

Background: Dysregulation of mechanosensitive gene regulatory pathways is associated with muscular dystrophies.
Results: Genome-wide microRNA analyses revealed dysregulation of pathways controlled by let-7e-5p and miR-98–5p in dystrophic muscle.
Conclusion: Impaired mechanomiR-controlled pathways may contribute to muscular dystrophies.
Significance: MechanomiR-dependent pathways regulating extracellular matrix proteins and myogenesis are potential therapeutic targets for ameliorating muscle fibrosis and regeneration.
Keywords: fibrosis, gene regulation, mechanotransduction, muscular dystrophy, skeletal muscle, diaphragm muscle, mdm mice
Abstract
Muscular dystrophies (MDs) are a heterogeneous group of genetic and neuromuscular disorders, which result in severe loss of motor ability and skeletal muscle mass and function. Aberrant mechanotransduction and dysregulated-microRNA pathways are often associated with the progression of MD. Here, we hypothesized that dysregulation of mechanosensitive microRNAs (mechanomiRs) in dystrophic skeletal muscle plays a major role in the progression of MD. To test our hypothesis, we performed a genome-wide expression profile of anisotropically regulated mechanomiRs and bioinformatically analyzed their target gene networks. We assessed their functional roles in the advancement of MD using diaphragm muscles from mdm (MD with myositis) mice, an animal model of human tibial MD (titinopathy), and their wild-type littermates. We were able to show that ex vivo anisotropic mechanical stretch significantly alters the miRNA expression profile in diaphragm muscles from WT and mdm mice; as a result, some of the genes associated with MDs are dysregulated in mdm mice due to differential regulation of a distinct set of mechanomiRs. Interestingly, we found a contrasting expression pattern of the highly expressed let-7 family mechanomiRs, let-7e-5p and miR-98–5p, and their target genes associated with the extracellular matrix and TGF-β pathways, respectively, between WT and mdm mice. Gain- and loss-of-function analysis of let-7e-5p in myocytes isolated from the diaphragms of WT and mdm mice confirmed Col1a1, Col1a2, Col3a1, Col24a1, Col27a1, Itga1, Itga4, Scd1, and Thbs1 as target genes of let-7e-5p. Furthermore, we found that miR-98 negatively regulates myoblast differentiation. Our study therefore introduces additional biological players in the regulation of skeletal muscle structure and myogenesis that may contribute to unexplained disorders of MD.
Introduction
Skeletal muscle is a highly plastic tissue that has the ability to adapt to physiological and non-physiological conditions in response to mechanical stress. Unfortunately, this plasticity is completely absent in muscular dystrophies (MDs),3 a heterogeneous group of genetic and neuromuscular disorders. MD causes severe loss of motor ability, skeletal muscle mass, strength, and function, mostly due to the loss of function of structural proteins such as dystrophin (Becker and Duchenne MDs) (1, 2) or due to the mutation of cytoskeletal genes such as titin (tibial distal MD and limb girdle MD) (3).
Mechanotransduction is a ubiquitous biophysical process by which cells sense their physical environment by translating physical forces and deformations into biochemical signals such as changes in intracellular calcium concentration and/or the activation of diverse signaling pathways (4–7). Such signaling pathways are essential for a wide range of cellular functions such as apoptosis, proliferation, differentiation, and migration, which are vital for organ development and cellular homeostasis. Defects in mechanotransduction are implicated in the development of many diseases, ranging from MDs, cardiomyopathies, and loss of hearing to cancer progression and metastasis (8). In Duchenne MD (DMD), the most common form of MD in children whose life expectancy extends only to the teens or early 20s, mutations in the dystrophin gene disrupt the force transmission between the cytoskeleton and the extracellular matrix (ECM) proteins resulting in progressive muscle degeneration (9). We have shown previously that disruption of cytoskeleton-ECM coupling due to loss of dystrophin causes aberrant activation of the MAPK extracellular signal-regulated kinase 1/2 (ERK1/2) signaling and increased expression of the inflammatory cytokines interleukin-1β and tumor necrosis factor-α in response to mechanical stretch in diaphragm muscle from mdx mouse, an animal model of DMD (5, 10). Furthermore, loss of dystrophin in the mdx mouse causes mechanical stress-induced rupture of the plasma membrane, which allows influx of extracellular calcium. This leads to abnormal muscle contraction and physical damage of the cytoskeleton, subsequently resulting in the loss of muscle cells (11). In addition to dystrophin, other cytoskeletal proteins also play a major role in mechanotransduction. For example, we have shown previously that loss of desmin in the desmin null mouse or mutation of the titin gene in mdm mouse (MD with myositis), an animal model of human tibial MD (titinopathy), causes disturbed cellular mechanics, including reduced force generation and altered (passive) cytoskeletal stiffness, which can decrease the relaxation dynamics of myocytes in the diaphragm (12, 13). Alteration in the mechanotransduction pathways in the mdm mouse, caused by a small in-frame deletion within the titin gene, results in an induction of aberrant signaling pathways (7) and impaired myogenic differentiation (14). Other studies from our laboratory have shown that the α7-integrin and sarcoglycan complex serve important mechanical functions in the diaphragm by contributing to passive compliance, viscoelasticity, and modulation of its muscle contractile properties (15, 16). Overall, these studies suggest the roles of cytoskeletal proteins in force transmission and mechanotransduction and their implication in MDs. Therefore, investigating the relative contributions of mechanotransduction to muscular dysfunction may have important clinical implications, because defects in mechanotransduction pathways could potentially be attenuated with genetic and/or pharmacological reagents. However, information on the genome-wide mechanosensitive gene regulatory networks is essentially lacking. Identification of changes in the global gene-regulatory network in response to normal and defective mechanotransduction could potentially lead to new paths to therapeutic approaches to MDs.
MicroRNAs (miRNAs) are the most abundant and well studied small noncoding RNAs, which act as “master switches” of the genome to regulate the expression of diverse proteins and orchestrate multiple cellular pathways (17). Changes in miRNA expression profiles are often associated with skeletal muscle diseases including MDs (Refs. 18–20 and references therein). Therefore miRNAs are not only used as diagnostic and prognostic markers, but they also may reveal deep insights into disease pathology. Several miRNAs are ubiquitously expressed, whereas some are tissue-specific and others are enriched in specific tissues. For example, miR-1, miR-133a, miR-133b, miR-206, miR-208, miR-208b, miR-486, and miR-499 are skeletal muscle-enriched miRNAs. These specific miRNAs play crucial roles in skeletal muscle functions in health and disease, and therefore they are called myomiRs (18, 21, 22). Using microarray technology, we pioneered the discovery of several mechanosensitive microRNAs (hereafter, we use the term mechanomiRs) in human airway smooth muscle cell. For example, we uncovered a novel mechanosensing role of miR-26a in regulating an airway smooth muscle phenotype (23). Subsequently, others have identified and demonstrated the role of individual mechanomiR in the pathogenicity of human diseases (24–31). Interestingly, we have shown previously that loss of desmin protein in mice alters the miRNA expression profile and induces airway smooth muscle cell hypertrophy as a consequence of miR-26a up-regulation (32), suggesting a potential key role for the cytoskeletal protein in regulating miRNAs. It is important to note however that information on the genome-wide expression profile of mechanomiRs and their target gene regulatory networks associated with human diseases, including MDs, is completely lacking.
Force transmission in skeletal muscles varies greatly with muscle fiber architecture (33). Whereas the in vivo skeletal muscles of either the hindlimb or forelimb are essentially mechanically loaded along the direction of the muscle fiber, the diaphragm muscle is mechanically loaded with trans-diaphragmatic pressure. Therefore, the diaphragm muscles are subjected to mechanical forces not only in the direction of the muscle fibers but also in a direction transverse to the fibers (33). The diaphragm, in particular, is the principal pressure-generating skeletal muscle that drives ventilation. Alteration of its mechanical properties could contribute to compromised respiratory muscle function. It is therefore important to determine how mechanotransduction is executed in the diaphragm muscle fibers in response to longitudinal and transverse directional forces. In agreement with this, we have shown previously the activation of two distinct signaling pathways, which are dependent on the direction of applied mechanical stretch in mouse diaphragm (4, 7). In particular, stretching the diaphragm along the muscle fibers up-regulates Ankrd2 gene expression via the Akt/NF-κB pathway, whereas stretching of the diaphragm transverse to the muscle fibers up-regulates the same gene, Aknrd2, via the Ras/Raf-1/ERK-1/2/AP-1 pathway, strongly suggesting anisotropic regulation of gene expression in the diaphragm (7). Therefore, it was important to uncover potential genome-wide anisotropically regulated mechanomiRs and their target gene regulatory networks in the diaphragm muscle in response to directional mechanical stretches. To explore this, in the present study, we used mdm mice and their WT littermates. Mdm is an early-onset autosomal recessive disease caused by a complex rearrangement mutation in the mouse titin gene that leads to an in-frame 83-amino acid deletion in the N2A region of titin (34). The physiological importance of the titin N2A region is underscored by the severe disease it causes in the mdm mouse. It is important to recognize that the mdm mouse model exhibits a clinical phenotype similar to that of DMD. In particular, the mdm mouse has an early-onset, rapidly progressing, muscle-wasting disease that ends in premature death, likely due to respiratory insufficiency. In addition, the m-line and N2A titin mutations abolish one of many putative binding sites for calpain-3, a muscle-specific protease, and the loss of calpain-3 is linked to limb girdle MD 2A (35). Histopathology of the tibial muscle in human MD has been reported to be similar to that in skeletal muscles of the mdm mouse (3). Therefore, there is a strong rationale to utilize mdm mice to study the molecular mechanisms that cause the progression and severity of MDs in human.
In the present study, using miRNA microarray followed by bioinformatics analyses, we identified differentially regulated mechanomiRs and their predicted target genes and associated biological pathways. We found that mechanomiRs from the let-7 family members, especially let-7e-5p and miR-98–5p, were highly dysregulated in the diaphragm of the mdm mouse. Using an in vitro validation method, we identified Col1a1, Col1a2, Col3a1, Col24a1, Col27a1, Itga1, Itga4, Scd1, and Thbs1 as target genes of let-7e-5p. Furthermore, we identified a role for miR-98–5p as a negative regulator of myoblast differentiation. We also found stretch-induced up-regulation of the TGF-β1 signaling pathway in diaphragm muscle from the mdm mouse. Finally, we were able to revoke the stretch-induced up-regulation of ECM proteins by restoring let-7e-5p and the stretch-induced inhibition of myoblast differentiation by reinstating miR-98–5p in primary myoblasts. Overall, we uncovered a role for mechanomiR in skeletal muscle function and propose that dysregulation of mechanomiRs may be associated with the development of human skeletal muscle diseases including MD.
Experimental Procedures
Mice and ex Vivo Mechanical Loading of Diaphragms
The C57BL/6JWT and B6.B6C3Fe-Ttnmdm J/Cx strain mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the approved animal protocols of the Institutional Animal Care and Use Committee of Baylor College of Medicine. In all experiments, we used diaphragms from 2-week-old mdm mice and their WT littermates. Each experiment was repeated at least three times in three different mice. Tissue preparation and ex vivo stretch were performed as described earlier (4, 7, 13). Briefly, mechanical stretch was applied to the entire costal muscles of the left hemidiaphragm (the right hemidiaphragm muscle was treated as an unstretched control with an environment similar to that of the left hemidiaphragm) by passively stretching the muscle in the longitudinal or transverse direction to the muscle fibers with a passive tension of ∼0.4 newton/cm. This application was equivalent to a passive stress of ∼11 newtons/cm2. A mechanical stretch of nearly 50% from the unstressed state was achieved in the normal diaphragm muscle. It is important to recognize that the unstressed length or the length of the excised muscle is the shortest muscle length and is equivalent to diaphragm muscle length at a lung volume of total lung capacity. The optimal length is about 125% of this unstressed length. Therefore, our stretch of 50% placed the diaphragm at a length equivalent to 120% of optimal muscle length. In each of these two stretch protocols, the diaphragm muscle was held in the stretched state for 15 min and then immediately processed for RNA or cell lysate extraction.
MicroRNA Microarray Analysis
Total RNA samples were isolated from unstretched or stretched hemidiaphragms by TRIzol reagent according to the manufacturer's protocol (Invitrogen). Ten micrograms of total RNA was size-fractionated with a YM-100 Microcon centrifugal filter (Millipore, Billerica, MA) and then used for miRNA expression analysis with a miRNA microarray using their proprietary μParaFlo microfluidic chip containing 700 mouse standard mature miRNA probes (Sanger miRBase 14.0). The fold difference values of altered mechanomiRs were converted into log2 scale.
Gene Functional Analysis
To identify the predicted-biological pathways regulated by mechanomiRs (≥2.0-fold), we used the DIANA miRPath v2.0 Web-based computational tool with a threshold p value of 0.05 and a MicroT threshold value of 0.8 (36). MechanomiRs and their predicted targets were identified using the miRWalk Web-based computational tool, which provides miRNA targets from at least eight established miRNA prediction programs (37).
Real-time PCR Expression Analysis
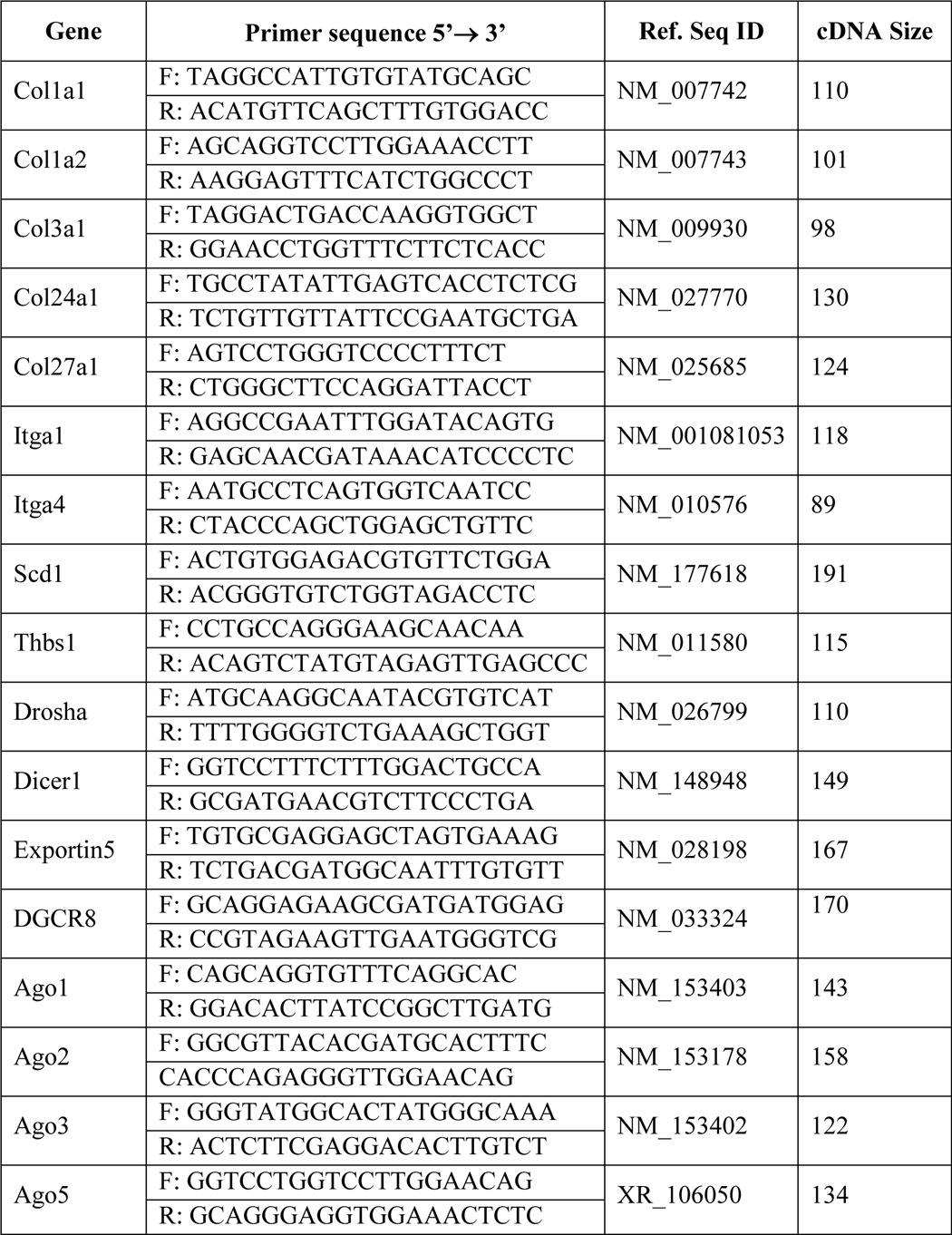
For miRNA expression analysis, we used a TaqMan advanced miRNA cDNA synthesis kit and TaqMan advanced miRNA assays to quantify mature miRNAs according to the manufacturer's protocol (Applied Biosystems, Grand Island, NY). mRNA expression levels were quantified by real-time analysis as mentioned earlier (23). Primer sequences are given in Table 1. All PCR products were verified on an agarose gel stained with ethidium bromide to discriminate between the correct amplification products and the potential primer dimers.
