

Background: MicroRNAs play important roles in regulating AKT pathway.
Results: miR-542-3p suppresses glioblastoma cell invasion through directly inhibiting AKT1, ILK, and PIK3R1.
Conclusion: miR-542-3p down-regulation contributes to aberrant activation of the AKT signaling, and miR-542-3p acts as a negative regulator in astrocytoma progression.
Significance: Learning how miRNAs participate in AKT pathway is crucial for understanding its regulators and cancer therapy.
Keywords: Akt PKB, cancer biology, cell invasion, neurobiology, signal transduction, microRNA-542-3p, AKT signaling, β-catenin, astrocytoma, invasiveness
Abstract
The molecular mechanism underlying constitutive activation of AKT signaling, which plays essential roles in astrocytoma progression, is not fully characterized. Increasing numbers of studies have reported that microRNAs are involved in the malignant behavior of astrocytoma cells via directly targeting multiple oncogenes or tumor suppressors. Here, we found that microRNA (miR)-542-3p expression was decreased in glioblastoma cell lines and astrocytoma tissues, and reduced levels of miR-542-3p expression correlated with high histopathological grades and poor prognosis of astrocytoma patients. Exogenous miR-542-3p suppressed glioblastoma cell invasion through not only targeting AKT1 itself but also directly down-regulating its two important upstream regulators, namely, integrin-linked kinase and PIK3R1. Notably, overexpressing miR-542-3p decreased AKT1 phosphorylation and directly and indirectly repressed nuclear translocation and transactivation activity of β-catenin to exert its anti-invasive effect. Furthermore, the miR-542-3p expression level negatively correlated with AKT activity as well as levels of integrin-linked kinase and PIK3R1 in human astrocytoma specimens. These findings suggest that miR-542-3p acts as a negative regulator in astrocytoma progression and that miR-542-3p down-regulation contributes to aberrant activation of AKT signaling, leaving open the possibility that miR-542-3p may be a potential therapeutic target for high grade astrocytoma.
Introduction
Astrocytoma is the most common primary malignant neoplasm of brain in adults. Patients with high grade astrocytoma with a median overall survival time of only 14.6 months have much worse outcome than those with the low grade disease (1). Currently available therapies against high grade astrocytoma, including surgery, radiotherapy, and chemotherapy, remain mainly unsuccessful generally due to high invasiveness of the tumor cells. Moreover, it is nearly impossible to remove the tumor cells infiltrating surrounding brain tissues, resulting in tumor recurrence and progression shortly after surgery. Of note, the patients with glioblastoma, which is the most aggressive type of astrocytoma, suffer an over 90% recurrence rate, and the recurrent patients have a median survival time as low as 4–6 months (2, 3). Therefore, understanding the molecular basis and identifying the key molecule(s) responsible for astrocytoma invasiveness will help to provide potentially applicable therapeutic agents and improve the therapeutic effect on management of patients with astrocytoma.
Cumulative evidence has revealed that the protein kinase B (AKT) pathway plays a crucial role in the invasive behavior of astrocytoma cells. AKT, a serine/threonine kinase, is constitutively activated in astrocytoma and tightly correlates with poor outcome of patients with the disease (4, 5). Increased AKT activity promotes malignant behavior of invasive glioblastoma cells (6, 7), and down-regulation of AKT importantly inhibits invasion of glioblastoma cells (8). Notably, not only increased phospho-AKT but also up-regulated total AKT expression levels contribute to the development and progression of astrocytoma (9). Mounting evidence has shown that increased or activated AKT governs several downstream signaling pathways, such as the glycogen synthesis kinase-3β (GSK-3β)4 pathway, β-catenin pathway, and NF-κB pathway (10, 11), to stimulate expression of matrix metalloproteases (MMPs), which are known to be essential for degradation of extracellular matrix, consequently exerting a proinvasive effect (12).
As a critical indicator in the progression of astrocytoma, AKT activation can be stimulated by distinct routes of protein kinases, including phosphatidylinositol 3-kinase (PI3K) and integrin-linked kinase (ILK). PI3K is a crucial oncogenic kinase consisting of catalytic subunit p110 and regulatory subunit p85 encoded by PIK3CA and PIK3R1 genes, respectively. It activates AKT through production of phosphatidylinositol 3,4,5-trisphosphate, which binds to the pleckstrin homology domain of AKT to recruit it to the cell membrane, and induces conformational change in AKT, revealing its two critical amino acids, Thr-308 and Ser-473, for phosphorylation (13–15). PTEN is able to inactivate AKT via directly dephosphorylating phosphatidylinositol 3,4,5-trisphosphate and thus acts as an important negative regulator of the PI3K/AKT signaling. Although mutations in PIK3CA and PTEN genes have been extensively observed to be contributive to various malignancies, only 7 and 10% of astrocytoma tumors, respectively, show PIK3CA mutation and PTEN mutation (16, 17). Therefore, the reason why AKT is constitutively activated in the vast majority of human astrocytoma is still unknown. In addition, we have previously demonstrated that ILK is widely overexpressed in astrocytoma, and its aberrantly high expression level significantly correlates with the histological grading and poor prognosis of patients with the disease (18). However, the mechanism underlying up-regulation of ILK in astrocytoma remains unclear. Thus, further clarification of how these oncogenic upstream kinases are dysregulated in astrocytoma will provide new insight into aberrant activation of AKT signaling and might lead to the development of targeting therapies against the disease.
Recently, microRNAs have been recognized to play important roles in the regulation of protein expression through directly binding to 3′-UTRs of target mRNAs, resulting in their post-transcriptional repression. Aberrant expression of microRNAs has been identified to play roles of oncogenes or suppressors in various human cancers (19–21), showing promising therapeutic potential for anticancer strategies (22, 23). It has been reported that miR-542-3p, which is located in Xq26.3, is down-regulated in non-small cell lung cancer and breast cancer and directly targets survivin or angiopoietin-2, respectively, leading to growth arrest and angiogenesis inhibition (24, 25). In the current study, we report that in astrocytoma down-regulation of miR-542-3p was significantly associated with histopathological grading and poor patient prognosis of the disease. Mechanistic investigations revealed that miR-542-3p inhibited invasive abilities of glioblastoma cells through directly repressing protein expression of AKT1, ILK, and PIK3R1 and strongly suppressing AKT signaling. Furthermore, the clinical relevance of the expression level of miR-542-3p with AKT activity as well as levels of ILK and PIK3R1 was evidenced in a cohort of human astrocytoma specimens.
Experimental Procedures
Cell Lines and Culture
Normal human astrocytes were purchased from Sciencell Research Laboratories (Carlsbad, CA) and cultured under the conditions recommended by the manufacturer. Glioblastoma cell lines, including U87MG, T98G, U118MG, A172, U251MG, and LN464, were kindly provided by Dr. Shiyuan Cheng (Northwestern University Feinberg School of Medicine, Martinez, CA), and cultured in DMEM supplemented with 10% fetal bovine serum (HyClone, Logan City, UT) and 1% penicillin/streptomycin (Invitrogen) at 37 °C in a humidified air atmosphere containing 5% carbon dioxide. All cell lines were authenticated by short tandem repeat fingerprinting at the Medicine Laboratory of Forensic Medicine Department of Sun Yat-Sen University (Guangzhou, China).
