

Background: lpg1496 is a member of the SidE family of Legionella pneumophila and involved in bacterial virulence.
Results: The three domains of lpg1496 have been crystallized and characterized in nucleotide-bound and free states.
Conclusion: The effector lpg1496 is likely involved in nucleotide metabolism.
Significance: This is the first structural characterization of KLAMP and SidE domains.
Keywords: nuclear magnetic resonance (NMR), pneumonia, protein structure, protein-nucleic acid interaction, structural biology, virulence factor, x-ray crystallography
Abstract
Pathogenic Gram-negative bacteria use specialized secretion systems that translocate bacterial proteins, termed effectors, directly into host cells where they interact with host proteins and biochemical processes for the benefit of the pathogen. lpg1496 is a previously uncharacterized effector of Legionella pneumophila, the causative agent of Legionnaires disease. Here, we crystallized three nucleotide binding domains from lpg1496. The C-terminal domain, which is conserved among the SidE family of effectors, is formed of two largely α-helical lobes with a nucleotide binding cleft. A structural homology search has shown similarity to phosphodiesterases involved in cleavage of cyclic nucleotides. We have also crystallized a novel domain that occurs twice in the N-terminal half of the protein that we term the KLAMP domain due to the presence of homologous domains in bacterial histidine kinase-like ATP binding region-containing proteins and S-adenosylmethionine-dependent methyltransferase proteins. Both KLAMP structures are very similar but selectively bind 3′,5′-cAMP and ADP. A co-crystal of the KLAMP1 domain with 3′,5′-cAMP reveals the contribution of Tyr-61 and Tyr-69 that produces π-stacking interactions with the adenine ring of the nucleotide. Our study provides the first structural insights into two novel nucleotide binding domains associated with bacterial virulence.
Introduction
Legionella pneumophila is a Gram-negative bacterium and is the causative agent of Legionnaires disease, an acute form of pneumonia (1). Pathogenic Gram-negative bacteria use specialized secretion systems that translocate bacterial proteins, termed effectors, directly into host cells where they interact with host proteins and hijack eukaryotic biochemical processes for the benefit of the pathogen. These secretion machineries are highly conserved among different bacterial species. L. pneumophila uses a Dot/Icm type IV (T4SS) secretion system to inject effector proteins into the host cells (2). The secreted effectors allow the bacterium to escape the host lysosomal pathway after phagocytosis.
Currently, about 300 Dot/Icm-dependent effectors of L. pneumophila have been identified using methods such as interaction with Dot/Icm components (3, 4), the presence of a C-terminal secretion signal (5), and a machine-learning approach where effectors were identified based on shared features (6). However, the functions of most remain unknown. lpg1496 is one such experimentally validated effector protein (6).
From sequence alignment, lpg1496 was found to contain a conserved domain from the SidE family. An interbacterial transfer assay in 2004 led to the identification of this family, which includes SdeA, SdeB, SdeC, and SidE (7). These four members were grouped based on their location on the chromosome, their interactions with IcmS, a putative chaperone for effectors, and sequence similarity (3, 8). SidE proteins are secreted during infection and localize to the poles of the bacterium, where they may interact with nearby Dot/Icm substrates such as LidA (3, 9). A member of this family, SdeA, is a paralog of LaiA, which contains homology to an integrin analogue gene of Saccharomyces cerevisiae (10, 11). LaiA has been shown to be required for adherence and entry into alveolar epithelial cells (10). In addition, SidE family secretion peaks 30 min post infection of mouse bone marrow macrophages, and SdeA-transfected cells fragment the Golgi apparatus, suggesting a role in the early events of intracellular growth, such as modification of the Legionella-containing vacuole (3, 12). A deletion strain of this family results in an approximate 100-fold less growth than wild type in Acanthamoeba castellanii, which can be complemented by the expression of SdeA on a plasmid (3). Recently, it has been reported that overexpression of SdeA in a SidJ mutant completely inhibits the growth of intracellular Legionella to levels similar to a translocation deficient DotA mutant (12).
No molecular characterization of lpg1496 has been done previously. Here, we identified three independently folded domains in the protein and determined their high resolution crystal structures. The two N-terminal domains are a novel fold that we call KLAMP3 domains. The C-terminal domain is found in all SidE family proteins but had not been characterized structurally. We showed that all the domains bind to nucleotides and revealed molecular determinants of their binding specificity.
Experimental Procedures
Cloning, Protein Expression, and Purification
The gene lpg1496 from L. pneumophila strain Philadelphia was cloned into pLR652 as a N-terminal GST-tagged fusion protein and expressed in a BL21 Star Escherichia coli strain. The cells were grown at 37 °C in Luria broth to an optical density of 0.8, and expression was induced with 1 mm isopropyl β-d-1-thiogalactopyranoside at 30 °C for 4 h or 16 °C overnight. After centrifuging the cells, the pellets were resuspended in phosphate-buffered saline (PBS) (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 2 mm KH2PO4, pH 7.4) containing 1 mm phenylmethylsulfonyl fluoride, 0.1 mg/ml lysozyme, and 80 units of deoxyribonuclease and lysed by sonication. Cell debris was removed by centrifugation, and the GST fusion protein was purified using glutathione-Sepharose affinity columns (GE Healthcare). After eluting the protein in PBS containing 20 mm glutathione, the middle domain of lpg1496 was obtained by a 2 h of trypsin cleavage in PBS and further purified on a Superdex75 gel filtration column (GE Healthcare) in buffer A (10 mm HEPES, pH 7.0, 100 mm NaCl) before crystallization trials.
The N-terminal domain of lpg1496 (residues 1–138) was cloned into pET29a using the primers 5′-agatatacatatggttacgaaaataatttgggtttc-3′ and 5′-ctagctcgagttttgttacgggaacaataacaggtg-3′ as a C-terminal His-tagged fusion and transformed into a BL21 E. coli strain. Mutagenesis for Y61A and Y69A was then performed using 5′-ctgatagaagcttcaaatgccccaattaatccttgtgg-3′ and 5′-taatccttgtggttgtgctatatccccaggtggg-3′, respectively. The expression conditions were the same as for the full-length protein. The fusion protein was bound to nickel-nitrilotriacetic acid-agarose (Qiagen) beads, washed with buffer B (50 mm HEPES, pH 7.6, 0.5 m NaCl, 5% (v/v) glycerol) containing 30 mm imidazole, and eluted with buffer B containing 250 mm imidazole. The protein was further purified by size-exclusion chromatography on a Superdex75 gel filtration column (GE Healthcare) in buffer A.
