

Background: Pertussis toxin (PTx) affects the blood-brain barrier in different models.
Results: PTx activates host cell signaling pathways identical to those exploited by E. coli K1 RS218 in brain-derived microvascular endothelial cells.
Conclusion: PTx sensitizes brain-derived microvascular endothelial cells (HBMEC/TY10) cells for invasion of E. coli K1 RS218.
Significance: Knowledge of host cell signaling pathways could help inhibiting neurological complications in whooping cough.
Keywords: beta-catenin (B-catenin), Bordetella pertussis, endothelial dysfunction, Escherichia coli (E. coli), translocation, pertussis toxin, VE-cadherin
Abstract
Pertussis toxin (PTx), an AB5 toxin and major virulence factor of the whooping cough-causing pathogen Bordetella pertussis, has been shown to affect the blood-brain barrier. Dysfunction of the blood-brain barrier may facilitate penetration of bacterial pathogens into the brain, such as Escherichia coli K1 (RS218). In this study, we investigated the influence of PTx on blood-brain barrier permissiveness to E. coli infection using human brain-derived endothelial HBMEC and TY10 cells as in vitro models. Our results indicate that PTx acts at several key points of host cell intracellular signaling pathways, which are also affected by E. coli K1 RS218 infection. Application of PTx increased the expression of the pathogen binding receptor gp96. Further, we found an activation of STAT3 and of the small GTPase Rac1, which have been described as being essential for bacterial invasion involving host cell actin cytoskeleton rearrangements at the bacterial entry site. In addition, we showed that PTx induces a remarkable relocation of VE-cadherin and β-catenin from intercellular junctions. The observed changes in host cell signaling molecules were accompanied by differences in intracellular calcium levels, which might act as a second messenger system for PTx. In summary, PTx not only facilitates invasion of E. coli K1 RS218 by activating essential signaling cascades; it also affects intercellular barriers to increase paracellular translocation.
Introduction
Bordetella pertussis is the causative agent of the respiratory disease whooping cough, which in young infants may occasionally be associated with neurological disorders (1–3). It has been shown in several studies that pertussis toxin (PTx),2 a decisive and secreted virulence factor of B. pertussis, affects cerebral vascular barriers and transiently increases the permeability of the blood-brain barrier (1, 3–8). PTx belongs to the class of AB toxins with a catalytically active monomeric A-subunit, which transfers ADP-ribose from NAD to a cysteine residue of Gi proteins (9, 10). This transfer decouples the Gαi proteins from their corresponding receptors and also alters Gi-mediated signaling. Depending on the specific cell type, also alterations in cAMP levels have been observed, further disturbing the homeostatic signaling of the cell. The pentameric B-subunit (S2, S3, 2xS4, S5) mediates binding and uptake of PTx. Apparently, it interacts with a NeuNAc-Gal motif displayed by several sialoglycoproteins on various cell types as well as glycolipids, such as ganglioside GD1a (11–13). More recent studies also implicate an interaction with the TLR4 receptor, although the function of this interaction is still unclear (14–16).
E. coli K1 is one of the leading causes of bacterial meningitis for newborns and infants in both developed and developing countries (17–20). E. coli K1 employs a complex pathogenic mechanism to evade the host immune defense and invade the brain endothelium (21–23). Initial binding of bacterial OmpA, FimH, and CNF1 to the host receptors gp96, CD48, and 37LRP, respectively, triggers various intracellular signaling cascades that facilitate invasion (24–30). On the molecular level it was shown that bacterial binding via OmpA up-regulated the expression of gp96 via the production of NO by inducible nitric-oxide synthase, which promotes a positive feedback loop for enhanced bacterial invasion (29, 31, 32). Additionally, recruitment and activation of STAT3 at the intracellular gp96 domain results in loading of the small GTPase Rac1 with GTP, which in concert with RhoA rearranges actin filaments to the bacterial invasion site (24, 27, 29, 33). Moreover, Ca2+ influx induced by bacterial binding to the host cell activates PKCα (34, 35). PKCα phosphorylates IQGAP1, which dissociates β-catenin from VE-cadherin and thereby weakens adherens junctions. This in turn facilitates the paracellular translocation route of bacterial and immune cells into the brain (36).
Previously, we showed that PTx transiently affects the permeability of human brain-derived endothelial cell layers in different in vitro systems, although the molecular mechanisms for this effect were still unclear (4–6, 13). To gain further insight into the mode of action of PTx, we investigated which host cell signaling cascades might be affected and whether the toxin alters the same signaling pathways as E. coli K1 RS218 in brain-derived endothelial cells.
Experimental Procedures
Chemicals, Antibodies, and Bacterial Strains
All chemicals were purchased from Sigma unless stated otherwise. Antibodies were acquired from Cell Signaling with the exception of anti-β-catenin (Sigma), Alexa Fluor 488 (Sigma), Alexa Fluor 594 (Sigma), phospho-β-catenin (Thr-41/Ser-45) (Santa Cruz Biotechnology, Inc.), and VE-cadherin (C-19 and H1) (Santa Cruz Biotechnology). Pertussis toxin was purchased from Calbiochem. E. coli K1 RS218 is a clinical isolate obtained from a newborn with meningitis (24). E. coli HB101 is a non-pathogenic laboratory strain (E. coli strain collection, Institute of Infectiology, Center for Molecular Biology of Inflammation, Westfälische Wilhelms-Universität Münster).
Cell Culture
TY10 cells (37, 38) were maintained in EGM-2 medium (Lonza) with 20% FBS (Sigma) at 33 °C for proliferation and 37 °C for differentiation (96 h). HBMEC cells were maintained in RPMI medium (Sigma) with 10% FCS (Sigma), 10% Nu-Serum (BD Biosciences), 2 mm glutamine, 1 mm pyruvate, 1% non-essential amino acids, 1% minimal Eagle's medium vitamins, 100 units/ml penicillin, and 100 μg/ml streptomycin. Both cell lines were subcultured up to 80% confluence before passaging.
Translocation Assay
Translocation assays were carried out with minor adjustments as described previously (4). Confluent TY10 or HBMEC cells were treated with PTx (200 ng/ml) for different time periods before infection with E. coli RS218 or E. coli HB101 at a multiplicity of infection of 100 for 90 min. For quantification of translocated bacteria, different dilutions of the basolateral media were plated on LB-agar plates, and bacterial colonies were counted the next day.
Invasion Assay
Invasion assays were performed as described previously (4). Confluent TY10 or HBMEC cells were treated with PTx (200 ng/ml) for different time periods before infection with E. coli RS218 or E. coli HB101 at a multiplicity of infection 100 for 90 min. Cells were washed three time with PBS and incubated for another 1 h with infection medium containing 200 μg/ml gentamicin. Afterward, cells were washed once with PBS and lysed with 1% Triton-X. Different dilutions were plated on LB-agar plates, and bacterial colonies were quantified the next day.
Coimmunoprecipitations
TY10 cells were grown in 60-mm dishes to confluence and differentiated for 96 h. Following stimulation with PTx (200 ng/ml, 6 h), the cells were washed with ice-cold PBS and lysed in radioimmune precipitation assay buffer (50 mm Tris/HCl, pH 7.4, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, phosphatase, and protease inhibitors). 250 μg of protein lysate were incubated with Protein-A-Plus agarose (Santa Cruz Biotechnology) and the primary antibody overnight at 4 °C. The immune complexes were washed three times with radioimmune precipitation assay buffer, separated by SDS-PAGE, and analyzed by Western blotting.