TABLE 1.
Sequences of mouse primers used in real-time PCR
Solution Hybridization Detection Analysis
The expression levels of mature miRNAs were measured by a solution hybridization detection method using mirVana miRNA and BrightStar BioDetect kits (Ambion) as described previously (23).
Immunoblots
Immunoblots were performed as documented earlier (14). Col1a1, Col1a2, Col3a1, Col24a1, Col27a1, Itga1, Itga4, Scd1, Thbs1, and GAPDH antibodies were purchased from Santa Cruz Biotechnology, Inc., Dallas, TX. TGF-β1, SMAD, and pSMAD antibodies were purchased from Cell Signaling, Danvers, MA.
Primary Myoblast Isolation and in Vitro Experiments
Primary myoblasts from the diaphragm muscles of WT and mdm mice were isolated and cultured as described previously (14).
Transfection
Transfection of myocytes was performed as described previously (38), except 2.5 μg of expression vector bearing mmu-let-7e-5p or mmu-miR-98–5p precursor, 400 ng of mmu-let-7e-5p or mmu-miR-98–5p miRCURY LNA knockdown probe (antagomir), or scrambled probe (Exiqon Inc., Woburn, MA) was used.
Statistical Analysis
The results of our experiments are expressed as means ± S.E. Comparisons among different groups were performed by one-way analysis of variance followed by a Bonferroni test. A p value of <0.05 was considered statistically significant. Each experiment was repeated at least three times in three different mice.
Results
Identification of Genome-wide Anisotropically Regulated MechanomiRs in Diaphragm from WT Mouse
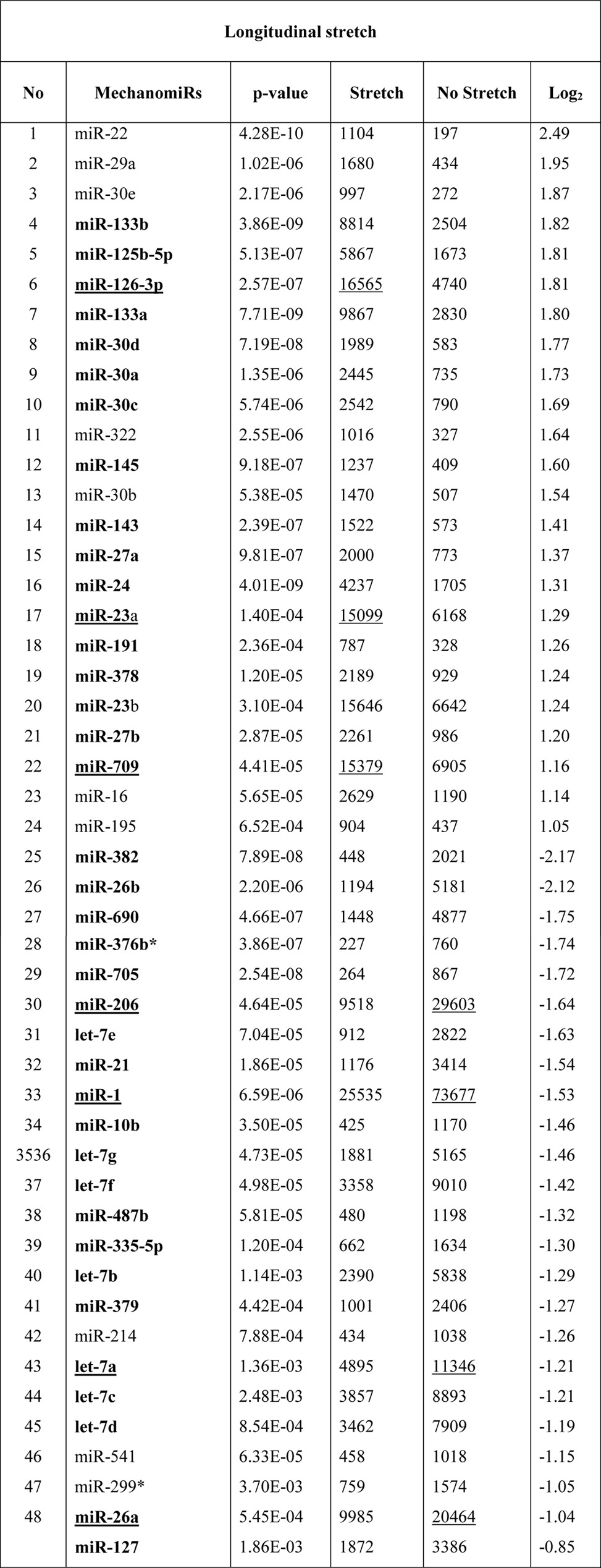
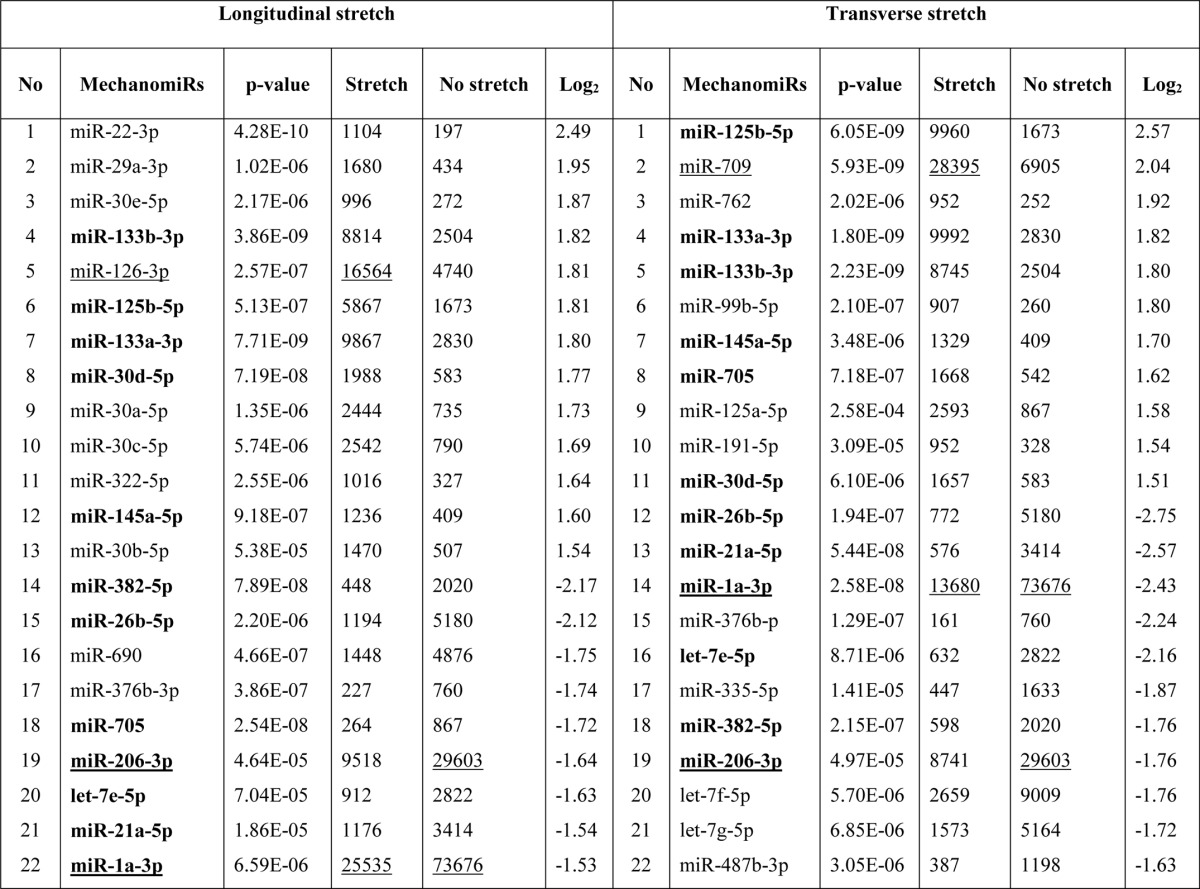
To identify the genome-wide expression of anisotropically regulated mechanomiRs in nondystrophic diaphragm, we performed miRNA microarray analyses using total RNA isolated from either longitudinally stretched or transversely stretched diaphragm from WT mouse. To eradicate false positives and identify miRNAs that had the highest chance of being mechanomiRs, we focused on those miRNAs with potential biological significance (signal intensity levels > 500) (39). Nearly 7.3% of an array of 700 miRNAs expressed a signal intensity > 500. MechanomiRs that were expressed with the highest signal intensity (>10,000) represented only 1.4% of the 700 analyzed miRNAs. Some of the highly expressed mechanomiRs in diaphragms from WT mice were myomiRs (i.e. miR-1, miR-133a, and miR-206), with miR-1 being the most highly expressed mechanomiR (59,747) followed by miR-206 (43,613) and miR-133a (10,374). Non-myomiRs such as miR-26a (20,475) and let-7 family members (>10,000) also displayed high signal intensities (Table 2). Of the 700 miRNA probes examined in the stretched or unstretched diaphragm muscle, the array uncovered the induction of 51 differentially regulated mechanomiRs (18 mechanomiRs were identified in response to only longitudinal stretch (LS), 15 mechanomiRs were identified in response to only transverse stretch (TS), and 18 mechanomiRs were identified in response to either LS or TS) (Fig. 1, A–D, and Table 2). Only 29 mechanomiRs, however, were differentially expressed at >1.5-fold (Table 3 and Fig. 1E) (nine mechanomiRs were identified in response to only LS, 19 mechanomiRs were identified in response to only TS, and a single mechanomiR was identified in response to either LS or TS). Of 29 mechanomiRs, LS and TS differentially regulated 10 (five up-regulated and five down-regulated) and 20 (10 up-regulated and 10 down-regulated) mechanomiRs, respectively, based on a p value of 0.01. By comparison, LS and TS altered 2.6 and 2.1% of the mechanomiRs (LS 35.3% and TS 29.4% of 51), respectively, among the total miRNAs in the array (Fig. 2F). Interestingly, both LS and TS altered 18 mechanomiRs (2.6%, which is 35.3% of 51) either in a similar manner or in an opposite manner (Table 2). For example, whereas LS and TS either up- or down-regulated miR-125a, miR-127, miR-199a, miR-24, miR-26a, miR-26b, miR-379, miR-541, and miR-690, both directional stretches oppositely regulated miR-126, miR-133a, miR-133b, miR-16, miR-206, miR-23a, miR-27a, miR-27b, and miR-382. Of note, some of the differentially regulated mechanomiRs are the previously identified mechanomiRs such as miR-23b, miR-25, miR-30b, miR-126, miR-143, miR-145a, miR-214, and miR-21a in non-skeletal muscle tissues (28). Taken together, using miRNA array, we identified new sets of mechanomiRs from WT mouse diaphragm; the regulation of some of these mechanomiRs is dependent on the direction of stretch. We also identified a few mechanomiRs that are independent of the direction of applied stretch. With regard to highly regulated mechanomiRs (>1.5-fold), compared with LS, TS induced a higher number of mechanomiRs that are entirely different from the mechanomiRs induced by LS (except for miR-382).
TABLE 2.
Differentially regulated mechanomiRs in diaphragm from WT mouse
Bold indicates differentially regulated mechanomiRs by longitudinal and transverse stretches. Underlines indicate differentially regulated mechanomiRs at signal intensities > 10,000.
