

Background: Phytaspase is a plant cell death-promoting protease capable of hydrolyzing a range of caspase substrates.
Results: Phytaspase possesses an exclusive Asp specificity of hydrolysis and prefers a cleavage motif that is strikingly hydrophobic.
Conclusion: Substrate specificities of plant and animal death proteases display both similarity and important distinction.
Significance: Knowledge of the phytaspase specificity provides insight toward possible targets of the enzyme.
Keywords: aspartate (aspartic acid), cell death, peptide hormone, proteolysis, proteolytic enzyme, substrate specificity, cholecystokinin, gastrin, phytaspase
Abstract
Plants lack aspartate-specific cell death proteases homologous to animal caspases. Instead, a subtilisin-like serine-dependent plant protease named phytaspase shown to be involved in the accomplishment of programmed death of plant cells is able to hydrolyze a number of peptide-based caspase substrates. Here, we determined the substrate specificity of rice (Oryza sativa) phytaspase by using the positional scanning substrate combinatorial library approach. Phytaspase was shown to display an absolute specificity of hydrolysis after an aspartic acid residue. The preceding amino acid residues, however, significantly influence the efficiency of hydrolysis. Efficient phytaspase substrates demonstrated a remarkable preference for an aromatic amino acid residue in the P3 position. The deduced optimum phytaspase recognition motif has the sequence IWLD and is strikingly hydrophobic. The established pattern was confirmed through synthesis and kinetic analysis of cleavage of a set of optimized peptide substrates. An amino acid motif similar to the phytaspase cleavage site is shared by the human gastrointestinal peptide hormones gastrin and cholecystokinin. In agreement with the established enzyme specificity, phytaspase was shown to hydrolyze gastrin-1 and cholecystokinin at the predicted sites in vitro, thus destroying the active moieties of the hormones.
Introduction
Programmed cell death (PCD)3 in plants, as well as in animals, serves to eliminate excessive and potentially dangerous cells. PCD is instrumental in the course of plant development and in response to environmental abiotic stresses and pathogen invasion (1–8). Proteolytic enzymes play essential roles in the implementation of PCD in plants (9). Although plants lack direct homologues of animal caspases, several types of plant proteases (metacaspases (10–15), vacuolar processing enzyme (16–18), phytaspase (19–21)) were reported to be essential for accomplishment of various forms of PCD in plants.
Among these proteases, only phytaspase, which is structurally very different from caspases and belongs to a family of serine-dependent subtilisin-like proteases, appears to display stringent “caspase-like” aspartate specificity of hydrolysis (20, 22). In the single known phytaspase target, the VirD2 protein of a plant pathogenic bacterium Agrobacterium tumefaciens, phytaspases of various plant organisms introduce a single break after the Asp residue within the TATD motif (19, 23), thus behaving as processing rather than digestive enzymes. Phytaspase-mediated hydrolysis of peptide-based caspase substrates showed that amino acid motifs preceding the Asp residue can influence efficiency of hydrolysis markedly (20, 24), thus providing a rationale for the high selectivity of the enzyme.
Phytaspase is involved in PCD triggered by abiotic insults, such as oxidative and salt stresses, as well as by biotic stresses. In the latter case, infection with tobacco mosaic virus of certain tobacco cultivars was used to induce a specific form of plant PCD known as the hypersensitive response, which leads to the death of infected cells and limits virus spread. All of these types of cell death were shown to be proportional to the level of phytaspase activity in plants (19, 20).
Despite the similarities with caspases in cleavage specificity and a role in PCD, regulation of the phytaspase activity is different. Being synthesized as preproenzyme, phytaspase is autocatalytically and constitutively processed to generate a mature proteolytically active enzyme, which is secreted out of healthy plant cells. Upon induction of PCD, phytaspase is re-imported from the intercellular space into the dying cells to get access to intracellular proteins (20).
In this study we defined the substrate specificity of rice (Oryza sativa) phytaspase using the positional scanning substrate combinatorial library (PS-SCL) approach (25). The preferred phytaspase cleavage sites identified turned out to be notably hydrophobic. Kinetic analysis of the cleavage of optimized peptide substrates revealed their efficient hydrolysis by phytaspase. The acquired knowledge of the amino acid sequence of the preferred phytaspase cleavage site allowed us to identify phytaspase substrates among human gastrointestinal peptide hormones.
Experimental Procedures
Purification of Rice Phytaspase
Fresh rice O. sativa cv. “Leader” leaves (100 g) were ground in liquid nitrogen and extracted twice with 250- and 100-ml portions of B1 buffer (20 mm MES, 2 mm DTT, 0.1% Tween 20, and 5% glycerol), pH 5.7, containing 50 mm NaCl and 1 mm PMSF. The debris was eliminated by 20-min centrifugations at 14,000 × g at +4 °C, and the supernatant exhibiting pH 6.7 was acidified at +4 °C to pH 5.3 by adding 3 n acetic acid dropwise. After centrifugation, the supernatant was applied onto a DE53 column (Whatman, 100 ml) equilibrated with B1 buffer, pH 5.7. Flow-through from this column was collected and fractionated by ammonium sulfate precipitation. The protein fraction that precipitated within the 30–60% interval of (NH4)2SO4 saturation was dissolved in B1 buffer, pH 5.7, and dialyzed against the same buffer.
The sample was then subjected to cation exchange chromatography on a HiTrap CM FF1 column (CM Sepharose Fast Flow, 1 ml) equilibrated with buffer B1, pH 5.7, using AKTA Purifier10 (GE Healthcare). Elution was performed with a 0–1.0 m NaCl gradient in the same buffer. Peak fractions containing phytaspase activity (0.25–0.45 m NaCl) were combined and brought to 0.4 m NaCl. The obtained sample was loaded onto a HiTrap Blue HP column (5 ml) equilibrated with B1 buffer, pH 5.7. Elution was performed with a 0–2.5 m NaCl gradient in the same buffer, and peak fractions containing phytaspase activity (1.3–2.2 m NaCl) were combined and concentrated using a YM-30 Centricon (Amicon).
Phytaspase sample was then incubated with 0.4 mm biotin-TATD-CHO phytaspase inhibitor (Bachem) in B1 buffer, pH 5.5, at 27 °C for 30 min, an excess of the unreacted inhibitor was removed using a Sephadex G10 (GE Healthcare) mini spin column, and the reaction mixture was applied onto a SoftLink Soft Release Avidin resin (Promega) at 0 °C. Biotinylated proteins eluted with 5 mm biotin were analyzed by SDS-PAGE and visualized by silver staining. Purified enzyme was dialyzed against B1 buffer, pH 6.5, and stored at −80 °C. Enzyme concentration was determined using BSA as a standard.
