol extraction and ethanol precipitation, 1% of input chromatin and the precipitated DNA were analyzed by qPCR with the primers listed in Table 1. The ChIP data are presented in the histograms as percent binding relative to input value.
Animals, Challenge and Evaluation of Inflammation
Eight-month-old WT and Neil2-null mice (n = 6, male and female; 50–50%) were challenged with lipopolysaccharide (LPS, 100 ng/lung) or glucose oxidase (GOx, 1 milliunit/lung) or TNF-α (20 ng/lung) intranasally (i.n.) as we described previously (33). Sixteen hours after challenge, mice were euthanized, and bronchoalveolar lavage was performed by cannulating the trachea and lavaging the lungs with two 0.7-ml aliquots of ice-cold Dulbecco's PBS (Sigma). The bronchoalveolar lavage cells were pelleted, washed, cyto-spun, and stained with Wright-Giemsa. The numbers of neutrophils, lymphocytes, and macrophages were determined by microscopic examination of a minimum of 400 cells/slide on each stained cytocentrifuge preparation (34).
Results
Generation of Neil2-KO Mice
We constructed a gene-targeting vector that would introduce two loxP sites in intron 1 and intron 2 of Neil2 so that we could inactivate Neil2 conditionally using the Cre-loxP recombination system (Fig. 1A). The vector was electroporated into B6129F1 hybrid G4 embryonic stem (ES) cells (35). We used Southern blot analysis (Fig. 1B) to test 164 ES cell clones that had survived drug selection, and we identified 36 correctly targeted clones. Multiple clones were injected into C57BL/6J blastocysts, and one clone (number 164) transmitted the Neil2 conditional allele (Neil2floxed) into the mouse germ line. Mice homozygous for the Neil2floxed allele were overtly normal. To obtain mice that carry a Neil2 mutant allele with an exon 2 deletion (Neil2KO), we crossed Neil2floxed/+ mice with the Zp3-Cre transgenic mice, which express Cre in developing oocytes (36). Crosses between wild-type (WT) C57BL/6J males and Neil2floxed/+ females that also carried a Zp3-Cre transgene resulted in Neil2KO/+ mice. Mice homozygous for the Neil2KO allele were viable and were indistinguishable from Neil2KO/+ or Neil2+/+ mice. We examined Neil2/NEIL2 expression levels by RT-PCR (Fig. 1C) and Western blot analysis (Fig. 1D) in kidney tissues of 24-month-old mice. Neil2 transcript levels and NEIL2 protein levels were reduced in Neil2KO/+ mice and completely absent in Neil2KO/KO mice. This indicates that Neil2KO is a null allele.
Age-dependent Increase of Oxidative DNA Damage in Neil2-null Mice
We have shown earlier by in vitro analysis that human NEIL2 preferentially repairs oxidized bases from the transcribed genes (18). Hence, to examine whether Neil2-null animals accumulate oxidative base damage in their transcribed genomes, we have selected a pair of genes as transcribed (DNA pol β and β-globin) and another pair as nontranscribed (neurogenic differentiation factor (NeuroD, expressed mostly in the brain) (37) and NanoG (expressed mostly in pluripotent stem cells) (38)). Expression of β-globin is generally presumed to be restricted; however, it has recently been shown that β-globin is expressed in mouse lungs (39). Our RT-PCR data further confirmed that pol β and β-globin are well transcribed in the lung, kidney, and liver (Fig. 2), whereas NeuroD and NanoG were found to have undetectable expression levels in the kidney, lung, and liver tissues (Fig. 2).
FIGURE 2.
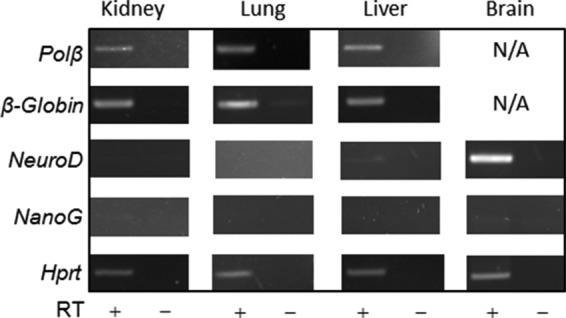
RT-PCR profiling of various mouse genes. RT-PCR-mediated expression profiling of pol β, β-globin, NeuroD, and NanoG in various tissues (kidney, lung, liver, and whole brain, as indicated) of wild-type mice. RT + and − represent samples with and without treatment with RT. Amplification of constitutively expressing mouse Hprt was used as control. N/A means not applicable, as the assay was not carried out for the respective genes from the corresponding tissues.
The levels of endogenous base damage in the pol β, β-globin, NeuroD, and NanoG genes in WT versus Neil2KO/KO mice were compared using LA-qPCR. LA-qPCR conditions for amplification of a long region (∼6–9 kb) for mouse pol β and β-globin genes with the proper set of primers had already been well standardized by Van Houten's group (23). We should mention here that designing specific primers for LA-qPCR is a task with a very low success rate. We made a continuous effort to amplify many genes; finally, we successfully standardized PCR conditions for two other mouse genes, NeuroD and NanoG. For LA-PCR analysis, the cellular DNA was initially isolated from the kidneys of mice of different age groups (2, 8, or 24 months) as described under “Experimental Procedures.” The genomic DNA was then treated with E. coli Fpg/Nei before analysis of the damage by LA-qPCR to excise oxidized bases and to generate DNA single strand breaks after excision of the damaged bases, thereby preventing PCR amplification by the polymerase. A decrease in the PCR product reflects a higher DNA damage level, and we indeed consistently found accumulation of a higher level of oxidative DNA damage in the region encompassing essentially the transcribing pol β and β-globin genes in Neil2KO/KO mice than in the same region of the WT (Fig. 3A) when a long region (∼6–9 kb) was amplified. Conversely, the level of oxidative DNA damage in the nontranscribing genes (NeuroD and NanoG) did not increase in Neil2-null versus WT animals. Amplification of a smaller fragment for each gene was similar between the samples, because the probability of damage in a small fragment is low. The data (expressed in terms of base lesion/10 kb using Poisson's distribution) indicate a role for NEIL2 in preferential repair of oxidized DNA bases in vivo for actively transcribing regions of the genome, which is consistent with our earlier observations in a cell culture model and our in vitro repair studies (18). Our data also reflect an enhancement of endogenous oxidized DNA damage with the progression of age, as the level of DNA damage is minimal in 2-month-old Neil2KO/KO mice, but is significant in 24-month-old Neil2KO/KO mice.
FIGURE 3.

Age-dependent accumulation of oxidized DNA bases in the transcribed versus nontranscribed genome of Neil2-null mice. A, LA-qPCR was used to evaluate oxidized base-specific genomic DNA damage levels in kidney tissue of WT versus Neil2-null mice of different age groups (2, 8, and 24 months old). Representative gels showing PCR-amplified fragments encompassing mouse pol β/β-globin and NeuroD/NanoG as transcribed and nontranscribed gene pairs, respectively. Amplification of each large fragment (upper panels) was normalized to that of a small fragment of the corresponding gene (lower panels), and the data were expressed as lesion frequency/10 kb of DNA as described under “Experimental Procedures.” Histograms represent the DNA damage quantitation for WT versus Neil2-null mice in each case (n = 3, **, p < 0.01). Error bars indicate standard error of the mean. B, LA-qPCR for DNA damage analysis for two additional tissue samples (lung and liver) from WT and Neil2-null mice of the 24-month age group (n = 3, **, p < 0.01). C, analysis of the accumulation of oxidized DNA bases in whole brain tissue samples from WT and Neil2-null mice with NeuroD as the transcribing gene and NanoG as the nontranscribed gene (n = 3, **, p < 0.01).