Three constructs of the SidE domain of lpg1496 were cloned. lpg1496 (293–580) was cloned into pET29a using the primers 5′-agatatacatatggaaatagagaaaaatgattatctactatc-3′ and 5′-ctagctcgagctttagacactcattgggatc-3′ as a C-terminal His-tagged fusion protein. lpg1496 (293–598) was cloned into pET15b using the primers 5′-agatatacatatggaaatagagaaaaatgattatctactatc-3′ and 5′-cgcggatccttaaataccatattgatttgccaag-3′ as an N-terminal His-tagged fusion protein. lpg1496 (154–598) was cloned into the pET29a vector using 5′-agatatacatatggattcttcgatttctattagtgc-3′ and 5′-ctagctcgagaataccatattgatttgccaag-3′. Constructs were verified by DNA sequencing. E. coli BL21 star cells were transformed with plasmids encoding the three constructs. The expression and purification conditions were the same as for the N-terminal domain, except size-exclusion chromatography was performed using buffer C (10 mm HEPES, pH 7.5, 100 mm NaCl). The His tag in the pET15b construct was cleaved with thrombin before injecting the protein into a size-exclusion column.
For 15N-labeling, the cells were grown in M9 minimal medium supplemented with [15N]ammonium chloride as the sole source of nitrogen. For 13C,15N double-labeling, the cells were grown in M9 minimal medium supplemented with [15N]ammonium chloride and d-[13C6]glucose as the sole sources of nitrogen and carbon.
For selenomethionine labeling, the plasmid was transformed into a methionine-auxotroph DL41 (DE3) E. coli strain, and the cells were grown in LeMaster medium supplemented with selenomethionine. The expression and purification protocols were the same as for the native protein.
Crystallization, Data Collection, and Processing
Crystallization was performed by the hanging drop vapor diffusion method at 293 K using the Classics II and JCSG+ Suite commercial screens (Qiagen). Crystals of the middle domain of lpg1496 were obtained in approximately 1 week from a 1:1 mixture of the protein solution (16.6 mg/ml) and the reservoir solution (0.1 m HEPES, pH 7.5, 25% (w/v) PEG 3350). Crystals of the N-terminal domain were obtained by equilibrating a drop consisting of 0.6 μl of lpg1496 (residues 1–138) (15 mg/ml) and 0.6 μl of 0.2 m sodium formate and 20% (w/v) PEG 3350. Crystals of lpg1496 (1–138) used for soaking experiments were obtained in a 1:1 mixture of protein at 37.8 mg/ml and the mother liquor (0.2 m sodium chloride, 0.1 m Bis-Tris, pH 6.5, 25% (w/v) PEG 3350). Soaking experiments with 3′,5′-cAMP were performed by dipping the crystal into a solution of mother liquor containing 15 mm 3′,5′-cAMP for 20 min. Crystals of lpg1496 (293–580) were obtained overnight from a 1:1 mixture of the protein solution (11 mg/ml) and the reservoir solution (1.1 m sodium malonate, 0.1 m HEPES, pH 7.0, 0.5% (v/v) Jeffamine ED-2001). Crystals of lpg1496 (293–598) at 7.1 mg/ml were obtained in condition #15 of the JCSG+ Suite (0.1 m Bicine, pH 9.0, 20% (w/v) PEG 6000) in approximately 1 week. Crystals of the seleno-l-methionine-labeled lpg1496 (293–598) were obtained in one week from a 1:1 mixture of protein at 10 mg/ml with the mother liquor (0.1 m Bis-Tris, pH 6.5, 20% (w/v) monomethyl ether PEG 5000). lpg1496 (154–598) was crystallized in 0.1 m HEPES, pH 7.5, 25% (w/v) PEG 3350, and 5 mm ADP.
The crystals were cryoprotected with 10–25% glycerol or ethylene glycol and flash-cooled in a N2 cold stream. X-ray diffraction data were collected using an ADSC Quantum 210 CCD detector (Area Detector Systems Corp.) on beamline A1 at the Cornell High-Energy Synchrotron Source (CHESS) at 0.9770 Å. Data processing and scaling were performed with HKL-2000 (Tables 1 and 2) (13).
TABLE 1.
Data collection and refinement statistics for the SidE domain
| Data collection | 293–580 | 293–598 | 154–598/ADP |
| Space group | P212121 | P212121 | P1 |
| Cell dimensions | |||
| a, b, c (Å) | 60.89, 71.47, 77.00 | 71.98, 77.23, 109.38 | 56.87, 69.71, 77.03 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 89.87, 72.30, 70.31 |
| Resolution (Å) | 50-1.60 (1.63-1.60)a | 50-2.35 (2.39-2.35) | 50-2.10 (2.14-2.10) |
| Rsym | 0.093 (0.465) | 0.169 (0.446) | 0.098 (0.435) |
| I /σI | 34.2 (8.5) | 42.5 (11.4) | 11.3 (1.7) |
| Completeness (%) | 99.9 (99.9) | 99.8 (100) | 95.0 (78.3) |
| Redundancy | 14.4 (14.2) | 13.0 (12.7) | 2.6 (2.5) |
| Refinement | |||
| Resolution (Å) | 29.6-1.60 | 37.9-2.35 | 72.9-2.11 |
| No. reflections | 42,595 | 25,991 | 54,209 |
| Rwork/Rfree | 0.161/0.191 | 0.239/0.293 | 0.229/0.273 |
| No. atoms | |||
| Protein | 2306 | 4653 | 9242 |
| Water | 414 | 220 | 121 |
| Nucleotide | 141 | ||
| B-factors | |||
| Protein | 15.32 | 27.23 | 37.57 |
| Water | 30.84 | 28.18 | 35.17 |
| Nucleotide | 44.66 | ||
| r.m.s.d. | |||
| Bond lengths (Å) | 0.017 | 0.002 | 0.007 |
| Bond angles (°) | 2.019 | 0.651 | 1.212 |
| Ramachandran statistics (%) | |||
| Most favored regions | 96.3 | 96.2 | 96.0 |
| Additional allowed regions | 3.7 | 3.8 | 4.0 |
a The highest resolution shell is shown in parentheses.