Phytaspase Activity Assays
Phytaspase activity was determined by assessing fragmentation of the GFP-VirD2Ct protein and by hydrolysis of synthetic fluorogenic peptide substrates. In the former case, His6-tagged GFP-VirD2Ct protein containing, in successive order, a His tag, a GFP moiety, and an 86-amino acid-long C-terminal region of the A. tumefaciens VirD2 protein, was isolated from Escherichia coli cells overproducing the recombinant protein by means of nickel nitrilotriacetic acid-agarose (Qiagen) chromatography as described (19). Aliquots of the enzyme were incubated with the GFP-VirD2Ct protein (3 μg) in B1 buffer, pH 5.7, at 27 °C for 1.5 h, reaction mixtures were fractionated by 15% SDS-PAGE, and substrate fragmentation was visualized by Coomassie Blue R-250 staining.
Individual fluorogenic peptide substrates (from American Peptide Co., Anaspec, Bachem, Calbiochem, California Peptide Research, Enzo Life Sciences, MP Biomedicals, and those synthesized in this work; see below) were tested at the concentration of 20 μm. Kinetic measurements of fluorescence intensities were performed in B1 buffer, pH 6.5, containing 0.5 m NaCl at 27 °C or 30 °C in triplicate using FLUOstar OPTIMA reader (BMG Labtech) equipped with 405-nm excitation and 520-nm emission filters (for peptides with an 7-amino-4-trifluoromethylcoumarin (AFC) fluorescent tag or with 355-nm excitation and 460-nm emission filters for 7-amino-4-carbamoylmethylcoumarin (ACC) derivatives). In the case of ACC derivatives, Tween 20 was omitted from B1 buffer to closely mimic reaction conditions used for library screenings (see below).
To determine the pH and salt dependence for phytaspase hydrolysis, the enzyme samples were diluted 10-fold with buffers of pH 3.0–8.0 with or without 0.5 m NaCl and incubated with fluorogenic peptide substrate at 30 °C for up to 5 h or with GFP-VirD2Ct substrate protein (in buffers lacking NaCl only) for 2.5 h. Sodium citrate, sodium acetate, MES, and HEPES were used at 20 mm to obtain buffers covering pH intervals 3.0–3.5, 4.0–5.0, 5.5–6.5, and 7.0–8.0, respectively. All buffers contained 2 mm DTT, 0.1% Tween 20, and 5% glycerol.
Synthesis of Combinatorial Peptide Library
The fluorogenic substrate library (P2, P3, and P4 positions) was synthesized similarly to the previously published protocols (26, 27). In the first step Fmoc-ACC-OH fluorophore was attached to 6 g of Rink Amide resin according to the procedure described elsewhere (28). After the Fmoc group deprotection (20% piperidine in N,N-dimethylformamide (DMF)), Fmoc-Asp(tBu)-OH (2.5 eq) was attached to the NH2-ACC-resin using 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5]pyridinium 3-oxide hexafluorophosphate (HATU, 2.5 eq) as a coupling agent and 2,4,6-trimethylpyridine (collidine, 2.5 eq) as a base. The reaction was carried out overnight. Next, the coupling of Fmoc-Asp(tBu)-OH was repeated with 1.5 eq of each reagent(overnight). The Fmoc-Asp(tBu)-OH substitution level was determined using HPLC analysis. A small portion of resin was subjected to TFA/triisopropylsilane (TIPS)/H2O cleavage, and the crude product was analyzed on HPLC; the only peak detected was from Fmoc-Asp(tBu)-ACC. After the Fmoc group deprotection the resin was washed with DMF (3×), dichloromethane (DCM, 3×), and MeOH (3×) and dried over P2O5 overnight. Next, the resin was divided into 60 portions (20 portions for each position). For the synthesis of the P2 library, 20 portions of resin were placed into a MultiChem 48-well synthesis apparatus (FlexChem from SciGene, Sunnyvale, CA) and swollen in DCM for 15 min and then washed with DMF (3×). For coupling fixed P2 position, each of 20 Fmoc-protected amino acids (19 natural and 1 unnatural, susceptible to oxidation cysteine was replaced with norleucine) (3 eq) was preactivated with N,N′-diisopropylcarbodiimide (3 eq) and N-hydroxybenzotriazole (3 eq) in DMF for 3 min and poured into the resin. The P2 coupling reaction was carried out for 3 h. After this time, the ninhydrin test confirmed the completion of coupling. The Fmoc group was deprotected as described previously. For coupling P3 and P4 positions of the P2 library, an isokinetic mixture of 19 Fmoc-protected amino acids was prepared (18 natural and 1 unnatural cysteine was omitted, and norleucine was used instead of methionine). Then, 5 eq of the isokinetic mixture was preincubated with 5 eq of N-hydroxybenzotriazole and 5 eq of N,N′-diisopropylcarbodiimide in DMF for 3 min and poured onto the resin. The coupling reaction was carried out for 3 h (monitoring by ninhydrin test). Finally, after P4 Fmoc group deprotection the N-terminal amine group was acetylated using a mixture of acetic acid (5 eq), HBTU, 5 eq, and N,N-diisopropylethylamine (5 eq) in DMF. The acetylation reaction was performed for 30 min to obtain Ac-mix-mix-P2-Asp(tBu)-ACC-resin. Next, the resin was washed with DMF (3×), DCM (3×), MeOH (3×), and dried over P2O5 overnight. The P3 and P4 sublibraries were synthesized in a similar manner. Finally, each of the 20-member sublibraries was cleaved from the resin with a mixture of TFA/TIPS/H2O (v/v/v 95:2.5:2.5, 2 ml per well) for 2 h. The solution from each well was collected separately, and diethyl ether was used to precipitate compounds. After centrifugation and decantation, the white crude product was dissolved in a mixture of water and acetonitrile (1:2, v/v) and lyophilized. The final product was dissolved in peptide grade DMSO to the final concentration of 20 mm and stored at −80 °C until use.