To further confirm our results in other tissues, we performed similar LA-qPCR-mediated DNA damage analyses in the lung and liver tissues of 24-month-old WT versus Neil2KO/KO mice. The profile of DNA damage accumulation in the aforementioned transcribed versus nontranscribed genes, which had a similar transcription status in these two tissues (Fig. 2), followed a similar pattern (Fig. 3B), thereby confirming our initial observation in kidney tissue. Unlike the kidney, liver, and lung tissue, NeuroD is a highly transcribed gene in brain tissue (Fig. 2) (37). We thus performed a reciprocal experiment to measure the accumulation of oxidized bases in the mouse NeuroD gene in the whole brain from WT and Neil2KO/KO mice, and we indeed found the accumulation of DNA damage in NeuroD in the Neil2KO/KO mice (Fig. 3C). However, the LA-qPCR profile for NanoG (nontranscribed in brain tissue, Fig. 2 (38)) showed no significant differences between WT and Neil2KO/KO mice (Fig. 3C). These data thus further confirmed the critical in vivo role of NEIL2 in repairing oxidized bases in the transcribed genes.
NEIL2 Associates with RNAP II and Several TCR-related Proteins
DNA replication, transcription, repair, and many other cellular processes are all accomplished via the action and regulation of dynamic multiprotein complexes. Hence, identification of proteins in the complexes helps to elucidate sequential steps and their role in those processes. In an effort to understand the mechanistic basis of NEIL2's physiological role in TC-BER, we performed co-IP analysis from the freshly prepared nuclear extract of mouse liver tissues with an anti-NEIL2 or Lig IIIα or RNAP II Ab. It was found that RNAP II and other critical TCR-related proteins (CSB and TFIIH) are present in both NEIL2 and Lig IIIα immunocomplexes (Fig. 4, A and B, lane 4). The reverse IP using RNAP II Ab (N20, Santa Cruz Biotechnology) showed the presence of NEIL2, Lig IIIα, and other proteins (TCR and BER) in the RNAP II immunocomplex as well (Fig. 4C, lane 4). The presence of these proteins in each other's complex indicates stable association of these proteins with NEIL2, Lig IIIα, and RNAP II under physiological conditions. Importantly, co-IP using the anti-NEIL2 Ab in Neil2-null tissue extract clearly shows the absence of their partners in the complex (Fig. 4A, compare lane 6 with 4), indicating the specificity of the NEIL2 Ab and the association of the proteins in the complex. Co-IP with Lig IIIα and RNAP II in Neil2-null tissue extracts can pull down various other interacting partners (Fig. 4B and C, lane 6), except NEIL2. This also indicates that interactions of RNAP II and Lig IIIα with other proteins are not mediated via NEIL2. Furthermore, the absence of APE1 (involved in OGG1/NTH1-mediated but not NEIL2-mediated repair) (32, 40) in the Neil2 IP (Fig. 4A) and XRCC4 (involved in DNA double strand break repair) in the Lig IIIα IP (Fig. 4B) also showed the specificity of our co-IP experiments.
FIGURE 4.
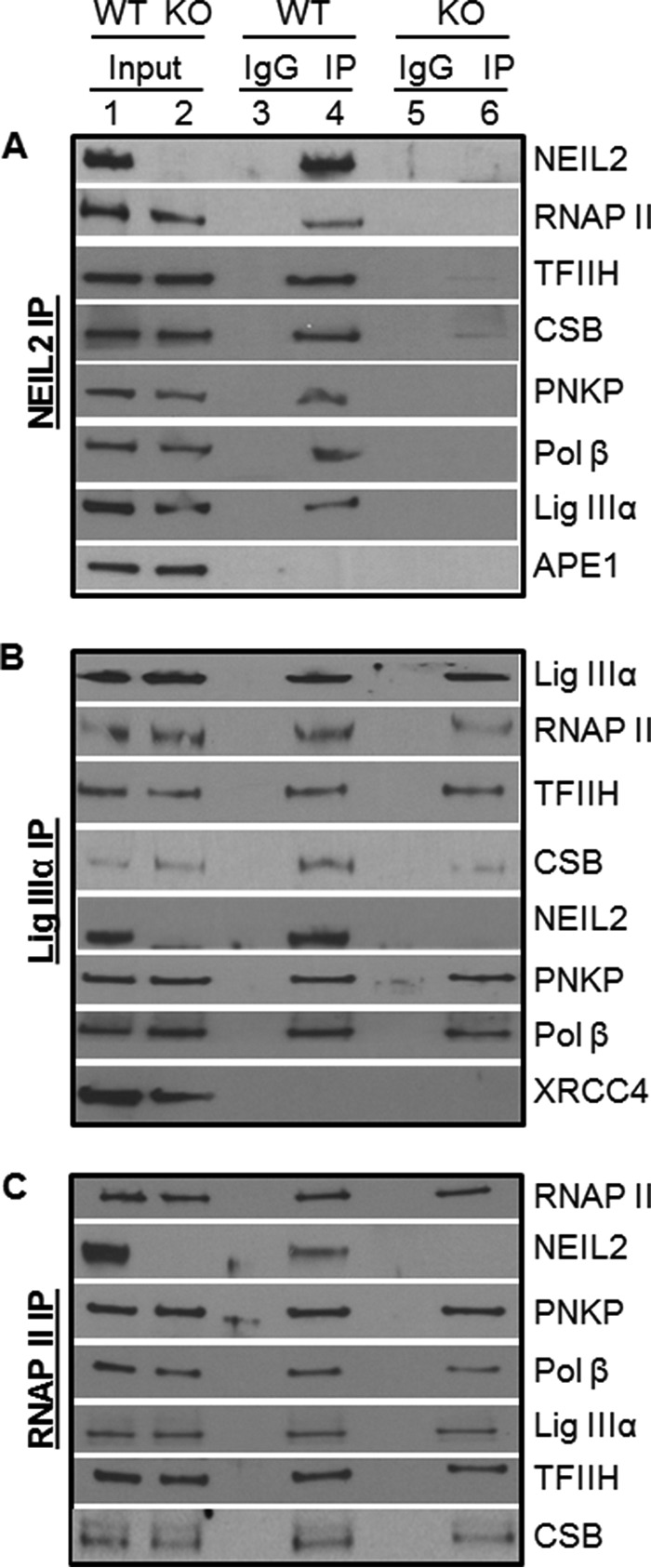
Co-IP analysis. Nuclear extracts (1 mg, benzonase and/or EtBr-treated) prepared from fresh liver tissue of WT or Neil2-null mice were immunoprecipitated with anti-NEIL2 (A) or Lig IIIα (B) or RNAP II (C) and tested for the presence of associated proteins with specific Abs.