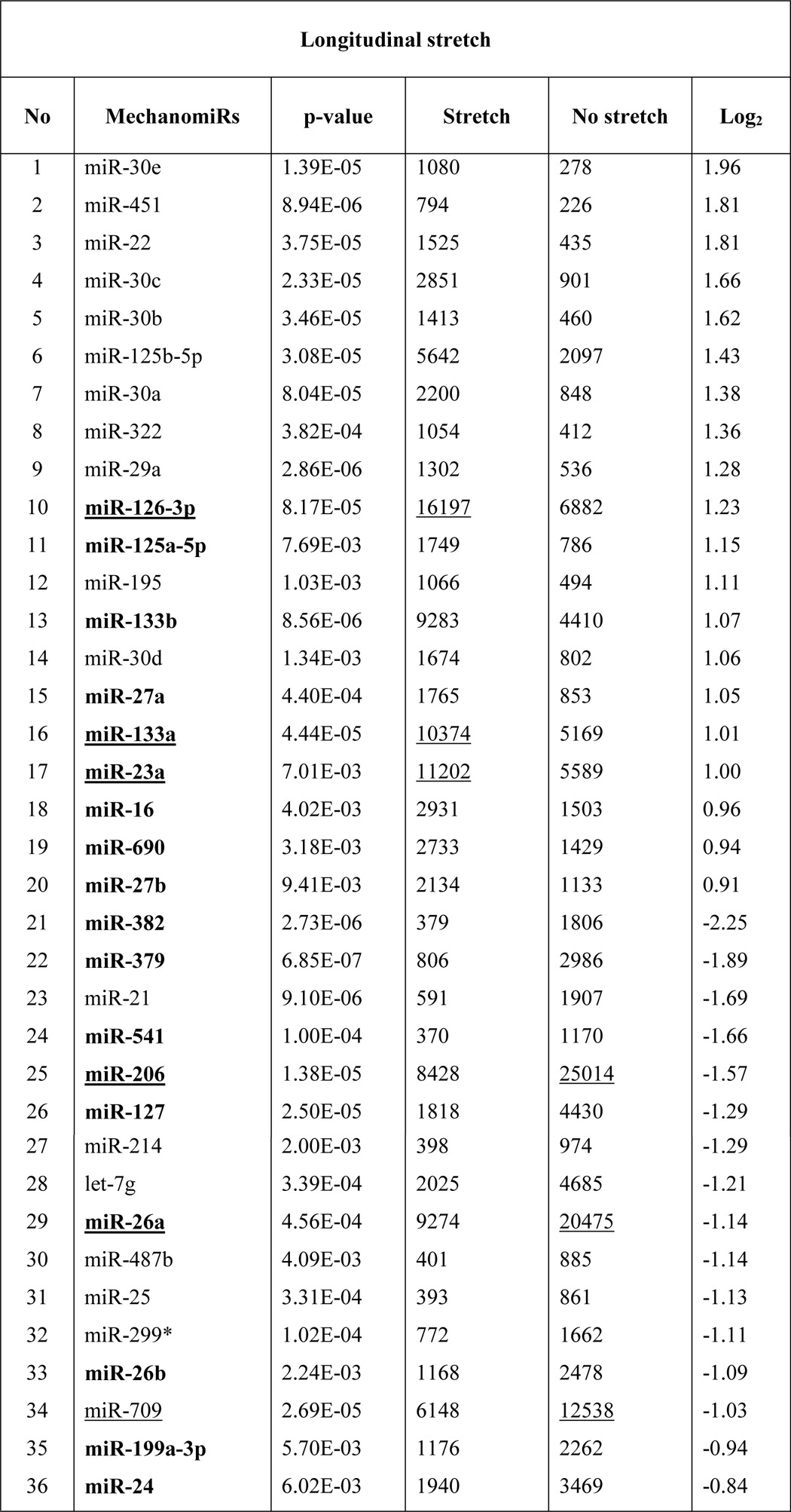
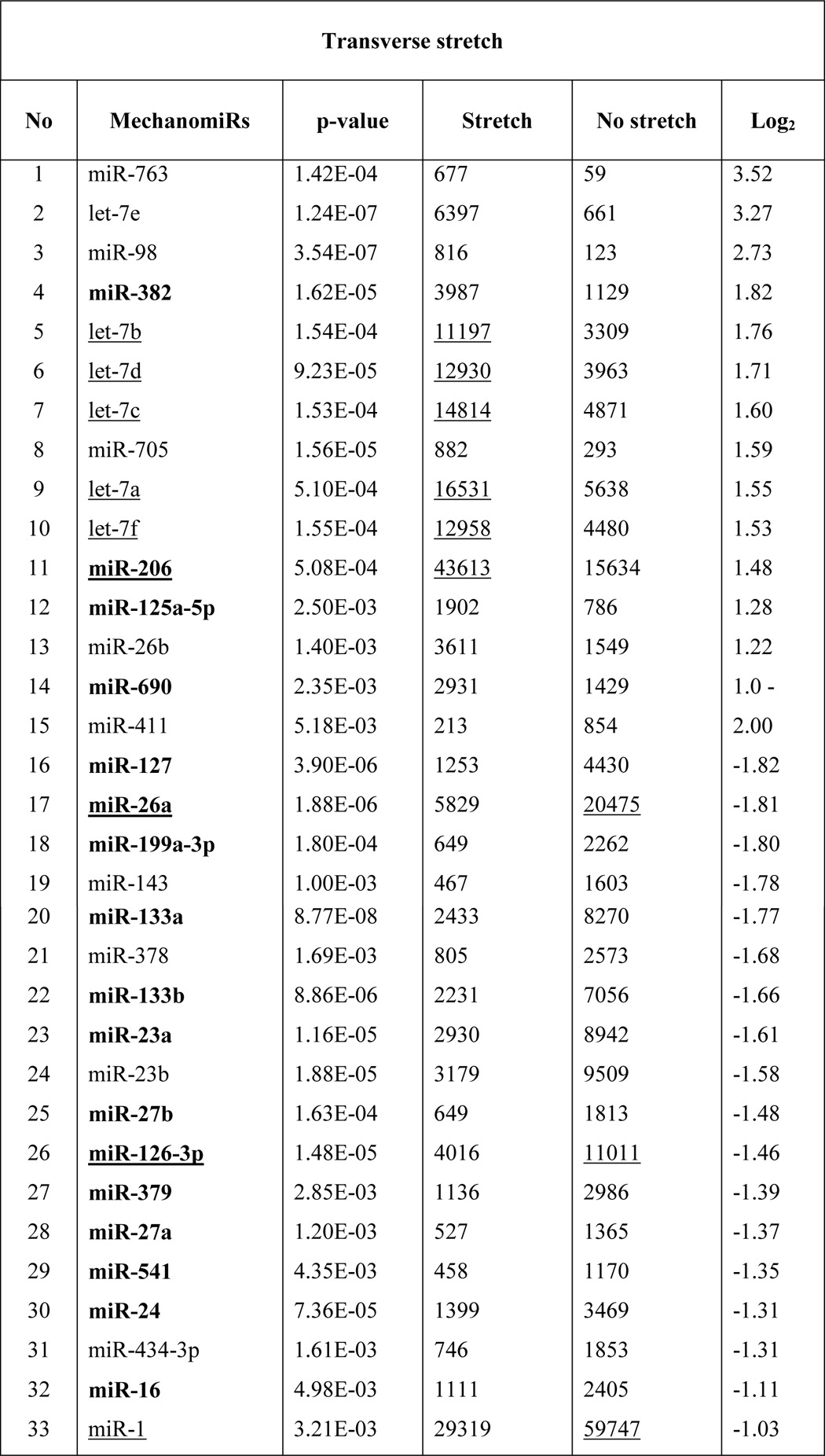
FIGURE 1.

Genome-wide expression profile of mechanomiRs in diaphragm from WT mouse. Total RNA was isolated from either longitudinally or transversely stretched hemidiaphragm from WT mouse and used in miRNA microarray analyses to determine differentially regulated mechanomiRs. A and B, the scatter plot shows log10-transformed signal intensities for each probe labeled with Cy3 for unstretched (control) and Cy5 for LS (A) or TS (B) diaphragm. Each dot represents one miRNA probe. C and D, data on the heat map show differentially expressed mechanomiRs in diaphragm in response to LS (C) or TS (D). E, Venn diagram shows up (red)- and down (blue)-regulated mechanomiRs (>1.5-fold) after corresponding stretch. F, percentage of differentially expressed mechanomiRs to the total number of miRNAs in the array. G, percentage of differentially expressed mechanomiRs based on their genomic location.
TABLE 3.
Differentially regulated mechanomiRs (>1.5-fold) in diaphragm from WT mouse
Bold indicates differentially regulated mechanomiRs by longitudinal and transverse stretches. Underlines indicate differentially regulated mechanomiRs at signal intensities > 10,000.
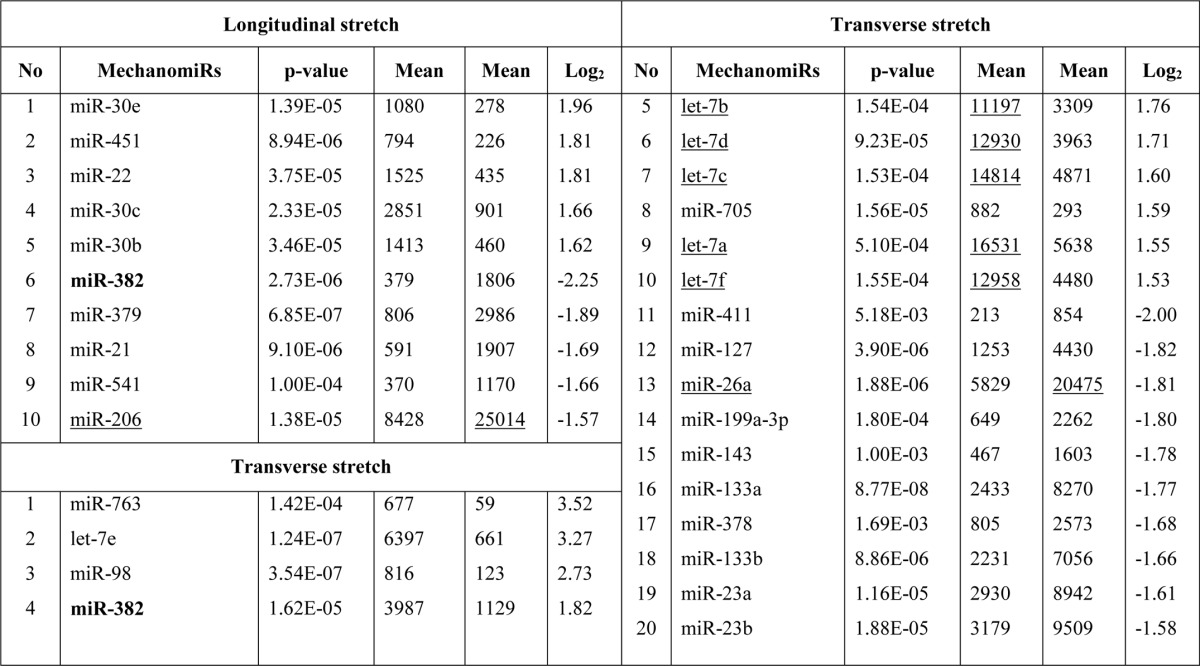
FIGURE 2.

Genome-wide expression profile of mechanomiRs in diaphragm from mdm mouse. Total RNA was isolated from longitudinally or transversely stretched hemidiaphragm from mdm mouse and used in miRNA microarray analyses to determine differentially regulated mechanomiRs. A and B, the scatter plot shows log10-transformed signal intensities for each probe labeled with Cy3 for unstretched (control) and Cy5 for LS (A) or TS (B) diaphragm. Each dot represents one miRNA probe. C and D, data on the heat map show mechanomiRs differentially expressed in diaphragm in response to LS (C) or TS (D). E, Venn diagram shows up (red)- and down (blue)-regulated mechanomiRs (>1.5-fold) after stretch. F, percentage of differentially expressed mechanomiRs to the total number of miRNAs in the array. G, percentage of differentially expressed mechanomiRs based on their genomic location.
Next, we grouped the mechanomiRs based on their genomic locations. We found that 25 (69.4%) and 26 (78.7%) mechanomiRs induced by LS and TS, respectively, were intragenic miRNAs, which are the highest number of mechanomiRs in the present study (Fig. 1G). On the other hand, 11 (15.9%) and seven (10.1%) mechanomiRs induced by LS and TS, respectively, were intergenic miRNAs, suggesting global regulation of mechanomiRs. However, a small number of mechanomiRs induced by LS (miR-2132 and miR-2142) or TS (miR-2142, miR-1224 and miR-805) were not included due to their prediction as “dead” miRNAs according to miRBase.
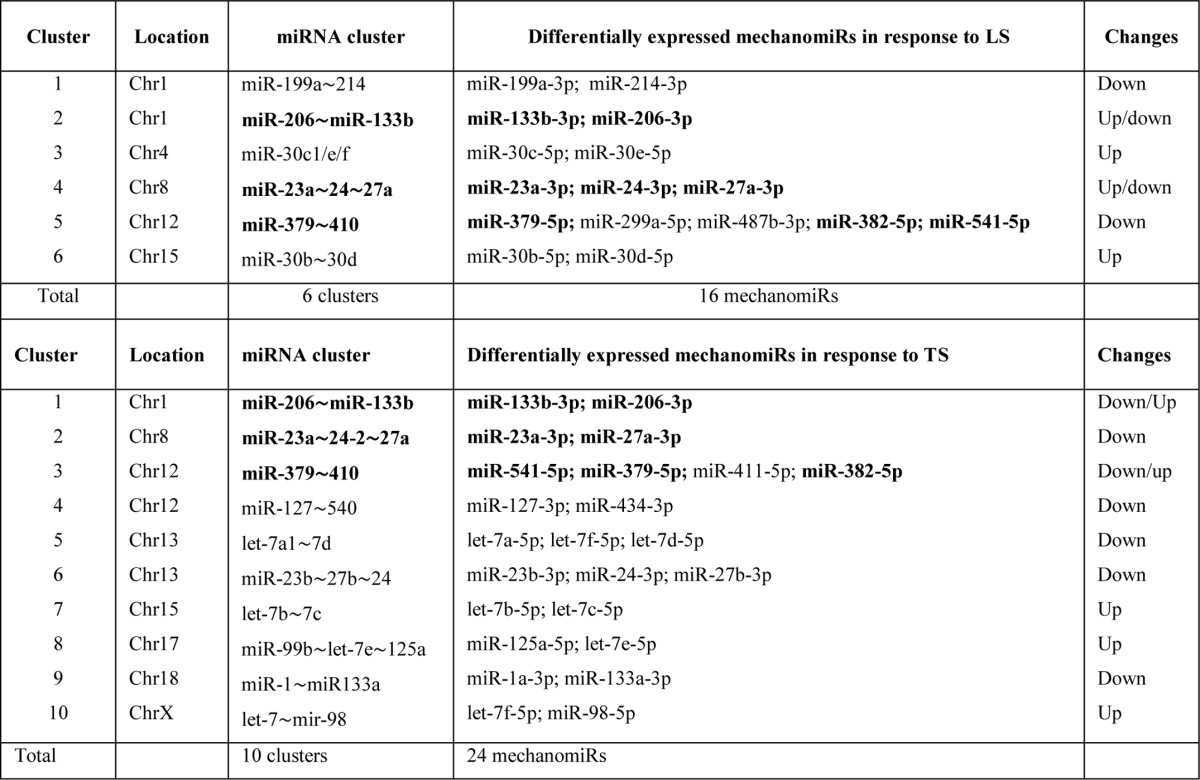
In addition, a significant number of mechanomiRs were derived from close genomic loci (<10 kb), encoded by bi- or polycistronic transcriptional units, which generate multiple miRNAs called miRNA clusters. Interestingly, these miRNA clusters have two different modes of transcription, yet their coordinated regulation during stretch indicated that they shared common regulatory features. In the present study, we found transcription of 40 mechanomiRs as clusters. LS induced the transcription of six clusters, which contained 16 mechanomiRs, whereas TS induced the transcription of 10 clusters, which contained 24 mechanomiRs (Table 4). Interestingly, some of the clusters of mechanomiRs were similar for LS and TS, suggesting that these clusters of mechanomiRs may have similar and/or coordinated biological functions.
TABLE 4.
MechanomiRs and their corresponding miRNA gene clusters in diaphragm from WT mouse
Bold indicates differentially regulated mechanomiRs by longitudinal and transverse stretches.