Screening of Combinatorial Peptide Libraries
Enzyme activity toward libraries was measured as a relative fluorescence units per second using a plate reader (Molecular Devices) according to the previously described method (28, 29). Kinetic analysis was performed at 30 °C in 20 mm MES, 2 mm DTT, 5% glycerol, 500 mm NaCl buffer. The final substrate library concentration in each well was 100 μm, and final enzyme concentration was 12.6 nm. The substrate library (stock solution 10 mm in DMSO) was added to a 96-well Corning Plate (opaque). The enzyme was preincubated in buffer for 30 min at 30 °C followed by addition to the plate wells. Relative fluorescence values were measured at 355/460 nm (cutoff 455 nm), and the obtained results were transformed to percentages by comparison of the highest value from each library separately (set as 100%) to relative fluorescence units per second for each library component. The result was presented as a bar graph made using Microsoft Excel.
Synthesis of Optimized Peptide Substrates
Substrates were synthesized according to the previously described method (28). 1 eq of Fmoc-Rink Amide resin (0.7 mmol/g, 200–300 mesh) was swollen for 30 min in DCM in a glass peptide synthesis vessel. Resin was washed 3 times with DMF (3 × 5 ml) followed by Fmoc-protecting group removal with 20% piperidine/DMF solution (3 times for 5, 5, and 25 min). The resin was washed 3 times with DMF and to confirm amine group deprotection, a ninhydrin test was conducted. In the next step 2.5 eq of Fmoc-ACC-OH was activated for 2 min with 2.5 eq of N-hydroxybenzotriazole and 2.5 eq of N,N′-diisopropylcarbodiimide in DMF in an Eppendorf tube followed by pouring onto the resin. The mixture was agitated for 24 h at room temperature, and then the supernatant was removed, the resin was washed 3 times with DMF, and Fmoc-ACC-OH coupling was repeated using 1.5 eq of reagents. After 24 h, the resin was washed 3 times with DMF (3 × 5 ml), and a ninhydrin test was carried out to confirm coupling of Fmoc-ACC-OH. Subsequently, the Fmoc-protecting group was removed using the previously described method, and 2.5 eq of Fmoc-Asp-OH was preactivated with 2.5 eq of HATU and 2.5 eq of 2,4,6-trimethylpyridine (collidine) in DMF for 2 min in an Eppendorf tube followed by pouring onto the resin. After gentle agitation for 24 h at room temperature,the resin was washed with DMF (3 × 5 ml), and coupling was repeated with 1.5 eq of reagents. Next, the solution was removed, and the resin was washed with DMF (3 × 5 ml). The Fmoc protecting group was removed as previously described, and 2.5 eq of Fmoc-P2-OH was preactivated in the solution of 2.5 eq of N-hydroxybenzotriazole and 2.5 eq of N,N′-diisopropylcarbodiimide in DMF and poured onto the resin. After 3 h of agitation, the resin was washed with DMF (3 × 5 ml), and after Fmoc deprotection, Fmoc-P3-OH and Fmoc-P4-OH amino acids were attached to the resin in the same way. The P4 amino acid was then acetylated with 5 eq of AcOH, 5 eq of HBTU, and 5 eq of N,N-diisopropylethylamine in DMF for 45 min at room temperature. The resin was washed with DMF (3 × 5 ml), DCM (2 × 5 ml), and with MeOH (2 × 5 ml) and dried over P2O5 overnight. Peptides were cleaved from the resin using a standard procedure. A solution of TFA:TIPS:H2O (95:2.5:2.5, v/v/v) was added to the resin, and the mixture was shaken once every 10 min for 2 h. The filtrate was then collected in conical Falcon tubes (15 ml), resin was washed with solution of TFA:TIPS:H2O (95:2.5:2.5, v/v/v), and filtrates were combined. Substrates were precipitated with cold Et2O in −20 °C, centrifuged, washed once with cold Et2O, and dried at room temperature. The crude products were then dissolved in DMSO, purified on HPLC (Waters, column: Spherisorb®, 5-μm particle size, length 25-cm, inner diameter 4.6 mm) in an H2O:acetonitrile gradient and lyophilized. Purified substrates were dissolved in DMSO and stored at −80 °C until use.
Kinetic Analysis of Phytaspase Activity
For phytaspase-mediated cleavage of fluorogenic peptide substrates carried out at 1 μm (ΑCC derivatives) and 5 μm (AFC derivatives) concentrations, the level of hydrolysis was determined fluorimetrically using either varying enzyme concentrations (10–100 pm) and a fixed time or using a constant enzyme concentration of 250 pm for measuring the time course of hydrolysis leading to complete substrate digestion. For fragmentation of GFP-VirD2Ct (0.2–0.4 μm), aliquots of reaction mixtures withdrawn at 5-min intervals were subjected to gel electrophoresis. Coomassie Blue-stained gels were scanned, the images were converted into black-and-white format, and the degree of protein fragmentation was determined using Image Quant software. From the obtained data, kcat/Km values were determined assuming first order reaction kinetics ([S] ≪ Km).
Kmvalues were determined by measuring the initial rates of substrate cleavage within the linear part of the plot at various substrate concentrations (15–200 μm for AFC derivatives, 1–15 μm for ACC derivatives, and 0.5–5.0 μm for GFP-VirD2Ct). Lineweaver-Burk plots drawn using the data obtained were used to derive values for Km.
Phytaspase-mediated Fragmentation of Gastrin-1 and CCK-8
Gastrin-1 (GenScript) and sulfated cholecystokinin (CCK-8, American Peptide Co.) peptides at the concentration of 40 μm were treated with phytaspase (1.9 nm) in 16 mm MES buffer, pH 5.5, containing 0.5 m NaCl at 37 °C for 18 h. At several time points, 1-μl aliquots of reaction mixtures were withdrawn and diluted 10-fold with 0.5% trifluoroacetic acid, and 0.5-μl aliquots were mixed with an equal volume of 2,5-dihydroxybenzoic acid (Aldrich, 20 mg/ml, in 20% acetonitrile and 0.5% trifluoroacetic acid) and dried on the matrix-assisted laser desorption/ionization (MALDI) target plate. Peptide masses were measured using an UltrafleXtreme MALDI-TOF/TOF mass spectrometer (Bruker Daltonics, Germany) equipped with an neodymium-doped (Nd) laser in positive and negative (for sulfated CCK-8 peptides) ion modes with a reflector. Spectra were obtained in the 600–4500 m/z range. The accuracy of monoisotopic mass measurements was within 0.1 Da. Fragmentation ion spectra were obtained in a tandem mode with the accuracy within 1 Da.