Patients and Tissue Samples
A cohort of 59 paraffin-embedded astrocytoma specimens was collected and diagnosed at the First Affiliated Hospital of Sun Yat-Sen University from 2000 to 2005. The fresh-frozen paired astrocytoma and adjacent non-cancerous brain tissues were obtained from the same hospital. The samples were classified as low grade astrocytoma, including pilocytic (grade I) and diffuse (grade II) astrocytoma, and high grade astrocytoma, including anaplastic astrocytoma (grade III) and glioblastoma multiforme (grade IV) according to World Health Organization histopathological grading. For the use of these clinical materials for research purposes, prior patients' consents and approval from the Institutional Research Ethics Committee were obtained.
Plasmid Construction
The total region of human AKT1-3′-UTR, ILK-3′-UTR, or PIK3R1-3′-UTR, respectively, was generated by PCR amplification with flanking PstI and SacII restriction enzyme digestion sites. The oligonucleotides were cloned into the PstI-SacII sites of the modified pGL3-Basic luciferase reporter plasmid (Promega, Madison, WI). Mutations were introduced into the seed region in AKT1-3′-UTR, ILK-3′-UTR, or PIK3R1-3′-UTR for miR-542-3p binding sites using the QuikChange site-directed mutagenesis kit (Stratagene, Wilmington, DE). The open reading frame (ORF; without 3′-UTR) of AKT1, PIK3R1, or ILK generated by PCR amplification was cloned into mammalian expression vector pcDNA (Invitrogen). The mutant plasmid ΔN47 β-catenin expressing constitutively active β-catenin and the pBabe-puro-myr-Akt1 plasmid expressing constitutively active AKT1 were purchased from Addgene Inc. (Cambridge, MA). The reporter plasmids containing wild-type (cctttgatc; TOPflash) or mutated (cctttggcc; FOPflash) T cell factor/lymphoid enhancer factor (TCF/LEF) DNA binding sites were purchased from Upstate Biotechnology (Lake Placid, NY).
Transient Transfection
U87MG and T98G cells were cultured in 6-well plates and then transfected with miR-542-3p mimic or control oligonucleotides individually with Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. miR-542-3p mimic and pcDNA-AKT1, pcDNA-ILK, or pcDNA-PIK3R1 were co-transfected using Lipofectamine 2000 reagent in the restoration experiments. 36 h after transfection, cells were collected for Western blotting and RNA extraction. miR-542-3p mimic oligonucleotides and negative control oligonucleotides were commercially synthesized by RiboBio (Guangzhou, China).
RNA Extraction and Real Time Quantitative PCR
Total microRNA of cultured cells, fresh-frozen astrocytoma and paired non-cancerous brain tissues, and paraffin-embedded, archived clinical astrocytoma specimens was extracted using the mirVana microRNA isolation kit (Ambion, Austin, TX) and RecoverAllTM total nucleic acid isolation kit (Ambion) according to the manufacturer's instructions. Detection of miR542-3p and U6 utilized their specific primers separately. All primers were synthesized by Invitrogen. Expression of miR-542-3p was analyzed by real time PCR using the TaqMan microRNA assay kit (Applied Biosystems, Life Technologies). The expression of miR542-3p was assessed based on the threshold cycle (Ct), and relative expression level was calculated as 2-((Ct of miR542-3p) − (Ct of U6)). U6 snRNA was used as an internal control.
Detection of mRNA was performed as described previously (26). The sequences of the primers were as follows: TCF4 forward, 5′-TCTCCATAGTTCCTGGACGG-3′; TCF4 reverse, 5′-CCAACTTCTTTGGCAAGTGG-3′; LEF1 forward, 5′-CACTGTAAGTGATGAGGGGG-3′; LEF1 reverse, 5′-TGGATCTCTTTCTCCACCCA-3′; MMP2 forward, 5′-CAGGGAATGAGTACTGGGTCTATT-3′; MMP2 reverse, 5′-ACTCCAGTTAAAGGCAGCATCTAC-3′; Axin2 forward, 5′-CTGGTGCAAAGACATAGCC-3′; Axin2 reverse, 5′-AGTGTGAGGTCCACGGAAAC-3′; and GAPDH forward, 5′-GACTCATGACCACAGTCCATGC-3′; GAPDH reverse, 5′-AGAGGCAGGGATGATGTTCTG-3′.
Western Blotting
The procedure was performed as described previously (27). Primary antibodies included anti-AKT1, anti-ILK, anti-phospho-GSK-3β (Ser-9), anti-phospho-β-catenin (Ser-552), anti-phospho-β-catenin (Thr-41/Ser-45), anti-MMP-2, and anti-β-actin antibodies (Cell Signaling Technology, Danvers, MA); anti-PIK3R1 (p85), anti-phospho-AKT1 (Ser-473), and anti-p84 antibodies (Abcam, Cambridge, MA); and anti-β-catenin antibody (BD Pharmingen). Horseradish peroxidase-conjugated secondary antibody was obtained from Promega. An enhanced chemiluminescence (ECL) kit was purchased from Thermo (Rockford, IL). Nuclear protein was extracted using Nuclear Extract kit (Active Motif, Carlsbad, CA) according to the manufacturer's instructions.
Matrigel Invasion Assay
The Transwell filters were coated with 10% Matrigel diluted in fetal bovine serum-free DMEM on the upper surface of polycarbonate membrane (BD Pharmingen; pore size, 8 μm). The filters were incubated at 37 °C for 3 h until Matrigel became solidified. Cells were harvested 24 h after transfection. 2 × 104 U87MG and 2.5 × 104 T98G cells in 200 μl of fetal bovine serum-free DMEM were added into the upper chamber, respectively. The complete DMEM with 20% fetal bovine serum was placed in the lower chamber. After 24 h of incubation at 37 °C with 5% CO2, the non-invaded cells on the upper chamber were wiped out. The cells that had infiltrated from Matrigel into the pores of the inserted filter were fixed and stained. The number of cells invading through the Matrigel was counted using five randomly selected visual fields using an inverted microscope at 200× magnification. Each assay was repeated in triplicates.
Three-dimensional Invasion Assay
Cells were harvested 48 h after transfection. 24-well plates coated with Matrigel were incubated at 37 °C for 30 min until the Matrigel became solidified. The cells (1.25 × 104) in 100 μl of complete DMEM with 10% serum mixed with 100 μl of Matrigel were seeded in pretreated 24-well plates. The growth of the indicated cells in Matrigel was imaged under a microscope at 5 days after three-dimensional overlay culture.
Gelatin Zymography Assay
The culture medium was replaced by DMEM without serum when cells were transfected by miR-542-3p for 24 h. 48 h later, the medium was collected and clarified by centrifugation. The total protein concentration was determined using the Bradford (45) assay, and 40 μg of total protein in supernatant were loaded and separated in a 7.5% Tris-glycine gel with 0.1% gelatin. The gel was renatured in a 2.5% Triton X-100 buffer to remove traces of SDS, Triton X-100 was removed using wash buffer, and then the gel was incubated in developing buffer for 42 h (37 °C). Finally, the gel was stained in 0.05% Coomassie Brilliant Blue and destained, respectively, for 3 h. Proteolytic bands from the zymography were quantified by scanning densitometry (Quantity One, Bio-Rad).
Luciferase Assay
Cells (1.5 × 105) were seeded in 24-well plates and allowed to settle for 24 h. miR-542-3p, pGL3-AKT1-3′-UTR, pGL3-ILK-3′-UTR, or pGL3-PIK3R1-3′-UTR (wild or mutated type) reporter plasmid and pRL-TK plasmid (Promega) were co-transfected in the indicated cells using the Lipofectamine 2000 reagent according to the manufacturer's recommendations. Luciferase activities of these reporters in whole cell lysate were measured 48 h after transfection and normalized to corresponding luciferase activities of pRL-TK using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's protocols. The β-catenin/TCF/LEF transcription reporter (TOPflash/FOPflash reporter) assay was performed as described (29).