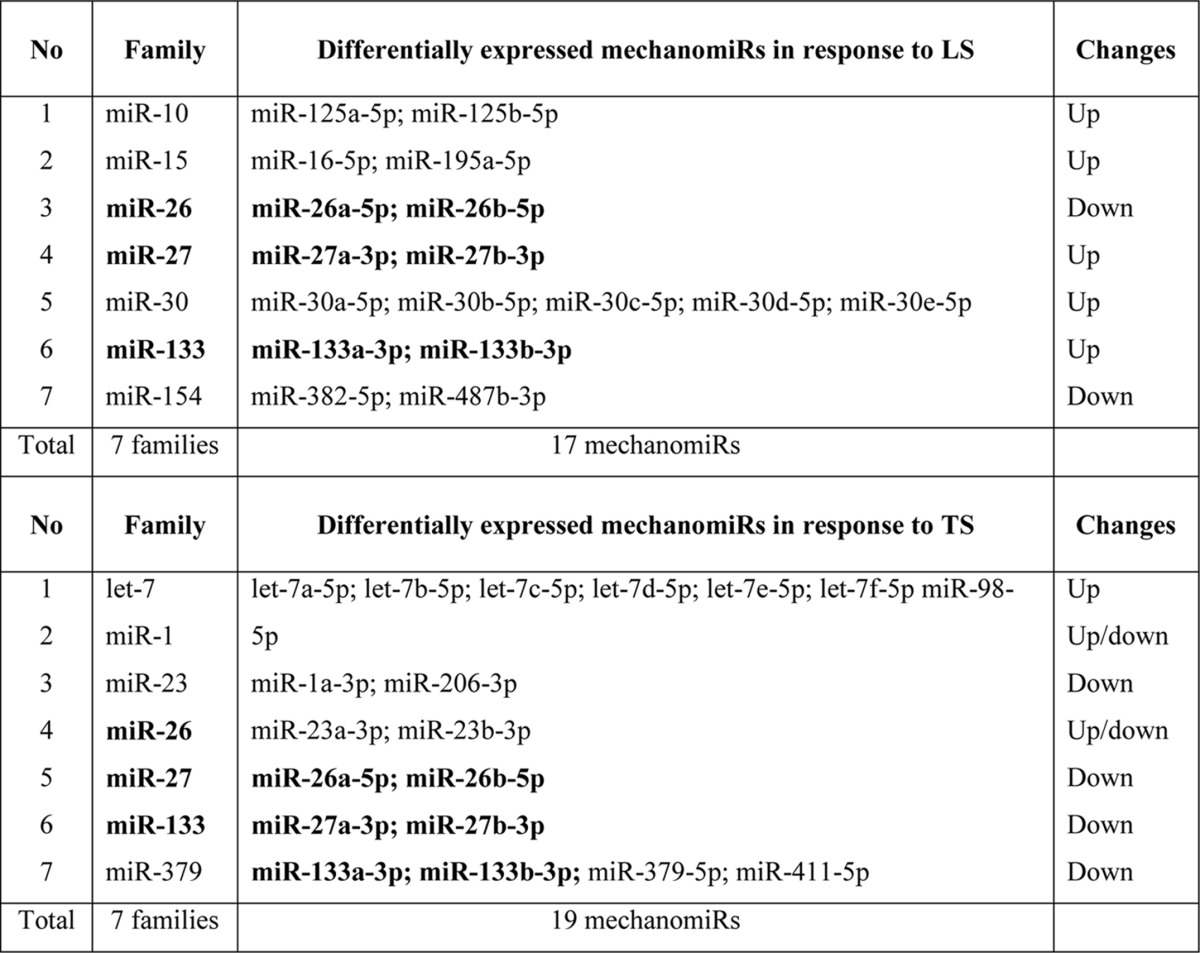
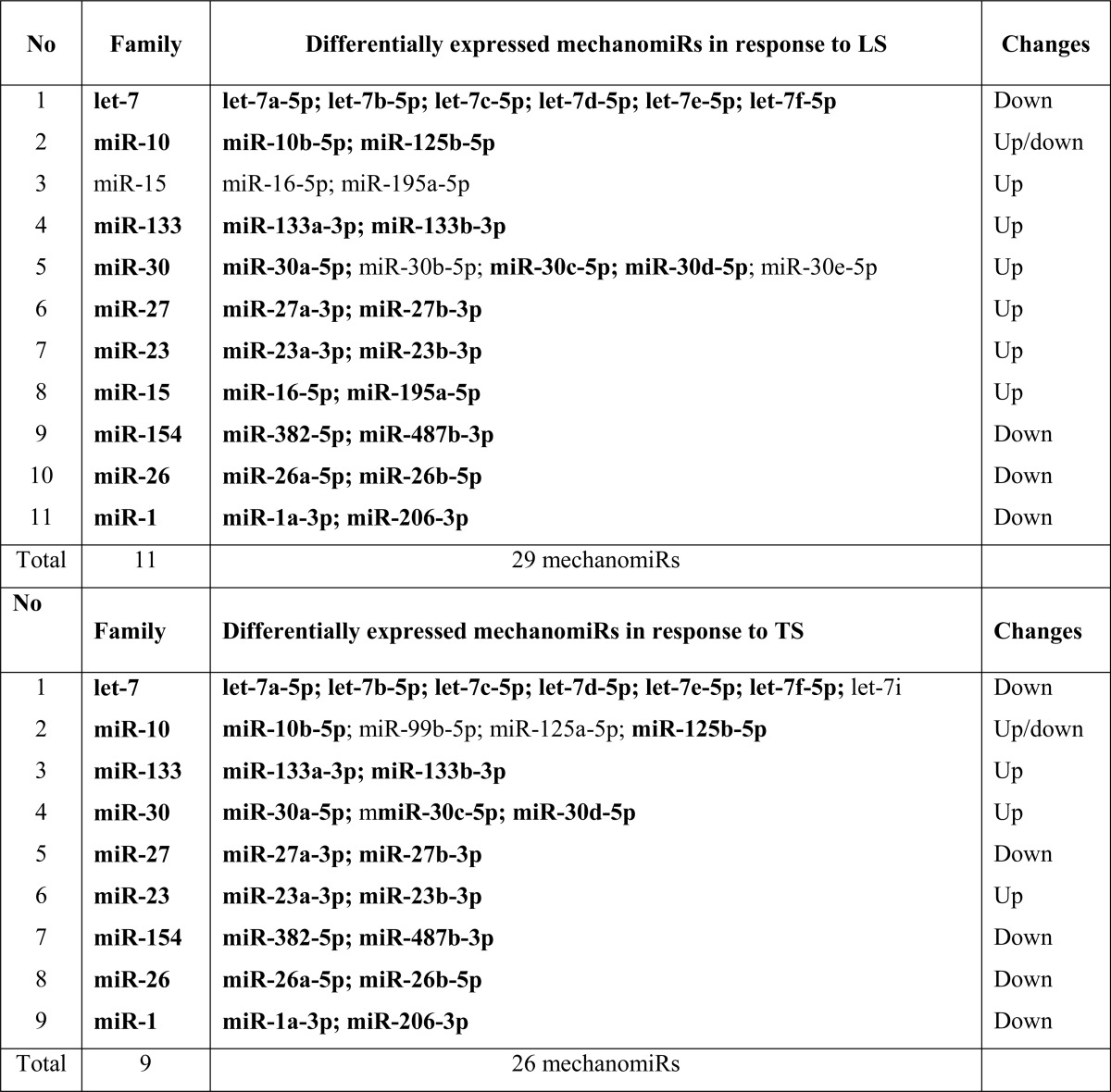
Several differentially regulated mechanomiRs also fell into the same miRNA family. For example, LS and TS induced 17 and 19 mechanomiRs, respectively, which fell into 14 families. Interestingly, whereas LS and TS together induced the miR-26, miR-27, and miR-133 families, mechanomiRs belonging to the miR-10, miR-15, miR-30, and miR-154 families were induced only by LS, and let-7, miR-1, miR-23 and miR-379 were induced only by TS (Table 5). Because members within the same miRNA family share 5′-seed sequences, which are highly conserved 7- or 8-mer sequences within miRNAs (e.g. let-7 family), the identification of mechanomiRs in the same family may have target specificity and similar and/or coordinated biological functions. These findings suggest that the alteration in the mechanomiR expression profile occurred in a regulated manner.
TABLE 5.
MechanomiRs and their families in diaphragm from WT mouse
Bold indicates differentially regulated mechanomiRs by longitudinal and transverse stretches.
Identification of Genome-wide MechanomiRs in Dystrophic Skeletal Muscle
Although other investigators uncovered a role for miRNAs in MDs in both humans (40–43) and mice (44–46), no studies have been designed specifically to uncover a role for mechanomiRs in MDs. In the current study we determined the pattern of regulation of mechanomiRs in dystrophic skeletal muscle. We utilized diaphragm muscle from mdm mouse and followed the same protocol that we had used to study diaphragm muscle from the WT mouse. As in the WT mouse, the mechanomiRs, expressed at a signal intensity of >500, were 7.8% of the total miRNAs in the array. MechanomiRs that were expressed at the highest signal intensity (>10,000) represented a much smaller fraction of the total miRNAs analyzed (1%). Some of the highly expressed mechanomiRs in mdm mice were also myomiRs (i.e. miR-1, miR-133a/b, and miR-206), with miR-1 being the most highly expressed miRNA (73,677) followed by miR-206 (29,603). Non-myomiRs such as miR-709 (28,395) and miR-26a (20,464) were also expressed at high signal intensities (Table 6). Of the 700 miRNA probes examined in the stretched and unstretched diaphragms from mdm mice (Fig. 2, A–D), the array uncovered the induction of 55 differentially regulated mechanomiRs with signal intensity levels of >500 (Table 6). In particular, we identified 10 mechanomiRs in response to LS only, and seven mechanomiRs were identified in response to TS only. In addition, we identified 38 mechanomiRs in response to either LS or TS). Unlike in the WT mouse, LS and TS differentially regulated equal number of mechanomiRs (22 each) at >1.5-fold (Table and Fig. 2E). Of 22 mechanomiRs, LS up-regulated 13 and down-regulated nine mechanomiRs and TS up- and down-regulated 11 mechanomiRs independently based on a p value of 0.01. By comparison, LS and TS altered 1.4 and 1% of the mechanomiRs, respectively, among the total miRNAs in the array, suggesting that both directional stretches altered nearly the same number of mechanomiRs in the mdm mouse diaphragm (Fig. 2F). Interestingly, both LS and TS altered 38 mechanomiRs (5.4%, which is 69% of 55) either in a similar manner or in an opposite manner (Table 6). As in the WT mouse, although some mechanomiRs were regulated by a similar pattern by stretch in both directions, a few mechanomiRs were oppositely regulated by LS and TS. Of note, a subset of mechanomiRs identified previously by other investigators (28) (miR-23b-3p, miR-25, miR-30b-5p, miR-126–3p, miR-143–3p, miR-145a-5p, miR-214, and miR-21a-5p) were also differentially regulated by LS and TS in the mdm mouse diaphragm. Taken together, although the diaphragms from WT and mdm mice differentially regulated a similar number of mechanomiRs (signal intensity > 500), the mdm mouse diaphragms showed a higher number of mechanomiRs; these were regulated >1.5-fold and included most of the previously identified mechanomiRs.
TABLE 6.
Differentially regulated mechanomiRs in diaphragm from mdm mouse
Bold indicates differentially regulated mechanomiRs by longitudinal and transverse stretches. Underlines indicate differentially regulated mechanomiRs at signal intensities > 10,000.
Next, we determined the genomic location of the mechanomiRs in mdm mouse diaphragm. We identified that 36 mechanomiRs distinctly induced by LS (70%) and TS (65%) were intragenic miRNAs (Fig. 2G), whereas 12 (23%) and nine (16%) mechanomiRs induced by LS and TS, respectively, were intergenic miRNAs (Fig. 2G). We also found 21 clusters of mechanomiR that were transcribed by LS and TS together, in which LS induced the transcription of 12 clusters (containing 30 mechanomiRs) and TS induced the transcription of nine clusters (containing 23 mechanomiRs) (Table 8). Furthermore, LS and TS induced 29 and 26 mechanomiRs, respectively, which fell into 11 and nine families, respectively. Interestingly, except for the miR-15 family, most of the mechanomiR families were induced by both LS and TS (Table 9).
TABLE 8.
MechanomiRs and their corresponding miRNA gene clusters in diaphragm from mdm mouse
Bold indicates differentially regulated mechanomiRs by longitudinal and transverse stretches. Underlines indicate differentially regulated mechanomiRs at signal intensities > 10,000.
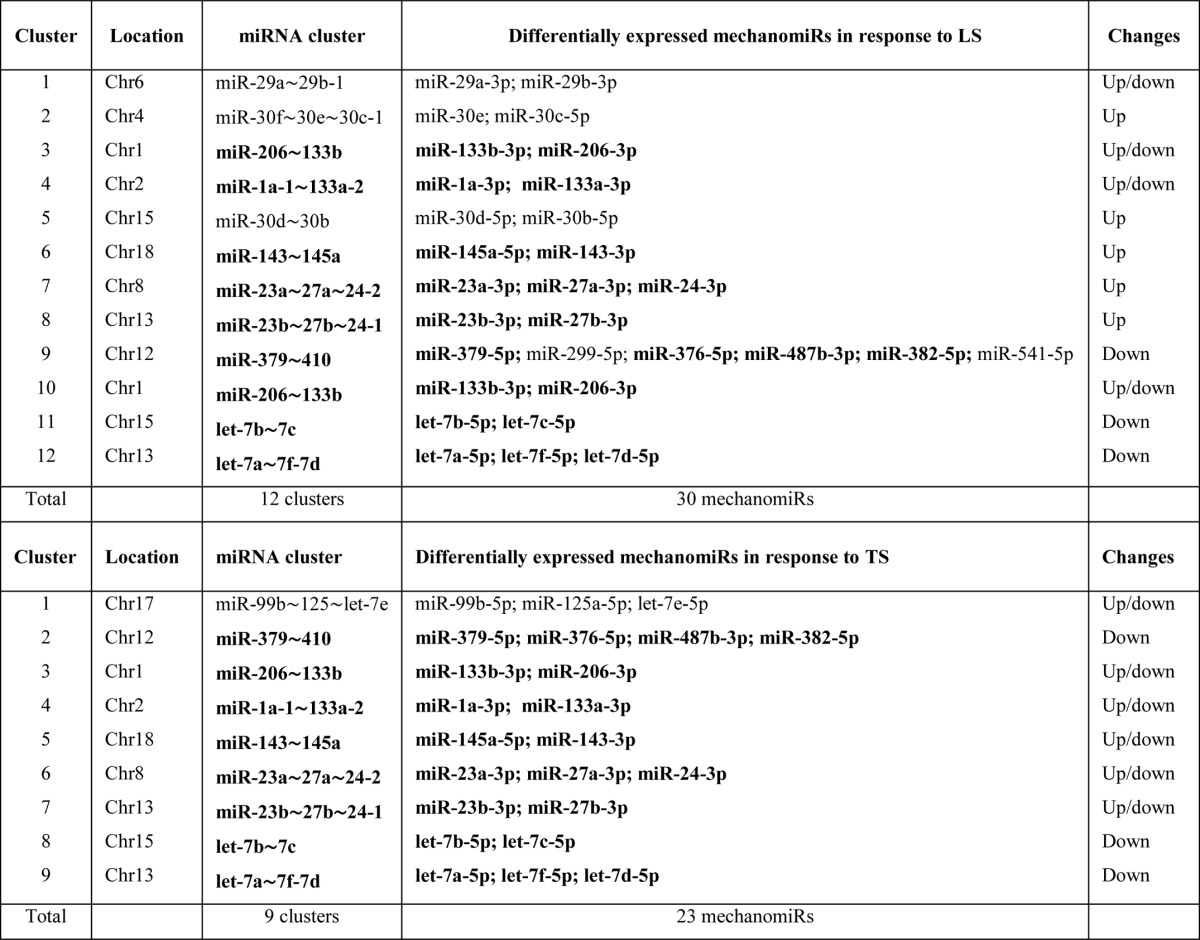
TABLE 9.
MechanomiRs and their families in diaphragm from mdm mouse
Bold indicates differentially regulated mechanomiRs by longitudinal and transverse stretches.
To validate the miRNA microarray data, we measured the expression levels of more than 2.0-fold differentially regulated mechanomiRs by real-time PCR, which confirmed the levels of miR-30e-5p, miR-382–5p, miR-379–5p, miR-763, let-7e-5p, miR-98–5p, miR-22–3p, miR-29a-3p, miR-382–5p, miR-26b-5p, miR-125b-5p, and miR-709 mechanomiRs as more than 2.0-fold (Fig. 3).
FIGURE 3.

Quantification of mechanomiRs by real-time PCR. A portion of RNA used in the microarray was used in real-time PCR to confirm the expression level of mechanomiRs that were differentially regulated >1.9-fold in the microarray. Shown is the expression level of mechanomiRs in diaphragms from WT (A) and mdm (B) mice. Each bar represents mean ± S.E. (n = 3). *, p < 0.05.
Mechanical Stretch Alters miRNA Synthesis Machinery
To explore the mechanism involved in the regulation of mechanomiRs, we investigated the effect of stretch on the expression of key components of the miRNA synthesis machinery. To this end, using real-time PCR we designed primers for selected genes involved in miRNA biogenesis to quantify their expression levels in diaphragms. We found that stretch significantly up-regulated the nuclear protein Drosha, the cytoplasmic protein Dicer1, and the miRNA export protein exportin-5 in diaphragms from WT and mdm mice. In contrast, stretch did not affect the expression of DGCR8 in diaphragm from WT mouse, whereas a similar stretch increased DGCR8 in diaphragm from mdm mouse (Fig. 4A). Next, we determined the expression of the Argonaute proteins, components of the RNA-induced silencing complex (RISC). Although the mRNA levels of Ago2 and Ago5 were significantly higher in diaphragm from WT mouse, diaphragm from mdm mouse showed an increased level of Ago1, Ago2, Ago3, and Ago5 in response to stretch (Fig. 4B). Interestingly, the overall levels of expression of components of the miRNA synthesis machinery were significantly higher in diaphragm from mdm mouse than those in diaphragm from WT mouse. These data suggest that the higher number of expression of mechanomiRs in diaphragm from mdm mouse could be due to the enhanced activation of number of specific players of the miRNA synthesis machinery.
FIGURE 4.