Rac Pull-down
TY10 or HBMEC cells were grown in 100-mm dishes to confluence and differentiated for 96 h. Before stimulation with PTx (200 ng/ml for 3, 4, or 6 h), the cells were washed three times with PBS and maintained in serum-free medium for 1 h. 20 μg of GST-PAK1-RBD were incubated for 90 min with GSH-Sepharose at 4 °C and washed three times with immunoprecipitation buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.5 mm MgCl2, protease inhibitor mixture (Roche Applied Science)). 500 μg of protein lysate were added and incubated for 45 min. The immune complexes were washed three times with immunoprecipitation buffer and subjected to SDS-PAGE and Western blotting for analysis.
Quantitative RT-PCR
Confluent TY10 or HBMEC cells were treated with PTx (200 ng/ml, 6 h). Total RNA was isolated using the RNeasy minikit (Qiagen) according to the manufacturer's instructions. 1 μg of RNA was reverse transcribed with the Primescript RT reagent kit (Takara) and oligo(dT) primers. Quantitative RT-PCR was performed using the SYBR Premix Ex TaqII kit (Takara). Real-time PCR conditions were as follows: 95 °C for 5 min, 40 cycles; 95 °C for 10 s; 60 °C for 30 s. The -fold increase in expression of mRNA was normalized by using the expression of hypoxanthine phosphoribosyltransferase. Quantitative real-time experiments were always run six times in duplicate with the following primers: HPRT-fwd, GCCAGACTTTGTTGGATTTG; HPRT-rev, CTCATCTTAGGCTTTGTATTTTG; gp96-fwd, CTGGAAATGAGGAACTAACAGTCA; gp96-rev, TCTTCTCTGGTCATTCCTACACC.
Intracellular Calcium Measurements
Intracellular calcium levels were measured according to the protocol of the manufacturer of the Fluoro-4AM kit. Briefly, TY10 cells were grown to confluence in 96-well plates and differentiated for an additional 96 h at 37 °C. Cells were washed once with PBS and maintained in EGM-2 medium without FCS. PTx (200 ng/ml) or histamine (100 μm) was applied for various time periods, followed by incubation of the sample at 37 °C for 1 h with the Fluoro-4AM kit solutions (Promega). To measure intracellular Ca2+ levels, the fluorescence of the samples was quantified (excitation/emission, 490 nm/520 nm).
Immunofluorescence
HBMEC and TY10 cells were grown on collagen-coated coverslips to confluence before experiments were carried out. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton X-100. The samples were blocked with 5% goat serum for 1 h at room temperature and incubated with the corresponding primary antibodies overnight. After extensive washing, the samples were incubated with Alexa Fluor-conjugated secondary antibodies, washed with PBS, and incubated with DAPI for 10 min. The samples were washed with PBS and mounted with fluorescent mounting medium. The slides were imaged with an LSM confocal system mounted on an Axio Observer.Z1 microscope equipped with differential interference contrast optics and a plan-apochromat ×63/1.4 numerical aperture oil immersion objective lens (Carl Zeiss MicroImaging, Thornwood, NY). Images were generated with Zen software version 2011.
Electron Microscopy
TY10 cells were fixed with 2% paraformaldehyde, 0.25% glutaraldehyde in 0.1 m phosphate buffer and processed using the method of Tokuyasu (39). Ultrathin (50-nm) frozen sections were cut and immunogold-labeled using anti-β-catenin and anti-VE-cadherin antibodies and detected with Protein A Gold (Department of Cell Biology, University Medical Center, Utrecht, The Netherlands) with gold particles of 10- or 15-nm diameter, respectively. Labeling with Protein A Gold (gold particles 15 or 10 nm) alone, without first antibodies was included as a control of specificity of the immunogold staining. The samples were analyzed at 80 kV on an FEI-Tecnai 12 electron microscope (FEI, Eindhoven, The Netherlands). Photographs of selected areas were documented with imaging plates (Ditabis, Pforzheim, Germany).
Results
PTx Increases E. coli RS218 Invasion and Translocation
To assess whether PTx enhances invasion and translocation of the pathogenic E. coli K1 RS218, we used transwell filter systems in our experiments. HBMEC as well as TY10 cells were grown to confluence on filter membranes and stimulated with PTx 6 h before infection with the pathogenic E. coli K1 RS218 or the non-pathogenic E. coli HB101 strain (Fig. 1, A and B). Quantification of the translocation rates showed that PTx increases the translocation of E. coli K1 RS218 (up to 6.72 ± 0.55-fold) as well as of E. coli HB101 (up to 3.52 ± 0.13-fold) in HBMEC cells. These findings are in agreement with previous studies in different in vitro systems (4, 5). In contrast, quantification of the invasion assays revealed that PTx significantly enhanced the invasion of E. coli K1 RS218 up to 4.11 ± 0.59-fold, whereas it had no effect on the invasion of the E. coli HB101 strain. Hence, it appears that the enhanced translocation observed for the non-pathogenic HB101 strain is not likely to be due to transcytosis following invasion but might be attributed to an enhanced paracellular penetration. Similar translocation results were obtained with our second human brain-derived microvascular endothelial cell line, TY10 (data not shown). Invasion, however, could not be quantified because no viable bacterial cells were obtained in this case, but we confirmed invasion with DAPI staining (data not shown). Because PTx selectively enhanced the invasion of pathogenic E. coli K1 RS218, we took a closer look at the underlying mechanisms.
FIGURE 1.
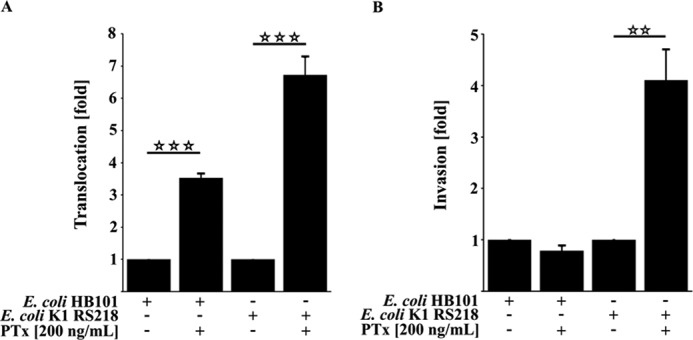
PTx increases invasion and translocation of E. coli K1 RS218. A, pretreatment of HBMEC with PTx (200 ng/ml) for 6 h significantly increases the translocation rate of E. coli HB101 by 3.52 ± 0.13-fold and E. coli K1 RS218 by 6.72 ± 0.55-fold, respectively. B, 6-h PTx pretreatment (200 ng/ml) of HBMEC significantly increases the invasion rate of the pathogenic E. coli K1 RS218 strain by 4.11 ± 0.59-fold compared with untreated HBMEC. No significant changes were observed for the nonpathogenic E. coli HB101 strain. Bars, mean ± S.E. (error bars) of three independent experiments performed in triplicate. **, p < 0.01; ***, p < 0.001 (determined by ANOVA followed by Bonferroni post hoc test).