Immunohistochemistry
Immunohistochemistry assays wereperformed and quantified as described previously (28). The degree of immunostaining of the indicated proteins in 59 cases of formalin-fixed, paraffin-embedded sections was evaluated and scored by two independent observers; both the proportions of positive staining tumor cells and the staining intensities were scored. Scores representing the proportion of positive stained tumor cells was graded as follows: 0 (no positive tumor cells), 1 (<10%), 2 (10–50%), and 3 (>50%). The intensity of staining was determined as follows: 0 (no staining), 1 (weak staining = light yellow), 2 (moderate staining = yellow brown), and 3 (strong staining = brown). The staining index was calculated as Staining intensity × Percentage of positive tumor cells, resulting in scores of 0, 1, 2, 3, 4, 6, and 9. Cutoff values for high and low expression of the protein of interest were chosen based on a measurement of heterogeneity using the log rank test with respect to overall survival. The optimal cutoff was identified as follows: a staining index score of ≥4 was considered as high expression, and a score of ≤3 was considered as low expression. Mean optical density (OD) (=Integrated optical density/Area) was analyzed using Image-Pro Plus 6.0 software.
Statistical Analysis
Statistical analysis was carried out using SPSS 17.0 statistical software. Spearman analysis was used to analyze the correlation between miR-542-3p expression and the clinicopathological characteristics. Survival curves were plotted using the Kaplan-Meier method and compared with the log rank test. p < 0.05 in all cases was considered statistically significant.
Results
MiR-542-3p Is Down-regulated in Astrocytoma Cell Lines and Tissue
To evaluate the expression status of miR-542-3p in astrocytoma, quantitative real time PCR was performed. As shown in Fig. 1A, the expression level of miR-542-3p in a panel of glioblastoma cell lines, including U87MG, T98G, U118MG, A172, U251MG, and LN464, was remarkably lower than that in normal human astrocytes. Notably, expression of miR-542-3p in A172 cells was decreased by more than 7 times as compared with that in normal human astrocytes. Next, we measured expression of miR-542-3p in five pairs of astrocytoma and adjacent non-cancerous brain tissue with each pair obtained from the same patient and found that miR-542-3p was consistently down-regulated in all five examined astrocytoma samples compared with their paired non-cancerous brain tissues, presenting at least a 2.6-fold reduction (Fig. 1B). These data suggest that miR-542-3p is widely down-regulated in astrocytoma and might be a negative regulator in the development and progression of the disease.
FIGURE 1.
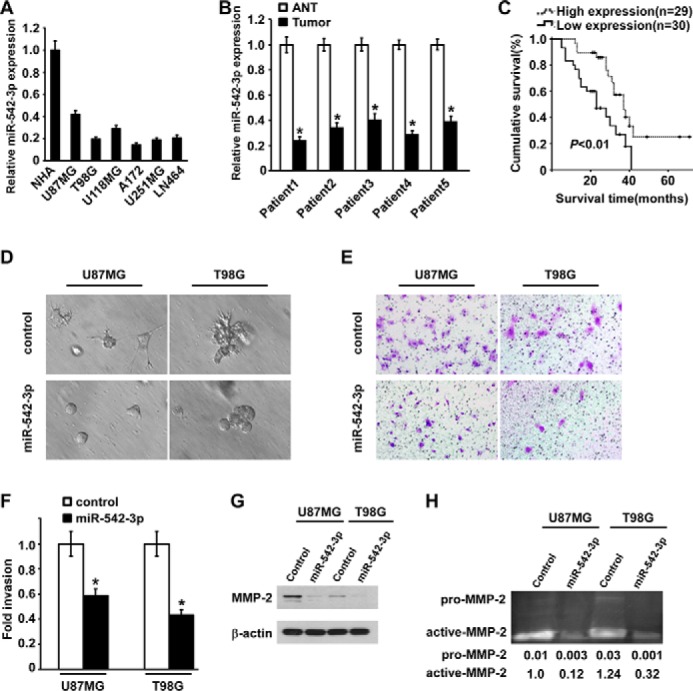
miR-542-3p is down-regulated in astrocytoma and inhibits invasion of glioblastoma cells. A, relative expression of miR-542-3p in a panel of human astrocytoma cell lines, including U87MG, T98G, U118MG, A172, U251MG, and LN464, and normal human astrocytes (NHA) were quantified by real time RT-PCR. B, relative expression of miR-542-3p in five pairs of astrocytoma tissues (Tumor) and matched adjacent non-cancerous brain tissues (ANT). The average miR-542-3p expression was normalized by U6 expression. C, Kaplan-Meier analysis of overall survival of astrocytoma patients divided into the low miR-542-3p expression group (less than the median; n = 30) and the high miR-542-3p expression group (greater than the median; n = 29). D, three-dimensional invasion assays revealed that miR-542-3p-overexpressing cells form space frame structure with fewer and shorter projections than those formed by control cells grown in Matrigel for 5 days. E, representative micrographs show the effect of miR-542-3p on glioblastoma cell invasion as evaluated by Matrigel invasion assay. F, the relative percentages of the indicated infiltrated cells comparing miR-542-3p-overexpression with control transfection. G, Western blotting analysis of protein expression of MMP-2 in the indicated miR-542-3p-overexpressing cells and control cells. H, gelatin zymography assay and quantitative data of the proteolytic bands show differential MMP-2 activity in supernatants derived from the indicated cells. Error bars represent mean ± S.D. derived from three independent experiments. *, p < 0.05.
Down-regulation of MiR-542-3p Is Associated with Progression and Poor Prognosis of Patients with Astrocytoma
To further assess the potential clinical significance of down-regulated miR-542-3p in astrocytoma, 59 paraffin-embedded, archived astrocytoma tissues, including 21 cases of low grade (I and II) and 38 cases of high grade (III and IV) astrocytomas, were examined for the expression levels of miR-542-3p. Based on the median value of miR-542-3p expression, the patients were divided into low level (less than the median; n = 30) and high level (greater than the median; n = 29) miR-542-3p expression groups. The correlation between the miR-542-3p expression level and clinicopathological characteristics was analyzed (Table 1). Although no significant correlation was found between the expression level of miR-542-3p and gender of patients (p = 0.514) or age (p = 0.093), the level of miR-542-3p expression negatively correlated with the histopathological grades of astrocytoma (p < 0.05), suggesting that decreased expression of miR-542-3p probably contributes to the progression of astrocytoma from low grade to high grade.
TABLE 1.
Correlation between miR-542-3p expression level and clinicopathological characteristics in 59 patients with astrocytoma
Spearman analysis demonstrated significant correlation between miR-542-3p expression level and histopathologic grade. No significant correlation was found between miR-542-3p expression level and gender or age of patients.
| Factors | Low level miR-542-3p expression (n = 30) | High level miR-542-3p expression (n = 29) | p value |
|---|---|---|---|
| Gender | |||
| Male | 23 | 20 | 0.514 |
| Female | 7 | 9 | |
| Age (median) | |||
| ≤41 | 12 | 18 | 0.093 |
| >41 | 18 | 11 | |
| Histopathologic grade | |||
| Low grade (I + II) | 7 | 14 | 0.034 |
| High grade (III + IV) | 23 | 15 | |
Subsequently, the clinical relevance of miR-542-3p expression to the prognosis of these astrocytoma patients was evaluated using Kaplan-Meier analysis. As shown in Fig. 1C, the median and mean overall survival times of the high miR-542-3p expression group were 37 and 41.2 months, respectively, in contrast to only 23 and 24.2 months, respectively, for the low miR-542-3p expression group. Of specific note, the cumulative 3-year survival rate for the patients with low miR-542-3p expression was significantly lower than that for the high miR-542-3p expression group (33.3 versus 48.3%) (p < 0.01). Collectively, these data indicate that down-regulation of miR-542-3p might represent a risk factor of poor prognosis of astrocytoma patients.