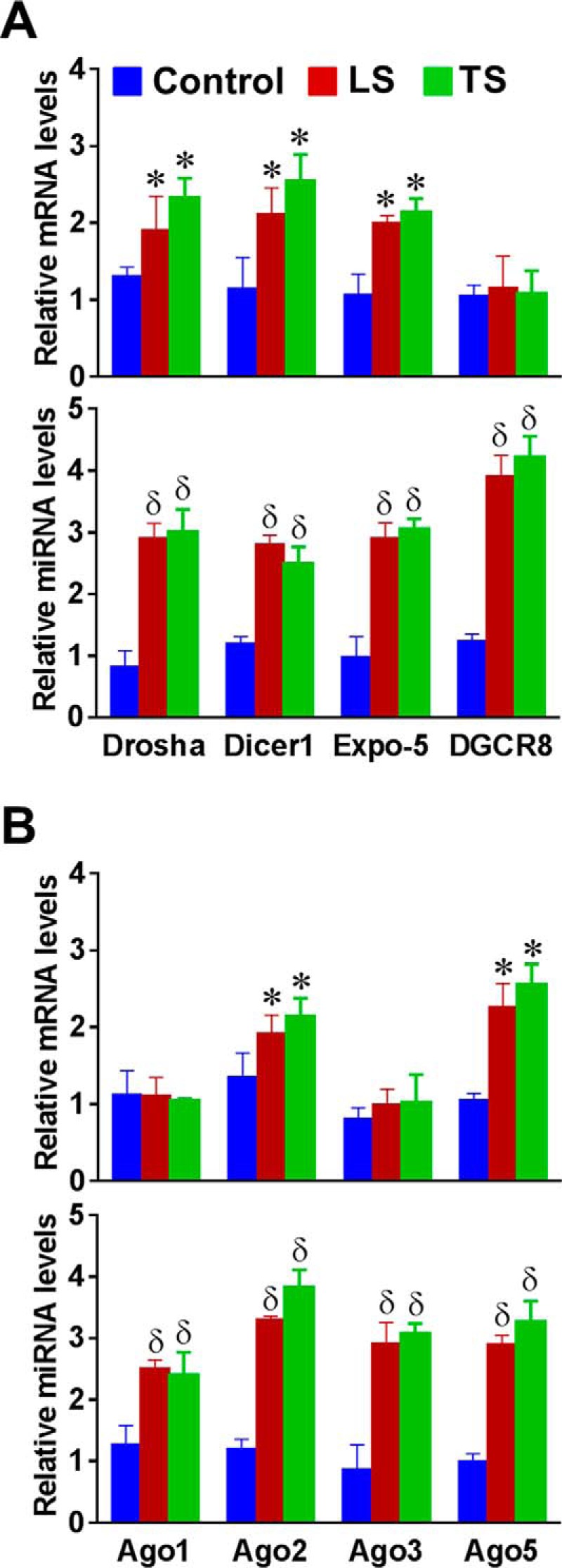
Mechanical stretch up-regulates miRNA synthesis machinery. Total RNA was isolated from control (no stretch), longitudinally stretched or transversally stretched hemidiaphragms from WT or mdm (bottom panels in A and B) mice and used in real-time PCR to determine the expression levels of selected genes that control miRNA biogenesis. Each bar represents mean ± S.E. (n = 4). *, p < 0.05 versus control diaphragm from WT mouse. δ, p < 0.05 versus control diaphragm from mdm mouse.
Identification of MechanomiR-regulated Predicted Biological Pathways in WT and mdm Mice
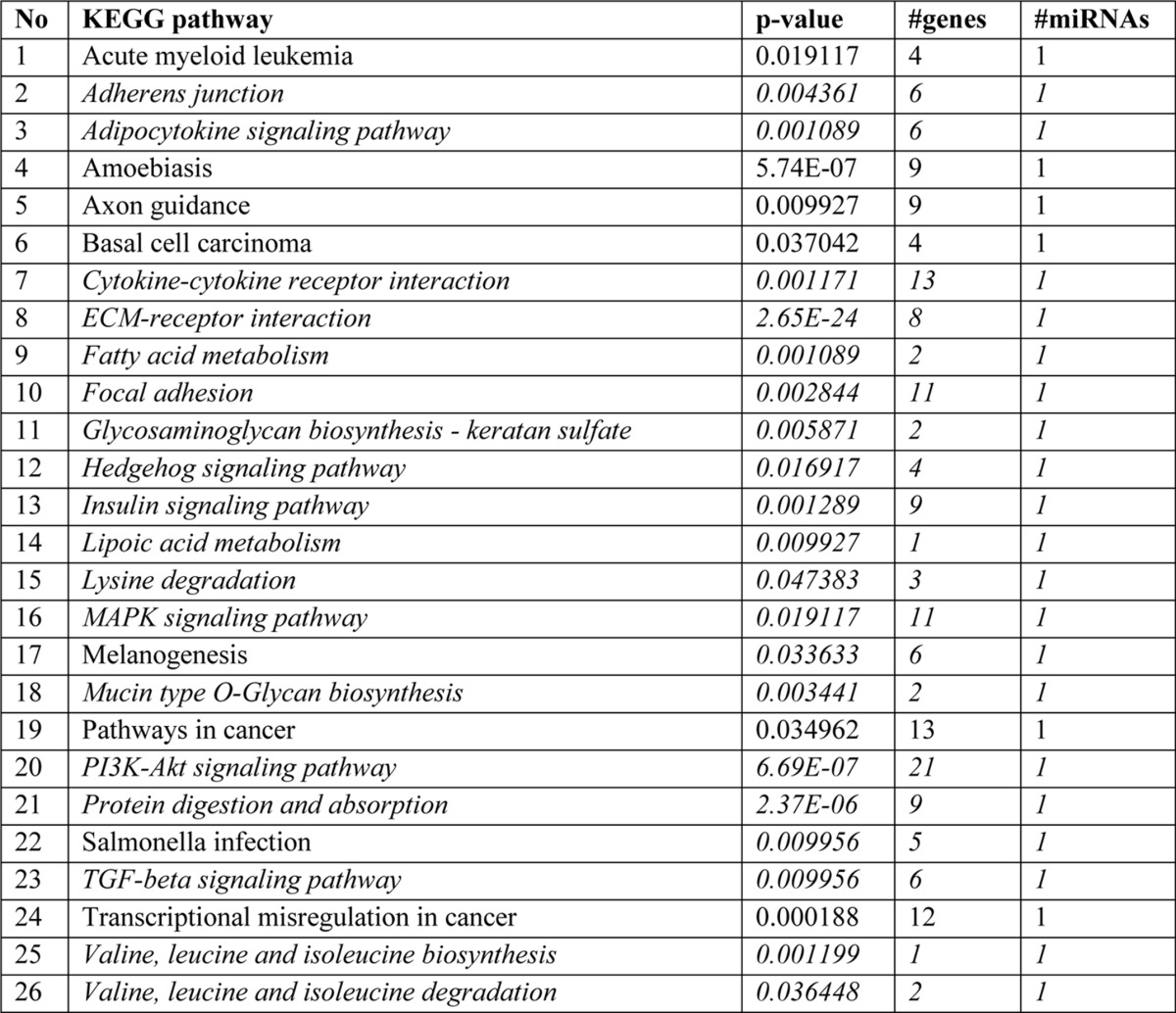
To determine the genome-wide mechanomiR-regulated predicted biological pathways in diaphragms from WT and mdm mice, we first combined both the LS- and TS-regulated mechanomiRs of WT or mdm mice. As a result, the WT and mdm mice totally regulated 51 and 55 mechanomiRs, respectively, at a signal intensity of >500 (Fig. 5A). However, with regard to highly differentially expressed mechanomiRs (>1.5-fold), WT and mdm mice regulated 29 and 31 mechanomiRs, respectively (Fig. 5B). As the mechanomiR numbers were still higher, we selected >2.0-fold differentially regulated mechanomiRs from WT and mdm mice for further analysis. As a result, we found that diaphragm muscle from WT mouse differentially regulated five mechanomiRs at >2.0-fold (three up-regulated and two down-regulated), whereas diaphragm from mdm mouse differentially regulated nine mechanomiRs at >2.0-fold (three up-regulated and six down-regulated). However, only two mechanomiRs (let-7e and miR-382) were found to be commonly regulated in both WT and mdm mice (Fig. 5C). To determine the mechanomiR-regulated biological pathways, we used DIANA miRPath v 2.0 software. The threshold p values for the pathway and MicroT were set to 0.05 and 0.8, respectively, and the KEGG pathway in DIANA miRPath identified 41 (Table 10) and 74 (Table 11) mechanomiR-regulated biological pathways for WT and mdm mice, respectively. In addition, 28 pathways were common for both WT and mdm mice (Fig. 5D). Among those pathways, 19 (WT) and 32 (mdm) pathways were associated with skeletal muscle functions. In addition, our data showed only five and 18 skeletal muscle-associated pathways that were unique to WT and mdm mice, respectively, and 14 pathways were common for both WT and mdm mice (Fig. 5D). According to gene ontology, there are 36 genes are involved in the five pathways in WT mouse (Fig. 5E), whereas 251 gene are involved in 18 pathways in mdm mouse (Fig. 5F). Although both WT and mdm mice shared 14 different pathways, the involvement and appearance of genes in each pathway were higher in diaphragm muscle from mdm mouse (Fig. 6, A and B). For example, the PI3-Akt and MAP kinase signaling pathways involve nearly twice the number of genes in the mdm mouse compared with those in the WT mouse (Fig. 6A). Next, we determined the let-7e-5p-predicted biological pathways in WT and mdm mice. DIANA miRPath provided 24 predicted pathways (Table 12), including 18 skeletal muscle-specific pathways (Fig. 6C). Interestingly, miR-98–5p was highly up-regulated in diaphragm muscle from WT mouse, but its mechanosensitivity was lost in diaphragm muscle from mdm mouse, which led us to explore miR-98–5p-predicted biological pathways. DIANA miRPath provided 26 predicted pathways (Table 13), among which were 18 pathways that are skeletal muscle-specific (Fig. 6D). Finally, we compared the differentially regulated mechanomiRs of mdm diaphragm with the differentially regulated miRNAs of the mdx skeletal muscle. As shown in Fig. 6E, nine differentially regulated miRNAs in mdx skeletal muscle were mechanomiRs that were identified in both WT and mdm mice. However, most of the mechanomiRs identified in the present study were regulated in an opposite manner to that in the skeletal muscles of mdx mice.
FIGURE 5.

Genome-wide mechanomiR-regulated predicted biological pathways in WT and mdm mice. A and B, Venn diagram shows number of mechanomiR-regulated biological pathways. Numbers outside the circles indicate the number of pathways regulated by mechanomiRs at signal intensity >500 (A) or ≥1.5-fold (B) in WT or mdm mice. Similarly, the numbers inside the circles indicate the total number of pathways regulated by mechanomiRs unique to WT or mdm mice or in both phenotypes. C and D, number of mechanomiRs regulated by LS and TS at ≥2.0-fold in WT or mdm mice or in both phenotypes (C) and their biological pathways (D). Numbers outside the circles indicate the number of pathways, and numbers inside the circles indicate the number of pathways unique to WT or mdm mice or in both phenotypes. The numbers in parentheses indicate the number of skeletal muscle-specific pathways. E and F, gene ontology of mechanomiR-regulated pathways in WT (E) and mdm (F) mice. Numbers outside and inside the parenthesis indicate the number of genes and mechanomiRs, respectively, associated with each pathway.
TABLE 10.
Biological pathways regulated by mechanomiRs (>2.0-fold) in diaphragm from WT mouse
Bold indicates pathways commonly regulated by mechanomiRs between WT and mdm mice; italics indicate skeletal muscle-specific pathways; and underlines indicate pathways unique to WT mouse diaphragm.
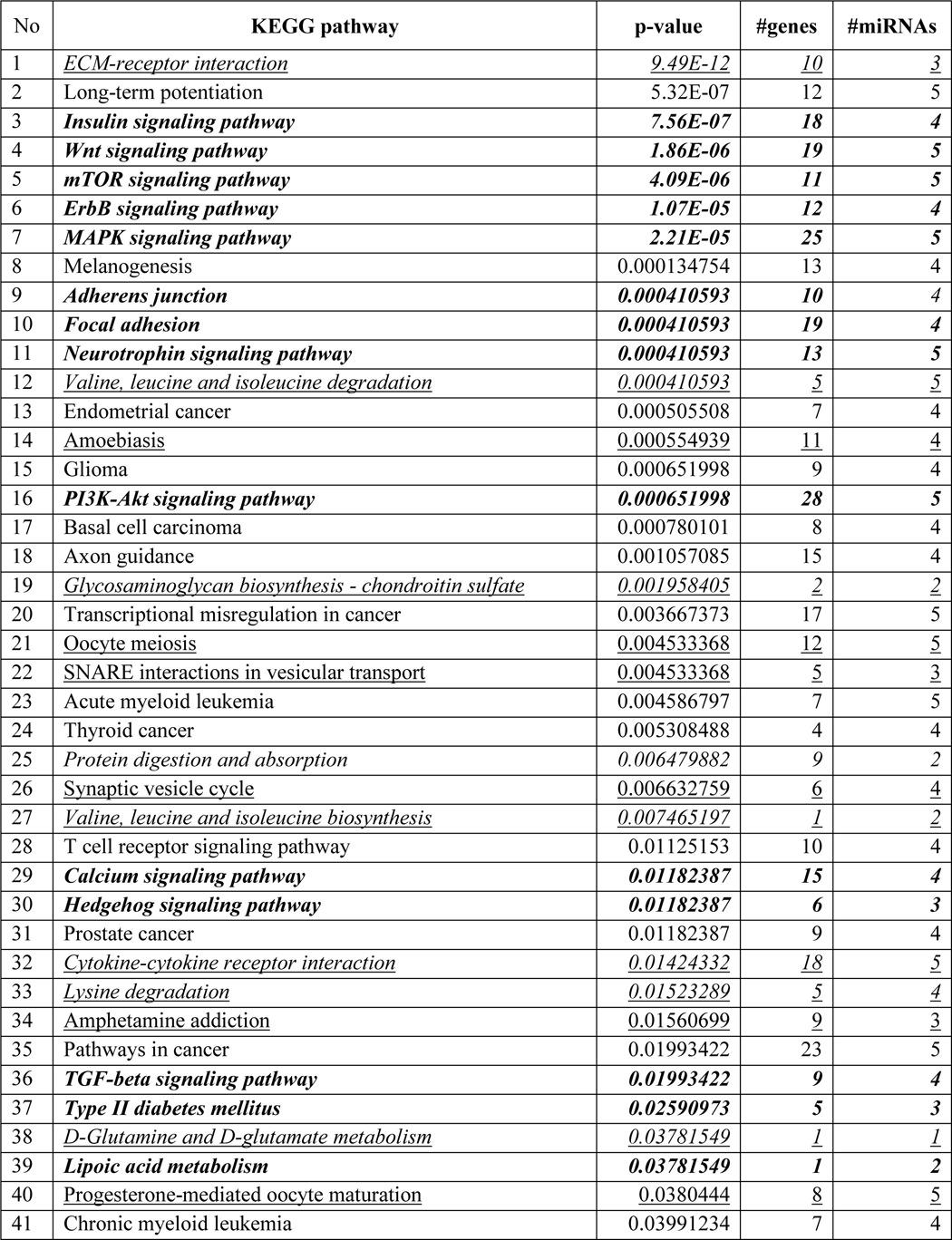
TABLE 11.
Biological pathways regulated by mechanomiRs (>2.0-fold) in diaphragm from mdm mouse
Bold indicates pathways commonly regulated by mechanomiRs of WT and mdm mice; italics indicate skeletal muscle-specific pathways; and underlines indicate pathways unique to mdm mouse diaphragm.