PTx Up-regulates gp96 mRNA Expression
Recently, it has been reported that the invasion of E. coli K1 RS218 depends on the interaction of OmpA, IbeA, CNF1, and FimH with their corresponding host cell receptors (24, 26, 27, 40–42). Specifically, the interactions with gp96 and 37LRP were described to be essential for the invasion. To further elucidate whether PTx might up-regulate the expression of these two receptors to enhance invasion, we performed quantitative RT-PCR of PTx-treated HBMEC and TY10 cells (Fig. 2, A–E). In both cell lines, PTx was able to increase the expression of gp96 but not of 37LRP. Although we found a significant increase for gp96 expression in both cell lines (1.79 ± 0.34-fold in HBMEC (Fig. 2A) and 3.83 ± 1.35-fold in TY10 cells (Fig. 2B)), the increased expression of 37LRP was inconsistent and therefore not significant after application of PTx (HBMEC cells (Fig. 2C) and TY10 cells (Fig. 2D)).
FIGURE 2.
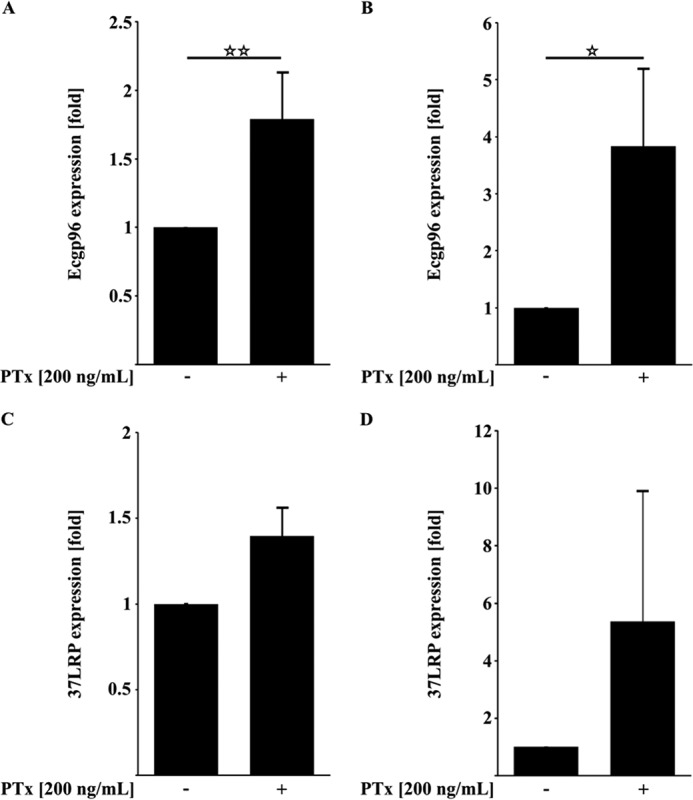
PTx up-regulates the expression of the E. coli-binding receptors gp96 but not 37LRP. A and B, quantitative RT-PCR analysis of PTx-treated HBMEC and TY10 cells (200 ng/ml, 6 h) showed significantly increased gp96 mRNA levels. C and D, application of PTx to HBMEC and TY10 cells (200 ng/ml, 6 h) led to no significant changes in 37LRP mRNA levels. Bars, mean ± S.E. of at least six independent experiments performed in duplicate. **, p < 0.01; ***, p < 0.001 (determined by ANOVA).
PTx Activates STAT3 and Rac1
Based on the quantitative RT-PCR results, we further investigated PTx effects on host cell signaling events downstream of gp96. Upon binding of OmpA to gp96, several signaling cascades are activated. One of those is the recruitment and activation of STAT3 by phosphorylation (24, 29). We investigated whether PTx is also able to activate STAT3 independently of E. coli K1 RS218. As shown in Fig. 3A, 6 h of PTx treatment led to a significant increase in STAT3 phosphorylation at Tyr-705 (1.39 ± 0.09-fold). Phosphorylation of STAT3 was shown to recruit Vav2 and subsequently activate the small GTPase Rac1 via the exchange of bound GDP to GTP (24). As described earlier, Rac1 activation peaked around 30 min after E. coli K1 RS218 infection (Fig. 3B). PTx application in HBMEC and TY10 for various time intervals led to an increased amount of activated Rac1 (Fig. 3, C and D), in which Rac1 activation was significant after 6 h of PTx treatment in both cell lines (HBMEC (Fig. 3C) and TY10 cells (Fig. 3D)).
FIGURE 3.
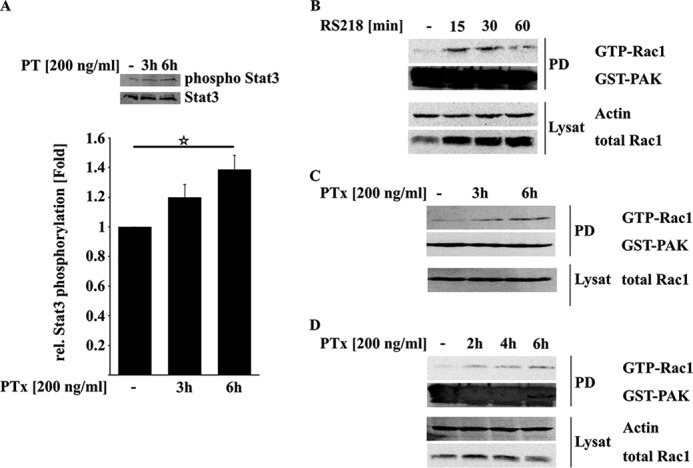
PTx increases STAT3 and Rac1 activity. A, treatment of serum-deprived HBMEC with PTx (200 ng/ml, 3 or 6 h) led to an increased phosphorylation of Stat3 Tyr-705. B–D, infection of HBMEC with E. coli K1 RS218 activates Rac1 within the first 30 min (B). In contrast, PTx activates Rac1 in HBMEC (C) and TY10 cells (D) significantly after 6 h of stimulation. Bars, mean ± S.E. (error bars) of at least six independent experiments performed in duplicate. Western blots of Rac1 pull-downs give representative results of three independent experiments. **, p < 0.01; ***, p < 0.001 (determined by ANOVA).
PTx Disrupts β-Catenin and VE-cadherin Membrane Localization
To further investigate how PTx is able to enhance the translocation of bacteria, we had a closer look at the adherens junction proteins VE-cadherin and β-catenin. It has been shown that different pathways induced by E. coli K1 RS218 binding affect the integrity of this complex (32, 36). As depicted in Fig. 4A, application of PTx led to increased amounts of phosphorylated β-catenin (Thr-41/Ser-45) concomitant with a strong loss of membrane localization, gap-forming structures between cells, and an enhanced appearance of stress fibers at cellular membranes (Fig. 4B). Interestingly, infection with E. coli K1 RS218 alone showed the same phenotypes in our immunofluorescence studies (Fig. 4B, bottom).
FIGURE 4.