Exogenous MiR-542-3p Represses Invasion of Glioblastoma Cells
To investigate the potential inhibitory effect of miR-542-3p on invasiveness of glioblastoma cells, gain-of-function experiments were performed using two glioblastoma cell lines. As shown in Fig. 1D, miR-542-3p-overexpressing U87MG cells formed space frame spheroid structures with much fewer and shorter projections than those formed by control cells cultured in a three-dimensional setting. In agreement, miR-542-3p-overexpressing T98G cells formed spheroid structures with rare projections, whereas control cells grew quite aggressive spheroid structures. Moreover, the invasive abilities of both U87MG and T98G cells were obviously repressed in response to transfection of miR-542-3p by 41.6 and 56.8%, respectively, compared with corresponding transfection of scrambled oligonucleotides (Fig. 1, E and F). In support of the inhibitory effect of miR-542-3p on invasiveness, overexpression of miR-542-3p drastically reduced the protein expression of MMP-2 and secretion of extracellular enzymatic MMP-2 as assessed by gel zymography (Fig. 1, G and H). Taken together, these data illustrate that overexpression of miR-542-3p apparently represses the invasive capabilities of glioblastoma cells.
MiR-542-3p Reduces Expression of AKT1 via Directly Targeting Its 3′-UTR
We then aimed to uncover the underlying mechanism of the inhibitory effect of miR-542-3p on invasiveness of glioblastoma cells. Notably, AKT1, one of three major isoforms of AKT and known for its widely important roles in malignant behavior of astrocytoma, is predicted to be a putative target of miR-542-3p using three bioinformatics algorithms (miRBase, Pictar, and TargetScan) (Fig. 2A). For validating this prediction, as shown in Fig. 2B, the relative luciferase activity of pGL3-AKT1-3′-UTR reporter was significantly inhibited by transfection of miR-542-3p, which, however, hardly showed any inhibitory effect on the luciferase activity when the predicted miR-542-3p-binding seed sequence in AKT1-3′-UTR was mutated, suggesting the importance of appropriate binding of miR-542-3p to AKT1-3′-UTR. Furthermore, Western blotting analysis showed that protein levels of endogenous AKT1 in miR-542-3p-overexpressing glioblastoma cells were markedly decreased compared with those in control cells and accompanied by reduced phosphorylation of AKT1 (Fig. 2C). Therefore, these data suggest that miR-542-3p inhibits AKT1 expression via direct interaction with its 3′-UTR in glioblastoma cells.
FIGURE 2.

miR-542-3p directly inhibits AKT1 expression in astrocytoma. A, predicted binding sites of miR-542-3p in human wild-type AKT1-3′-UTR. Mutations in the seed sequence of full-length AKT1-3′-UTR are shown in red. mut, mutant. B, relative luciferase activity of wild-type or mutated pGL3-AKT1-3′-UTR reporter in the indicated miR-542-3p-overexpressing cells and control cells. C, Western blotting analysis of total AKT1 and phospho-AKT1 (p-AKT1) (Ser-473) in the indicated miR-542-3p-overexpressing and control cells. β-Actin was used as a loading control. D, Matrigel invasion assays show that the invasive abilities of the indicated miR-542-3p-overexpressing cells were slightly and completely revived, respectively, by restoration of WT-AKT1 and constitutively active AKT1 (myr-AKT1). Error bars represent mean ± S.D. derived from three independent experiments. *, p < 0.05; NS, not significant.
To further elucidate whether down-regulating AKT1 expression essentially mediates miR-542-3p-suppressed invasiveness, AKT1 ORF without 3′-UTR was co-transfected with miR-542-3p in U87MG and T98G cells, and a Matrigel invasion assay was performed. As shown in Fig. 2C, although AKT1 protein expression was completely restored after wild-type AKT1 (WT-AKT1) ORF transfection in miR-542-3p-overexpressing U87MG and T98G cells, the phospho-AKT level was only slightly revived. In parallel, WT-AKT1 restoration weakly enhanced the invasive capability of miR-542-3p-overexpressing glioblastoma cells (Fig. 2D). By contrast, overexpression of myristoylated AKT1 (myr-AKT1) (ORF without 3′-UTR), a constitutively active form of AKT1, was able to completely revive the phospho-AKT level and the inhibitory effect of miR-542-3p in miR-542-3p-overexpressing glioblastoma cells (Fig. 2, C and D). These discrepancies imply that some other upstream regulatory molecules of AKT1 phosphorylation may be simultaneously repressed by miR-542-3p.
ILK and PIK3R1 Are Also the Targets of MiR-542-3p and Mediate MiR-542-3p-induced Anti-invasion in Glioblastoma Cells
Accordingly, we hypothesized that two important upstream contributors of AKT1 phosphorylation, namely ILK and PIK3R1, may be the putative candidate targets of miR-542-3p based on the prediction by TargetScan algorithm (Fig. 3A). Indeed, luciferase assays showed that the relative luciferase activities of reporters of pGL3-ILK-3′-UTR and pGL3-PIK3R1-3′-UTR were obviously decreased by miR-542-3p transfection, whereas those of reporters containing mutant ILK-3′-UTR or PIK3R1-3′-UTR underwent no significant change in response to miR-542-3p transfection (Fig. 3B). Furthermore, expression levels of endogenous ILK and PIK3R1 protein in addition to total AKT1 and phospho-AKT1 in miR-542-3p-overexpressing cells were both dramatically decreased compared with those in control cells (Fig. 3, C and D). Taken together, these results indicate that ILK and PIK3R1 as well as AKT1 are bona fide targets of miR-542-3p in glioblastoma cells.
FIGURE 3.

ILK and PIK3R1 are two other targets of miR-542-3p and mediate the inhibitory effect of miR-542-3p on invasion of glioblastoma cells. A, predicted binding sites of miR-542-3p in human ILK- and PIK3R1-3′-UTR. Mutations in the seed sequence of full-length ILK- and PIK3R1-3′-UTR are shown in red. mut, mutant. B, relative luciferase activity of wild-type or mutated pGL3-ILK-3′-UTR or pGL3-PIK3R1-3′-UTR reporter in the indicated miR-542-3p-overexpressing cells and control cells. C and D, Western blotting analysis of protein levels of ILK, PIK3R1, total AKT1, and phospho-AKT1 (p-AKT1) (Ser-473) in the indicated miR-542-3p-overexpressing and control cells. Restored expression of ILK or PIK3R1 protein was validated. Each significantly but not fully revived phospho-AKT1 levels when pcDNA-ILK-ORF (without 3′-UTR) or pcDNA-PIK3R1-ORF (without 3′-UTR) and miR-542-3p were co-transfected into U87MG and T98G cells. β-Actin was used as a loading control. E and F, Matrigel invasion assays showed that the invasive abilities of miR-542-3p-overexpressing cells were significantly, although only partly, revived after restoration of ILK or PIK3R1 and were almost completely revived by the combination of WT-AKT1 and ILK or PIK3R1. Error bars represent mean ± S.D. derived from three independent experiments. *, p < 0.05.