Results
Defining the Optimum Phytaspase Cleavage Site
To define the cleavage specificity of rice phytaspase, native enzyme was purified to homogeneity by a series of chromatography fractionations of leaf extracts, including an affinity chromatography step (see “Experimental Procedures”). The resultant phytaspase displayed a single protein band of ∼80 kDa upon gel electrophoresis (Fig. 1A). The proteolytic activity of the isolated enzyme was manifested by specific fragmentation of the protein substrate GFP-VirD2Ct at the TATD motif (Fig. 1B) as well as by its ability to hydrolyze fluorogenic peptide substrates Ac-VEID-AFC, Ac-VNLD-AFC, and Ac-STATD-AFC but not Ac-DEVD-AFC (Fig. 1C).
FIGURE 1.

Properties of native phytaspase from O. sativa leaves (OsPhyt) used to determine substrate specificity. A, purified rice phytaspase displays a single band of ∼80 kDa upon SDS-gel electrophoresis. Positions of Mr protein markers (M) are indicated on the left. Silver-stained gel is shown. B, phytaspase hydrolyzes wild type GFP-VirD2Ct protein in vitro after the Asp residue in the TATD motif. Mutating this residue (D39A mut) abrogates the cleavage. The gel was stained with Coomassie Blue. C, phytaspase-mediated hydrolysis of fluorogenic peptide substrates. Relative rates of peptide-AFC (20 μm) hydrolysis are expressed as a change of relative fluorescence units per hour (ΔRFU/h, mean values from three experiments).
To determine the optimum phytaspase hydrolysis motif, the PS-SCL approach was used (25). Tetrapeptides in P2-P4 libraries contained an aspartic acid residue in the P1 position linked to a fluorogenic ACC group. In three subsets of peptide libraries, a residue in the P2, P3, or P4 positions was fixed (susceptible to oxidation cysteine was replaced with norleucine), whereas the amino acid residues in the other positions were represented by equimolar mixtures of 19 amino acids (cysteine was omitted, and norleucine was used instead of methionine) (Fig. 2). The total number of peptide variants represented by the libraries was 361 (19 × 19) per sublibrary in P2-P4 positions and 20 individual sequences in the P1 position.
FIGURE 2.

Phytaspase substrate specificity based on peptide library screenings using P4-P1 libraries. Structures of the peptide libraries containing the ACC reporter group are depicted schematically on the left. Right, results of proteolytic activity measurements. The x axes represent the amino acids depicted using the three-letter code. The y axes represent relative enzymatic activity. The highest activity in each assay was set as 100%; all other activities are shown relative to this. All assays were repeated at least three times.
Peptides from the combinatorial libraries were treated with phytaspase, and the rate of hydrolysis was monitored fluorimetrically. The results of these experiments are presented in Fig. 2 (P2-P4 libraries). It was shown that amino acids in positions P2-P4 markedly affect the rate of phytaspase-mediated hydrolysis. In position P2, the best residue is Leu followed by norleucine and His. In position P3, the preferred residue is Trp followed by Tyr and Phe. A preference for an aromatic amino acid residue in this position is remarkable. In the P4 position of the substrate, branched and hydrophobic Ile and Val are by far better than other residues.
Interestingly, acidic amino acid residues (Asp, Glu) are poorly tolerated in all the positions except for Asp at P1. In accord with this observation, a VNLD-based substrate was hydrolyzed more efficiently by the enzyme than the VEID-based substrate (Fig. 1C). Moreover, Pro residues in P2 and P3 positions strongly attenuate the rate of hydrolysis. Overall, the optimum phytaspase cleavage motif conforms to the sequence Ile-Trp-Leu-Asp, which is remarkably hydrophobic.
We then assessed whether phytaspase is able to cleave the peptide bond after any residue other than Asp. Our previous data obtained with a limited set of commercially available peptide substrates as well as with protein substrates made such a possibility unlikely (19, 20, 24) but could not exclude it completely. To obtain compelling evidence, a defined tetrapeptide library Ac-Leu-Arg-Ser-Aaa-ACC (where Aaa is one of the 20 amino acids) was used. The kinetic analysis demonstrated that none of the substrates, except Ac-LRSD-ACC, was hydrolyzed by the phytaspase (Fig. 2, P1 library). We thus concluded that phytaspase possesses an absolute specificity for hydrolysis after the Asp residue.
Kinetic Parameters of Phytaspase-mediated Hydrolysis
To verify the conclusions derived from the analysis of combinatorial peptide libraries, several fluorogenic tetrapeptides matching the optimized phytaspase cleavage motif were synthesized: Ac-IWLD-ACC, Ac-IYLD-ACC, Ac-IFHD-ACC, Ac-IYHD-ACC, and Ac-VYnLD-ACC (nL is norleucine). The kinetic analysis revealed that these newly designed substrates are efficiently hydrolyzed by the enzyme with kcat/Km values in the range of 105 m−1 s−1 (Table 1 and Fig. 3). Because the corresponding value for one of the best peptide substrates found so far (Ac-VEID-AFC; see Ref. 20 and Fig. 1C) was 2.7 × 104 m−1s−1, a significant improvement of potency (up to 8-fold), and of a repertoire of phytaspase peptide substrates was achieved through the use of combinatorial peptide libraries. The improvement appears to be largely due to tighter binding characteristics (lower Km values) of the novel substrates. With the protein substrate GFP-VirD2Ct, which encompasses a region of the A. tumefaciens VirD2 protein cleaved by phytaspase, kcat/Km ∼ 5.48 × 106 m−1 s−1 was obtained (Table 1), testifying to the efficient fragmentation of this protein substrate by phytaspase.
TABLE 1.
kcat/Km and Km values for phytaspase-mediated hydrolysis of peptide and protein substrates
| Substrate | kcat/Km | Km |
|---|---|---|
| m−1s−1 | μm | |
| Ac-VEID-AFC | 27,141 ± 169 | 527 ± 101 |
| Ac-VNLD-AFC | 58,121 ± 100 | 194 ± 44 |
| Ac-IYLD-ACC | 224,556 ± 16,958 | 12.74 ± 6.1 |
| Ac-IWLD-ACC | 202,976 ± 9,463 | 8.3 ± 2.2 |
| Ac-IFHD-ACC | 187,510 ± 1,078 | 15.37 ± 2.65 |
| Ac-IYHD-ACC | 154,141 ± 10,899 | 15.24 ± 7.15 |
| Ac-VYnLD-ACC | 132,745 ± 1,351 | 11.43 ± 1.1 |
| GFP-VirD2Ct | 5,480,000 ± 2,560,000 | 3.02 ± 1.36 |
FIGURE 3.