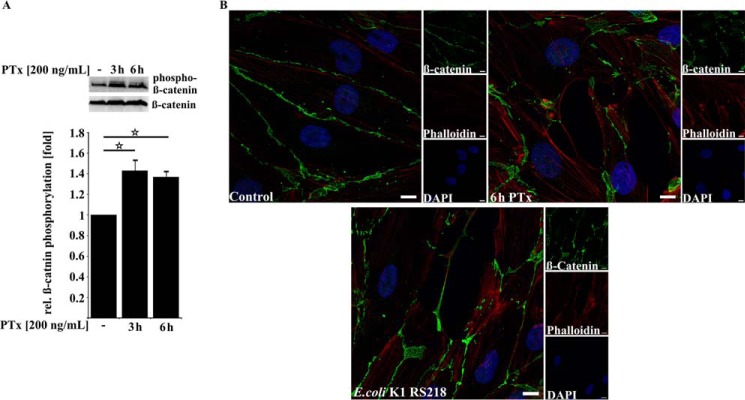
PTx increases β-catenin phosphorylation and alters its membrane localization. A, application of PTx (200 ng/ml for 3 or 6 h) significantly increased β-catenin phosphorylation at Thr-41/Ser-45. B, confocal image of TY10 cells stained for β-catenin (Alexa Fluor 488), phalloidin (Alexa Fluor 594), and DAPI either without treatment or with PTx treatment (200 ng/ml, 6 h) or infected with E. coli K1 RS218 (multiplicity of infection of 100, 90 min) shows that β-catenin localization at the cellular membrane is disturbed in the case of PTx application (top right) and E. coli K1 RS218 infection (bottom). In addition, gaplike structures appear in PTx-stimulated and E. coli K1 RS218-infected samples. Scale bar, 10 μm. Bars, mean ± S.E. (error bars) of at least three independent experiments performed in duplicate. *, p < 0.05 (determined by ANOVA).
A reduced localization of β-catenin at the cellular membrane should be accompanied by a reduced binding to VE-cadherin. To support this assumption, we performed co-immunoprecipitation experiments (Fig. 5A). We found a significant decrease in β-catenin bound to precipitated VE-cadherin after application of PTx for 4 and 6 h (4-h PTx, 88 ± 2%; 6-h PTx, 77.54 ± 11.90%). Immunofluorescence staining of VE-cadherin and β-catenin with and without PTx application further confirmed the results obtained by immunoprecipitation experiments (Fig. 5B). β-Catenin and VE-cadherin exhibited a strong colocalization under non-stimulating conditions. Application of PTx for 6 h disrupts the membrane localization of β-catenin as well as of VE-cadherin to some degree. The same result was obtained with immunogold labeling of VE-cadherin and β-catenin in ultra-cryo-sections (Fig. 5C). The amount of VE-cadherin at the cellular membrane, visualized by antibodies labeled with 15-nm gold particles, and β-catenin (labeled with 10-nm gold particles) is significantly reduced after application of PTX.
FIGURE 5.

PTx decreases β-catenin/VE-cadherin interaction and disturbs VE-cadherin membrane localization. A, PTx decreases the amount of β-catenin bound to precipitated VE-cadherin in TY10 after 4–6 h significantly. B, immunofluorescence double staining of β-catenin (Alexa Fluor 594) and VE-cadherin (Alexa Fluor 488) followed by confocal imaging shows a strong colocalization of both proteins at the cellular membrane, which is disrupted after application of PTx (200 ng/ml, 6 h). Scale bar, 10 μm. C, double immunogold labeling of ultra-cryo-sections of TY10 cells shows in control cells a strong localization of VE-cadherin (15-nm gold) and β-catenin (10-nm gold) at outer cellular membranes (left). Application of PTx (200 ng/ml, 6 h) decreases the amount of detectable VE-cadherin and β-catenin at the outer cellular membranes. Bars, mean ± S.E. of at least three independent experiments performed in duplicate. *, p < 0.05; **, p < 0.01 (determined by ANOVA).
PTx Influences Intracellular Ca2+ Levels
Previous studies implicated a role of PTx in regulating the activity of Ca2+ channels by its inhibitory function toward heterotrimeric G-proteins (15, 44–46). Because intracellular Ca2+ levels are strictly regulated to ensure homeostasis of host cell signaling, we investigated whether an application of PTx in TY10 cells may influence intracellular Ca2+ levels to induce the observed cellular effects. As shown in Fig. 6A, application of PTx had a small but significant effect on intracellular Ca2+ levels. 2 h after application, intracellular Ca2+ concentrations were significantly decreased to 78.9 ± 3.2% compared with untreated cells. Interestingly, Ca2+ levels returned to normal control levels after 4 h (92 ± 6.5%) and were increased 6 h after PTx application (115.8 ± 6.1%) However, in comparison with the alterations in Ca2+ levels induced, for example, by histamine, PTx-induced changes in Ca2+ levels are clearly less prominent (Fig. 6B). As an example, we investigated whether these changes might influence the activity of signaling proteins, which are known to decrease the interaction of β-catenin and VE-cadherin. One of those proteins is the Src kinase. Src is activated following Ca2+ influx and was recently described as displacing β-catenin from VE-cadherin by phosphorylation and binding to VE-cadherin (47). However, in our model systems, we could not detect a significant activation of Src after application of PTx, in contrast to the effects observed after stimulation with histamine (Fig. 6C). Moreover, the amounts of Src bound to VE-cadherin were unchanged with or without the presence of PTx (data not shown).
FIGURE 6.
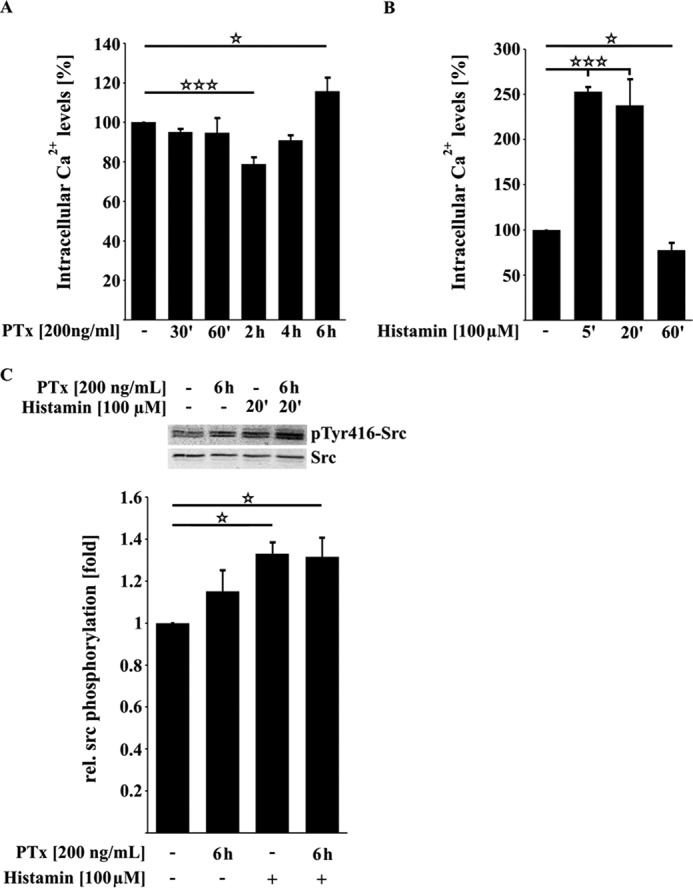
PTx influences Ca2+ levels but is not able to activate the protein kinase Src in TY10 cells. A, upon application of PTx (200 ng/ml), Ca2+ levels are significantly decreased after 2 h before they rise again to be significantly increased after 6 h of toxin application. B, compared with the PTx-induced changes in intracellular Ca2+ levels, histamine induces a fast and strong increase of Ca2+ in TY10 cells (5 min, 100 μm). C, Western blotting studies showed that PTx (200 ng/ml, 6 h) has no influence on the activation phosphorylation of the protein kinase Src in contrast to histamine (100 μm, 20 min). Bars, mean ± S.E. (error bars) of at least three independent experiments performed in duplicate. *, p < 0.05; ***, p < 0.001 (determined by ANOVA)
In addition, PKCα and, further downstream, IQGAP1 have been shown to be responsible factors for the dissociation of β-catenin from VE-cadherin subsequent to an infection with E. coli K1 RS218 and Ca2+ influx (35, 36). However, we were unable to find any evidence that PTx increases the interaction of IQGAP1 with β-catenin (data not shown). Therefore, we conclude that the observed PTx-induced alterations in Ca2+ levels do not affect the interactions of VE-cadherin or IQGAP1 with β-catenin.