Co-occupancy of RNAP II and NEIL2 on Transcribed Genes
ChIP/re-ChIP assays will allow us to determine whether the two proteins are close together in the context of chromatin, presumably forming a complex within a specific genomic region. Hence, to confirm co-occupancy of NEIL2 with RNAP II on the transcribed genes, we performed ChIP followed by re-ChIP from soluble chromatin preparations of liver tissue of WT mice. The chromatin fractions, derived from cross-linked single-cell suspensions of liver tissue, were first immunoprecipitated with anti-RNAP II Ab (N20). After stringent washes, the bound immune DNA complexes were eluted and prepared for a second immuno-pulldown (re-ChIP) with anti-NEIL2 Ab or IgG as control. After reverse cross-linking, the extracted DNA was subjected to qPCR using gene-specific primers corresponding to two transcribing (Hprt and pol β) and two nontranscribing (NanoG and NeuroD) genes (Fig. 5). Fig. 5A indeed shows co-occupancy of NEIL2 and RNAP II with the transcribed genes but not with nontranscribed genes (p < 0.01). Furthermore, in an attempt to examine any possible differences in the association of RNAP II with the transcribed genome in WT versus Neil2 KO mice, RNAP II ChIP was also performed with liver tissue. Fig. 5B shows a moderate increase in the association of RNAP II with both these transcribed genes in the Neil2 KO mice as compared with WT mice (p < 0.05). Accumulation of more oxidative damage in the transcribed genome of Neil2 KO mice and subsequent attempts at TC-BER of such lesions (which cannot succeed in the absence of NEIL2) are the likely causes for enhanced association of RNAP II with those genes.
FIGURE 5.
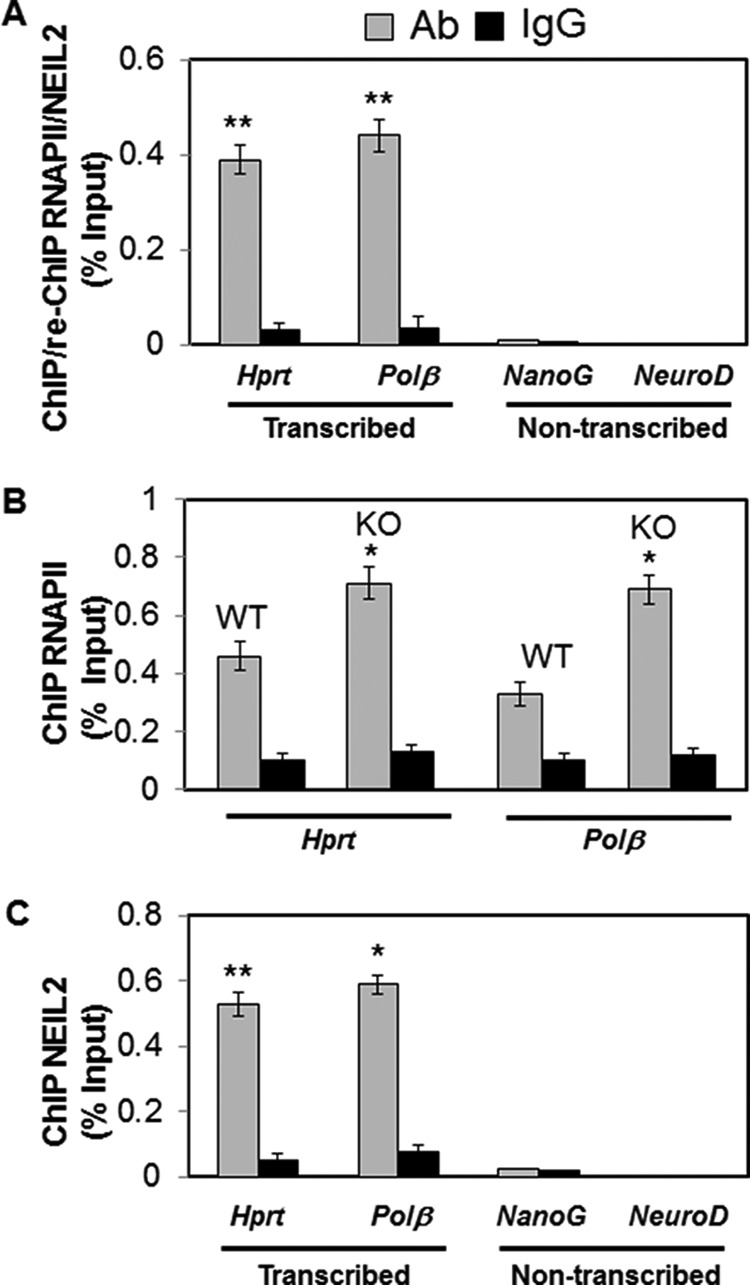
Quantitative ChIP assays. A, ChIP/Re-ChIP analysis (first IP with RNAP II Ab and the second IP with NEIL2 Ab or control IgG) of liver tissue of WT mice, showing co-occupancy of RNAP II and NEIL2 on the transcribed genome. B, Q-ChIP analysis of liver tissue of WT versus Neil2 KO mice showing differential association of RNAP II with the transcribed genome. C, Q-ChIP analysis involving NEIL2 and liver tissue of WT mice showing preferential association of NEIL2 with the transcribed genes. Mouse Hprt/pol β and NanoG/NeuroD were used as transcribed and nontranscribed gene pairs, respectively. For Q-ChIP analysis, genes of interest were amplified from immunoprecipitated DNA by SYBR Green-based qPCR. Each sample was assayed in triplicate, and the amount of immunoprecipitated DNA was calculated as the percentage of input sample. (*, p < 0.05; **, p < 0.01). Other details are provided under “Experimental Procedures.”
NEIL2 Associates with Transcribed Genes
To further validate NEIL2's role in TC-BER, a separate ChIP experiment involving NEIL2 was also conducted with the chromatin fraction from liver tissue. Fig. 5C clearly shows that NEIL2 indeed preferably associated with transcribed but not with nontranscribed genes. Collectively, these findings are consistent with our earlier physiological characterization of an association of NEIL2 with RNAP II, and here we provide the first in vivo evidence of NEIL2's preferential association with the transcribing genes.
NEIL2 Is Required for Maintaining Telomere Length Homeostasis and Genome Stability
Because Neil2-null mice accumulate oxidative damage in their transcribed genomes, which can lead to genomic instability, including telomere dysfunction, we examined metaphase spreads of primary MEFs derived from WT versus Neil2-null mouse embryos (E13.5) (41). Primary MEFs were grown to passage 6 in 20% oxygen but in the absence of any damaging agent treatment. Telomeres in metaphase spreads were detected by fluorescent in situ hybridization (FISH) with a telomere-specific probe as described previously (42, 43). Interestingly, we found that Neil2-null MEFs had 0.81 chromosomal aberrations (gaps + breaks + radials) per metaphase versus 0.06 in WT cells (Fig. 6A, compare panels a and b with panels c and d). The frequency of chromosomal aberrations in Neil2-null MEFs is thus significantly higher than that in WT control cells as assessed by χ2 analysis (p < 0.05). Furthermore, we found a higher frequency (Fig. 6B, compare panels a and b with panels c and d) of undetectable telomere signals in Neil2−/− cells, which is statistically significant compared with Neil2+/+ cells as assessed by χ2 analysis (p < 0.05). These data clearly indicate the requirement for NEIL2 in maintaining genomic integrity.