To evaluate the importance of suppressing ILK and PIK3R1 for miR-542-3p-mediated inhibition of invasion in glioblastoma cells, re-expression of ILK or PIK3R1 was performed in miR-542-3p-overexpressing U87MG and T98G cells followed by Matrigel invasion assay. As shown in Fig. 3 (C and D), ILK or PIK3R1 protein expression was correspondingly markedly recovered by their respective transfection in miR-542-3p-overexpressing glioblastoma cells, causing remarkable, but not full, revival in phosphorylation of AKT1 as compared with negative control microRNA-transfected glioblastoma cells. Accordingly, restoration of ILK or PIK3R1 expression partially, but significantly, revived the invasive abilities of miR-542-3p-overexpressing cells and showed an almost complete revival effect when combined with transfection of WT-AKT1. In contrast, WT-AKT1 overexpression weakly increased the invasive capability of miR-542-3p-overexpressing glioblastoma cells (Fig. 3, E and F). Taken together, these data strongly demonstrate that miR-542-3p suppresses glioblastoma cell invasion through targeting the AKT pathway by directly inhibiting AKT1, ILK, and PIK3R1 simultaneously.
Down-regulated Expression of MiR-542-3p Is Clinically Associated with Up-regulated Expression of ILK and PIK3R1 in Astrocytoma
Although aberrant activation of AKT signaling in astrocytoma is well documented and we have previously reported that up-regulated expression of ILK, presenting clinical prognostic significance and tight correlation with histological grading of astrocytoma, partly contributes to aberrant AKT activity, the expression profile of PIK3R1 in astrocytoma has remained undefined. Thus, protein expression of PIK3R1 was measured in the aforementioned cohort of 59 cases of astrocytoma specimens using an immunohistochemistry assay. As shown in Fig. 4A, PIK3R1 staining was positive in 86.4% (51 of 59) of astrocytoma samples and was widely expressed in distinct grades of astrocytoma, whereas its expression in three cases of normal brain tissues was hardly detectable. Quantitative analysis of average mean OD of PIK3R1 staining verified higher expression of PIK3R1 in astrocytoma tissues than that in normal brain tissues (all p < 0.01) (Fig. 4B). Moreover, the expression level of PIK3R1 significantly correlated with histopathological grading of astrocytoma (r = 0.570, p = 0.000) (Fig. 4, A–C). We subsequently evaluated whether up-regulated expression of ILK and PIK3R1 was clinically associated with down-regulated expression of miR-542-3p in astrocytoma specimens. Of specific note, many more cases of specimens with high miR-542-3p expression showed low levels of ILK and PIK3R1 than did those expressing low miR-542-3p, whereas more cases with low miR-542-3p expression displayed high levels of ILK and PIK3R1 (r = −0.288, p = 0.027 and r = −0.267, p = 0.041, respectively) (Fig. 4D). Consistently, high expression of PIK3R1 and ILK and strong staining of active phospho-AKT1 were uniformly observed in the set of specimens from the same astrocytoma patient with low miR-542-3p expression, whereas the astrocytoma specimens with high miR-542-3p expression showed weak staining of PIK3R1, ILK, and phospho-AKT1 (Fig. 4E). Therefore, down-regulation of miR-542-3p expression probably contributes to up-regulated expression of PIK3R1 and ILK and consequently aberrant activation of AKT signaling in human astrocytoma specimens. Notably, it appears that the inverse correlation between expression levels of miR-542-3p and ILK/PIK3R1 is not observed in all human astrocytoma specimens (Fig. 4D), implying that expression of ILK/PIK3R can also be regulated by other factors and that miR-542-3p may target additional protein(s) in human astrocytoma with distinct cellular context.
FIGURE 4.
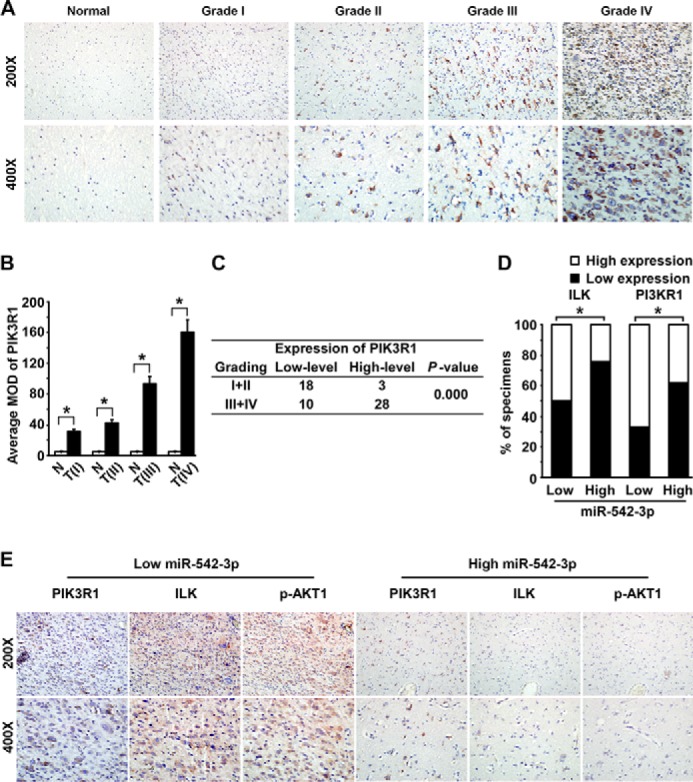
Clinical association of miR-542-3p down-regulation with up-regulation of PIK3R1 and ILK in astrocytoma specimens. A, representative images of PIK3R1 staining by an immunohistochemistry assay in normal brain tissues and 59 cases of archived astrocytoma specimens. B, quantitative analysis of the average mean OD (MOD) of PIK3R1 staining between normal brain tissues (N) and astrocytoma specimens (T) of different World Health Organization grades (I–IV). Data are presented as mean ± S.D. (error bars). *, p < 0.01. C, expression level of PIK3R1 positively correlates with histopathological grading of astrocytoma specimens. D, percentages of specimens showing low or high PIK3R1 or ILK expression in patient specimens with low or high miR-542-3p expression. *, p < 0.05. E, representative micrographs of immunostaining of PIK3R1, ILK, and phospho-AKT1 (p-AKT1) in astrocytoma specimens with low or high miR-542-3p expression level.