TABLE 2.
Data collection and refinement statistics for KLAMP1 and KLAMP2
| Data collection | KLAMP1 | KLAMP1/3′5′-cAMP | KLAMP2 | KLAMP2 |
| Space group | P21 | P21 | P21 | P212121 |
| Cell dimensions | ||||
| a, b, c (Å) | 32.41, 56.92, 60.18 | 32.62, 57.45, 63.78 | 41.80, 35.91, 43.66 | 36.14, 44.07, 77.53 |
| α, β, γ (°) | 90, 91.18, 90 | 90, 93.32, 90 | 90, 104.7, 90 | 90, 90, 90 |
| Resolution (Å) | 50-1.20 (1.22-1.20)a | 50-1.75 (1.78-1.75) | 50-1.15 | 50-1.60 (1.63-1.60) |
| Rsym | 0.056 (0.567) | 0.076 (0.497) | 0.068 (0.437) | 0.057 (0.406) |
| I /σI | 43.9 (2.8) | 39.0 (5.3) | 43.2 (3.1) | 49.7 (4.9) |
| Completeness (%) | 92.7 (84.3) | 89.2 (92.3) | 92.0 (57.9) | 99.9 (100.0) |
| Redundancy | 7.7 (6.0) | 6.6 (6.4) | 7.4 (5.3) | 8.6 (8.7) |
| Refinement | ||||
| Resolution (Å) | 60.17-1.20 | 63.67-2.10 | 40.5-1.15 | 38.8-1.60 |
| No. reflections | 60,366 | 12,544 | 39,061 | 16,100 |
| Rwork/Rfree | 0.158/0.180 | 0.238/0.292 | 0.169/0.192 | 0.199/0.231 |
| No. atoms | ||||
| Protein | 2,138 | 2,087 | 1,071 | 1,043 |
| Water | 207 | 107 | 200 | 130 |
| Nucleotide | 44 | |||
| B-factors | ||||
| Protein | 14.0 | 21.3 | 11.5 | 13.0 |
| Water | 21.0 | 34.8 | 22.8 | 33.4 |
| Nucleotide | 38.6 | |||
| r.m.s.d. | ||||
| Bond lengths (Å) | 0.012 | 0.011 | 0.005 | 0.006 |
| Bond angles (°) | 1.46 | 1.64 | 1.12 | 1.03 |
| Ramachandran statistics (%) | ||||
| Most favored regions | 95.1 | 94.4 | 99.2 | 97.6 |
| Additional allowed regions | 4.9 | 5.6 | 0.8 | 2.4 |
a The highest resolution shell is shown in parentheses.
Structure Determination and Refinement
The diffraction data of the middle domain were phased using an anomalous signal from selenium atoms by the single-wavelength anomalous dispersion method, with the program SHELX (14). The initial model, consisting of >90% of the residues, was built with ARP/wARP (15) and refined with Refmac5 (16).
The N-terminal domain structure (residues 1–138) was determined by molecular replacement (MR) using the middle domain of lpg1496 as the search model. The initial model was built by ARP/wARP (15) using phases from the MR solution. Model building was completed with the help of the program Coot (17) and was improved by several cycles of refinement using Refmac5 (16).
The N-terminal domain structure in complex with 3′,5′-cAMP was determined by MR using lpg1496 (1–138) as the search model. The model was built by PhaserMR (18) and completed with Coot (17). Refmac5 was used to improve density (16). The water molecules were added in the last stage of refinement. The refinement statistics for the N-terminal half are in Table 2.
Diffraction data of the 293–598 construct were phased using single-wavelength anomalous dispersion with the program PHENIX (19). The initial model was built using PHENIX (19), improved using Coot (17), and further refined using PHENIX (19).
Data from the 293–580 and 154–598 constructs were phased by MR using the lpg1496 (293–598) structure as the search model. The initial model was built by ARP/wARP (15) and further built with Coot (17). The final model was improved by several cycles of refinement using Refmac5 (17). The refinement statistics are shown in Table 1.
The final models have no outliers in the Ramachandran plot computed using PROCHECK (20) and MolProbity (12). Coordinates of the middle domain in 2 space groups (P21 and P212121) and the N-terminal domain without and with 3′,5′-cAMP have been deposited in the Protein Data Bank with the accession codes 5BTY, 5BTZ, 5BTW, and 5BTX, respectively. Coordinates of the native SidE domain (293–580, 293–598) and its ADP-bound structure have been deposited in the Protein Data Bank with the accession codes 5BU1, 5BU0, and 5BU2, respectively.
NMR Spectroscopy
NMR resonance assignments of the 13C,15N-labeled middle domain of lpg1496 were determined using HNCACB and HN(CO)CACB experiments. The three-dimensional heteronuclear experiments were recorded at 298 K on a Bruker 800 MHz spectrometer. The samples were prepared as 400 μm in 90% buffer A and 10% D2O. NMR spectra were processed with NMRPipe (21) and analyzed with SPARKY (22).
NMR titration experiments were performed at 303 K on a Bruker 600 MHz spectrometer. For titrations with nucleotides, NMR samples of the 15N-labeled lpg1496 (1–138) construct were prepared as 0.17–0.20 mm in 90% buffer A and 10% D2O. Samples of the middle domain of 15N-labeled lpg1496 were prepared as 135 μm in 90% buffer A and 10% D2O. 1H,15N heteronuclear single quantum correlation spectroscopy (HSQC) titrations were performed by the stepwise addition of 2′,3′-cAMP, 3′,5′-cAMP, 3′,5′-cGMP, ATP, ADP, and AMP to a final molar ratio of 1:10 of N-terminal lpg1496 to ligand. The N-terminal mutants were titrated with 3′,5′-cAMP only. 1H,15N HSQC titrations were performed by stepwise addition of ATP, ADP, AMP, and GDP to the middle domain to a final molar ratio of 1:10 (50 for AMP). Minimal changes in volume and pH were ensured throughout the sample preparations. The NMR spectra were processed by NMRPipe (21) and analyzed using SPARKY (22).