FIGURE 6.
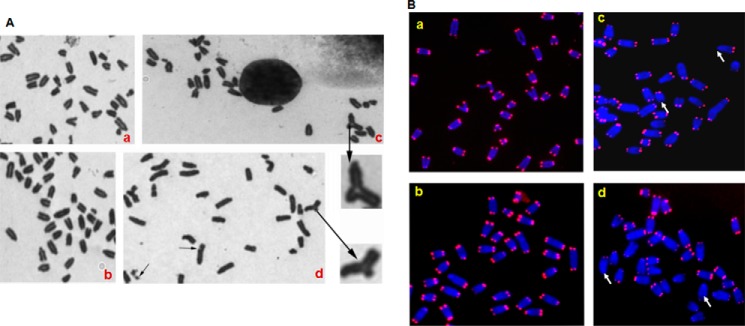
Detection of chromosomal abnormalities. A, Giemsa-stained metaphase spread of Neil2+/+ (panels a and b) and Neil2−/− (panels c and d) mouse embryonic fibroblast cells. Note radials and chromosome gaps as indicated by arrows. B, segments of metaphases from Neil2+/+ (panels a and b) and Neil2−/− (panel c and d) cells showing telomere FISH signals. Metaphases were analyzed by FISH with a telomere-specific probe. Note the higher frequency of telomere signal loss in Neil2−/− cells, as indicated by arrows.
Increased Susceptibility of Neil2-null Mice to Inflammation
Previous reports have indicated that Ogg1-null mice have decreased susceptibility to LPS-induced and oxidative stress-induced inflammation (44, 45) and also to allergic immune responses (46, 47). To address the susceptibility of Neil2-null mice to inflammation, animals were challenged intranasally with LPS (100 ng/per lung) or TNF-α (20 ng/lung) or GOx (1 milliunit/lung). In the lungs of mock-treated WT or Neil2-KO mice, only resident macrophages along with 3–11 neutrophils/ml were observed (Fig. 7, A and B). However, LPS challenge to WT mice increased neutrophil numbers to 2.1 × 105/ml but in KO mice neutrophil numbers were increased further to 9.6 ± 1.9 × 105/ml (Fig. 7A, left panel), which was unexpectedly high. To visually illustrate the robust inflammatory response to LPS in KO mice, we selected representative microscopic fields of stained cells (Fig. 7B).
FIGURE 7.
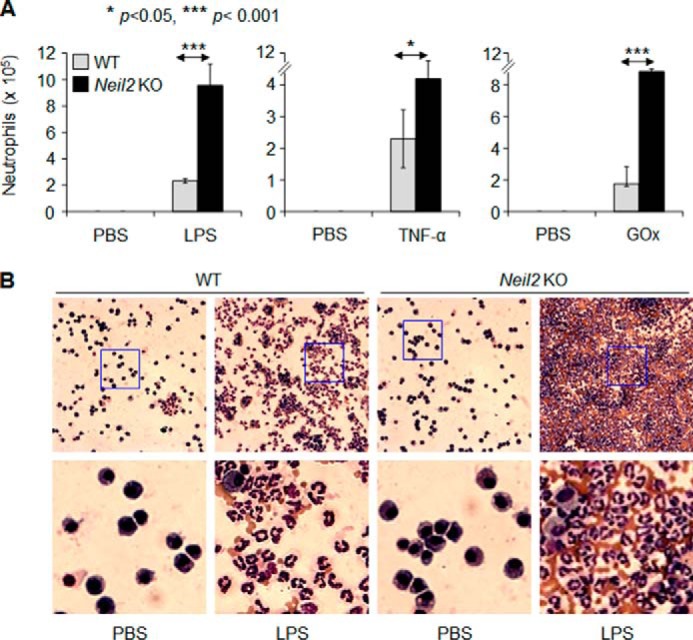
Increased susceptibility of Neil2 KO mice to inflammation. Wild-type and Neil2-null mice (8 months old) were challenged intranasally with LPS (100 ng/lung) or TNF-α (20 ng/lung) or GOx (1 milliunit/lung). A, 16 h after challenge, bronchoalveolar lavage fluid was derived, and inflammatory cells were counted and expressed as the number of cells/ml. B, representative microscopic images of cells visible in saline-challenged (left panels) and LPS-challenged mouse lung tissue (right panels). Magnification: upper panels, ×32; lower panels, ×134 (representing blue-boxed portions of the upper panel).
TNF-α is a potent pro-inflammatory cytokine that induces an innate inflammatory response (48, 49). We found that TNF-α challenge to WT mice increased the neutrophil numbers to 2.28 ± 1.35 × 105/ml, and in Neil2 KO animals it was increased further to 4.45 ± 0.8 × 105/ml (>2-fold, Fig. 7A, middle panel).
We have previously shown that the induction of oxidative stress in the lungs after challenge with GOx (50) recruits neutrophils to the lungs. Here, we show that GOx challenge recruited 1.82 ± 0.45 × 105/ml neutrophils in WT mice; however, in Neil2 KO mice, the neutrophil numbers were increased further, to 9.7 × 105/ml (>4-fold, Fig. 7A, right panel). We observed an insignificant increase in the numbers of lymphocytes and macrophages in LPS-, TNFα-, or GOx-challenged lungs of KO animals compared with WT controls. All these data suggest that Neil2-null animals are highly susceptible to a variety of inflammatory agents.
Discussion
Five oxidized base-specific DNA glycosylases with overlapping substrate specificities are involved in the repair of approximately 24 oxidized DNA bases via the BER pathway in mammalian cells. To examine the in vivo role of these DNA glycosylases, gene knock-out mice for four DNA glycosylases (Ogg1, Nth1, Neil1, and Neil3) have already been generated (51–54). We report here for the first time the generation of a Neil2-null mouse strain that lacks the NEIL2 DNA glycosylase. Our strategy involved Cre-mediated targeted disruption of exon 2 of mNeil2, which is critical for its enzymatic activity. Our data showed that the transcripts and NEIL2 protein are absent in the homozygous mutant animals. Neil2-null mice, like the other four DNA glycosylase loss-of-function mouse models, did not show any obvious abnormality or spontaneous tumorigenesis. Surprisingly, knock-out animal models of the other components in the BER pathways, downstream of DNA glycosylases (such as APE1, pol β, and Lig IIIα), are embryonic lethal (55–58). This suggests that DNA glycosylase-mediated repair intermediates (AP sites or strand breaks) are lethal to the whole organism.