Ectopic MiR-542-3p Inhibits AKT1/β-Catenin Pathway to Exert Anti-invasive Effect
Based on the repression of AKT signaling and the anti-invasive activity of miR-542-3p, we next examined crucial downstream effectors tightly associated with proinvasion in astrocytoma. As shown in Fig. 5A, accompanying decreased activity of AKT1, phosphorylation of GSK-3β, one of the substrates of AKT1, was apparently decreased in miR-542-3p-overexpressing glioblastoma cells, resulting in an increase of phospho-β-catenin (Thr-41/Ser-45), which is directly phosphorylated by GSK-3β and could attenuate β-catenin activity. Moreover, the phosphorylation of β-catenin on site Ser-552, which is reported to be directly phosphorylated by active AKT to promote β-catenin activity, was also greatly suppressed by ectopic miR-542-3p. Because activation of β-catenin signaling requires accumulation of β-catenin in the nucleus to interact with the TCF/LEF transcription factors and thereby activate transcription of downstream genes, a subcellular fractionation assay of β-catenin and TOPflash/FOPflash reporter assay indicative of the transactivation activity of β-catenin were performed to further validate the inhibitory effect of miR-542-3p on β-catenin signaling. Indeed, nuclear accumulation of β-catenin and its subsequent transactivation activity were markedly diminished in miR-542-3p-overexpressing glioblastoma cells compared with control cells (Fig. 5, B and C). In parallel, mRNA expression levels of several downstream genes of β-catenin signaling, including MMP-2, Axin2, TCF4, and LEF1, were reduced in response to ectopic miR-542-3p, supporting the inhibitory effect of miR-542-3p on the AKT1/β-catenin pathway (Fig. 5D). Furthermore, the invasive activities of miR-542-3p-overexpressing U87MG and T98G cells were revived to 92.44 ± 9.12 and 87.13 ± 8.06%, respectively, when β-catenin signaling was reactivated by overexpression of a constitutively active β-catenin mutant with deletion of amino acids 1–47 in the N terminus (ΔN47 β-catenin) (Fig. 5E). In addition, MMP-2 transcription was increased when β-catenin signaling was reactivated in miR-542-3p-overexpressing glioblastoma cells (Fig. 5F), and MMP-2 re-expression by transfection of MMP-2-expressing plasmid in miR-542-3p-overexpressing glioblastoma cells markedly revived their invasive abilities (Fig. 5G), suggesting the critical role of β-catenin/MMP-2 in the miR-542-3p-mediated anti-invasion effect in glioblastoma cells. Taken together, these data support a model in which miR-542-3p down-regulation in glioblastoma directly causes increased levels of AKT1, ILK, and PIK3R1 and the resultant aberrant activation of AKT signaling, which could directly and indirectly activate the β-catenin/MMP-2 signaling cascade to promote invasion of glioblastoma cells (Fig. 5H).
FIGURE 5.

Up-regulated miR-542-3p inhibits AKT1/β-catenin signaling pathway in glioblastoma cells to exert anti-invasive effect. A, Western blotting analysis of expression of phospho-AKT1 (p-AKT1) (Ser-473), phospho-GSK-3β (p-GSK-3β) (Ser-9), phospho-β-catenin (Thr-41/Ser-45), and phospho-β-catenin (Ser-552) in the indicated miR-542-3p-overexpressing cells and control cells. B, subcellular fractionation assay of nuclear β-catenin distribution in the indicated cells. p84 was used as loading control for extracted nuclear proteins. C, luciferase activity assays of TOPflash and FOPflash reporters in the indicated miR-542-3p-overexpressing cells and control cells. D, real time PCR quantification of several downstream genes of β-catenin signaling, including MMP-2, Axin2, TCF4, and LEF1, in the indicated miR-542-3p-overexpressing cells and control cells. E, the anti-invasive effect of ectopic miR-542-3p could be mostly reversed by overexpression of the constitutively active form of β-catenin (Δβ-catenin). F, real time PCR quantification of MMP-2 expression in the indicated miR-542-3p-overexpressing cells and control cells. G, MMP-2 re-expression by transfecting MMP-2-expressing plasmid in miR-542-3p-overexpressing glioblastoma cells markedly revived their invasive abilities. H, schematic model for miR-542-3p-mediated down-regulation of its target genes, namely ILK, PIK3R1, and AKT1; aberrant inhibition of the AKT/β-catenin/MMP-2 signaling cascade; and resultant suppression of astrocytoma invasiveness. Error bars represent mean ± S.D. derived from three independent experiments. *, p < 0.05.
Discussion
In the current study, we have presented the first demonstration that miR-542-3p expression was down-regulated in astrocytoma cell lines and tissues and that reduced miR-542-3p expression level correlated with high histopathological grades and poor prognosis of patients with astrocytoma. Ectopic expression of miR-542-3p inhibited the aggressive behavior of glioblastoma cells through not only targeting AKT1 itself but also directly down-regulating its two important upstream regulators, namely ILK and PIK3R1, to fully inhibit the AKT signaling. Furthermore, down-regulation of miR-542-3p was clinically associated with aberrantly active AKT and increased protein levels of ILK and PIK3R1 in human astrocytoma specimens. Finally, we found that miR-542-3p repressed the AKT1/β-catenin signaling cascade directly and indirectly to exert an anti-invasive effect. Collectively, our study revealed a novel microRNA-based novel mechanism for multiple layer regulation of oncogenic AKT signaling and astrocytoma cell invasion.
High aggressiveness is a crucial factor in failed therapy of high grade astrocytoma, and constitutive activation of AKT signaling has been described as an essential mediator for the aggressive behavior of the disease (29, 30). As mutation of upstream factors such as PIK3CA and PTEN accounts for only 7 and 14%, respectively, of astrocytoma cases (16, 17), the genetic variation of this signaling cannot fully recapitulate its importance in the development and progression of this malignancy, making it necessary to clarify other mechanisms of regulation. The current study identified that miR-542-3p in astrocytoma cell lines not only down-regulated the total level of AKT1 via directly binding to its 3′-UTR but also simultaneously reduced the phosphorylation level of AKT1 via directly targeting ILK and PI3KR1, two crucial upstream oncogenic kinases promoting phosphorylation of AKT1. Thus, the miR-542-3p-induced anti-invasive effect could only be slightly revived by restored expression of total WT-AKT1 because AKT1 phosphorylation was still inhibited but was significantly, although only partly, reversed by restoration of ILK or PIK3R1. Notably, constitutively active AKT1 or the combination of WT-Akt and ILK or PIK3R1 was able to almost completely reverse the miR-542-3p effect. It has been documented that ILK facilitates cancer cell invasion through mediating the interaction with extracellular matrix components (31–33), and ILK inhibitor QLT0267 reduces tumor growth and angiogenesis of glioblastoma xenografts (34), indicating the therapeutic potential of miR-542-3p for glioblastoma progression. Although knockdown of PIK3R1 has been reported to inhibit glioblastoma cell proliferation and invasion (35, 36) and abrogating the activation of PIK3R1 increases the sensitization of glioblastoma cells to tamoxifen-induced apoptosis (37), unlike ILK, the clinical significance of PI3KR1 in astrocytoma has not been defined. Our study is the first report that up-regulated expression of PIK3R1 protein significantly correlated with histopathological grades of astrocytoma. Furthermore, the clinical relevance of miR-542-3p down-regulation to up-regulation of PI3KR1 and ILK was consistently confirmed in human astrocytoma specimens. Therefore, our study strongly suggests that miR-542-3p down-regulation in astrocytoma contributes to increased expression of crucial positive regulators of the AKT signaling, including PI3KR1, ILK, and AKT1 itself, and thus leads to aberrant activation of AKT signaling in astrocytoma. The tumor-suppressive function of miR-542-3p largely results from its synergistic effect on simultaneous modulation of multiple targets. Notably, our data also showed that not all samples with high miR-542-3p expression had low expression of ILK/PIK3R, implying that miR-542-3p targets additional protein(s) in human astrocytoma specimens. However, whether miR-542-3p targets additional proteins to mediate invasion of astrocytoma remains a potentially interesting area of investigation to completely underscore the importance of miR-542-3p-mediated regulation in astrocytomaprogression and provide miR-542-3p-targeted therapeutic strategies.