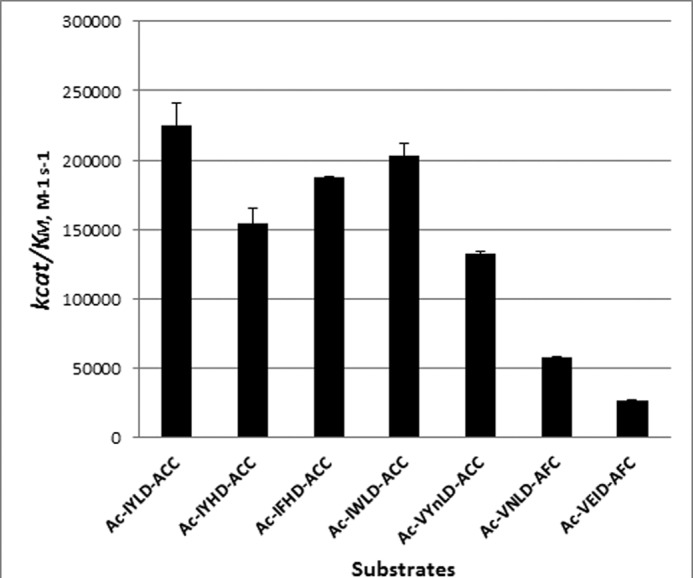
Comparison of the kcat/Km values for phytaspase-mediated hydrolysis of the newly synthesized phytaspase substrates with those for hydrolysis of caspase substrates. Experimental details are listed under “Experimental Procedures.”
Heterologous Targets of Phytaspase
We then performed a database search for proteins containing amino acid motifs resembling the best phytaspase cleavage sites identified in this study (Trp-X-Asp, in particular) and, hence, representing potential phytaspase targets. The search was not limited to plant proteins. Among numerous candidate proteins obtained through this search, two human peptide hormones, gastrin and cholecystokinin (CCK), attracted our attention. These peptide hormones share a Gly-Trp-Met-Asp-Phe-NH2 motif (amidated Phe) at their C termini (Fig. 4A), whose integrity is known to be essential for manifestation of biological activities of these peptides (30–33).
FIGURE 4.

Sites of phytaspase hydrolysis in human gastrointestinal peptide hormones, CCK and gastrin. A, amino acid sequences of sulfated octapeptide CCK-8 and of the C-terminal region of gastrins are shown. FNH2 stands for the amidated C-terminal phenylalanine residue. The active moiety shared by both hormones is underlined. An arrow points to the predicted phytaspase cleavage site. B, time course (0–18 h) of the phytaspase-mediated hydrolysis of gastrin-1 (a 17-amino acid-long peptide) assessed by mass spectrometry analysis. A peak with m/z 2119.9 corresponds to the non-digested peptide (sodium salt). A peak with m/z 1973.8 corresponds to the reaction product that has lost the FNH2 residue. The y axes represent signal intensities in arbitrary units (a.u.). C, time course of the phytaspase-mediated hydrolysis of CCK-8. A peak with m/z 1141.5 corresponds to non-digested CCK-8. A peak with m/z 995.4 corresponds to the reaction product, i.e. truncated CCK-8 lacking the FNH2 residue, as was further confirmed by the MS/MS sequencing. Peaks with m/z 1061.5 and 915.5 belong to non-sulfated forms of CCK-8 and its truncated derivative, respectively.
Although the sequence found in these hormones does not perfectly match the optimum phytaspase cleavage site (IWLD), we have focused on these peptides for several reasons. Gastrin and CCK precursor proteins are synthesized in the alimentary tract (stomach and gut, respectively), and thus it is possible for them to encounter phytaspase ingested with plant food. In line with this argument, mechanical damage of plant tissues (such as chewing) is known to result in the release of active phytaspase (34). Furthermore, cleavage with phytaspase is predicted to detach the amidated Phe from a conserved and functionally important region of these hormones. Such a truncation is known to inactivate these hormones and even convert them into antagonists (31, 35).
To test whether phytaspase could indeed hydrolyze human peptide hormones, human gastrin-1, a synthetic non-sulfated 17-amino acid long peptide, was treated in vitro with phytaspase, and the reaction products were analyzed by mass spectrometry. The predicted processing of the initial peptide (m/z 2119.9) after the Asp residue did occur, as manifested by the formation of a truncated peptide lacking the C-terminal amidated Phe residue (a peak with m/z 1973.8) (Fig. 4B). Hormone fragmentation was apparently complete upon prolonged incubation. Because complete (>95%) hydrolysis of Ac-VEID-AFC by phytaspase was achieved under the identical reaction conditions after 18 h incubation as well (data not shown), we estimated that gastrin-1 is cleaved by phytaspase with an efficiency comparable with that of Ac-VEID-AFC hydrolysis.
Likewise, when a sulfated octapeptide CCK-8 was treated in vitro with phytaspase, it was processed in a similar manner, as evidenced by the decrease in the amount of the initial peptide (m/z 1141.5) and by formation of the product with m/z 995.4 (Fig. 4C). MS/MS sequencing of this peptide product has confirmed that the amidated Phe was removed from the hormone by phytaspase. Moreover, phytaspase appeared to cleave both the sulfated and non-sulfated forms (present in minor amounts in the peptide preparation) of CCK-8, as manifested by the appearance of sulfated (m/z 995.4) and non-sulfated (m/z 915.4) products of hydrolysis.
We concluded from these studies that the PS-SCL approach produced reliable data on the substrate specificity of phytaspase. Furthermore, due to the peculiarity of the optimum phytaspase cleavage sequence, valid predictions on the potential phytaspase targets can be made.
Phytaspase-mediated Hydrolysis at Low pH and Low Salt Conditions
We observed previously that efficient phytaspase hydrolysis of peptide substrates required specific reaction conditions, such as mildly acidic pH (5.0–6.5) and a high NaCl concentration (0.5 m) (see Ref. 20 and Fig. 5A). Therefore, these conditions were used for peptide hydrolysis throughout the current study. However the “assay optimal” conditions are different from those in gastrointestinal tract, where hydrolysis of gastrin and CCK is proposed to occur. We, therefore, aimed to determine whether phytaspase can accomplish peptide hydrolysis under acidic conditions and in the absence of a high NaCl concentration, which mimics the gastrointestinal environment.
FIGURE 5.