Using a cell culture model, we have reported earlier that NEIL2 initiates the repair of oxidized bases preferentially from the transcribed genes, and also characterized the NEIL2-mediated TC-BER biochemically using an in vitro reconstituted repair system (18). In this study, we further evaluated the age-dependent accumulation of spontaneously generated oxidative genome damage in various tissues of Neil2-null and WT mice. Gene-specific analysis of oxidative genome damage by LA-qPCR clearly demonstrated that young (2 months) Neil2-null mice do not show a significant amount of DNA damage accumulation. However, middle age (8 months) and old age (24 months) animals do accumulate significant amounts of oxidative DNA lesions, mostly in the transcribed but not in the nontranscribed genes of various tissues, further implicating NEIL2's biological role in TC-BER. Two other reports also have indicated transcription-coupled repair of oxidized bases in mammalian cells, as well as in yeast (59, 60). To our knowledge, ours is the first in vivo evidence for such repair of oxidized bases in mammals.
Given the role of NEIL2 in TC-BER, we have analyzed physiologically relevant protein-protein interactions/associations using mouse tissue extracts, and we have shown NEIL2's preferential association with the transcribed genes and the co-association of RNAP II and several critical TCR-related proteins in a complex with NEIL2 and Lig IIIα. Our partial characterization of NEIL2 and Lig IIIα immunocomplexes demonstrates that NEIL2 and Lig IIIα co-opt CSB and TFIIH, two critical TC-nucleotide excision repair proteins. A recent study by Aamann et al. (15) reported that CSB physically interacts with and stimulates NEIL2's activity in transcription bubble-mimic DNA. Several studies have also shown that CSB cooperates in enhancing RNAP II-mediated transcription, and TFIIH helps remodel stalled RNAP II for allowing repair proteins to access the DNA lesion during TCR (61–63). Hence, the association of TCR-related proteins with both NEIL2 and Lig IIIα, respectively, the first and last enzyme in the BER pathway, is consistent with NEIL2's role in TC-BER. TC-BER is obviously a complex process, and many more proteins are likely to be involved with NEIL2 forming a multiprotein complex. Identification of additional proteins and their role in TC-BER thus warrants further investigation.
Several studies have indicated that BER deficiency and/or persistent oxidative DNA base accumulation interfere with telomere length homoeostasis and overall genomic integrity (64, 65). We have shown here that primary Neil2-null MEFs undergo severe telomere loss at chromosome ends, indicating NEIL2's important role in telomere maintenance. Notably, several recent studies have demonstrated that RNAP II transcribes the chromosomal ends into a variety of noncoding RNA species, including telomeric repeat-containing RNA constituting a “telomeric transcriptome” (66, 67). Therefore, it is likely that NEIL2-mediated TC-BER plays a critical role therein as well. Importantly, the majority of somatic human cells express a low level of telomerase; hence, inactive or low levels of NEIL2 could have a significant impact on telomere maintenance and genome stability in human tissues.
Despite NEIL2's role in the repair of the transcribing genome, why the KO animals do not show any apparent phenotype is not clear to us at present. Although the mouse has been extensively used as a model organism in the study of human biology and diseases, it has been found that >20% of essential human genes have nonessential mouse orthologs (68). These discrepancies may be caused by adaptive evolution for the prolonged life span in humans. The age-dependent increase in the accumulation of oxidatively damaged bases in the transcribed genes of Neil2-null mice indicates NEIL2's critical role in long term genomic maintenance. Hence, caution should be used in extrapolating the animal data while addressing the physiological importance of the human gene based on the animal data.
Oxidative DNA damage levels are elevated in many pathological conditions; however, no incidence of carcinogenesis has been reported in many cases (69). It is important to mention here that Ogg1−/− mice, despite the accumulation of ∼250-fold higher amounts of mutagenic 8-oxoG compared with WT mice in their genomes due to KBrO3 (an oxygen radical-forming agent) exposure, did not show any tumor formation (70). This suggests that DNA damage in the genome alone is not sufficient for tumor formation, a promoting process or impairment of another parallel/back-up pathway is necessary. Recently, an international consortium comprehensively analyzed germ line mutation carriers (∼24,000) in the BRCA1 and BRCA2 genes and their correlation to a lifetime risk of developing breast and ovarian cancer. They found that the age of disease onset is highly variable, and not all BRCA carriers develop cancer, indicating the involvement of other genetic factors or modifier genes. Surprisingly, one minor allele of NEIL2 was found to be associated with breast cancer risk in BRCA2 mutation carriers and an OGG1 SNP with the risk of ovarian cancer in BRCA1 carriers (71). Because the Neil2-null mice are living in a stress-free environment, the other genetic or environmental factors may modify the risk of pathogenic development. We thus postulate that double-mutant (Neil2 and Brca2) animals will develop aggressive breast cancer at a very early stage.
Several recent studies have indicated that in addition to its primary function in BER, OGG1 plays a role in cell signaling, gene expression, and modulating allergic inflammatory responses. Specifically, we have shown that OGG1 in complex with 8-oxoG base (repair product) induces an inflammatory response in the lungs via K-RAS-MAPK, PI3K, and MS kinase and the NF-κB pathway (45, 48). These observations are consistent with the earlier observation that Ogg1-null mice are resistant to innate and allergic airway inflammation (34) and LPS-induced organ dysfunction and inflammatory cell infiltration (44). By contrast, this study demonstrates that Neil2-null mice are extremely susceptible to inflammation induced by pro-inflammatory agents such as LPS, TNFα, and oxidative stress that utilize distinct signaling pathways. LPS activates inflammatory signaling via a TLR4-MD2 complex (72, 73), whereas TNF-α has been shown to signal via distinct cell surface receptors TNFR-1 and TNFR-2 (74, 75). In contrast, GOx primarily generates superoxide anions to activate inflammatory signaling via redox-reactive kinases (45, 50). To our surprise, Neil2 KO mice were highly susceptible to LPS- and GOx-induced inflammation, although their sensitivity to TNF-α (one of the most potent inflammatory cytokines) was modest (76). The mechanism of differential susceptibility of Neil2-null mice to innate inflammation requires extensive study. Nonetheless, taken together, all these data imply that OGG1 is involved in a pro-inflammatory but NEIL2 in an anti-inflammatory response. How these two DNA glycosylases maintain and balance the cellular inflammatory response will be an exciting area of research in the future.
A linkage of inflammation and cancer is well established (77, 78). Samson and co-workers (79) recently demonstrated how chronic inflammation could contribute to carcinogenesis and the protective role of several DNA repair proteins, such as methyl purine DNA glycosylase, ALKBH2 and ALKBH3. Consistent with these findings, this study indicates that depletion of NEIL2 causes genomic instability, and it simultaneously induces innate inflammation. These combinatorial effects of genomic damage and innate inflammation due to NEIL2 deficiency could play a critical role in the development of breast cancer in BRCA2 carriers but also in other diseases as well. Notably, we and others have reported the association of some NEIL2 SNPs with lung and oropharyngeal cancer risk (19, 80). Therefore, understanding the molecular mechanisms of such combinatorial responses using Neil2-null mice as an experimental tool may help to explain the previously proposed link of oxidative stress and inflammation to various pathologies, and the knowledge gained from such studies should ultimately benefit human health.