PTx Reduces p42/44 MAPK Activity
We screened for additional kinases with different activation patterns after PTx application to identify a possible candidate that induces the reduction of β-catenin bound to VE-cadherin. PTx led to a significant and strong decrease in the activation phosphorylation of p42/44 MAPK up to 50% (Fig. 7A). We confirmed down-regulation of MAPK activity by investigating the phosphorylation of its downstream effector ELK-1, which was also reduced by more than 50% (Fig. 7B). In addition to inhibiting basal activation levels of p42/44 MAPK, PTx also reduced the activation of p42/44 MAPK after stimulation with serum (data not shown). Although there is not much evidence in the literature of a possible role of reduced MAPK activity in negatively regulating the interaction between VE-cadherin and β-catenin, we investigated whether reduction of MAPK activity leads to a reduced VE-cadherin/β-catenin interaction. As shown in Fig. 7C, mimicking the PTx-mediated down-regulation of MAPK activity by the pharmacological inhibitor U0126 led to a decreased amount of β-catenin bound to VE-cadherin of about 40%.
FIGURE 7.
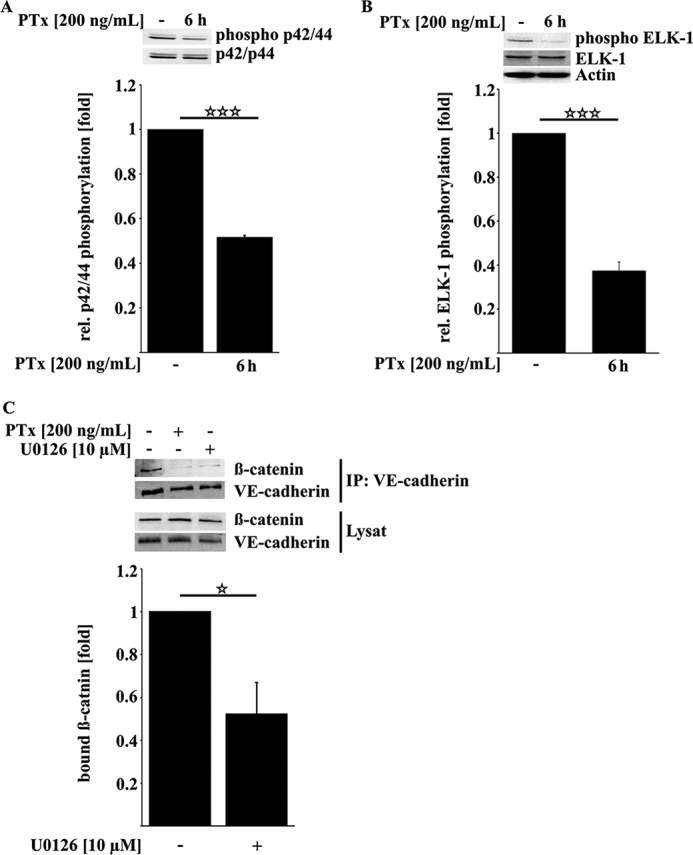
PTx decreases p42/44 MAPK activity, which is sufficient to reduce β-catenin/VE-cadherin interaction. A and B, PTx (200 ng/ml, 6 h) significantly decreased p42/44 MAPK activation phosphorylation at Thr-202/Tyr-204 (A) as well as activation phosphorylation of the p42/44 MAPK downstream target ELK-1 at Ser-383 (B). C, inhibition of p42/44 MAPK by U0126 (10 μm, 6 h) reduced the amount of β-catenin bound to VE-cadherin. Bars, mean ± S.E. (error bars) of at least three independent experiments performed in duplicate. *, p < 0.05; ***, p < 0.001 (determined by ANOVA).
In summary, we showed that PTx activates and inhibits specific signaling pathways, similar to those induced or inhibited by E. coli K1 RS218 infection. Activation of STAT3 and Rac1 (Figs. 2 and 3) is likely to facilitate bacterial uptake, whereas disruption of membrane localization of VE-cadherin and β-catenin and their interaction (Figs. 4, 5, and 7) might facilitate enhanced paracellular penetration (Fig. 8).
FIGURE 8.

Schematic representation of PTx-affected pathways in transcellular versus paracellular translocation. Based on the data obtained in this study, we hypothesize that PTx preactivates essential signaling pathways for E. coli K1 RS218 invasion and translocation. Up-regulation of gp96 and activation of STAT3 and Rac1, respectively, increase invasive processes. Dissociation of β-catenin and VE-cadherin weakens adherens junctions and enhances paracellular translocation.
Discussion
The exotoxin PTx is a major virulence factor of the Gram-negative bacterium B. pertussis, the causative agent of whooping cough. Especially in infants, severe complications, such as encephalopathy, are observed occasionally, which lead to neurological disorders. These sequelae have been associated with PTx, which is able to disrupt the integrity of the blood-brain barrier (3–6, 8, 48–50). However, the exact molecular mechanism underlying this complication is not yet known. As a result of this PTx-induced breach in endothelial barrier integrity, bacterial penetration into the central nervous system might be facilitated. This issue was investigated with the pathogenic E. coli K1 strain (51–54). Recently, it has been shown that the E. coli K1 strain (RS218) binds to a defined set of host receptors (24, 29, 40–42, 55, 56). Upon binding of the pathogen, several signaling cascades of the target host cell are activated, leading to a variety of cellular effects ranging from rearrangements of actin filaments by STAT3, Rac1, PKCα, and RhoA to an influx of Ca2+ and the production of NO to facilitate further bacterial uptake (24, 26, 27, 29, 31, 32, 40, 41, 55, 56). Additionally, activation of the protein kinases PKCα and Src destabilize the adherens junction complex of VE-cadherin·β-catenin, thereby also promoting paracellular translocation (36, 47).