The affinities were measured by the calculations of the dissociation constant (Kd). Chemical shift changes were fitted to Equation 1,
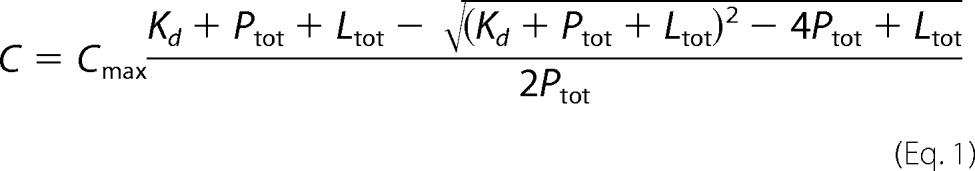 |
where C is the chemical shift perturbation, Cmax is the chemical shift perturbation at saturation, Kd is the dissociation constant, Ptot is the total concentration of the labeled protein, and Ltot is the total ligand concentration.
Malachite Green Assay
Phosphodiesterase assays were performed in 96-well plates in 10 mm HEPES, pH 7.0, 100 μm nucleotide (3′-AMP, 5′-AMP, ADP, ADP-ribose, 2′,3′-cAMP, 3′,5′-cAMP, and 3′,5′-cGMP) and 2.5 μm full-length lpg1496 to a total reaction volume of 75 μl. When necessary, 6 units of alkaline phosphatase was used to release the phosphate group for detection. The reaction was incubated at 37 °C for 15 min and stopped by the addition of 43 μl of 28 mm ammonium molybdate in 2.1 m H2SO4 and 32 μl of 0.76 mm malachite green in 0.35% polyvinyl alcohol (Mr ∼ 16,000). Free phosphate was determined by measuring absorbance at 610 nm.
Results
The SidE Domain
Sequence analysis of the C terminus of lpg1496 reveals sequence similarity to the N-terminal region of the original members of the SidE family (SdeA, SdeB, SdeC, and SidE) (Fig. 1, A and B). This domain is also found in other strains of L. pneumophila such as Shakespearei and Longbeachae (Fig. 1C). Some proteins containing the SidE domain contained the Vip2 domain, which is present in an actin-ADP-ribosylating toxin family. These proteins confer virulence by modifying monomeric actin, resulting in depolymerization of the actin cytoskeleton and eventually leading to cell death (23).
FIGURE 1.

SidE sequence and structure. A, domain architecture of lpg1496 (KLAMP1 in green, KLAMP2 in wheat, SidE in yellow). B, the occurrence of the SidE domain in Legionella proteins. C, sequence alignment of the SidE domain of lpg1496 with other bacterial proteins. The secondary structure of the lpg1496 SidE domain is overlaid on top (cylinder = α-helix; arrow = β-sheet).
The C-terminal domain of lpg1496 (293–580) was crystallized, yielding a high resolution structure of the SidE domain. This construct diffracted to 1.6 Å resolution with one SidE domain in the asymmetric unit from Asp-298 to Lys-580. A longer 293–598 construct was also crystallized and refined to 2.34 Å (Table 1). Phasing was performed using the single-wavelength anomalous dispersion method with selenomethionine-labeled crystals. The structure contains two SidE domains in the asymmetric unit with interpretable density for Asp-298–Leu-592 and Asp-298–Ala-589, respectively, with an r.m.s.d. of 0.24 Å over 264 Cα atoms. The structures of both constructs are very similar, yielding an r.m.s.d. of 0.58 Å over 245 Cα atoms.
The general fold of the SidE domain consists of two mostly α-helical lobes with a cleft in-between. The larger lobe comprises 11 α-helices, 1 α-helical turn, and 2 antiparallel β-strands. The smaller lobe contains three α-helices (Fig. 2A).
FIGURE 2.

SidE ADP binding. A, structural similarity of the SidE domain of lpg1496 (yellow) and LmjPDEB1 (PDB code 2R8Q, purple). B, superposition of the active site of LmjPDEB1 (purple) and lpg1496 (yellow), with important catalytic residues labeled for lpg1496. C, surface charge representation of the SidE domain (blue = positive, red = negative) with the bound ADP (gray). D, the SidE domain forms the most consistent polar contacts with the ribose ring of ADP. Two conserved histidines with LmjPDEB1 and human PDEs are involved in hydrogen bonds (His-370 and His-366).
Putative Substrate Binding Site
A structural homology search using the SidE domain of lpg1496 against the DALI database (24) led to the identification of LmjPDEB1, with a Z-score of 9.2 and r.m.s.d. of 3.3 Å. LmjPDEB1 is a cAMP-specific cyclic nucleotide phosphodiesterase (PDE) found in Leishmania (PDB code 2R8Q) (Fig. 2A) (25). It requires the binding of two divalent metal ions (zinc and magnesium) for activity. Half of the residues involved in metal coordination are conserved between LmjPDEB1 (His-685, His-721, Asp-722, Asp-835) and lpg1496 (His-370, Ser-412, Val-413, His-504). The presence of serine and valine instead of histidine and aspartate explains why there are no metal ions in the lpg1496 structure (Fig. 2B). Of the two histidines involved in catalysis in LmjPDEB1, one is conserved in lpg1496 (His-366), whereas the other is substituted with arginine (Arg-416). These two conserved histidines, His-370 for metal coordination and His-366 for catalysis, are also conserved among the human PDEs (25).
Overall, the SidE domain is structurally a member of a superfamily of metal-dependent phosphohydrolases, named HD for the presence of a conserved histidine and aspartate involved in coordination of divalent cations. HD domains have demonstrated phosphatase, nucleotide triphosphatase, phosphodiesterase, and ribonuclease activities (26). In the case of lpg1496, His-504 and Asp-505 are part of the potential active site that was identified by alignment with LmjPDEB1 (Asp-835 and Val-836). To test for putative phosphatase and phosphodiesterase activity, malachite green assays were performed against 3′-AMP, 5′-AMP, ADP, ADP-ribose, 2′,3′-cAMP, 3′,5′-cAMP, and 3′,5′-cGMP. lpg1496 did not show significant activity against any nucleotide (data not shown). This may be explained by the lack of metal binding in addition to other differences in active sites between lpg1496 and known PDEs.