For further investigation at the molecular level, we demonstrated that miR-542-3p suppressed the β-catenin pathway due to its specific inhibitory effect on AKT signaling. Serving as a vital downstream effector of AKT signaling, β-catenin importantly participates in activating the invasive ability of tumor cells (11, 38). A high level of β-catenin is associated with histopathological grades of astrocytoma and promotes the invasive capacity of glioblastoma cells (39), whereas inhibition of β-catenin activity results in the opposite effects (40). Because of the crucial roles of β-catenin in the development and progression of astrocytoma, targeting aberrant activity of β-catenin signaling in astrocytoma is considered to be an applicable approach to anticancer therapy (41–43). Our demonstration that ectopic miR-542-3p not only decreased nuclear translocation of β-catenin through enhancing the activity of tumor suppressor GSK-3β due to inhibition of AKT activity but also repressed the transactivation activity of β-catenin via reducing AKT-mediated phosphorylation at the Ser-552 site to exert an anti-invasive effect on glioblastoma cells suggests that miR-542-3p might be a promising molecular therapeutic target for high grade astrocytoma.
Consistent with our results, miR-542-3p has been reported to be down-regulated in colon cancer cell lines and to inhibit colon cancer cell proliferation and invasion via targeting ILK (44). Moreover, overexpression of miR-542-3p induces G1 and G2/M arrest in multiple cancer cell lines, including lung cancer, breast cancer, and cervical cancer (24). Our present study indicates that miR-542-3p down-regulation contributes to the progression of astrocytoma and might represent a novel prognostic biomarker for the disease. These findings together suggest an extensively suppressive role of miR-542-3p in various cancer types. However, the underlying molecular basis for the decline of miR-542-3p expression in astrocytoma remains mysterious, making it of great interest to explore for further development of miR-542-3p-based therapeutic strategies.
Author Contributions
H. Z., J. W., and J. C. conceived and coordinated the study and wrote the paper. J. C., X. X., and R. L. designed, performed, and analyzed the experiments. J. C. and J. Z. designed, performed, and analyzed the experiments in the revision. Y. Y., L. F., and L. Z. provided technical assistance and contributed to the preparation of the figures. N. Z. collected the human astrocytoma samples and analyzed the clinical data. M. L. provided help in revising the manuscript. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by the National Natural Science Foundation of China (Grants 81472351, 81001121, 81472574, 81330058, and 81370072), National Science and Technique Major Project (Grants 201305017, 2012ZX10004213, and 311030), the Fundamental Research Funds for the Central Universities (Grant 11ykzd06), the Natural Science Foundation of Guangdong Province for Distinguished Young Scholars (Grant S2013050014535), The Key Project Supported by the Science and Technology Planning Project of Guangdong Province (Grant S2012020006147), and the Pearl River New Star Science and Technology Program of Guangzhou City (Grant 2013J2200022). The authors declare that they have no potential conflicts of interest.
- GSK-3β
- glycogen synthesis kinase-3β
- miR
- microRNA
- ILK
- integrin-linked kinase
- MMP
- matrix metalloprotease
- PTEN
- phosphatase and tensin homolog
- TCF
- T cell factor
- LEF
- lymphoid enhancer factor
- myr
- myristoylated.
References
- 1. Arko L., Katsyv I., Park G. E., Luan W. P., Park J. K. (2010) Experimental approaches for the treatment of malignant gliomas. Pharmacol. Ther. 128, 1–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balmaceda C., Peereboom D., Pannullo S., Cheung Y. K., Fisher P. G., Alavi J., Sisti M., Chen J., Fine R. L. (2008) Multi-institutional phase II study of temozolomide administered twice daily in the treatment of recurrent high-grade gliomas. Cancer 112, 1139–1146 [DOI] [PubMed] [Google Scholar]
- 3. Wong E. T., Hess K. R., Gleason M. J., Jaeckle K. A., Kyritsis A. P., Prados M. D., Levin V. A., Yung W. K. (1999) Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J. Clin. Oncol. 17, 2572–2578 [DOI] [PubMed] [Google Scholar]
- 4. Wang H., Wang H., Zhang W., Huang H. J., Liao W. S., Fuller G. N. (2004) Analysis of the activation status of Akt, NFκB, and Stat3 in human diffuse gliomas. Lab. Invest. 84, 941–951 [DOI] [PubMed] [Google Scholar]
- 5. Antonelli M., Massimino M., Morra I., Garrè M. L., Gardiman M. P., Buttarelli F. R., Arcella A., Giangaspero F. (2012) Expression of pERK and pAKT in pediatric high grade astrocytomas: correlation with YKL40 and prognostic significance. Neuropathology 32, 133–138 [DOI] [PubMed] [Google Scholar]
- 6. Molina J. R., Hayashi Y., Stephens C., Georgescu M. M. (2010) Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia 12, 453–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Holland E. C., Celestino J., Dai C., Schaefer L., Sawaya R. E., Fuller G. N. (2000) Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat. Genet. 25, 55–57 [DOI] [PubMed] [Google Scholar]
- 8. Zhang B., Gu F., She C., Guo H., Li W., Niu R., Fu L., Zhang N., Ma Y. (2009) Reduction of Akt2 inhibits migration and invasion of glioma cells. Int. J. Cancer 125, 585–595 [DOI] [PubMed] [Google Scholar]
- 9. Sonoda Y., Ozawa T., Aldape K. D., Deen D. F., Berger M. S., Pieper R. O. (2001) Akt pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of glioma. Cancer Res. 61, 6674–6678 [PubMed] [Google Scholar]
- 10. Zhang J., Han L., Zhang A., Wang Y., Yue X., You Y., Pu P., Kang C. (2010) AKT2 expression is associated with glioma malignant progression and required for cell survival and invasion. Oncol. Rep. 24, 65–72 [DOI] [PubMed] [Google Scholar]
- 11. Han L., Yang Y., Yue X., Huang K., Liu X., Pu P., Jiang H., Yan W., Jiang T., Kang C. (2010) Inactivation of PI3K/AKT signaling inhibits glioma cell growth through modulation of β-catenin-mediated transcription. Brain Res. 1366, 9–17 [DOI] [PubMed] [Google Scholar]
- 12. Kwiatkowska A., Kijewska M., Lipko M., Hibner U., Kaminska B. (2011) Downregulation of Akt and FAK phosphorylation reduces invasion of glioblastoma cells by impairment of MT1-MMP shuttling to lamellipodia and downregulates MMPs expression. Biochim. Biophys. Acta 1813, 655–667 [DOI] [PubMed] [Google Scholar]
- 13. Matheny R. W., Jr., Adamo M. L. (2009) Current perspectives on Akt Akt-ivation and Akt-ions. Exp. Biol. Med. 234, 1264–1270 [DOI] [PubMed] [Google Scholar]
- 14. Quayle S. N., Lee J. Y., Cheung L. W., Ding L., Wiedemeyer R., Dewan R. W., Huang-Hobbs E., Zhuang L., Wilson R. K., Ligon K. L., Mills G. B., Cantley L. C., Chin L. (2012) Somatic mutations of PIK3R1 promote gliomagenesis. PLoS One 7, e49466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murga C., Zohar M., Teramoto H., Gutkind J. S. (2002) Rac1 and RhoG promote cell survival by the activation of PI3K and Akt, independently of their ability to stimulate JNK and NF-κB. Oncogene 21, 207–216 [DOI] [PubMed] [Google Scholar]