In this study, we investigated whether PTx would increase the invasion and translocation of E. coli K1 RS218 in human brain-derived microvascular endothelial barrier model systems (HBMEC/TY10 cell lines). Furthermore, we determined that host cell signaling pathways in response to PTx might mimic those induced by E. coli K1 RS218 in such a way as to promote bacterial invasion in and translocation across brain endothelial barriers. First, we showed that incubation of endothelial barriers with PTx significantly increased the translocation of both non-pathogenic E. coli HB101 and the pathogenic E. coli K1 RS218 (Fig. 1A). Interestingly, only the invasion of the pathogenic E. coli K1 RS218 strain was enhanced (Fig. 1B), whereas uptake of E. coli HB101 was not affected. Our group showed earlier that PTx had no effect on the invasion of the E. coli K1 strain in HBMEC cells (4). This change of behavior is most probably due to the use of a different E. coli K1 strain in the current study (O1:K1 versus O18ac:K1:H7). Next, we found that PTx significantly increases the mRNA levels of the pathogen-binding receptor gp96 in the two human microvascular endothelial cell lines (Fig. 2, A and B). An increase of gp96 within the cellular membrane is shown to be mediated by an induction of NO production (29, 31, 32). Increased NO levels are induced by activation of the STAT3-PLCγ-PKCα pathway via gp96 binding as well as by the MyD88-NFκB pathway via gp96·TLR2 complex binding. Our results showed that STAT3 was also activated when we applied PTx alone (Fig. 3A). However, elevated NFκB activation levels could not be observed employing Western blotting (data not shown). In addition, we could not find increased NO levels by indirect measurements via nitrite after PTx application. There seems to be another so far unknown pathway for up-regulation of gp96 expression in these two cell lines, which must be addressed in further studies. Activation of STAT3 not only promotes enhanced NO production but also activates the small GTPase Rac1 via recruitment of the Rac-GEF Vav2 (24). The increase in the amount of GTP-Rac1 due to the application of PTx followed the same time course as STAT3 activation, which indicates that Rac1 activation is mediated by STAT3, as described by Maruvada and Kim (24). Compared with Rac1 activation induced by E. coli K1 RS218 (Fig. 3B), activation by PTx was rather low but significant. Active Rac1 is necessary to induce host cell actin cytoskeleton rearrangements to the bacterial entry site to facilitate bacterial uptake by the host cell. Taken together, these data could explain how PTx is able to increase the invasion of E. coli K1 RS218 in HBMEC cells, because it increases the expression of host-binding receptors in the cellular membrane and activates host cell signaling pathways that are shown to relevant to E. coli K1 RS218 invasion. This would explain why the PTx-induced increase in invasion is only observed with the pathogenic E. coli strain.
In addition to its support of invasion, we investigated how PTx is able to increase cellular permeability to enhance translocation via the paracellular route that was previously described by our group and others (4–6, 48). We demonstrated that PTx drastically influences the membrane localization of β-catenin and VE-cadherin (Figs. 4B and 5B). Moreover, VE-cadherin immunoprecipitation experiments showed lower amounts of bound β-catenin, which is in agreement with the findings of a previous study (36). Although the cellular effect of PTx is quite remarkable, it has remained enigmatic how PTx achieves the dissociation of β-catenin and VE-cadherin and induces the redistribution from their membrane localization. To be able to rationalize these findings, we focused in the beginning on intracellular Ca2+ levels as a possible second messenger system utilized by PTx, which was able to influence intracellular Ca2+ levels. One possible way in which PTx could be able to increase Ca2+ levels is by enhanced angiotensin II type 1 receptor (AT1R) expression, which was shown recently (15). PTx could not only up-regulate AT1R expression but also enhance AT1R-induced Ca2+ influx. Interestingly, AT1R was shown to be involved in E. coli K1 RS218 invasion because inhibition of AT1R by telmisartan or siRNA-mediated knockdown abolished E. coli K1 RS218 invasion (43). Furthermore, AT1R seems to interact with gp96 and, upon binding of E. coli K1 RS218, induces Ca2+ influx.
We then focused on the Ca2+-sensitive protein kinase Src, which was shown to phosphorylate and/or bind VE-cadherin, thereby displacing β-catenin to increase vascular permeability (47). Under our experimental conditions, we were unable to detect an influence of PTx on Src activity (Fig. 6C) or on VE-cadherin phosphorylation, respectively. Also, IQGAP1-mediated β-catenin dissociation appeared not to be affected. Investigations employing laser scanning confocal microscopy were unable to detect any significant differences in co-localization of IQGAP1 and β-catenin.
After screening for additional kinases affected by PTx, we found p42/44 MAPK activity to be drastically reduced. We mimicked p42/44 MAPK activity reduction using U0126 and could show that less β-catenin is bound to VE-cadherin. These results indicate that reduction of p42/44 MAPK activity by PTx or U0126 is involved in the disruption of VE-cadherin/β-catenin interaction. Because MAPK signaling is quite complex and depending on spatial and temporal aspects, including different scaffolding complexes, further studies are needed to address the mechanism of this observed effect.
Taken together, our study provides evidence that PTx partially mimics E. coli K1 RS218-induced effects with respect to activation of host cell signaling pathways needed for invasion and translocation (Fig. 8). Based on these results, we believe that PTx is preactivating signal transduction pathways that are essential for invasive processes, thereby facilitating a secondary infection by E. coli K1 RS218 in the course of an ongoing B. pertussis infection. Further studies are needed to define how PTx is able to activate and inhibit those pathways. This information may help in developing possible strategies to reduce neurological complications during an ongoing B. pertussis infection.
Author Contributions
S. K. and M. A. S. designed the study and wrote the paper. S. K., L. S., and J. S. designed, performed, and evaluated the experiments shown in Figs. 1–7. L. G. performed the experiment shown in Fig. 5C. K. K., Y. S., and T. K. contributed to the experiments and to the writing of the paper.
Acknowledgment
We thank Rolf Heumann (Universität Bochum) for kindly providing the GST-Pak1-RBD construct.
This work was supported by Deutsche Forschungsgemeinschaft Grants DFG SFB629 B03 and GRK1409 and by the Cells-in-Motion Cluster of Excellence (EXC 1003−CiM). The authors declare that they have no conflicts of interest with the contents of this article.
This publication is dedicated to the memory of Johanna Solbach who tragically left us in her prime.
- PTx
- pertussis toxin
- AT1R
- angiotensin II type 1 receptor
- ANOVA
- analysis of variance
- GD1a
- Neu5Acα3Galβ3GalNAcβ4(Neu5Acα3)Galβ4GlcCer.