The SidE Domain of lpg1496 Binds Nucleotides
To determine whether the C-terminal SidE domain nonetheless binds nucleotides, we set up crystallization screens for lpg1496 (154–598) in the presence of ADP or 3′,5′-cAMP. Crystallization trials with ADP produced crystals in the P1 space group that diffracted to 2.1 Å with interpretable density for the conserved C-terminal SidE domain (Table 1). No density was observed for residues 154–297. There were four molecules in the asymmetric unit, all of which contained bound nucleotide. Three molecules showed the complete ADP nucleotide, whereas one only had interpretable density for the ribose moiety (Fig. 2C). Although lpg1496 does not have phosphatase activity, its SidE domain is capable of binding ADP. However, the binding was not detected by isothermal titration calorimetry measurements.
An overlay of the four chains shows that this domain forms the most consistent polar contacts with the ribose ring. Arg-510 and His-366 form hydrogen bonds with O2′, His-370 with O3′, and Arg-416 with O4′ (Fig. 2D). The phosphates of ADP are generally stabilized by interactions with the main chain amides. The ADP-bound C-terminal domain structure shows that the putative substrate binding site of lpg1496 is in fact shifted closer toward the metal binding sites of previously characterized PDEs, adding to the possibility that lpg1496 is involved in ADP-ribosylation rather than in the cleavage of phosphoester bonds.
lpg1496 Contains Two Homologous Domains in the N-terminal Half
To further understand lpg1496, we looked into the previously uncharacterized ∼300-residue N-terminal half of the protein (Fig. 1A). Sequence similarity searches identified two repeats of ∼120 residues (residues 8–116 and 162–277) displaying 32% sequence identity (Fig. 3A). Limited proteolysis experiments showed that these regions form independently folded domains. Trypsin digestion of the C-terminal fragment of lpg1496 containing residues 154–598 generated two stable fragments of ∼16 and 35 kDa (data not shown). The fragments could be separated by size-exclusion chromatography. The 35-kDa fragment corresponds to the SidE domain, whereas the 16-kDa fragment was later characterized as the middle domain of lpg1496.
FIGURE 3.

KLAMP sequence and structure. A, sequence alignment of lpg1496 domains with predicted ATPase from Streptomyces pratensis (WP_014153402) and precorrin-6y methyltransferase CbiE from Beijerinckia indica (WP_012386418). Secondary structure is shown according to lpg1496 domain structures. Residues involved in binding of 3′,5′-cAMP by KLAMP1 of lpg1496 are marked with asterisks. B, schematic representation of the KLAMP1 domain structure, color-coded from the N terminus (blue) to C terminus (red). C, overlay of structures of KLAMP2 (wheat) and KLAMP1 (faint green) shows high similarity of secondary structure regions with differences in loop conformations.
A BLAST sequence similarity search using the N-terminal sequence of lpg1496 found a number of other proteins, notably a previously uncharacterized region of histidine kinase-like ATP binding region-containing bacterial proteins and the C-terminal domain of bacterial S-adenosylmethionine-dependent methyltransferases (Fig. 3A). We propose to term the domain family as KLAMP domains for their presence in histidine kinase-like ATP binding region-containing proteins and S-adenosylmethionine-dependent methyltransferase proteins. Thus, lpg1496 contains two KLAMP domains (KLAMP1 and KLAMP2) and a C-terminal SidE domain (Fig. 1A).
Crystallization trials with the second KLAMP domain of lpg1496 produced crystals in the P21 space group that diffracted to 1.15 Å. The structure was solved using single-wavelength anomalous dispersion (Table 2). The structure contains one KLAMP molecule with density from Asp-154 to Thr-288. Crystals were also obtained in the P212121 space group with nearly identical structure (r.m.s.d. of 0.2 Å over 117 Cα atoms), confirming that the observed structures were not influenced by crystal contacts.
We also determined the structure of the first KLAMP domain. lpg1496 (residues 1–138) yielded crystals that diffracted to 1.2 Å using synchrotron radiation (Table 2). The asymmetric unit contains two molecules consisting of residues Met-1–Val-132 and Met-1–Pro-131, respectively. The structures of KLAMP1 and KLAMP2 domains are very similar, displaying r.m.s.d. of 1.2 Å over 90 Cα atoms (Fig. 3C).
The structure of the KLAMP domain consists of two anti-parallel α-helices (α1-α2) flanked by a five-stranded anti-parallel β-sheet (β9-β1-β2-β4-β3) on one side and a four-stranded anti-parallel β-sheet on the other side (β5-β7-β6-β8) (Fig. 3B). This arrangement leaves the opposite sides of both β-sheets solvent-exposed. The biggest difference between the two KLAMP structures is the conformation of two loops between α1 and β3 and between β7 and β8, which are longer in KLAMP2. Three cis-proline residues are found in KLAMP2: Pro-202 in the loop between α1 and β3, Pro-208 in the loop between β3 and β4, and Pro-219 in the loop between β4 and β5. This is an interesting feature of the structure, as cis-proline residues are not very common in protein structures. In KLAMP1, Pro-62 and Pro-79 are conserved and correspond to Pro-202 and Pro-219, respectively, but only Pro-62 is in cis conformation. The conformation of the β4-β5 loop is different between the two domains, which can be partly attributed to the trans-conformation of Pro-79.
Structural similarity search with the program DALI (24) using the middle domain of lpg1496 shows that the KLAMP fold is relatively novel. The highest similarity hit was to a mixed α-β protein, pterin dehydratase-like protein (PDB code 4LOW) with a low Z-score of 4.7.
KLAMP Domains of lpg1496 Bind Nucleotides
The presence of KLAMP-like sequences in proteins predicted to bind ATP suggested that we test the KLAMP2 domain for binding to nucleotides. We used 15N-labeled middle domain of lpg1496 and obtained its 1H,15N correlation spectrum. The spectrum showed well dispersed signals for backbone amides, characteristic of a well folded protein. NMR titrations were performed by a stepwise addition of potential nucleotide ligands monitored by HSQC experiments (Fig. 4A).
FIGURE 4.