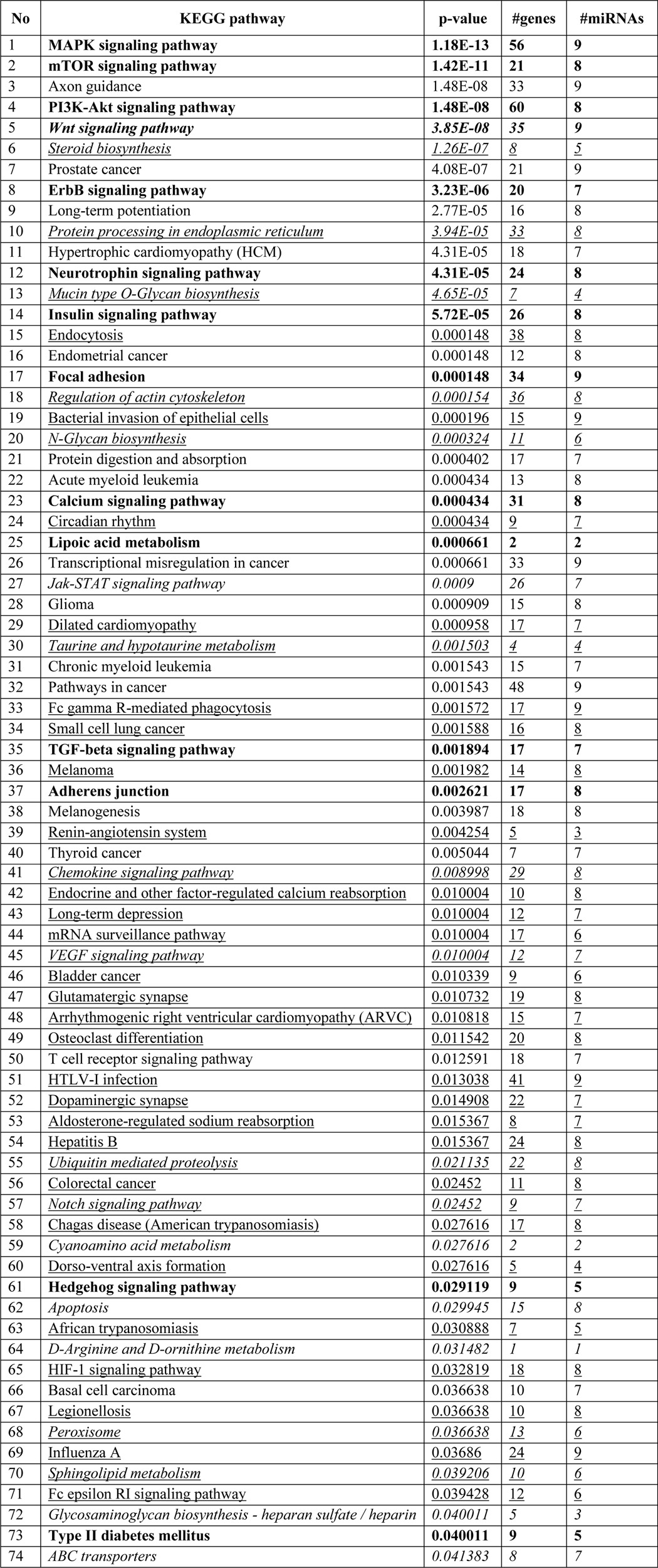
FIGURE 6.

MechanomiR-regulated biological pathways in WT and mdm mice. A, mechanomiR-regulated skeletal muscle-specific pathways in both WT and mdm mice. B, number of genes involved in each pathway (from A) in WT (blue) or mdm (red) mice or in both phenotypes. C and D, let-7e- and miR-98-regulated pathways. E, Venn diagram showing a comparison among the differentially regulated mechanomiRs of WT, mdm, and mdx mice. F, pMBwt were stably transfected with either a pEGP empty vector or a pEGP-let-7e-5p vector. Total RNA was isolated after 24 h of transfection and used in real-time PCR and ribonuclease protection assay to measure let-7e-5p levels. U6 was used as a loading control. Gel pictures are representative of four independent experiments. Each bar represents mean ± S.E. (n = 4). *, p < 0.05.
TABLE 12.
Biological pathways regulated by let-7e-5p
Italics indicate skeletal muscle-specific pathways.
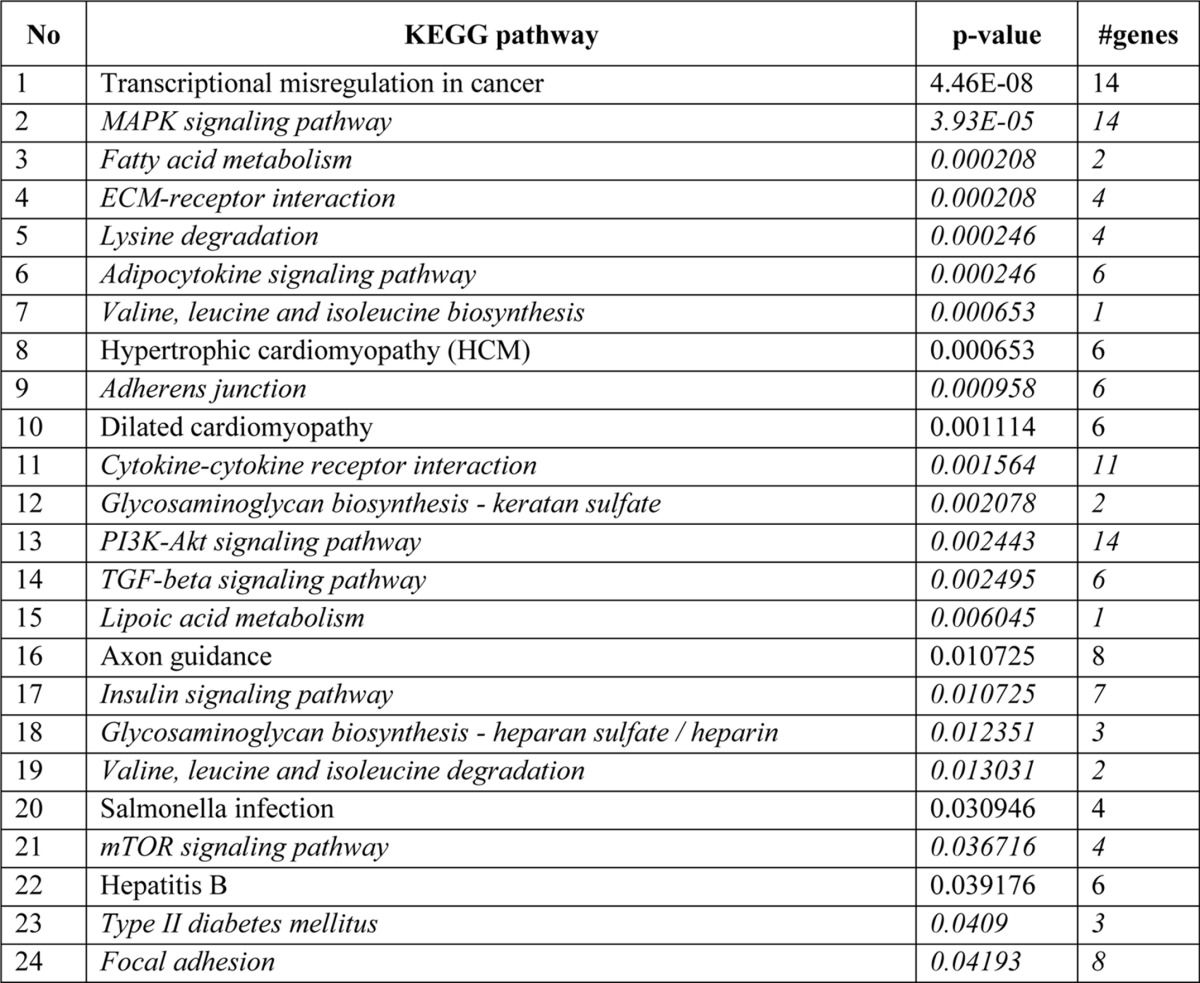
TABLE 13.
Biological pathways regulated by miR-98-5p
Italics indicate skeletal muscle-specific pathways.
Dysregulation of the MechanomiR-let-7e-5p in mdm Mouse Increases ECM Proteins
Interestingly, let-7e-5p is the only mechanomiR that is highly regulated in an opposite manner between the diaphragms of WT and mdm mice. Therefore, we focused our study on determining whether let-7e-5p is potentially involved in the progression of MD in mdm mouse. Although the DIANA miRPath identified 18 pathways related to skeletal muscle functions, ECM-receptor interaction was ranked first (Table 10). Many lines of evidence have shown that the up-regulation of ECM proteins in dystrophic skeletal muscle plays a key role in the development of fibrosis, a hallmark of dystrophic skeletal muscle (47). In the present study, we found that let-7e-5p was highly down-regulated in diaphragm from mdm mouse. These data are consistent with our hypothesis that fibrosis in the skeletal muscles from the mdm mice is caused, at least in part, by let-7e-5p-mediated up-regulation of ECM proteins. To further test our hypothesis, we first determined whether let-7e-5p transcriptionally or post-transcriptionally suppressed endogenous ECM gene expressions. To do this, we isolated primary myoblasts from WT mouse diaphragm (pMBwt) and stably transfected them with either a pEGP empty vector or a pEGP-let-7e-5p vector. pMBwt carrying pEGP-let-7e-5p showed nearly a 3-fold overexpression of let-7e-5p (Fig. 6F). To determine the expression of ECM genes in pMBwt overexpressing let-7e-5p, we isolated total RNA from pEGP or pEGP-let-7e-5p-transfected pMBwt and used it in a custom-made PCR array having 87 genes related to ECM proteins. The array identified that overexpression of let-7e-5p significantly decreased expression of Col1a1, Col1a2, Col3a1, Col24a1, Col27a1, Itga1, Itga4, Scd1, and Thbs1 mRNAs by more than 2-fold (Fig. 7A), suggesting that let-7e-5p targets at least those nine mRNAs of ECM gene. Second, we examined whether stretch could alter the expression of let-7e-5p and its target ECM genes in vitro. To do this, we stretched pMBwt or pMBmdm as described under “Experimental Procedures.” Interestingly, although stretch significantly decreased Col1a1, Col1a2, Col3a1, Col24a1, Col27a1, Itga1, Itga4, Scd1, and Thbs1 mRNA levels in pMBwt, a similar stretch increased the mRNA levels of these genes in pMBmdm (Fig. 7B). Likewise, let-7e-5p expression was also regulated in an opposite manner between pMBwt and pMBmdm (Fig. 7C). These results provide the first experimental evidence of a negative correlation between let-7e-5p and ECM gene expressions in myocytes from WT and mdm mice. To confirm whether the stretch-induced down-regulation of ECM genes in pMBwt was due to the mechanomiR let-7e-5p, we inhibited let-7e-5p in pMBwt with let-7e-5p antagomir. Although the stretching of pMBwt significantly decreased ECM gene expressions, the transfection of pMBwt with let-7e-5p antagomir abolished the stretch-induced ECM gene expression (Fig. 8A). To further confirm that let-7e-5p-regulated ECM gene expressions, we overexpressed let-7e-5p in pMBmdm with a pEGP-let-7e-5p construct. Although stretch up-regulated ECM gene expression in pMBmdm, transfection of the let-7e-5p overexpression construct dramatically decreased the stretch-induced ECM gene expression profile (Fig. 8B). Finally, we sought to determine the expression of let-7e-5p targeting ECM proteins in WT and mdm mouse diaphragms. As expected, whereas stretch down-regulated the ECM proteins Col1a1, Col1a2, Col3a1, Col24a1, Col27a1, Itga1, Itga4, Scd1, and Thbs1 in diaphragm muscle from WT mouse, similar stretch up-regulated those ECM protein from mdm mouse. Overall, these data confirmed that mechanomiR let-7e-5p is accountable for the up-regulation of ECM proteins in the diaphragm muscle from mdm mouse.
FIGURE 7.

Identification of let-7e-5p target genes. pMBwt were stably transfected with pEGP or pEGP-let-7e-5p expression construct. A, ECM gene mRNA levels in let-7e-5p overexpressed in pMBwt were analyzed 24 h after transfection by real-time PCR array. B, expression level of each gene targeted by let-7e-5p was verified in freshly isolated RNA from stretched (ST) or unstretched WT and mdm mice diaphragms by real-time PCR. C, total RNA was isolated from stretched (ST) or unstretched pMBwt and pMBmdm and used in real-time PCR to quantify let-7e-5p levels. Each bar represents mean ± S.E. (n = 4). *, p < 0.05 versus WT or mdm.
FIGURE 8.

In vitro experimental validation of let-7e-5p target genes. A, pMBwt were transfected with let-7e-5p antagomir either in the presence or absence of stretch, and let-7e-5p target gene mRNA levels were determined by real-time PCR. B, pMBmdm were transfected with let-7e-5p overexpression construct and subjected to stretch, or no stretch, and let-7e-5p target gene mRNA levels were determined by real-time PCR. C, the protein levels of let-7e-5p target genes were determined by Western blot using cell lysate isolated from the unstretched (C, control) or stretched (ST) diaphragm of WT or mdm mouse. GAPDH was used as a loading control. Gel pictures are representative of four independent experiments. Each bar represents mean ± S.E. (n = 4). *, p < 0.05, versus NC (negative control); δ, p < 0.05 versus NC + ST.
Diaphragm from mdm Mouse Displays Up-regulation of TGF-β1 Pathway
Studies have shown an increased expression of TGF-β1 gene in skeletal muscles from mdx mice (48–50) and DMD patients (51). Other investigators have demonstrated that activated TGF-β signaling in dystrophic muscles in DMD patients is associated with impaired muscle regeneration and increased fibrosis (51, 52). However, the regulation of TGF-β1 signaling in skeletal muscles from mdm mice is unknown. In the current study, we determined whether stretch could alter the expression of TGF-β1 in diaphragm muscle. As shown in Fig. 9, although stretch reduced TGF-β1 levels in diaphragm muscle from WT mouse (Fig. 9A), similar stretch enhanced TGF-β1 levels in diaphragm muscle from mdm mouse (Fig. 9B). It is well documented that TGF-β1-induced gene transcription is mediated by intracellular substrates known as SMADs (53). Therefore, we sought to determine the expressions of SMAD2 and SMAD3 proteins in response to stretch. Stretching of the diaphragm decreased the phosphorylation of SMAD2/SMAD3 proteins in WT mouse (Fig. 9C). In contrast, stretch elevated the phosphorylation of SMAD2 and SMAD3 proteins in diaphragm muscle from mdm mouse (Fig. 9B), suggesting an opposite regulation of TGF-β1 and activation of SMAD proteins between WT and mdm mice.
FIGURE 9.
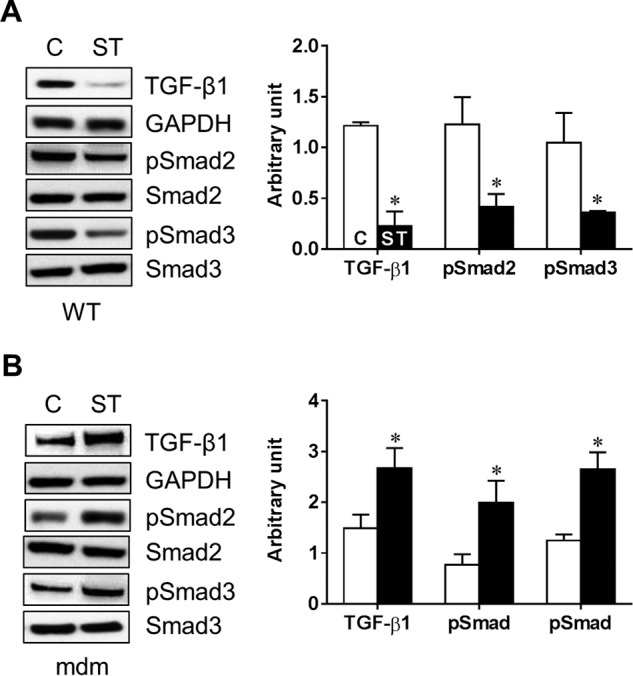
Mechanical stretch alters TGF-β1 signaling pathway. Diaphragm muscle from WT (A) or mdm (B) mouse were subjected to either LS (ST) or left unstretched (C, control). Cell lysate was isolated and used in Western blot to measure the expression levels of TGF-β1, SMAD2, and SMAD3 as well as phosphorylated SMAD2 and SMAD3 (pSMAD). GAPDH was used as a loading control. Gel pictures are representative of four independent experiments. Each bar represents mean ± S.E. (n = 4). *, p < 0.05 versus control.