Phytaspase is operative at low pH and low salt conditions. A, pH dependence of Ac-VEID-AFC (20 μm) hydrolysis by phytaspase in the presence or absence of 0.5 m NaCl. Mean values from three experiments are given. B, pH dependence of GFP-VirD2Ct hydrolysis by phytaspase. Note that hydrolysis was performed in the absence of NaCl, as salt is known to inhibit cleavage of the protein substrate (34). S, untreated substrate protein. Positions of Mr protein markers (M) are indicated on the right. The gel was stained with Coomassie Blue. ΔRFU/h, relative fluorescence units per hour.
To monitor the enzyme activity, Ac-VEID-AFC was used as a fluorogenic substrate. Reactions were performed in buffers covering the pH 3.0–8.0 range and with or without 0.5 m NaCl. Omission of NaCl from reaction buffers was found to drastically diminish phytaspase activity toward the peptide substrate at neutral, slightly acidic, and slightly basic pH (Fig. 5A). On the other hand, phytaspase activity in the absence of NaCl reached its peak at a lower pH (4.5–5.5). As a consequence, at pH 4.5 phytaspase activity in the absence of NaCl was only 2.5 times lower than in the presence of 0.5 m salt (Fig. 5A). Further acidification of the reaction medium gradually decreased enzymatic activity, although phytaspase-mediated hydrolysis could still be detected down to pH 3.5. Similar results regarding preservation of phytaspase activity at pH 3.5 in the absence of NaCl were obtained with a GFP-VirD2Ct protein substrate (Fig. 5B). The detailed analysis of the phytaspase activity dependence on the pH and NaCl concentration allowed us to assume that this enzyme, when ingested with plant food, could hydrolyze gastrin and CCK hormones.
Discussion
Caspases (cysteine-dependent death and inflammation-related proteases of animal origin) have for many years constituted the principal class of proteolytic enzymes with an absolute preference for cleavage of peptide bonds after an Asp residue. The only additional example was granzyme B, a serine-dependent animal protease of the trypsin type (36, 37). It, therefore, comes as a surprise that phytaspases belonging to the broad family of plant serine-dependent subtilisin-like proteases display a similar substrate specificity of hydrolysis. Whether by chance or otherwise, phytaspases are also death proteases implicated in dismantling of plant cells in response to various biotic (pathogen-induced) and abiotic (environmental) stresses (20, 22).
Here we examined the substrate specificity of rice phytaspase, a typical representative of phytaspases (20), by using a PS-SCL approach. We have extended our previous observations that amino acid residues in positions P2-P4 of a substrate strongly influence the efficiency of phytaspase-mediated hydrolysis. Similarly to caspases, recognition by phytaspase of a stretch of residues, rather than simply of an Asp residue in the P1 position, provides the basis for the exceptional selectivity of these proteases.
We have demonstrated that phytaspases efficiently hydrolyze many (but not all) synthetic caspase substrates. However, the optimal phytaspase recognition motif defined in this study (IWLD) differs significantly from classical caspase recognition sequences. The “ideal” phytaspase cleavage site is notably hydrophobic, with an aromatic amino acid residue preferred at the P3 position, whereas caspases usually prefer the negatively charged Glu residue there, which binds to the Arg-containing S3 pocket (38, 39). In general, phytaspase appears to poorly tolerate negatively charged residues in the P2-P4 region. This evidently accounts for an earlier observation that the DEVD-based caspase substrate represents a rare exception in being completely resistant to hydrolysis by phytaspases of various origins. Also of note, the Pro residue in positions P2 and P3 appears to form an unfavorable peptide scaffold for phytaspase hydrolysis. We found that the kinetic analysis of individual tetrapeptide substrates is in line with phytaspase substrate specificity obtained through PS-SCL profiling. The slight differences between individual substrates and combinatorial mixtures can be explained by phytaspase subsites cooperativity.
One could predict that the substrate binding pocket of phytaspase should be rather hydrophobic (S2-S4) but also contain a basic residue(s) to orient the P1 Asp in the correct position. Although a three-dimensional structure of phytaspase is not available, in silico modeling has suggested that a His residue in the substrate binding pocket of phytaspase is likely to be involved in the interaction with the side chain of the Asp residue in the P1 position of the substrate (40).
The sequence of the preferred phytaspase cleavage motif revealed here using peptide substrates is important for understanding enzyme-substrate interactions. Yet it should be critically considered when predicting potential phytaspase cleavage sites within proteins, as it may not precisely mimic the diversity of phytaspase cleavage sites in natural proteins. An instructive example is provided by the VirD2 protein of A. tumefaciens, a plant pathogen, which is fragmented by phytaspases (and human caspase-3) at the TATD site (19). The TATD motif appears to have little in common with the optimum phytaspase recognition motif, IWLD, and consistently, at peptide level, Ac-STATD-AFC is poorly hydrolyzed by phytaspase (Fig. 1C). Nevertheless at the protein level, hydrolysis of the VirD2 protein proceeds extremely efficiently, >24-fold faster than the hydrolysis of the best tetrapeptide substrate (Table 1). This discrepancy strongly suggests that additional interactions between the enzyme and the substrate protein, or perhaps some peculiarities within the VirD2 structure, could facilitate hydrolysis greatly. From a strategic point, it will be of importance to identify endogenous protein substrates of phytaspase and to determine the cleavage site preferences in the full-length natural proteins.
On the other hand, current knowledge of the preferred phytaspase recognition motif has enabled us to predict and prove that the human peptide hormones gastrin and CCK are phytaspase targets, at least in vitro. Phytaspase-mediated hydrolysis disrupts the active moiety of these hormones and should inactivate them and even convert them into antagonists (31, 35). The prediction of heterologous (human) proteins as phytaspase targets seems to be counterintuitive at a first glance, but perhaps it is not. Phytaspase, in sharp contrast to caspases, is constitutively and autocatalytically processed in healthy plant tissues, giving rise to an active enzyme that is secreted and stored in the intercellular fluid (apoplast) (20). Consistent with this mode of phytaspase behavior, mechanical damage of plant tissues releases active phytaspase (34). In turn, precursor proteins of gastrin and CKK are synthesized in the cells of the animal gastrointestinal tract (although, in the case of CCK, not only there) and secreted to stimulate digestive functions, such as gastric acid secretion and gallbladder contraction (for review see Refs. 32 and 33). In addition, gastrin stimulates cell proliferation and tumor cell growth (41), whereas CCK is also a neuropeptide causing hunger suppression and neuropsychiatric disorders, such as anxiety and panic attacks (42). We hypothesize that ingestion of plant food would provide phytaspases with access to gastrin and CCK (or to their precursors) in the alimentary tract (43), resulting in the hormone cleavage and inactivation. Inactivation of gastrin and CCK may be beneficial in many pathological conditions to prevent excessive gastric acid secretion, tumor cell growth, and behavioral disorders and is indeed commonly attempted through pharmaceutical interventions targeting receptors of these hormones (44). An alternative possibility, to modulate levels of active hormones by ingestion of plant food with high or low phytaspase content, may therefore deserve attention.