Author Contributions
T. K. H. conceived, designed, and coordinated the research. A. C. performed experiments in Figs. 1, C and D, and 2–5. M. W. generated Neil2-null mice, performed all mouse surgeries related to Figs. 1–5, and performed experiments in Fig. 1B. T. V. C. had a major contribution in designing and standardizing PCR conditions in Fig. 3 and contributed to the preparation of the final figures. R. K. P. and D. K. S. performed experiments in Fig. 6. L. A. A. and K. H. performed experiments in Fig. 7. A. H. S. and I. B. derived and characterized MEFs from WT and Neil2-KO mice. T. G. W. generated constructs shown in Fig. 1A. G. S., V. C., and P. S. S. provided valuable scientific inputs and technical support. T. K. H., S. S., T. K. P., and I. B. analyzed the data. T. K. H. wrote the paper with contributions from I. B., S. S., and T. K. P. All the authors read, reviewed and approved the final version of the manuscript.
Acknowledgments
We thank Dr. David Konkel for critically editing this manuscript and Justin Barr of IDT for designing long amplicon quantitative PCR primers for mouse NanoG.
This work was supported, in whole or in part, by National Institutes of Health Grant NS073976 from USPHS (to T. K. H.), RO1 CA129537 and RO1 CA154320 (to T. K. P.), and Grant P30 ES 06676 (to NIEHS Center Cell Biology Core and Molecular Genomics Core of UTMB's NIEHS Center for DNA sequencing). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as a Paper of the Week.
- BER
- base excision repair
- Ab
- antibody
- co-IP
- co-immunoprecipitation
- CSB
- Cockayne syndrome group B protein
- IP
- immunoprecipitate
- MEF
- mouse embryonic fibroblast
- TC-BER
- transcription-coupled base excision repair
- TCR
- transcription-coupled repair
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- qPCR
- quantitative PCR
- RNAP
- RNA polymerase
- LA-qPCR
- long amplicon-quantitative PCR
- GOx
- glucose oxidase
- oligo
- oligonucleotide
- Lig
- ligase.
References
- 1. Ames B. N., Shigenaga M. K., Hagen T. M. (1993) Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. U.S.A. 90, 7915–7922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kasai H., Nishimura S. (1991) in Oxidative Stress: Oxidants and Antioxidants (Sies H., ed) pp. 99–116, Academic Press, London [Google Scholar]
- 3. Halliwell B. (1994) Free radicals, antioxidants, and human disease: curiosity, cause, or consequence? Lancet 344, 721–724 [DOI] [PubMed] [Google Scholar]
- 4. Hazra T. K., Das A., Das S., Choudhury S., Kow Y. W., Roy R. (2007) Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair 6, 470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hegde M. L., Hazra T. K., Mitra S. (2008) Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 18, 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ikeda S., Biswas T., Roy R., Izumi T., Boldogh I., Kurosky A., Sarker A. H., Seki S., Mitra S. (1998) Purification and characterization of human hNTH1, a homolog of Escherichia coli endonuclease III: direct identification of Lys-212 as the active nucleophilic residue. J. Biol. Chem. 273, 21585–21593 [DOI] [PubMed] [Google Scholar]
- 7. Lu R., Nash H. M., Verdine G. L. (1997) A DNA repair enzyme that excises oxidatively damaged guanines from the mammalian genome is frequently lost in lung cancer. Curr. Biol. 7, 397–407 [DOI] [PubMed] [Google Scholar]
- 8. Hazra T. K., Izumi T., Boldogh I., Imhoff B., Kow Y. W., Jaruga P., Dizdaroglu M., Mitra S. (2002) Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 99, 3523–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bandaru V., Sunkara S., Wallace S. S., Bond J. P. (2002) A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair 1, 517–529 [DOI] [PubMed] [Google Scholar]
- 10. Takao M., Kanno S., Shiromoto T., Hasegawa R., Ide H., Ikeda S., Sarker A. H., Seki S., Xing J. Z., Le X. C., Weinfeld M., Kobayashi K., Miyazaki J., Muijtjens M., Hoeijmakers J. H., et al. (2002) Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. EMBO J. 21, 3486–3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu M., Bandaru V., Bond J. P., Jaruga P., Zhao X., Christov P. P., Burrows C. J., Rizzo C. J., Dizdaroglu M., Wallace S. S. (2010) The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 4925–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hazra T. K., Kow Y. W., Hatahet Z., Imhoff B., Boldogh I., Mokkapati S. K., Mitra S., Izumi T. (2002) Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 277, 30417–30420 [DOI] [PubMed] [Google Scholar]
- 13. Dou H., Mitra S., Hazra T. K. (2003) Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem. 278, 49679–49684 [DOI] [PubMed] [Google Scholar]
- 14. Krokeide S. Z., Laerdahl J. K., Salah M., Luna L., Cederkvist F. H., Fleming A. M., Burrows C. J., Dalhus B., Bjørås M. (2013) Human NEIL3 is mainly a monofunctional DNA glycosylase removing spiroimindiohydantoin and guanidinohydantoin. DNA Repair 12, 1159–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aamann M. D., Hvitby C., Popuri V., Muftuoglu M., Lemminger L., Skeby C. K., Keijzers G., Ahn B., Bjørås M., Bohr V. A., Stevnsner T. (2014) Cockayne Syndrome group B protein stimulates NEIL2 DNA glycosylase activity. Mech. Ageing Dev. 135, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dou H., Theriot C. A., Das A., Hegde M. L., Matsumoto Y., Boldogh I., Hazra T. K., Bhakat K. K., Mitra S. (2008) Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 283, 3130–3140 [DOI] [PubMed] [Google Scholar]
- 17. Hegde M. L., Hegde P. M., Bellot L. J., Mandal S. M., Hazra T. K., Li G. M., Boldogh I., Tomkinson A. E., Mitra S. (2013) Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. U.S.A. 110, E3090–E3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Banerjee D., Mandal S. M., Das A., Hegde M. L., Das S., Bhakat K. K., Boldogh I., Sarkar P. S., Mitra S., Hazra T. K. (2011) Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 286, 6006–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dey S., Maiti A. K., Hegde M. L., Hegde P. M., Boldogh I., Sarkar P. S., Abdel-Rahman S. Z., Sarker A. H., Hang B., Xie J., Tomkinson A. E., Zhou M., Shen B., Wang G., Wu C., Yu D., et al. (2012) Increased risk of lung cancer associated with a functionally impaired polymorphic variant of the DNA glycosylase NEIL2. DNA Repair 11, 570–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maiti A. K., Boldogh I., Spratt H., Mitra S., Hazra T. K. (2008) Mutator phenotype of mammalian cells due to deficiency of NEIL1 DNA glycosylase, an oxidized base-specific repair enzyme. DNA Repair 7, 1213–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Todaro G. J., Green H. (1963) Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17, 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee C. (2007) Protein extraction from mammalian tissues. Methods Mol. Biol. 362, 385–389 [DOI] [PubMed] [Google Scholar]
- 23. Santos. J. H., Meyer. J. N., Mandavilli. B. S., Van Houten. B. (2006) Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 314, 183–199 [DOI] [PubMed] [Google Scholar]