MechanomiR-98 Negatively Regulates Myoblast Differentiation
We have shown previously that mechanical stretch inhibits differentiation of C2C12 myoblasts (6). To determine whether let-7e-5p and/or miR-98–5p may play a role in controlling the stretch-induced inhibition of myoblast differentiation, we transfected pMBwt with let-7e-5p and/or miR-98–5p antagomirs and cultured them in differentiation medium with stretch for 1 h every 24 h for 5 days. First, we determined whether each antagomir effectively inhibited the respective endogenous mechanomiR. As shown in Fig. 10A, transfection of pMBwt with let-7e-5p and miR-98–5p antagomirs efficiently decreased the stretch-increased endogenous let-7e-5p and miR-98–5p levels, respectively, suggesting the specificity and efficacy of antagomirs. As expected, culture of pMBwt in differentiation medium for 5 days displayed the differentiation of most of the myoblasts into myotubes (Fig. 10B). In agreement with our previous findings (6), stretching pMBwt in differentiation medium for 5 consecutive days reduced the formation of myotubes (Fig. 10C). Interestingly, pMBwt transfected with both antagomirs or with miR-98–5p alone, induced myoblast differentiation in the presence of stretch (Fig. 10C), confirming that miR-98–5p, but not let-7e-5p, regulates the stretch-induced inhibition of myoblast differentiation. These studies also suggest that although let-7e-5p and miR-98–5p belong to the same family with almost identical sequences and synergistically up-regulated by stretch, their biological functions are distinct, at least in their roles in the myoblast differentiation program.
FIGURE 10.

MechanomiR-98–5p regulates myoblast differentiation. pMBwt were transfected with let-7e-5p antagomir and/or miR-98–5p antagomir. A, let-7e-5p and miR-98–5p levels were determined by real-time PCR after a 24-h transfection in either the presence or absence of stretch (ST). B and C, a portion of pMBwt from A were cultured in either GM or DM for 5 days and subjected to stretch (1 h for every 24 h) or no stretch over the 5-day period. Photomicrographs are representative of four independent experiments. Each bar represents mean ± S.E. (n = 4). *, p < 0.05 versus corresponding unstretched control. δ, p < 0.05 versus corresponding stretch.
Discussion
In this study, we tested the hypothesis that the dysregulation of mechanomiRs in dystrophic skeletal muscle plays key role in the progression of disease. We conducted a comprehensive miRNA analysis on mechanically stretched and unstretched diaphragm muscles from WT and mdm mice using miRNA microarrays, followed by bioinformatics analyses to determine the predicted target genes of mechanomiRs and their biological pathways. The predicted mechanomiR target genes were validated by gain-of-function and loss-of-function studies. We found that mechanical stretch significantly altered miRNA expression profile in diaphragms from WT and mdm mice compared with unstretched diaphragms. Some of the mechanomiRs were previously identified as mechanosensitive miRNAs in endothelial cells (27, 28). Interestingly, although stretch altered (> 500 signal intensity) almost equal number of mechanomiRs between WT and mdm mice, number of highly expressed mechanomiRs (>2-fold) were more in mdm mice than those in WT mice. Among the highly expressed mechanomiRs (> 2.0-fold), let-7e-5p was the only mechanomiR found to be regulated in an opposite manner between WT and mdm mice. Our data provide experimental evidence that let-7e-5p targets the ECM genes Col1a1, Col1a2, Col3a1, Col24a1, Col127a1, Itga1, Itga4, Scd1, and Thbs1 as evidenced by real-time PCR and Western blot analyses, and gain-of-function and loss-of-function experiments. In addition, stretch up-regulated mechanomiR-98–5p in diaphragm from WT mouse, but not in diaphragm from mdm mouse. We identified a novel role for miR-98–5p as a negative regulator of myoblast differentiation program. Finally, we showed overactivation of TGF-β1/SMAD signaling pathway in diaphragm from mdm mouse. These results provide the first experimental evidence that the dysregulation of mechanomiRs and their target gene regulatory networks in dystrophic skeletal muscle contributes, at least in part, to disease progression in MD, at least in the mdm mouse model of MD.
Controlled regulation of ECM proteins during skeletal muscle growth and repair is critical in providing a scaffold to build and structure new muscles. However, the excessive or uncontrolled deposition of ECM proteins leads to fibrosis, which progressively deteriorates locomotor capacity, posture maintenance, and the vital function of muscles (51, 55). Fibrosis plays a negative role in the potential treatment of DMD because it alters muscle function, reduces the amount of target muscle available for therapy and repair, and considerably reduces the ability of proliferation and differentiation of implanted cells (56, 57). It has been shown that dysregulated and/or disordered ECM proteins also contribute to disease progression in myopathies other than DMD (58, 59). A better understanding of the mechanism that causes muscle fibrosis is important for improving muscle repair and restoring muscle function, which could lead to a potential treatment for DMD. In the present study, we uncovered let-7e-5p-controlled regulation of ECM proteins and demonstrated that dysregulation of let-7e-5p in mdm mouse could trigger muscle fibrosis. More precisely, we identified the ECM genes Col1a1, Col1a2, Col3a1, Col24a1, Col27a1, Itga1, Itga4, Scd1, and Thbs1 as targets of let-7e-5p. The in vitro inhibition of let-7e-5p by its antagomir in pMBWT reversed the stretch-induced suppression of ECM gene expression. In contrast, overexpression of let-7e-5p in pMBmdm revoked the stretch-induced up-regulation of ECM gene expressions. These data confirmed Col1a1, Col1a2, Col3a1, Col24a1, Col27a1, Itga1, Itga4, Scd1, and Thbs1 as target genes of mechanomiR-let-7e-5p and support our hypothesis that dysregulation of let-7e-5p may contribute to the development of fibrosis in diaphragm muscle from mdm mouse. In agreement with these results, we have shown previously that diaphragm muscles from 2-week-old mdm mice exhibit moderate fibrosis when compared with severe fibrosis in diaphragm from 6-week-old mdm mouse (13). Other investigators have shown that when compared with hindlimb muscle, diaphragm muscle from mdx mouse exhibits a high grade fibrosis (47) that recapitulates clinical signs of human DMD (60). In DMD patients, endomysial fibrosis is the only myopathologic parameter that significantly correlates with poor motor outcome at the age of 10 years and at the time of ambulation loss (61). Overall, we uncovered a mechanomiR-mediated regulation of fibrosis in dystrophic skeletal muscle in which suppression of let-7e-5p contributed to the inhibition of proteins that trigger fibrosis in diaphragm from mdm mouse. However, further studies are warranted to explore the role of mechanomiRs other than let-7e-5p in regulating fibrosis in dystrophic skeletal muscles.
TGF-β1 is a multifunctional cytokine that plays major roles in many biological pathways including the initiation of fibrosis (49–51). Enhanced TGF-β1 expression has been shown in humans who suffer from MD (51, 62, 63) and mouse models of MDs (48, 50, 64, 65). In line with these findings, in the present study, we observed elevated levels of TGF-β1 in diaphragm muscle from mdm mouse. In general, the binding of TGF-β1 to type II receptors and recruitment of type I receptors (ALK4, ALK5, and ALK7) specifically phosphorylate SMAD2 and SMAD3, which produce a nuclear accumulation of active SMAD complexes with SMAD4 that directly regulate gene transcription in conjunction with transcription factors (66). Besides TGF-β1, in the present study, we also observed an increased phosphorylation of SMAD2 and SMAD3 in diaphragm muscle from mdm mouse in response to stretch. In contrast, stretch decreased the phosphorylation of SMAD2 and SMAD3 in diaphragm from WT mouse. These results suggest that the TGF-β1 signaling pathway is mechanosensitive and is dysregulated in diaphragm muscle from mdm mouse in response to stretch. In agreement with the present study, a previous study has shown that mice in which muscle lacking the dystrophin-associated protein γ-sarcoglycan (Sgcg null) were subjected to a lengthening protocol to produce maximal muscle injury, which then produced a rapid accumulation of nuclear phosphorylated SMAD2/3 (67).
Interestingly, the TGF-β1 receptor antagonist suramin attenuates fibrosis in the diaphragm, tibialis anterior, and biceps brachii muscles (47), suggesting that inhibition of the TGF-β1 signaling pathway may be a useful potential alternative therapy for the treatment of DMD. In the present study, we showed that mechanomiRs from WT and mdm mice potentially target the genes that are involved in the TGF-β1 signaling pathway (Fig. 6, A, C, and D). Interestingly, many of the predicted genes in the TGF-β1 signaling pathway are potentially targeted by mechanomiRs including let-7e-5p and miR-98, which were up-regulated in diaphragm from WT mouse. In contrast, the majority of the mechanomiRs that were down-regulated in diaphragm from mdm mouse appears to target most of the predicted genes in the TGF-β1 signaling pathway. In addition, the loss of mechanosensitivity to miR-98 in diaphragm from mdm mouse, along with the observation that miR-98 potentially targets most of the predicted TGF-β1 signaling pathway genes, provides a rationale that supports the possibility of activation of TGF-β1 pathway in mdm mice. Overall, patients with MD and animal models of MD, including the mdm mouse model, confirm the integral role of TGF-β1 and SMAD signaling in the progression and severity of muscle disease. Further studies are under way in our laboratory to identify the mechanomiR that targets TGF-β1 signaling and that may be beneficial for treating MD.
The let-7 family miRNAs were one of the first miRNAs identified in Caenorhabditis elegans (68) and later were found to be conserved across species including mouse and human (69). In the mouse and human, the let-7 family is composed of 10 members (let-7a, -7b, -7c,-7d, -7e, -7f, -7g, and -7i, miR-98, and -202), as recognized by an identical “seed” sequence across those two species (70). Interestingly, all let-7 family members, except miR-202, are mechanomiRs; particularly let-7e-5p (3.27-fold) and miR-98–5p (2.73-fold) were significantly elevated in response to stretch in WT mice. Although the levels of other let-7 family members (let-7a-5p, -7b-5p, -7c-5p,-7d-5p, and -7f-5p) were between 1.5- and 1.7-fold, their signal intensities in response to stretch were very high (>11,000) in the miRNA array. These results indicate that the let-7 family members are highly regulated mechanomiRs, especially in the diaphragm muscle. Surprisingly, diaphragm muscle from mdm mouse displayed down-regulation of the let-7 family members let-7a-5p, -7b-5p, -7c-5p,-7d-5p, -7e-5p, -7f-5p, -7g-5p, and -7i-5p, and especially let-7e-5p, -7f-5p, and -7g-5p, by more than 1.5-fold, indicating dysregulation of the let-7 family of mechanomiRs in dystrophic skeletal muscle. In line with our observation, other investigators have shown that regulation of let-7 is associated with aging and cancer progression. For example, skeletal muscles from elderly people display an elevated levels of let-7b and -7e when compared with that from younger people, and the most highly ranked cellular processes likely to be regulated by let-7b and let-7e miRNAs are associated with cell cycle regulation and cellular growth and proliferation (71). Another study has shown that the primary role of let-7 members is the possession of a conserved function as a tumor suppressor, and therefore they are able to down-regulate the expression of genes associated with cell proliferation, thereby promoting differentiation of human and mouse neuronal stem cells (20). A recent study has identified PABP2 (poly(A)-binding protein) as a genetic interaction partner of the let-7 miRNA (72); the phenotypes associated with human PABP2 mutation in oculopharyngeal MD (a late-onset, progressive disease) seem to affect selectively only muscle cells (73). These studies suggest that the elevated levels of let-7b and let-7e miRNAs in the skeletal muscle of elderly people may contribute to their attenuated skeletal muscle regenerative capacity (74). Taken together, our current findings suggest that the down-regulation of let-7 members, especially let-7e-5p and miR-98-5p in mdm mouse, may compromise the ability of these mechanomiRs to increase satellite cell proliferation to improve skeletal muscle regenerative capacity.
Furthermore, we have demonstrated previously that mechanical stretch inhibits differentiation and promotes the proliferation of C2C12 myoblasts (6), suggesting that the differentially regulated mechanomiR(s) in diaphragm muscle may regulate myogenesis. To explore this possibility, we transfected pMBWT with the antagomir of let-7e-5p and/or miR-98 and cultured them in differentiation medium with a 1-h stretch for 5 consecutive days. We found that miR-98–5p, but not miR-7e-5p, promoted myoblast differentiation by revoking the inhibitory effect of stretch on differentiation, suggesting a role for mechanomiR-98 in differentiation. In agreement with our study, a previous study has shown that inhibition of miR-98 increases both human (LHCN-M2) and C2C12 myoblast differentiation by directly altering the expression of the transcription factor E2F5, which in turn directly down-regulates the differentiation inhibitors HMOX1 and ID1 (75). Further studies are warranted to confirm the role of other mechanomiRs in myogenesis.
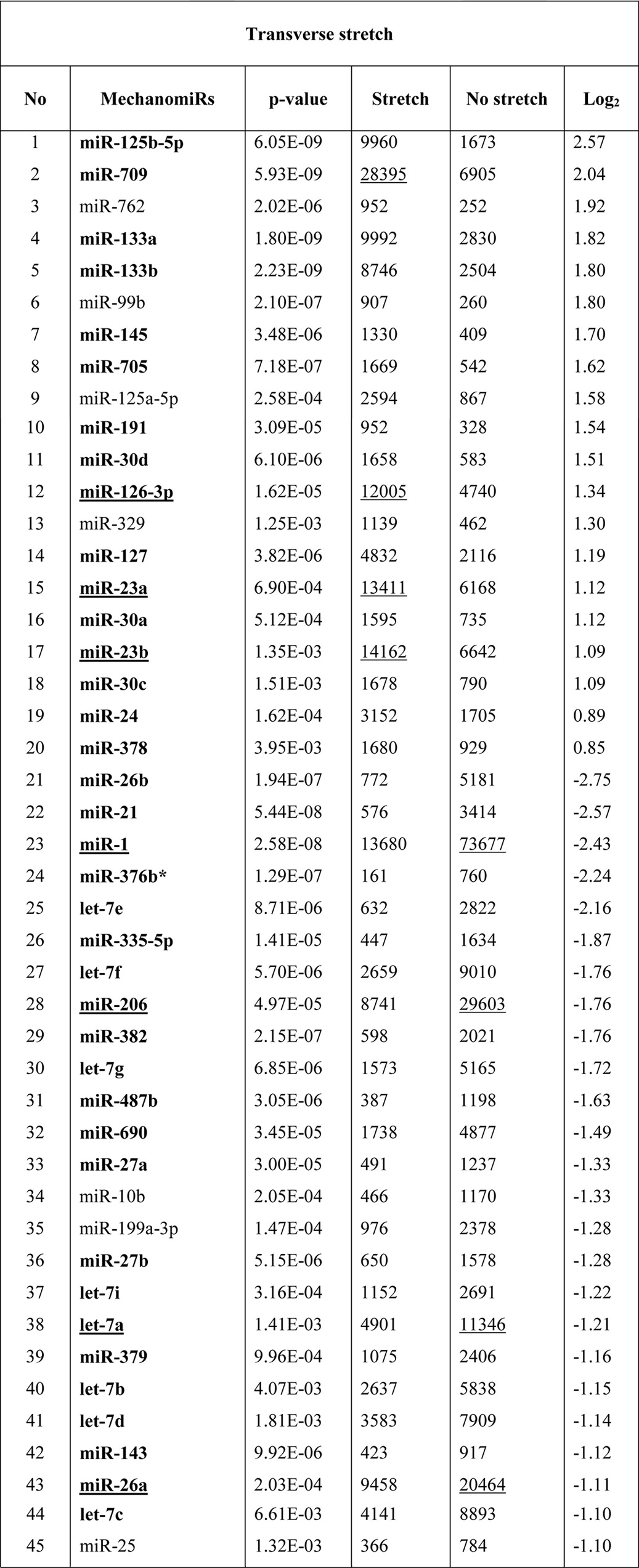
Biaxial loading, in which muscle fibers experience both transverse and longitudinal loads during each respiratory cycle, is the most obvious feature of the mechanical environment of the diaphragm (76). In this study, we applied mechanical stress in either the longitudinal or transverse direction to the muscle fibers, with the magnitude of the mechanical stress along the muscle fibers being essentially the same as that applied in the transverse direction to the muscle fibers. Stress-strain relationship data from our previous studies of the diaphragm clearly demonstrate that muscle compliance is small and extensibility is reduced in the transverse direction when compared with the longitudinal direction of the muscle fibers (15, 33, 54). In addition, muscle stiffness, a measure of the resistance to mechanical stretch, is greater in the transverse direction of the muscle fibers than in the direction of the fibers (33). In agreement with these studies, data from the present study show that TS differentially regulated a higher number of mechanomiRs (>1.5-fold) compared with LS in the WT mouse (Table 3 and Fig. 1E), suggesting that the mechanosensing ability of the WT mouse diaphragm was greater in the transverse direction due to greater transverse muscle stiffness compared with longitudinal stiffness. In contrast, diaphragm muscle from mdm mouse showed no difference in the number of mechanomiRs between LS and TS (Table 7 and Fig. 2E). However, the number of mechanomiRs that were commonly regulated in response to LS or TS were higher in mdm mouse than WT mouse. This suggests that diaphragm muscle from the mdm mouse senses greater stiffness in response to longitudinal or transverse directional stretch than diaphragm from age-matched WT mouse as shown previously (13). These data also suggest that the specificity in the mechanosensing ability of the mdm mouse diaphragm might be modified due to its dystrophic character. Further studies will be necessary to explore the mechanism by which mechanotransduction is altered in dystrophic skeletal muscle, especially in the diaphragm.