Author Contributions
A. B. V. and M. D. designed the study, analyzed data, and wrote the paper. R. A. G. isolated the enzyme and characterized its activity. M. P. designed, synthesized, and analyzed the peptide libraries. P. K. and A. S. carried out library screening, synthesis, and validation of synthetic substrates. N. V. C. identified human peptide hormones as phytaspase targets. M. V. S. performed mass spectrometry characterization of phytaspase cleavage products. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to Dr. Guy S. Salvesen for promoting collaboration between the authors' laboratories and to Dr. Christopher U. T. Hellen for critical reading of the manuscript.
This work was supported by Russian Foundation for Basic Research Grants 14-04-00232 (to N. V. C.) and 14-04-00256 and 14-04-91330 (to A. B. V.) and National Science Centre Grant 2011/03/B/ST5/01048 in Poland (to M. D.) and the Foundation for Polish Science. The work was also supported by a statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry at Wroclaw University of Technology. The authors declare that they have no conflicts of interest with the contents of this article.
- PCD
- programmed cell death
- ACC
- 7-amino-4-carbamoylmethylcoumarin
- AFC
- 7-amino-4-trifluoromethylcoumarin
- CCK
- cholecystokinin
- CCK-8
- cholecystokinin octapeptide
- DCM
- dichloromethane
- DMF
- N,N-dimethylformamide
- Fmoc
- fluorenylmethyloxycarbonyl protecting group
- HATU
- 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5]pyridinium 3-oxide hexafluorophosphate
- HBTU
- 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- PS-SCL
- Positional Scanning Substrate Combinatorial Library
- TIPS
- triisopropylsilane
- VirD2Ct
- C-terminal region of the VirD2 protein.
References
- 1. Danon A., Delorme V., Mailhac N., Gallois P. (2000) Plant programmed cell death: a common way to die. Plant Physiol. Biochem. 38, 647–655 [Google Scholar]
- 2. Jones A. M. (2001) Programmed cell death in development and defense. Plant Physiol. 125, 94–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoeberichts F. A., Woltering E. J. (2003) Multiple mediators of plant programmed cell death: interplay of conserved cell death mechanisms and plant-specific regulators. Bioessays 25, 47–57 [DOI] [PubMed] [Google Scholar]
- 4. Williams B., Dickman M. (2008) Plant programmed cell death: can't live with it; can't live without it. Mol. Plant Pathol. 9, 531–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reape T. J., Molony E. M., McCabe P. F. (2008) Programmed cell death in plants: distinguishing between different modes. J. Exp. Bot. 59, 435–444 [DOI] [PubMed] [Google Scholar]
- 6. Reape T. J., McCabe P. F. (2010) Apoptotic-like regulation of programmed cell death in plants. Apoptosis 15, 249–256 [DOI] [PubMed] [Google Scholar]
- 7. Cai Y. M., Yu J., Gallois P. (2014) Endoplasmic reticulum stress-induced PCD and caspase-like activities involved. Front. Plant Sci. 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rantong G., Gunawardena A. H. L. A. N. (2015) Programmed cell death: genes involved in signaling, regulation, and execution in plants and animals. Botany 93, 193–210 [Google Scholar]
- 9. Schaller A. (2004) A cut above the rest: the regulatory function of plant proteases. Planta 220, 183–197 [DOI] [PubMed] [Google Scholar]
- 10. Bozhkov P. V., Suarez M. F., Filonova L. H., Daniel G., Zamyatnin A. A., Jr., Rodriguez-Nieto S., Zhivotovsky B., Smertenko A. (2005) Cysteine protease mcII-Pa executes programmed cell death during plant embryogenesis. Proc. Natl. Acad. Sci. U.S.A. 102, 14463–14468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He R., Drury G. E., Rotari V. I., Gordon A., Willer M., Farzaneh T., Woltering E. J., Gallois P. (2008) Metacaspase-8 modulates programmed cell death induced by ultraviolet light and H2O2 in Arabidopsis. J. Biol. Chem. 283, 774–783 [DOI] [PubMed] [Google Scholar]
- 12. Coll N. S., Vercammen D., Smidler A., Clover C., Van Breusegem F., Dangl J. L., Epple P. (2010) Arabidopsis type I metacaspases control cell death. Science 330, 1393–1397 [DOI] [PubMed] [Google Scholar]
- 13. Watanabe N., Lam E. (2011) Arabidopsis metacaspase 2d is a positive mediator of cell death induced during biotic and abiotic stresses. Plant J. 66, 969–982 [DOI] [PubMed] [Google Scholar]
- 14. Tsiatsiani L., Van Breusegem F., Gallois P., Zavialov A., Lam E., Bozhkov P. V. (2011) Metacaspases. Cell Death Differ. 18, 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Minina E. A., Smertenko A. P., Bozhkov P. V. (2014) Vacuolar cell death in plants: metacaspase releases the brakes on autophagy. Autophagy 10, 928–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hatsugai N., Kuroyanagi M., Yamada K., Meshi T., Tsuda S., Kondo M., Nishimura M., Hara-Nishimura I. (2004) A plant vacuolar protease, VPE, mediates virus-induced hypersensitive cell death. Science 305, 855–858 [DOI] [PubMed] [Google Scholar]
- 17. Rojo E., Martín R., Carter C., Zouhar J., Pan S., Plotnikova J., Jin H., Paneque M., Sánchez-Serrano J. J., Baker B., Ausubel F. M., Raikhel N. V. (2004) VPEγ exhibits a caspase-like activity that contributes to defense against pathogens. Curr. Biol. 14, 1897–1906 [DOI] [PubMed] [Google Scholar]