- 24. Chatterjee A., Saha S., Chakraborty A., Silva-Fernandes A., Mandal S. M., Neves-Carvalho A., Liu Y., Pandita R. K., Hegde M. L., Hegde P. M., Boldogh I., Ashizawa T., Koeppen A. H., Pandita T. K., Maciel P., et al. (2015) The role of the mammalian DNA end-processing enzyme polynucleotide kinase 3′-phosphatase in spinocerebellar ataxia type 3 pathogenesis. PLoS Genet. 11, e1004749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suganya R., Chakraborty A., Miriyala S., Hazra T. K., Izumi T. (2015) Suppression of oxidative phosphorylation in mouse embryonic fibroblast cells deficient in apurinic/apyrimidinic endonuclease. DNA Repair 27, 40–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ayala-Torres S., Chen Y., Svoboda T., Rosenblatt J., Van Houten B. (2000) Analysis of gene specific DNA damage and repair using quantitative polymerase chain reaction. Methods 22, 135–147 [DOI] [PubMed] [Google Scholar]
- 27. Dignam J. D., Lebovitz R. M., Roeder R. G. (1983) Acute transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kudrycki K., Stein-Izsak C., Behn C., Grillo M., Akeson R., Margolis F. L. (1993) Olf-1-binding site: characterization of an olfactory neuron-specific promoter motif. Mol. Cell. Biol. 13, 3002–3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang J., Ding X. (1998) Identification and characterization of a novel tissue-specific transcriptional activating element in the 5′-flanking region of the CYP2A3 gene predominantly expressed in rat olfactory mucosa. J. Biol. Chem. 273, 23454–23462 [DOI] [PubMed] [Google Scholar]
- 30. Aygün O., Svejstrup J., Liu Y. (2008) A RECQ5-RNA polymerase II association identified by targeted proteomic analysis of human chromatin. Proc. Natl. Acad. Sci. U.S.A. 105, 8580–8584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sailaja B. S., Takizawa T., Meshorer E. (2012) Chromatin immunoprecipitation in mouse hippocampal cells and tissues. Methods Mol. Biol. 809, 353–364 [DOI] [PubMed] [Google Scholar]
- 32. Das A., Wiederhold L., Leppard J. B., Kedar P., Prasad R., Wang H., Boldogh I., Karimi-Busheri F., Weinfeld M., Tomkinson A. E., Wilson S. H., Mitra S., Hazra T. K. (2006) NEIL2-initiated, APE-independent repair of oxidized bases in DNA: evidence for a repair complex in human cells. DNA Repair 5, 1439–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boldogh I., Bacsi A., Choudhury B. K., Dharajiya N., Alam R., Hazra T. K., Mitra S., Goldblum R. M., Sur S. (2005) ROS generated by pollen NADPH oxidase provide a signal that augments antigen-induced allergic airway inflammation. J. Clin. Invest. 115, 2169–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bacsi A., Aguilera-Aguirre L., Szczesny B., Radak Z., Hazra T. K., Sur S., Ba X., Boldogh I. (2013) Down-regulation of 8-oxoguanine DNA glycosylase 1 expression in the airway epithelium ameliorates allergic lung inflammation. DNA Repair 12, 18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. George S. H., Gertsenstein M., Vintersten K., Korets-Smith E., Murphy J., Stevens M. E., Haigh J. J., Nagy A. (2007) Developmental and adult phenotyping directly from mutant embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 104, 4455–4460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lewandoski M., Wassarman K. M., Martin G. R. (1997) Zp3-cre, a transgenic mouse line for the activation or inactivation of loxP-flanked target genes specifically in the female germ line. Curr. Biol. 7, 148–151 [DOI] [PubMed] [Google Scholar]
- 37. Kawakami H., Maruyama H., Yasunami M., Ohkubo H., Hara H., Saida T., Nakanishi S., Nakamura S. (1996) Cloning and expression of a rat brain basic helix-loop-helix factor. Biochem. Biophys. Res. Commun. 221, 199–204 [DOI] [PubMed] [Google Scholar]
- 38. Hart A. H., Hartley L., Ibrahim M., Robb L. (2004) Identification, cloning and expression analysis of the pluripotency promoting Nanog genes in mouse and human. Dev. Dyn. 230, 187–198 [DOI] [PubMed] [Google Scholar]
- 39. Newton D. A., Rao K. M., Dluhy R. A., Baatz J. E. (2006) Hemoglobin is expressed by alveolar epithelial cells. J. Biol. Chem. 281, 5668–5676 [DOI] [PubMed] [Google Scholar]
- 40. Wiederhold L., Leppard J. B., Kedar P., Karimi-Busheri F., Rasouli-Nia A., Weinfeld M., Tomkinson A. E., Izumi T., Prasad R., Wilson S. H., Mitra S., Hazra T. K. (2004) AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 15, 209–220 [DOI] [PubMed] [Google Scholar]
- 41. Hunt C. R., Dix D. J., Sharma G. G., Pandita R. K., Gupta A., Funk M., Pandita T. K. (2004) Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol. Cell. Biol. 24, 899–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pandita R. K., Sharma G. G., Laszlo A., Hopkins K. M., Davey S., Chakhparonian M., Gupta A., Wellinger R. J., Zhang J., Powell S. N., Roti Roti J. L., Lieberman H. B., Pandita T. K. (2006) Mammalian Rad9 plays a role in telomere stability, S- and G2-phase-specific cell survival, and homologous recombinational repair. Mol. Cell. Biol. 26, 1850–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pandita R. K., Chow T. T., Udayakumar D., Bain A. L., Cubeddu L., Hunt C. R., Shi W., Horikoshi N., Zhao Y., Wright W. E., Khanna K. K., Shay J. W., Pandita T. K. (2015) Single-strand DNA-binding protein SSB1 facilitates TERT recruitment to telomeres and maintains telomere G-overhangs. Cancer Res. 75, 858–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mabley J. G., Pacher P., Deb A., Wallace R., Elder R. H., Szabó C. (2005) Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 19, 290–292 [DOI] [PubMed] [Google Scholar]
- 45. Aguilera-Aguirre L., Bacsi A., Radak Z., Hazra T. K., Mitra S., Sur S., Brasier A. R., Ba X., Boldogh I. (2014) Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-κB pathway. J. Immunol. 193, 4643–4653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li G., Yuan K., Yan C., Fox J., 3rd, Gaid M., Breitwieser W., Bansal A. K., Zeng H., Gao H., Wu M. (2012) 8-Oxoguanine-DNA glycosylase 1 deficiency modifies allergic airway inflammation by regulating STAT6 and IL-4 in cells and in mice. Free Radic. Biol. Med. 52, 392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bacsi A., Chodaczek G., Hazra T. K., Konkel D., Boldogh I. (2007) Increased ROS generation in subsets of OGG1 knockout fibroblast cells. Mech. Ageing Dev. 128, 637–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ba X., Bacsi A., Luo J., Aguilera-Aguirre L., Zeng X., Radak Z., Brasier A. R., Boldogh I. (2014) 8-Oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J. Immunol. 192, 2384–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brasier A. R. (2006) The NF-κB regulatory network. Cardiovasc. Toxicol. 6, 111–130 [DOI] [PubMed] [Google Scholar]
- 50. Das A., Hazra T. K., Boldogh I., Mitra S., Bhakat K. K. (2005) Induction of the human oxidized base-specific DNA glycosylase NEIL1 by reactive oxygen species. J. Biol. Chem. 280, 35272–35280 [DOI] [PubMed] [Google Scholar]