KLAMP2 ADP binding. A, downfield region of HSQC spectra of 15N-labeled KLAMP2 domain titrated with increasing amounts of ADP at 0 mm (red), 0.13 mm (yellow), 0.26 mm (green), 0.63 mm (blue), and 1.18 mm (purple). The spectra show specific chemical shift changes for a number of signals. B, Kd of the binding estimated from a fit of the 15N chemical shift changes for two assigned and one unassigned signal that show large chemical shift changes. C, mapping of the chemical shifts measured onto the structure of the KLAMP2 domain. Red indicates largest chemical shift changes; white indicates no change detected. The most affected surface is centered on the β4-β5 loop and strands β5 and β6. D, model of an ADP-bound KLAMP2 domain based on the 3′,5′-cAMP/KLAMP1 structure, with residues important for binding shown.
Titration of the KLAMP2 domain of lpg1496 with ATP resulted in chemical shift changes of roughly 20 backbone amides, indicating specific binding to the domain. The affinity of the interaction can be estimated by a fit of chemical shift changes of NMR signals versus ligand concentration. Using several signals with the biggest chemical shift changes, the dissociation constant (Kd) of ATP binding was estimated to be 800 ± 150 μm. Even larger chemical shift changes were observed upon the addition of ADP with a Kd of 109 ± 7 μm (Fig. 4, A and B), whereas titration with AMP produced much smaller spectral changes with a Kd of 1500 ± 200 μm. There is a preference for a diphosphate group in the nucleotide binding site of KLAMP2. To test the base specificity, we titrated the 15N-labeled middle domain of lpg1496 with GDP. The addition of GDP did not result in spectral changes (data not shown), indicating a clear preference of KLAMP2 for binding adenine nucleotides.
NMR binding studies can identify the ligand binding site on a protein via a residue-specific assignment of NMR signals and mapping the binding-induced spectral changes on the three-dimensional structure. We prepared 13C,15N-labeled KLAMP2 and assigned the backbone amides using standard heteronuclear NMR experiments. The residues showing the biggest chemical shift changes upon ADP addition are Leu-223 (0.80), Tyr-218 (0.77), Lys-217 (0.75), Gly-238 (0.42), Gly-224 (0.36), Tyr-226 (0.31), Ile-227 (0.30), and Ser-216 (0.28). Mapping of the chemical shift changes on the structure identifies a pocket formed by the β-sheet β5-β7-β6-β8 and the surrounding loops (β4-β5, β5-β6, and α2-β8) (Fig. 4C). Some of the biggest changes come from the residues in the β4-β5 loop, suggesting the loop is involved in nucleotide binding.
We hypothesized that the N-terminal KLAMP1 domain also binds nucleotides. NMR titrations using the same set of ligands (ATP, ADP, AMP, and GDP) revealed a preference of KLAMP1 for ADP, albeit with significantly lower affinity. The Kd of ADP binding was estimated to be 800 ± 250 μm, whereas the affinity toward other nucleotides was much lower and could not be reliably measured. Unexpectedly, titration of 15N-labeled KLAMP1 with 3′,5′-cAMP resulted in large chemical shift changes for a number of signals, showing binding with a Kd estimated to be 280 ± 32 μm (Fig. 5, A and B). Although sequence-specific signal assignments were not obtained for the KLAMP1 domain, the changes in the spectrum were similar, suggesting that KLAMP1 and KLAMP2 bind nucleotides in a similar fashion. The binding of the cyclic nucleotide is specific, as the NMR titration of KLAMP1 with 2′,3′-cAMP displayed no interaction. In addition, no binding was observed upon the addition of 3′,5′-cAMP or 2′,3′-cAMP to KLAMP2. These experiments show that KLAMP1 and KLAMP2 are nucleotide binding domains but with differing specificities: KLAMP1 for 3′,5′-cAMP and KLAMP2 for ADP.
FIGURE 5.

KLAMP1 3′,5′-cAMP binding. A, downfield region of HSQC spectra of the 15N-labeled KLAMP1 domain show specific changes upon titration with increasing amounts of 3′,5′-cAMP at 0 mm (red), 0.20 mm (yellow), 0.39 mm (green), 0.57 mm (blue), and 1.67 mm (purple). B, Kd of the binding estimated from a fit of the 15N chemical shift changes for several signals with the largest chemical shift changes. The NMR resonances of the KLAMP1 domain have not been assigned. C, surface charge representation of the KLAMP1 domain (blue = positive, red = negative) with the bound 3′,5′-cAMP (gray). D, principal contribution to the binding comes from π-stacking of Tyr-61 and Tyr-69 with the adenine ring of cyclic nucleotide. Interaction is stabilized by hydrogen bonds (dash lines) between 3′,5′-cAMP and the side chains of Ser-59, Tyr-61, Asn-79, His-106, and Thr-108.
Molecular Determinants of 3′,5′-cAMP Recognition by KLAMP1
To understand the molecular basis of nucleotide binding by the KLAMP domains, we soaked the crystals of KLAMP1 and KLAMP2 with 3′,5′-cAMP and ADP, respectively. The electron density map of the KLAMP1 crystal soaked with 3′,5′-cAMP showed easily interpretable density for the nucleotide, and the structure of the complex of KLAMP1 with 3′,5′-cAMP was refined to 2.1 Å (Table 2). The adenine ring fits snugly into a narrow ridge formed by the β4-β5 loop and the β-strand β5 (Fig. 5C). Three KLAMP1 residues are directly involved in the recognition of the adenine ring. Two tyrosine side chains (Tyr-61 and Tyr-69) provide π-stacking interactions with the adenine ring, whereas the N1 atom of adenine is hydrogen-bonded with the side chain of Ser-59 (Fig. 5D). Comparison of the nucleotide-bound and unliganded KLAMP1 structures reveals movement of the β4-β5 loop, which closes on the nucleotide upon binding and presents Tyr-61 as a major binding determinant. Three residues are engaged in polar contacts with the ribose ring of nucleotide. The side chain of Tyr-61 hydrogen bonds with the oxygen atom of the ring, whereas the side chains of Asn-79 and Thr-108, both, bind 2′OH. Finally, the O2 atom of the phosphate group forms a hydrogen bond with the side chain of His-106.
We mutated residues Tyr-61 and Tyr-69 to test their roles in nucleotide binding. NMR spectra of the Y61A and Y69A KLAMP1 mutants are very similar to the wild-type protein, confirming that they are still well folded. However, NMR titrations of the 15N-labeled mutants with 3′,5′-cAMP yielded no spectral changes, confirming that these mutations abolished binding (data not shown).