- 18. Hatsugai N., Yamada K., Goto-Yamada S., Hara-Nishimura I. (2015) Vacuolar processing enzyme in plant programmed cell death. Front. Plant Sci. 6, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chichkova N. V., Kim S. H., Titova E. S., Kalkum M., Morozov V. S., Rubtsov Y. P., Kalinina N. O., Taliansky M. E., Vartapetian A. B. (2004) A plant caspase-like protease activated during the hypersensitive response. Plant Cell 16, 157–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chichkova N. V., Shaw J., Galiullina R. A., Drury G. E., Tuzhikov A. I., Kim S. H., Kalkum M., Hong T. B., Gorshkova E. N., Torrance L., Vartapetian A. B., Taliansky M. (2010) Phytaspase, a relocalisable cell death promoting plant protease with caspase specificity. EMBO J. 29, 1149–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fomicheva A. S., Tuzhikov A. I., Beloshistov R. E., Trusova S. V., Galiullina R. A., Mochalova L. V., Chichkova N. V., Vartapetian A. B. (2012) Programmed cell death in plants. Biochemistry (Moscow) 77, 1452–1464 [DOI] [PubMed] [Google Scholar]
- 22. Chichkova N. V., Tuzhikov A. I., Taliansky M., Vartapetian A. B. (2012) Plant phytaspases and animal caspases: structurally unrelated death proteases with a common role and specificity. Physiol Plant. 145, 77–84 [DOI] [PubMed] [Google Scholar]
- 23. Reavy B., Bagirova S., Chichkova N. V., Fedoseeva S. V., Kim S. H., Vartapetian A. B., Taliansky M. E. (2007) Caspase-resistant VirD2 protein provides enhanced gene delivery and expression in plants. Plant Cell Rep. 26, 1215–1219 [DOI] [PubMed] [Google Scholar]
- 24. Chichkova N. V., Galiullina R. A., Beloshistov R. E., Balakireva A. V., Vartapetian A. B. (2014) Phytaspases: aspartate-specific proteases involved in plant cell death. Russ. J. Bioorg. Chem. 40, 606–611 [DOI] [PubMed] [Google Scholar]
- 25. Poręba M., Szalek A., Kasperkiewicz P., Drąg M. (2014) Positional scanning substrate combinatorial library (PS-SCL) approach to define caspase substrate specificity. Methods Mol. Biol. 1133, 41–59 [DOI] [PubMed] [Google Scholar]
- 26. Harris J. L., Backes B. J., Leonetti F., Mahrus S., Ellman J. A., Craik C. S. (2000) Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc. Natl. Acad. Sci. U.S.A. 97, 7754–7759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walters J., Pop C., Scott F. L., Drag M., Swartz P., Mattos C., Salvesen G. S., Clark A. C. (2009) A constitutively active and uninhibitable caspase-3 zymogen efficiently induces apoptosis. Biochem. J. 424, 335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poreba M., Kasperkiewicz P., Snipas S. J., Fasci D., Salvesen G. S., Drag M. (2014) Unnatural amino acids increase sensitivity and provide for the design of highly selective caspase substrates. Cell Death Differ. 21, 1482–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kasperkiewicz P., Poreba M., Snipas S. J., Parker H., Winterbourn C. C., Salvesen G. S., Drag M. (2014) Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc. Natl. Acad. Sci. U.S.A. 111, 2518–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gregory H., Hardy P. M., Jones D. S., Kenner G. W., Sheppard R. C. (1964) The antral hormone gastrin. Structure of gastrin. Nature 204, 931–933 [DOI] [PubMed] [Google Scholar]
- 31. Morley J. S., Tracy H. J., Gregory R. A. (1965) Structure-function relationships in the active C-terminal tetrapeptide sequence of gastrin. Nature 207, 1356–1359 [DOI] [PubMed] [Google Scholar]
- 32. Calatayud S., Alvarez A., Víctor V. M. (2010) Gastrin: an acid-releasing, proliferative and immunomodulatory peptide? Mini Rev. Med. Chem. 10, 8–19 [DOI] [PubMed] [Google Scholar]
- 33. Rehfeld J. F., Agersnap M. (2012) Unsulfated cholecystokinin: an overlooked hormone? Regul. Pept. 173, 1–5 [DOI] [PubMed] [Google Scholar]
- 34. Chichkova N. V., Galiullina R. A., Taliansky M. E., Vartapetian A. B. (2008) Tissue disruption activates a plant caspase-like protease with TATD cleavage specificity. Plant Stress 2, 89–95 [Google Scholar]
- 35. Spanarkel M., Martinez J., Briet C., Jensen R. T., Gardner J. D. (1983) Cholecystokinin-27–32-amide. A member of a new class of cholecystokinin receptor antagonists. J. Biol. Chem. 258, 6746–6749 [PubMed] [Google Scholar]
- 36. Fuentes-Prior P., Salvesen G. S. (2004) The protein structures that shape caspase activity, specificity, activation, and inhibition. Biochem. J. 384, 201–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Afonina I. S., Cullen S. P., Martin S. J. (2010) Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol. Rev. 235, 105–116 [DOI] [PubMed] [Google Scholar]
- 38. Thornberry N. A., Rano T. A., Peterson E. P., Rasper D. M., Timkey T., Garcia-Calvo M., Houtzager V. M., Nordstrom P. A., Roy S., Vaillancourt J. P., Chapman K. T., Nicholson D. W. (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B: functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272, 17907–17911 [DOI] [PubMed] [Google Scholar]
- 39. Talanian R. V., Quinlan C., Trautz S., Hackett M. C., Mankovich J. A., Banach D., Ghayur T., Brady K. D., Wong W. W. (1997) Substrate specificities of caspase family proteases. J. Biol. Chem. 272, 9677–9682 [DOI] [PubMed] [Google Scholar]
- 40. Vartapetian A. B., Tuzhikov A. I., Chichkova N. V., Taliansky M., Wolpert T. J. (2011) A plant alternative to animal caspases: subtilisin-like proteases. Cell Death Differ. 18, 1289–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fourmy D., Gigoux V., Reubi J. C. (2011) Gastrin in gastrointestinal diseases. Gastroenterology 141, 814–818 [DOI] [PubMed] [Google Scholar]
- 42. Crawley J. N., Corwin R. L. (1994) Biological actions of cholecystokinin. Peptides 15, 731–755 [DOI] [PubMed] [Google Scholar]
- 43. Smith G. P., Jerome C., Cushin B. J., Eterno R., Simansky K. J. (1981) Abdominal vagotomy blocks the satiety effect of cholecystokinin in the rat. Science 213, 1036–1037 [DOI] [PubMed] [Google Scholar]
- 44. Berna M. J., Jensen R. T. (2007) Role of CCK/gastrin receptors in gastrointestinal/metabolic diseases and results of human studies using gastrin/CCK receptor agonists/antagonists in these diseases. Curr. Top. Med. Chem. 7, 1211–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]