- 51. Klungland A., Rosewell I., Hollenbach S., Larsen E., Daly G., Epe B., Seeberg E., Lindahl T., Barnes D. E. (1999) Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. U.S.A. 96, 13300–13305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ocampo M. T., Chaung W., Marenstein D. R., Chan M. K., Altamirano A., Basu A. K., Boorstein R. J., Cunningham R. P., Teebor G. W. (2002) Targeted deletion of mNth1 reveals a novel DNA repair enzyme activity. Mol. Cell. Biol. 22, 6111–6121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vartanian V., Lowell B., Minko I. G., Wood T. G., Ceci J. D., George S., Ballinger S. W., Corless C. L., McCullough A. K., Lloyd R. S. (2006) The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc. Natl. Acad. Sci. U.S.A. 103, 1864–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Torisu K., Tsuchimoto D., Ohnishi Y., Nakabeppu Y. (2005) Hematopoietic tissue-specific expression of mouse Neil3 for endonuclease VIII-like protein. J. Biochem. 138, 763–772 [DOI] [PubMed] [Google Scholar]
- 55. Gu H., Marth J. D., Orban P. C., Mossmann H., Rajewsky K. (1994) Deletion of a DNA polymerase β gene segment in T cells using cell type-specific gene targeting. Science 265, 103–106 [DOI] [PubMed] [Google Scholar]
- 56. Sugo N., Aratani Y., Nagashima Y., Kubota Y., Koyama H. (2000) Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase β. EMBO J. 19, 1397–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Izumi T., Brown D. B., Naidu C. V., Bhakat K. K., Macinnes M. A., Saito H., Chen D. J., Mitra S. (2005) Two essential but distinct functions of the mammalian abasic endonuclease. Proc. Natl. Acad. Sci. U.S.A. 102, 5739–5743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Puebla-Osorio N., Lacey D. B., Alt F. W., Zhu C. (2006) Early embryonic lethality due to targeted inactivation of DNA ligase III. Mol. Cell. Biol. 26, 3935–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Reis A. M., Mills W. K., Ramachandran I., Friedberg E. C., Thompson D., Queimado L. (2012) Targeted detection of in vivo endogenous DNA base damage reveals preferential base excision repair in the transcribed strand. Nucleic Acids Res. 40, 206–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Guo J., Hanawalt P. C., Spivak G. (2013) Comet-FISH with strand-specific probes reveals transcription coupled repair of 8-oxoguanine in human cells. Nucleic Acids Res. 41, 7700–7712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sarker A. H., Tsutakawa S. E., Kostek S., Ng C., Shin D. S., Peris M., Campeau E., Tainer J. A., Nogales E., Cooper P. K. (2005) Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne syndrome. Mol. Cell 20, 187–198 [DOI] [PubMed] [Google Scholar]
- 62. Selby C. P., Sancar A. (1997) Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 94, 11205–11209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tantin D., Kansal A., Carey M. (1997) Recruitment of the putative transcription-repair coupling factor CSB/ERCC6 to RNA polymerase II elongation complexes. Mol. Cell. Biol. 17, 6803–6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vallabhaneni H., O'Callaghan N., Sidorova J., Liu Y. (2013) Defective repair of oxidative base lesions by the DNA glycosylase Nth1 associates with multiple telomere defects. PLoS Genet. 9, e1003639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang Z., Rhee D. B., Lu J., Bohr C. T., Zhou F., Vallabhaneni H., de Souza-Pinto N. C., Liu Y. (2010) Characterization of oxidative guanine damage and repair in mammalian telomeres. PLoS Genet. 6, e1000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bah A., Azzalin C. M. (2012) The telomeric transcriptome: from fission yeast to mammals. Int. J. Biochem. Cell Biol. 44, 1055–1059 [DOI] [PubMed] [Google Scholar]
- 67. Porro A., Feuerhahn S., Delafontaine J., Riethman H., Rougemont J., Lingner J. (2014) Functional characterization of the TERRA transcriptome at damaged telomeres. Nat. Commun. 5, 5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liao B. Y., Zhang J. (2008) Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc. Natl. Acad. Sci. U.S.A. 105, 6987–6992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cooke M. S., Evans M. D., Dizdaroglu M., Lunec J. (2003) Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 17, 1195–1214 [DOI] [PubMed] [Google Scholar]
- 70. Arai T., Kelly V. P., Minowa O., Noda T., Nishimura S. (2006) The study using wild-type and Ogg1 knockout mice exposed to potassium bromate shows no tumor induction despite an extensive accumulation of 8-hydroxyguanine in kidney DNA. Toxicology 221, 179–186 [DOI] [PubMed] [Google Scholar]
- 71. Osorio A., Milne R. L., Kuchenbaecker K., Vaclová T., Pita G., Alonso R., Peterlongo P., Blanco I., de la Hoya M., Duran M., Díez O., Ramon Y. C., Konstantopoulou I., Martínez-Bouzas C., Andrés Conejero R., et al. (2014) DNA glycosylases involved in base excision repair may be associated with cancer risk in BRCA1 and BRCA2 mutation carriers. PLoS Genet. 10, e1004256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nijland R., Hofland T., van Strijp J. A. (2014) Recognition of LPS by TLR4: potential for antiinflammatory therapies. Mar. Drugs 12, 4260–4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. da Silva Correia J., Soldau K., Christen U., Tobias P. S., Ulevitch R. J. (2001) Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex. Transfer from CD14 to TLR4 and MD-2. J. Biol. Chem. 276: 21129–21135 [DOI] [PubMed] [Google Scholar]
- 74. Jamaluddin M., Wang S., Boldogh I., Tian B., Brasier A. R. (2007) TNF-α-induced NF-κB/RelA Ser (276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell. Signal. 19, 1419–1433 [DOI] [PubMed] [Google Scholar]
- 75. Choudhary S., Kalita M., Fang L., Patel K. V., Tian B., Zhao Y., Edeh C. B., Brasier A. R. (2013) Inducible tumor necrosis factor (TNF) receptor-associated factor-1 expression couples the canonical to the non-canonical NFκB pathway in TNF stimulation. J. Biol. Chem. 288, 14612–14623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tian B., Nowak D. E., Brasier A. R. (2005) A TNF-induced gene expression program under oscillatory NF-κB control. BMC Genomics 6, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Balkwill F., Mantovani A. (2001) Inflammation and cancer: back to Virchow? Lancet 357, 539–545 [DOI] [PubMed] [Google Scholar]
- 78. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 79. Calvo J. A., Meira L. B., Lee C. Y., Moroski-Erkul C. A., Abolhassani N., Taghizadeh K., Eichinger L. W., Muthupalani S., Nordstrand L. M., Klungland A., Samson L. D. (2012) DNA repair is indispensable for survival after acute inflammation. J. Clin. Invest. 122, 2680–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhai X., Zhao H., Liu Z., Wang L. E., El-Naggar A. K., Sturgis E. M., Wei Q. (2008) Functional variants of the NEIL1 and NEIL2 genes and risk and progression of squamous cell carcinoma of the oral cavity and oropharynx. Clin. Cancer Res. 14, 4345–4352 [DOI] [PMC free article] [PubMed] [Google Scholar]