- 16. Mueller W., Mizoguchi M., Silen E., D'Amore K., Nutt C. L., Louis D. N. (2005) Mutations of the PIK3CA gene are rare in human glioblastoma. Acta Neuropathol. 109, 654–655 [DOI] [PubMed] [Google Scholar]
- 17. Broderick D. K., Di C., Parrett T. J., Samuels Y. R., Cummins J. M., McLendon R. E., Fults D. W., Velculescu V. E., Bigner D. D., Yan H. (2004) Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 64, 5048–5050 [DOI] [PubMed] [Google Scholar]
- 18. Li J., Zhang H., Wu J., Guan H., Yuan J., Huang Z., Li M. (2010) Prognostic significance of integrin-linked kinase1 overexpression in astrocytoma. Int. J. Cancer 126, 1436–1444 [DOI] [PubMed] [Google Scholar]
- 19. Schickel R., Boyerinas B., Park S. M., Peter M. E. (2008) MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 27, 5959–5974 [DOI] [PubMed] [Google Scholar]
- 20. Calin G. A., Croce C. M. (2006) MicroRNA signatures in human cancers. Nat. Rev. Cancer 6, 857–866 [DOI] [PubMed] [Google Scholar]
- 21. Ma L., Teruya-Feldstein J., Weinberg R. A. (2007) Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 449, 682–688 [DOI] [PubMed] [Google Scholar]
- 22. Hayes J., Peruzzi P. P., Lawler S. (2014) MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol. Med. 20, 460–469 [DOI] [PubMed] [Google Scholar]
- 23. Garofalo M., Croce C. M. (2013) MicroRNAs as therapeutic targets in chemoresistance. Drug Resist. Updat. 16, 47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoon S., Choi Y. C., Lee S., Jeong Y., Yoon J., Baek K. (2010) Induction of growth arrest by miR-542-3p that targets survivin. FEBS Lett. 584, 4048–4052 [DOI] [PubMed] [Google Scholar]
- 25. He T., Qi F., Jia L., Wang S., Song N., Guo L., Fu Y., Luo Y. (2014) MicroRNA-542-3p inhibits tumour angiogenesis by targeting angiopoietin-2. J. Pathol. 232, 499–508 [DOI] [PubMed] [Google Scholar]
- 26. Wang Y., Jia L., Wu C. Y. (2008) Triptolide inhibits the differentiation of Th17 cells and suppresses collagen-induced arthritis. Scand. J. Immunol. 68, 383–390 [DOI] [PubMed] [Google Scholar]
- 27. Li J., Zhang N., Song L. B., Liao W. T., Jiang L. L., Gong L. Y., Wu J., Yuan J., Zhang H. Z., Zeng M. S., Li M. (2008) Astrocyte elevated gene-1 is a novel prognostic marker for breast cancer progression and overall patient survival. Clin. Cancer Res. 14, 3319–3326 [DOI] [PubMed] [Google Scholar]
- 28. Li H., Du S., Yang L., Chen Y., Huang W., Zhang R., Cui Y., Yang J., Chen D., Li Y., Zhang S., Zhou J., Wei Z., Yao Z. (2009) Rapid pulmonary fibrosis induced by acute lung injury via a lipopolysaccharide three-hit regimen. Innate Immun. 15, 143–154 [DOI] [PubMed] [Google Scholar]
- 29. Manning B. D., Cantley L. C. (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bleeker F. E., Lamba S., Zanon C., van Tilborg A. A., Leenstra S., Troost D., Hulsebos T., Vandertop W. P., Bardelli A. (2009) Absence of AKT1 mutations in glioblastoma. PLoS One 4, e5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cortez V., Nair B. C., Chakravarty D., Vadlamudi R. K. (2011) Integrin-linked kinase 1: role in hormonal cancer progression. Front. Biosci. 3, 788–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Troussard A. A., McDonald P. C., Wederell E. D., Mawji N. M., Filipenko N. R., Gelmon K. A., Kucab J. E., Dunn S. E., Emerman J. T., Bally M. B., Dedhar S. (2006) Preferential dependence of breast cancer cells versus normal cells on integrin-linked kinase for protein kinase B/Akt activation and cell survival. Cancer Res. 66, 393–403 [DOI] [PubMed] [Google Scholar]
- 33. Filipenko N. R., Attwell S., Roskelley C., Dedhar S. (2005) Integrin-linked kinase activity regulates Rac- and Cdc42-mediated actin cytoskeleton reorganization via α-PIX. Oncogene 24, 5837–5849 [DOI] [PubMed] [Google Scholar]
- 34. Edwards L. A., Woo J., Huxham L. A., Verreault M., Dragowska W. H., Chiu G., Rajput A., Kyle A. H., Kalra J., Yapp D., Yan H., Minchinton A. I., Huntsman D., Daynard T., Waterhouse D. N., Thiessen B., Dedhar S., Bally M. B. (2008) Suppression of VEGF secretion and changes in glioblastoma multiforme microenvironment by inhibition of integrin-linked kinase (ILK). Mol. Cancer Ther. 7, 59–70 [DOI] [PubMed] [Google Scholar]
- 35. Fu Y., Zhang Q., Kang C., Zhang J., Zhang K., Pu P., Wang G., Wang T. (2009) Inhibitory effects of adenovirus mediated Akt1 and PIK3R1 shRNA on the growth of malignant tumor cells in vitro and in vivo. Cancer Biol. Ther. 8, 1002–1009 [DOI] [PubMed] [Google Scholar]
- 36. Weber G. L., Parat M. O., Binder Z. A., Gallia G. L., Riggins G. J. (2011) Abrogation of PIK3CA or PIK3R1 reduces proliferation, migration, and invasion in glioblastoma multiforme cells. Oncotarget 2, 833–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li C., Zhou C., Wang S., Feng Y., Lin W., Lin S., Wang Y., Huang H., Liu P., Mu Y. G., Shen X. (2011) Sensitization of glioma cells to tamoxifen-induced apoptosis by PI3-kinase inhibitor through the GSK-3β/β-catenin signaling pathway. PLoS One 6, e27053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Williams S. P., Nowicki M. O., Liu F., Press R., Godlewski J., Abdel-Rasoul M., Kaur B., Fernandez S. A., Chiocca E. A., Lawler S. E. (2011) Indirubins decrease glioma invasion by blocking migratory phenotypes in both the tumor and stromal endothelial cell compartments. Cancer Res. 71, 5374–5380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kahlert U. D., Maciaczyk D., Doostkam S., Orr B. A., Simons B., Bogiel T., Reithmeier T., Prinz M., Schubert J., Niedermann G., Brabletz T., Eberhart C. G., Nikkhah G., Maciaczyk J. (2012) Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 325, 42–53 [DOI] [PubMed] [Google Scholar]
- 40. Pu P., Zhang Z., Kang C., Jiang R., Jia Z., Wang G., Jiang H. (2009) Downregulation of Wnt2 and β-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Ther. 16, 351–361 [DOI] [PubMed] [Google Scholar]
- 41. MacDonald B. T., Tamai K., He X. (2009) Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang K., Park J. O., Zhang M. Q. (2013) Treatment of glioblastoma multiforme using a combination of small interfering RNA targeting epidermal growth factor receptor and β-catenin. J. Gene Med. 15, 42–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kahn M. (2014) Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 13, 513–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oneyama C., Morii E., Okuzaki D., Takahashi Y., Ikeda J., Wakabayashi N., Akamatsu H., Tsujimoto M., Nishida T., Aozasa K., Okada M. (2012) MicroRNA-mediated upregulation of integrin-linked kinase promotes Src-induced tumor progression. Oncogene 31, 1623–1635 [DOI] [PubMed] [Google Scholar]
- 45. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]