The structure explains similarities and differences in the nucleotide binding specificities of KLAMP domains. An overlay reveals the residues involved in adenosine recognition are strictly conserved, as Tyr-218, Tyr-226, and Ser-216 of KLAMP2 correspond to Tyr-61, Tyr-69, and Ser-59 of KLAMP1 (Fig. 3A). Even the KLAMP1 residues involved in forming hydrogen bonds with ribose (Asn-79, Thr-108, and Tyr-61) are conserved in KLAMP2 (Asn-236, Thr-269, and Tyr-218) (Fig. 4D). Furthermore, the complex structure explains why the KLAMP domains will not recognize guanine and pyrimidine rings. Compared with adenine, guanine has an extra NH2 group that will push the ring out of the binding site. NMR titrations experimentally verified that GDP and 3′,5′-cGMP do not interact with the KLAMP domains. On the other hand, pyrimidines (cytosine and thymine) possess smaller rings, which will not reach the conserved serine for hydrogen bonding and would also result in insufficient stacking with the tyrosines.
The specificity of KLAMP1 and KLAMP2 for different adenine-containing ligands results from two structural differences. One of them is a rather subtle substitution of Phe-110 in KLAMP1 for Tyr-271 in KLAMP2. A tyrosine residue in this position would clash with the phosphate of 3′,5′-cAMP, whereas a phenylalanine residue is in close contact with this phosphate (Figs. 5D and 4D). KLAMP2 also possesses a larger β7-β8 loop, which would also clash with the 3′,5′-cAMP phosphate group. The position of this phosphate group also explains the specificity of KLAMP1 for 3′,5′-cAMP as opposed to 2′,3′-cAMP, whose binding would be obstructed by the side chains of Phe-110, Thr-108, and Tyr-69.
KLAMP2 is highly selective against binding cyclic nucleotides, but among the non-cyclic adenosine phosphates there is increasing affinity for AMP, ATP, and ADP. Analysis of the structure suggests that the length of two phosphates may allow ADP to reach the β7-β8 loop and form a hydrogen bond with Thr-265. The third phosphate of ATP could be repelled by Glu-264, whereas AMP is too short to make any contact with the loop.
Discussion
Here, we have identified a novel KLAMP domain that is present in two copies in the N-terminal half of lpg1496, a L. pneumophila effector. Based on sequence similarity, a similar domain is also found in histidine kinase-like ATP binding region-containing proteins and S-adenosylmethionine-dependent methyltransferase proteins. The structures of the domains from lpg1496 are very similar to each other but do not display significant structural similarity to other known protein structures. More significantly, we demonstrate using NMR that both domains bind nucleotides, albeit with different specificity.
Sequence alignment of KLAMP1/2 of lpg1496, histidine kinase-like ATP binding region-containing protein, and S-adenosylmethionine-dependent methyltransferases highlight the importance of several conserved residues. Ser-59 of lpg1496 that hydrogen bonds with the adenine ring through N1 is generally conserved. Aromaticity is also conserved at the Tyr-69 position with either tyrosine or phenylalanine for π-stacking with adenine. Polar contacts with the 2′OH atom of the ribose ring is conserved in Thr-108. This suggests that all domains of the identified family may interact with adenosine-containing molecules.
Specificity of the KLAMP1 domain of lpg1496 for 3′,5′-cAMP is intriguing. Lipopolysaccharides (LPS) found on the cell wall of Gram-negative bacteria contribute to the activation of host inflammatory responses but also serve to promote survival of the bacterium (27). For example, LPS induces arachidonic acid release, which in turn is metabolized to prostaglandins and leukotrienes. Increased release of prostaglandin E2 has been detected after activation of macrophages with LPS (28). Prostaglandin E2 suppresses microbicidal activity of macrophages through GS-coupled receptors, increasing adenylyl cyclase activity and effectively increasing intracellular cAMP levels. cAMP functions as a secondary messenger influencing numerous cellular functions, acting through the downstream effectors, protein kinase A, and Epac-1 and -2. Through cAMP, prostaglandin E2 inhibits the microbicidal production of reactive oxygen intermediates by NADPH oxidase (29). In summary, elevated cAMP levels result in increased bacterial survival in macrophages.
Comparison with other protein structures in complex with 3′,5′-cAMP shows that in many cases adenine recognition elements involve π-stacking with a tyrosine or phenylalanine residue, whereas KLAMP1 simultaneously uses two tyrosines for π-stacking with adenine. To our knowledge there is only one other structure in the Protein Data Bank, where the adenine ring is sandwiched between two aromatic residues (both tyrosines) providing π-stacking interactions (PDB code 1LPC) (30). However, in KLAMP1 one of the tyrosines (Tyr-61) additionally hydrogen-bonds to the oxygen of the ribose ring, increasing its importance in ligand recognition.
We have also crystallized the conserved C-terminal SidE domain of lpg1496 that is found in the N-terminal region of members of the original SidE family. Bioinformatics analyses highlight a potential function for lpg1496, as this domain can be found in combination with the ADP-ribosylating domain, Vip2. In addition, we have crystallized the catalytic SidE domain in complex with ADP in a possible substrate binding site. An overlay of this structure with a structurally similar phosphodiesterase shows that this binding occurs in a shifted catalytic pocket, explaining the inactivity of lpg1496 against cyclic nucleotides.
Taken together, all three domains of lpg1496 are capable of binding nucleotides, pointing toward a connection with nucleotide metabolism (Fig. 6). The importance of nucleotide binding for lpg1496 function can be tested in future cell-based assays using mutations of key residues identified here: Tyr-61 and Tyr-69 for 3′,5′-cAMP/KLAMP1, Tyr-218 and Tyr-226 for ADP/KLAMP2, and His-366 and His-370 for ADP/SidE. The discovery of KLAMP domains, a novel nucleotide binding fold, will have implications for understanding the function of other KLAMP-domain containing proteins.
FIGURE 6.

lpg1496 structure and characterization. Schematic model of the arrangement of the KLAMP and SidE domains in lpg1496 with nucleotides bound. A model of ADP bound to KLAMP2 is shown. A summary of the binding affinities measured by NMR titration is presented for the KLAMP domains.