References
- 1. Geier D. A., Geier M. R. (2004) An evaluation of serious neurological disorders following immunization: a comparison of whole-cell pertussis and acellular pertussis vaccines. Brain Dev. 26, 296–300 [DOI] [PubMed] [Google Scholar]
- 2. Heininger U., Kleemann W. J., Cherry J. D., and Sudden Infant Death Syndrome Study Group (2004) A controlled study of the relationship between Bordetella pertussis infections and sudden unexpected deaths among German infants. Pediatrics 114, e9–e15 [DOI] [PubMed] [Google Scholar]
- 3. Grant C. C., McKay E. J., Simpson A., Buckley D. (1998) Pertussis encephalopathy with high cerebrospinal fluid antibody titers to pertussis toxin and filamentous hemagglutinin. Pediatrics 102, 986–990 [DOI] [PubMed] [Google Scholar]
- 4. Seidel G., Böcker K., Schulte J., Wewer C., Greune L., Humberg V., Schmidt M. A. (2011) Pertussis toxin permeabilization enhances the traversal of Escherichia coli K1, macrophages, and monocytes in a cerebral endothelial barrier model in vitro. Int. J. Med. Microbiol. 301, 204–212 [DOI] [PubMed] [Google Scholar]
- 5. Brückener K. E., el Bayâ A., Galla H. J., Schmidt M. A. (2003) Permeabilization in a cerebral endothelial barrier model by pertussis toxin involves the PKC effector pathway and is abolished by elevated levels of cAMP. J. Cell Sci. 116, 1837–1846 [DOI] [PubMed] [Google Scholar]
- 6. Kügler S., Böcker K., Heusipp G., Greune L., Kim K. S., Schmidt M. A. (2007) Pertussis toxin transiently affects barrier integrity, organelle organization and transmigration of monocytes in a human brain microvascular endothelial cell barrier model. Cell. Microbiol. 9, 619–632 [DOI] [PubMed] [Google Scholar]
- 7. Arimoto H., Tanuma N., Jee Y., Miyazawa T., Shima K., Matsumoto Y. (2000) Analysis of experimental autoimmune encephalomyelitis induced in F344 rats by pertussis toxin administration. J. Neuroimmunol. 104, 15–21 [DOI] [PubMed] [Google Scholar]
- 8. Amiel S. A. (1976) The effects of Bordetella pertussis vaccine on cerebral vascular permeability. Br. J. Exp. Pathol. 57, 653–662 [PMC free article] [PubMed] [Google Scholar]
- 9. Katada T., Ui M. (1982) Direct modification of the membrane adenylate cyclase system by islet-activating protein due to ADP-ribosylation of a membrane protein. Proc. Natl. Acad. Sci. U.S.A. 79, 3129–3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Katada T., Ui M. (1982) ADP ribosylation of the specific membrane protein of C6 cells by islet-activating protein associated with modification of adenylate cyclase activity. J. Biol. Chem. 257, 7210–7216 [PubMed] [Google Scholar]
- 11. Armstrong G. D., Clark C. G., Heerze L. D. (1994) The 70-kilodalton pertussis toxin-binding protein in Jurkat cells. Infect. Immun. 62, 2236–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brennan M. J., David J. L., Kenimer J. G., Manclark C. R. (1988) Lectin-like binding of pertussis toxin to a 165-kilodalton Chinese hamster ovary cell glycoprotein. J. Biol. Chem. 263, 4895–4899 [PubMed] [Google Scholar]
- 13. el Bayâ A., Linnemann R., von Olleschik-Elbheim L., Schmidt M. A. (1995) Identification of binding proteins for pertussis toxin on pancreatic beta cell-derived insulin-secreting cells. Microb. Pathog. 18, 173–185 [DOI] [PubMed] [Google Scholar]
- 14. Racke M. K., Hu W., Lovett-Racke A. E. (2005) PTX cruiser: driving autoimmunity via TLR4. Trends Immunol. 26, 289–291 [DOI] [PubMed] [Google Scholar]
- 15. Nishida M., Suda R., Nagamatsu Y., Tanabe S., Onohara N., Nakaya M., Kanaho Y., Shibata T., Uchida K., Sumimoto H., Sato Y., Kurose H. (2010) Pertussis toxin up-regulates angiotensin type 1 receptors through Toll-like receptor 4-mediated Rac activation. J. Biol. Chem. 285, 15268–15277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fan H., Peck O. M., Tempel G. E., Halushka P. V., Cook J. A. (2004) Toll-like receptor 4 coupled GI protein signaling pathways regulate extracellular signal-regulated kinase phosphorylation and AP-1 activation independent of NFκB activation. Shock 22, 57–62 [DOI] [PubMed] [Google Scholar]
- 17. Houdouin V., Bonacorsi S., Bidet P., de La Rocque F., Cohen R., Aujard Y., Bingen E. (2008) [Clinical outcome and bacterial characteristics of 99 Escherichia coli meningitis in young infants]. Arch. Pediatr. 15, S138–S147 [DOI] [PubMed] [Google Scholar]
- 18. Houdouin V., Bonacorsi S., Bidet P., Blanco J., De La Rocque F., Cohen R., Aujard Y., Bingen E. (2008) Association between mortality of Escherichia coli meningitis in young infants and non-virulent clonal groups of strains. Clin. Microbiol. Infect. 14, 685–690 [DOI] [PubMed] [Google Scholar]
- 19. Sunakawa K., Sakai F., Hirao Y., Hanaki H., Nonoyama M., Iwata S., Akita H., Sato Y. (2010) [Childhood bacterial meningitis trends in Japan from 2007 to 2008]. Kansenshogaku Zasshi 84, 33–41 [DOI] [PubMed] [Google Scholar]
- 20. Zaidi A. K., Khan H., Lasi R., Mahesar W., and Sindh Meningitis Group (2009) Surveillance of pneumococcal meningitis among children in Sindh, southern Pakistan. Clin. Infect. Dis. 48, S129–S135 [DOI] [PubMed] [Google Scholar]
- 21. Kim K. S. (2001) Escherichia coli translocation at the blood-brain barrier. Infect. Immun. 69, 5217–5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim K. S. (2002) Strategy of Escherichia coli for crossing the blood-brain barrier. J. Infect. Dis. 186, S220–S224 [DOI] [PubMed] [Google Scholar]
- 23. Xie Y., Kim K. J., Kim K. S. (2004) Current concepts on Escherichia coli K1 translocation of the blood-brain barrier. FEMS Immunol. Med. Microbiol. 42, 271–279 [DOI] [PubMed] [Google Scholar]
- 24. Maruvada R., Kim K. S. (2012) IbeA and OmpA of Escherichia coli K1 exploit Rac1 activation for invasion of human brain microvascular endothelial cells. Infect. Immun. 80, 2035–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Prasadarao N. V., Wass C. A., Weiser J. N., Stins M. F., Huang S. H., Kim K. S. (1996) Outer membrane protein A of Escherichia coli contributes to invasion of brain microvascular endothelial cells. Infect. Immun. 64, 146–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Teng C. H., Cai M., Shin S., Xie Y., Kim K. J., Khan N. A., Di Cello F., Kim K. S. (2005) Escherichia coli K1 RS218 interacts with human brain microvascular endothelial cells via type 1 fimbria bacteria in the fimbriated state. Infect. Immun. 73, 2923–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Y., Kim K. S. (2002) Role of OmpA and IbeB in Escherichia coli K1 invasion of brain microvascular endothelial cells in vitro and in vivo. Pediatr. Res. 51, 559–563 [DOI] [PubMed] [Google Scholar]
- 28. Wang Y., Wen Z. G., Kim K. S. (2004) Role of S fimbriae in Escherichia coli K1 binding to brain microvascular endothelial cells in vitro and penetration into the central nervous system in vivo. Microb. Pathog. 37, 287–293 [DOI] [PubMed] [Google Scholar]