Author Contributions
K. W. and G. K. designed the experiments. K. W., G. K., and Y. Z. performed the experiments. K. W. and G. K. analyzed the data. K. W., G. K., and K. G. wrote the manuscript.
Acknowledgments
We thank Dr. Miroslaw Cygler (University of Saskatchewan) for the initial construct of lpg1496, Dr. Irena Ekiel for helpful discussions, and the laboratory of Dr. Alexander Yakunin (University of Toronto) for screening potential substrates of lpg1496. Data were acquired at the Macromolecular Diffraction (MacCHESS) facility at the Cornell High Energy Synchrotron Source (CHESS). CHESS is supported by the National Science Foundation (NSF) and NIGMS, National Institutes of Health (NIH)/via NSF Award DMR-0225180, and the MacCHESS resource is supported by Center for Research Resources, NIH Award RR-01646.
This work was funded by Canadian Institutes of Health Research Genomics Grant GSP-48370. The authors declare that they have no conflicts of interest with the contents of this article.
- KLAMP
- kinase-like ATP binding region-containing proteins and S-adenosylmethionine-dependent methyltransferase protein
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- CHESS
- Cornell High-Energy Synchrotron Source
- MR
- molecular replacement
- HSQC
- heteronuclear single quantum correlation spectroscopy
- r.m.s.d.
- root mean square deviation
- PDE
- phosphodiesterase.
References
- 1. Fraser D. W., Tsai T. R., Orenstein W., Parkin W. E., Beecham H. J., Sharrar R. G., Harris J., Mallison G. F., Martin S. M., McDade J. E., Shepard C. C., Brachman P. S. (1977) Legionnaires' disease: description of an epidemic of pneumonia. N. Engl. J. Med. 297, 1189–1197 [DOI] [PubMed] [Google Scholar]
- 2. Vogel J. P., Andrews H. L., Wong S. K., Isberg R. R. (1998) Conjugative transfer by the virulence system of Legionella pneumophila. Science 279, 873–876 [DOI] [PubMed] [Google Scholar]
- 3. Bardill J. P., Miller J. L., Vogel J. P. (2005) IcmS-dependent translocation of SdeA into macrophages by the Legionella pneumophila type IV secretion system. Mol. Microbiol. 56, 90–103 [DOI] [PubMed] [Google Scholar]
- 4. Ninio S., Zuckman-Cholon D. M., Cambronne E. D., Roy C. R. (2005) The Legionella IcmS-IcmW protein complex is important for Dot/Icm-mediated protein translocation. Mol. Microbiol. 55, 912–926 [DOI] [PubMed] [Google Scholar]
- 5. Nagai H., Cambronne E. D., Kagan J. C., Amor J. C., Kahn R. A., Roy C. R. (2005) A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc. Natl. Acad. Sci. U.S.A. 102, 826–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burstein D., Zusman T., Degtyar E., Viner R., Segal G., Pupko T. (2009) Genome-scale identification of Legionella pneumophila effectors using a machine learning approach. PLoS Pathog. 5, e1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Luo Z. Q., Isberg R. R. (2004) Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl. Acad. Sci. U.S.A. 101, 841–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coers J., Kagan J. C., Matthews M., Nagai H., Zuckman D. M., Roy C. R. (2000) Identification of Icm protein complexes that play distinct roles in the biogenesis of an organelle permissive for Legionella pneumophila intracellular growth. Mol. Microbiol. 38, 719–736 [DOI] [PubMed] [Google Scholar]
- 9. Conover G. M., Derré I., Vogel J. P., Isberg R. R. (2003) The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol. Microbiol. 48, 305–321 [DOI] [PubMed] [Google Scholar]
- 10. Chang B., Kura F., Amemura-Maekawa J., Koizumi N., Watanabe H. (2005) Identification of a novel adhesion molecule involved in the virulence of Legionella pneumophila. Infect Immun. 73, 4272–4280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hostetter M. K., Tao N. J., Gale C., Herman D. J., McClellan M., Sharp R. L., Kendrick K. E. (1995) Antigenic and functional conservation of an integrin I-domain in Saccharomyces cerevisiae. Biochem. Mol. Med. 55, 122–130 [DOI] [PubMed] [Google Scholar]
- 12. Jeong K. C., Sexton J. A., Vogel J. P. (2015) Spatiotemporal regulation of a Legionella pneumophila T4SS substrate by the metaeffector SidJ. PLoS Pathog. 11, e1004695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 14. Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A 64, 112–122 [DOI] [PubMed] [Google Scholar]
- 15. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 18. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 [Google Scholar]
- 21. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 22. Goddard T. D., Kneller D. G. (2008) SPARKY 3. University of California, San Francisco [Google Scholar]
- 23. Han S., Craig J. A., Putnam C. D., Carozzi N. B., Tainer J. A. (1999) Evolution and mechanism from structures of an ADP-ribosylating toxin and NAD complex. Nat. Struct. Biol. 6, 932–936 [DOI] [PubMed] [Google Scholar]
- 24. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang H., Yan Z., Geng J., Kunz S., Seebeck T., Ke H. (2007) Crystal structure of the Leishmania major phosphodiesterase LmjPDEB1 and insight into the design of the parasite-selective inhibitors. Mol. Microbiol. 66, 1029–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aravind L., Koonin E. V. (1998) The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 23, 469–472 [DOI] [PubMed] [Google Scholar]
- 27. Morrison D. C., Ryan J. L. (1987) Endotoxins and disease mechanisms. Annu. Rev. Med. 38, 417–432 [DOI] [PubMed] [Google Scholar]
- 28. Rosenstreich D. L., Glode L. M., Mergenhagen S. E. (1977) Action of endotoxin on lymphoid cells. J. Infect Dis. 136, S239–S245 [DOI] [PubMed] [Google Scholar]
- 29. Serezani C. H., Chung J., Ballinger M. N., Moore B. B., Aronoff D. M., Peters-Golden M. (2007) Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. Am. J. Respir. Cell Mol. Biol. 37, 562–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kurinov I. V., Rajamohan F., Uckun F. M. (2004) High resolution x-ray structure and potent anti-HIV activity of recombinant dianthin antiviral protein. Arzneimittelforschung 54, 692–702 [DOI] [PubMed] [Google Scholar]