- 29. Krishnan S., Chen S., Turcatel G., Arditi M., Prasadarao N. V. (2013) Regulation of Toll-like receptor 2 interaction with Ecgp96 controls Escherichia coli K1 invasion of brain endothelial cells. Cell. Microbiol. 15, 63–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krishnan S., Prasadarao N. V. (2014) Identification of minimum carbohydrate moiety in N-glycosylation sites of brain endothelial cell glycoprotein 96 for interaction with Escherichia coli K1 outer membrane protein A. Microbes Infect. 16, 540–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mittal R., Gonzalez-Gomez I., Goth K. A., Prasadarao N. V. (2010) Inhibition of inducible nitric oxide controls pathogen load and brain damage by enhancing phagocytosis of Escherichia coli K1 in neonatal meningitis. Am. J. Pathol. 176, 1292–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mittal R., Prasadarao N. V. (2010) Nitric oxide/cGMP signalling induces Escherichia coli K1 receptor expression and modulates the permeability in human brain endothelial cell monolayers during invasion. Cell. Microbiol. 12, 67–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maruvada R., Argon Y., Prasadarao N. V. (2008) Escherichia coli interaction with human brain microvascular endothelial cells induces signal transducer and activator of transcription 3 association with the C-terminal domain of Ec-gp96, the outer membrane protein A receptor for invasion. Cell. Microbiol. 10, 2326–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim Y. V., Pearce D., Kim K. S. (2008) Ca2+/calmodulin-dependent invasion of microvascular endothelial cells of human brain by Escherichia coli K1. Cell Tissue Res. 332, 427–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sukumaran S. K., Prasadarao N. V. (2003) Escherichia coli K1 invasion increases human brain microvascular endothelial cell monolayer permeability by disassembling vascular-endothelial cadherins at tight junctions. J. Infect. Dis. 188, 1295–1309 [DOI] [PubMed] [Google Scholar]
- 36. Krishnan S., Fernandez G. E., Sacks D. B., Prasadarao N. V. (2012) IQGAP1 mediates the disruption of adherens junctions to promote Escherichia coli K1 invasion of brain endothelial cells. Cell. Microbiol. 14, 1415–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maeda T, Sano Y., Abe M., Shimizu F., Kashiwamura Y., Ohtsuki S., Terasaki T., Obinata M., Ueda M., Kanda T. (2013) Establishment and characterization of spinal cord microvascular endothelial cell lines. Clin. Exp. Neuroimmunol. 4, 326–338 [Google Scholar]
- 38. Sano Y., Kashiwamura Y., Abe M., Dieu L., Huwyler J., Shimizu F., Haruki H., Maeda T., Saito K., Tasaki A., Kanda T. (2012) Stable human brain microvascular endothelial cell line retaining its barrier-specific nature independent of the passage number. Clin. Exp. Neuroimmunol. 3, 1–12 [Google Scholar]
- 39. Tokuyasu K. T. (1980) Immunochemistry on ultrathin frozen sections. Histochem. J. 12, 381–403 [DOI] [PubMed] [Google Scholar]
- 40. Khan N. A., Wang Y., Kim K. J., Chung J. W., Wass C. A., Kim K. S. (2002) Cytotoxic necrotizing factor-1 contributes to Escherichia coli K1 invasion of the central nervous system. J. Biol. Chem. 277, 15607–15612 [DOI] [PubMed] [Google Scholar]
- 41. Khan N. A., Kim Y., Shin S., Kim K. S. (2007) FimH-mediated Escherichia coli K1 invasion of human brain microvascular endothelial cells. Cell. Microbiol. 9, 169–178 [DOI] [PubMed] [Google Scholar]
- 42. Shin S., Lu G., Cai M., Kim K. S. (2005) Escherichia coli outer membrane protein A adheres to human brain microvascular endothelial cells. Biochem. Biophys. Res. Commun. 330, 1199–1204 [DOI] [PubMed] [Google Scholar]
- 43. Krishnan S., Shanmuganathan M. V., Behenna D., Stoltz B. M., Prasadarao N. V. (2014) Angiotensin II receptor type 1: a novel target for preventing neonatal meningitis in mice by Escherichia coli K1. J. Infect. Dis. 209, 409–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen H., Lambert N. A. (1997) Inhibition of dendritic calcium influx by activation of G-protein-coupled receptors in the hippocampus. J. Neurophysiol. 78, 3484–3488 [DOI] [PubMed] [Google Scholar]
- 45. Jeong S. W., Ikeda S. R. (2000) Effect of G protein heterotrimer composition on coupling of neurotransmitter receptors to N-type Ca2+ channel modulation in sympathetic neurons. Proc. Natl. Acad. Sci. U.S.A. 97, 907–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jeong S. W., Ikeda S. R. (1998) G protein α subunit G α z couples neurotransmitter receptors to ion channels in sympathetic neurons. Neuron 21, 1201–1212 [DOI] [PubMed] [Google Scholar]
- 47. Wessel F., Winderlich M., Holm M., Frye M., Rivera-Galdos R., Vockel M., Linnepe R., Ipe U., Stadtmann A., Zarbock A., Nottebaum A. F., Vestweber D. (2014) Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol. 15, 223–230 [DOI] [PubMed] [Google Scholar]
- 48. Schellenberg A. E., Buist R., Del Bigio M. R., Toft-Hansen H., Khorooshi R., Owens T., Peeling J. (2012) Blood-brain barrier disruption in CCL2 transgenic mice during pertussis toxin-induced brain inflammation. Fluids Barriers CNS 9, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Richard J. F., Roy M., Audoy-Rémus J., Tremblay P., Vallières L. (2011) Crawling phagocytes recruited in the brain vasculature after pertussis toxin exposure through IL6, ICAM1 and ITGαM. Brain Pathol. 21, 661–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Toft-Hansen H., Buist R., Sun X. J., Schellenberg A., Peeling J., Owens T. (2006) Metalloproteinases control brain inflammation induced by pertussis toxin in mice overexpressing the chemokine CCL2 in the central nervous system. J. Immunol. 177, 7242–7249 [DOI] [PubMed] [Google Scholar]
- 51. Quagliarello V., Scheld W. M. (1992) Bacterial meningitis: pathogenesis, pathophysiology, and progress. N. Engl. J. Med. 327, 864–872 [DOI] [PubMed] [Google Scholar]
- 52. Croxen M. A., Finlay B. B. (2010) Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 8, 26–38 [DOI] [PubMed] [Google Scholar]
- 53. Kim K. S. (2003) Pathogenesis of bacterial meningitis: from bacteraemia to neuronal injury. Nat. Rev. Neurosci. 4, 376–385 [DOI] [PubMed] [Google Scholar]
- 54. Kim K. S. (2008) Mechanisms of microbial traversal of the blood-brain barrier. Nat. Rev. Microbiol. 6, 625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Prasadarao N. V., Wass C. A., Hacker J., Jann K., Kim K. S. (1993) Adhesion of S-fimbriated Escherichia coli to brain glycolipids mediated by sfaA gene-encoded protein of S-fimbriae. J. Biol. Chem. 268, 10356–10363 [PubMed] [Google Scholar]
- 56. Kim K. J., Chung J. W., Kim K. S. (2005) 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 280, 1360–1368 [DOI] [PubMed] [Google Scholar]