TABLE 7.
Differentially regulated mechanomiRs (>1.5-fold) in diaphragm from mdm mouse
Bold indicates differentially regulated mechanomiRs by longitudinal and transverse stretches. Underlines indicate differentially regulated mechanomiRs at signal intensities > 10,000.
Finally, we performed a comparative analysis between differentially regulated mechanomiRs in mdm mouse and previously identified miRNAs (>1.5-fold) in mdx mouse skeletal muscle. In the present study, WT and mdm mice diaphragms showed up-regulation of the mechanomiRs in let-7a-5p, miR-22–3p, and miR-30e-5p, whereas mdx mouse diaphragm showed a down-regulation of these mechanomiRs (Fig. 6E). Similarly, the down-regulated mechanomiRs in miR-21a-5p, miR-127–3p, miR-206–3p, miR-335–5p, and miR-379–5p in WT and mdm mice were up-regulated in mdx mouse skeletal muscle (44). Although mdx and mdm mice are mouse models of human MDs, the discrepancy in the regulation of miRNAs most likely is due to the stage of severity of the disease. For example, in the present study, we used 2-week-old mdm mice in which the histopathological signs in the diaphragm were largely absent when compared with the 6-week-old mdm mouse diaphragm (13). The mdx mice used by Greco et al. (44) were 2 and 14 months old. Therefore, our experiments utilizing 2-week-old mdm mice were conducted before onset of the disease, which may explain the discrepancy in the regulation of miRNAs between mdx and mdm mice. Moreover, the contrasting pathophysiology in skeletal muscles between mdx and mdm mice is most likely due to the dysfunction of different cytoskeletal proteins (mdx, dystrophin; mdm, titin), which may contribute to the distinct regulation of miRNAs in these two mouse models.
Author Contributions
J. S. M. designed and wrote the manuscript and performed most experiments. A. H. contributed to bioinformatics and statistical analyses. M. A. L. performed mouse breeding and ex vivo experiments. A. M. B. contributed to the design of the study and reviewed and edited the manuscript. All authors reviewed and take full responsibility for the contents of the manuscript. A. M. B. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
This work was supported in part by National Institutes of Health Grant HL-63134 from the NHLBI. This work was also supported by the National Science Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
- MD
- muscular dystrophy
- DMD
- Duchenne muscular dystrophy
- ECM
- extracellular matrix
- miRNA
- microRNA
- myomiR
- myogenic skeletal muscle-enriched microRNA
- mechanomiR
- mechanosensitive microRNA
- TS
- transverse stretch
- LS
- longitudinal stretch.
References
- 1. Hoffman E. P., Brown R. H., Jr., Kunkel L. M. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928 [DOI] [PubMed] [Google Scholar]
- 2. Koenig M., Monaco A. P., Kunkel L. M. (1988) The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 53, 219–228 [DOI] [PubMed] [Google Scholar]
- 3. Hackman P., Vihola A., Haravuori H., Marchand S., Sarparanta J., De Seze J., Labeit S., Witt C., Peltonen L., Richard I., Udd B. (2002) Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am. J. Hum. Genet. 71, 492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kumar A., Chaudhry I., Reid M. B., Boriek A. M. (2002) Distinct signaling pathways are activated in response to mechanical stress applied axially and transversely to skeletal muscle fibers. J. Biol. Chem. 277, 46493–46503 [DOI] [PubMed] [Google Scholar]
- 5. Kumar A., Boriek A. M. (2003) Mechanical stress activates the nuclear factor-κB pathway in skeletal muscle fibers: a possible role in Duchenne muscular dystrophy. FASEB J. 17, 386–396 [DOI] [PubMed] [Google Scholar]
- 6. Kumar A., Murphy R., Robinson P., Wei L., Boriek A. M. (2004) Cyclic mechanical strain inhibits skeletal myogenesis through activation of focal adhesion kinase, Rac-1 GTPase, and NF-κB transcription factor. FASEB J. 18, 1524–1535 [DOI] [PubMed] [Google Scholar]
- 7. Mohamed J. S., Lopez M. A., Cox G. A., Boriek A. M. (2010) Anisotropic regulation of Ankrd2 gene expression in skeletal muscle by mechanical stretch. FASEB J. 24, 3330–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jaalouk D. E., Lammerding J. (2009) Mechanotransduction gone awry. Nat. Rev. Mol. Cell Biol. 10, 63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heydemann A., McNally E. M. (2007) Consequences of disrupting the dystrophin-sarcoglycan complex in cardiac and skeletal myopathy. Trends Cardiovasc. Med. 17, 55–59 [DOI] [PubMed] [Google Scholar]
- 10. Kumar A., Khandelwal N., Malya R., Reid M. B., Boriek A. M. (2004) Loss of dystrophin causes aberrant mechanotransduction in skeletal muscle fibers. FASEB J. 18, 102–113 [DOI] [PubMed] [Google Scholar]
- 11. Claflin D. R., Brooks S. V. (2008) Direct observation of failing fibers in muscles of dystrophic mice provides mechanistic insight into muscular dystrophy. Am. J. Physiol. Cell Physiol. 294, C651–C658 [DOI] [PubMed] [Google Scholar]
- 12. Boriek A. M., Capetanaki Y., Hwang W., Officer T., Badshah M., Rodarte J., Tidball J. G. (2001) Desmin integrates the three-dimensional mechanical properties of muscles. Am. J. Physiol. Cell Physiol. 280, C46–C52 [DOI] [PubMed] [Google Scholar]
- 13. Lopez M. A., Pardo P. S., Cox G. A., Boriek A. M. (2008) Early mechanical dysfunction of the diaphragm in the muscular dystrophy with myositis (Ttnmdm) model. Am. J. Physiol. Cell Physiol. 295, C1092–C1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mohamed J. S., Lopez M. A., Cox G. A., Boriek A. M. (2013) Ankyrin repeat domain protein 2 and inhibitor of DNA binding 3 cooperatively inhibit myoblast differentiation by physical interaction. J. Biol. Chem. 288, 24560–24568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patel N. D., Jannapureddy S. R., Hwang W., Chaudhry I., Boriek A. M. (2003) Altered muscle force and stiffness of skeletal muscles in α-sarcoglycan-deficient mice. Am. J. Physiol. Cell Physiol. 284, C962–C968 [DOI] [PubMed] [Google Scholar]
- 16. Lopez M. A., Mayer U., Hwang W., Taylor T., Hashmi M. A., Jannapureddy S. R., Boriek A. M. (2005) Force transmission, compliance, and viscoelasticity are altered in the α7-integrin-null mouse diaphragm. Am. J. Physiol. Cell Physiol. 288, C282–C289 [DOI] [PubMed] [Google Scholar]
- 17. Bushati N., Cohen S. M. (2007) microRNA functions. Annu. Rev. Cell Dev. Biol. 23, 175–205 [DOI] [PubMed] [Google Scholar]
- 18. Sharma M., Juvvuna P. K., Kukreti H., McFarlane C. (2014) Mega roles of microRNAs in regulation of skeletal muscle health and disease. Front. Physiol. 5, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Winbanks C. E., Ooi J. Y., Nguyen S. S., McMullen J. R., Bernardo B. C. (2014) MicroRNAs differentially regulated in cardiac and skeletal muscle in health and disease: potential drug targets? Clin. Exp. Pharmacol. Physiol. 41, 727–737 [DOI] [PubMed] [Google Scholar]
- 20. Zacharewicz E., Lamon S., Russell A. P. (2013) MicroRNAs in skeletal muscle and their regulation with exercise, ageing, and disease. Front. Physiol. 4, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Callis T. E., Deng Z., Chen J. F., Wang D. Z. (2008) Muscling through the microRNA world. Exp. Biol. Med. (Maywood) 233, 131–138 [DOI] [PubMed] [Google Scholar]
- 22. McCarthy J. J., Esser K. A., Andrade F. H. (2007) MicroRNA-206 is overexpressed in the diaphragm but not the hindlimb muscle of mdx mouse. Am. J. Physiol. Cell Physiol. 293, C451–C457 [DOI] [PubMed] [Google Scholar]
- 23. Mohamed J. S., Lopez M. A., Boriek A. M. (2010) Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase-3β. J. Biol. Chem. 285, 29336–29347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guan Y. J., Yang X., Wei L., Chen Q. (2011) MiR-365: a mechanosensitive microRNA stimulates chondrocyte differentiation through targeting histone deacetylase 4. FASEB J. 25, 4457–4466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang Y., Crawford M., Higuita-Castro N., Nana-Sinkam P., Ghadiali S. N. (2012) miR-146a regulates mechanotransduction and pressure-induced inflammation in small airway epithelium. FASEB J. 26, 3351–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobus K., Kopycinska J., Kozlowska-Wiechowska A., Urasinska E., Kempinska-Podhorodecka A., Haas T. L., Milkiewicz P., Milkiewicz M. (2012) Angiogenesis within the duodenum of patients with cirrhosis is modulated by mechanosensitive Kruppel-like factor 2 and microRNA-126. Liver Int. 32, 1222–1232 [DOI] [PubMed] [Google Scholar]
- 27. Kumar S., Kim C. W., Simmons R. D., Jo H. (2014) Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler. Thromb. Vasc. Biol. 34, 2206–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marin T., Gongol B., Chen Z., Woo B., Subramaniam S., Chien S., Shyy J. Y. (2013) Mechanosensitive microRNAs-role in endothelial responses to shear stress and redox state. Free Radic. Biol. Med. 64, 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mendias C. L., Gumucio J. P., Lynch E. B. (2012) Mechanical loading and TGF-β change the expression of multiple miRNAs in tendon fibroblasts. J. Appl. Physiol. 113, 56–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Son D. J., Kumar S., Takabe W., Kim C. W., Ni C. W., Alberts-Grill N., Jang I. H., Kim S., Kim W., Won Kang S., Baker A. H., Woong Seo J., Ferrara K. W., Jo H. (2013) The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nat. Commun. 4, 3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wei F., Liu D., Feng C., Zhang F., Yang S., Hu Y., Ding G., Wang S. (2015) microRNA-21 Mediates stretch-induced osteogenic differentiation in human periodontal ligament stem cells. Stem Cells Dev. 24, 312–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mohamed J. S., Hajira A., Li Z., Paulin D., Boriek A. M. (2011) Desmin regulates airway smooth muscle hypertrophy through early growth-responsive protein-1 and microRNA-26a. J. Biol. Chem. 286, 43394–43404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boriek A. M., Zhu D., Zeller M., Rodarte J. R. (2001) Inferences on force transmission from muscle fiber architecture of the canine diaphragm. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R156–R165 [DOI] [PubMed] [Google Scholar]
- 34. Garvey S. M., Rajan C., Lerner A. P., Frankel W. N., Cox G. A. (2002) The muscular dystrophy with myositis (mdm) mouse mutation disrupts a skeletal muscle-specific domain of titin. Genomics 79, 146–149 [DOI] [PubMed] [Google Scholar]
- 35. Ono Y., Shimada H., Sorimachi H., Richard I., Saido T. C., Beckmann J. S., Ishiura S., Suzuki K. (1998) Functional defects of a muscle-specific calpain, p94, caused by mutations associated with limb-girdle muscular dystrophy type 2A. J. Biol. Chem. 273, 17073–17078 [DOI] [PubMed] [Google Scholar]
- 36. Vlachos I. S., Kostoulas N., Vergoulis T., Georgakilas G., Reczko M., Maragkakis M., Paraskevopoulou M. D., Prionidis K., Dalamagas T., Hatzigeorgiou A. G. (2012) DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 40, W498–W504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dweep H., Sticht C., Pandey P., Gretz N. (2011) miRWalk-database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 44, 839–847 [DOI] [PubMed] [Google Scholar]
- 38. Mohamed J. S., Hajira A., Pardo P. S., Boriek A. M. (2014) MicroRNA-149 inhibits PARP-2 and promotes mitochondrial biogenesis via SIRT-1/PGC-1α network in skeletal muscle. Diabetes 63, 1546–1559 [DOI] [PubMed] [Google Scholar]
- 39. Doench J. G., Sharp P. A. (2004) Specificity of microRNA target selection in translational repression. Genes Dev. 18, 504–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alexander M. S., Kawahara G., Motohashi N., Casar J. C., Eisenberg I., Myers J. A., Gasperini M. J., Estrella E. A., Kho A. T., Mitsuhashi S., Shapiro F., Kang P. B., Kunkel L. M. (2013) MicroRNA-199a is induced in dystrophic muscle and affects WNT signaling, cell proliferation, and myogenic differentiation. Cell Death Differ. 20, 1194–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alexander M. S., Casar J. C., Motohashi N., Myers J. A., Eisenberg I., Gonzalez R. T., Estrella E. A., Kang P. B., Kawahara G., Kunkel L. M. (2011) Regulation of DMD pathology by an ankyrin-encoded miRNA. Skelet. Muscle 1, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cacchiarelli D., Incitti T., Martone J., Cesana M., Cazzella V., Santini T., Sthandier O., Bozzoni I. (2011) miR-31 modulates dystrophin expression: new implications for Duchenne muscular dystrophy therapy. EMBO Rep. 12, 136–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eisenberg I., Eran A., Nishino I., Moggio M., Lamperti C., Amato A. A., Lidov H. G., Kang P. B., North K. N., Mitrani-Rosenbaum S., Flanigan K. M., Neely L. A., Whitney D., Beggs A. H., Kohane I. S., Kunkel L. M. (2007) Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. U.S.A. 104, 17016–17021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Greco S., De Simone M., Colussi C., Zaccagnini G., Fasanaro P., Pescatori M., Cardani R., Perbellini R., Isaia E., Sale P., Meola G., Capogrossi M. C., Gaetano C., Martelli F. (2009) Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 23, 3335–3346 [DOI] [PubMed] [Google Scholar]
- 45. Roberts T. C., Blomberg K. E., McClorey G., El Andaloussi S., Godfrey C., Betts C., Coursindel T., Gait M. J., Smith C. I., Wood M. J. (2012) Expression analysis in multiple muscle groups and serum reveals complexity in the microRNA transcriptome of the mdx mouse with implications for therapy. Mol. Ther. Nucleic Acids 1, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang L., Zhou L., Jiang P., Lu L., Chen X., Lan H., Guttridge D. C., Sun H., Wang H. (2012) Loss of miR-29 in myoblasts contributes to dystrophic muscle pathogenesis. Mol. Ther. 20, 1222–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Taniguti A. P., Pertille A., Matsumura C. Y., Santo Neto H., Marques M. J. (2011) Prevention of muscle fibrosis and myonecrosis in mdx mice by suramin, a TGF-β1 blocker. Muscle Nerve 43, 82–87 [DOI] [PubMed] [Google Scholar]
- 48. Zhou L., Porter J. D., Cheng G., Gong B., Hatala D. A., Merriam A. P., Zhou X., Rafael J. A., Kaminski H. J. (2006) Temporal and spatial mRNA expression patterns of TGF-β1, 2, 3 and TβRI, II, III in skeletal muscles of mdx mice. Neuromuscul. Disord. 16, 32–38 [DOI] [PubMed] [Google Scholar]
- 49. Vidal B., Serrano A. L., Tjwa M., Suelves M., Ardite E., De Mori R., Baeza-Raja B., Martínez de Lagrán M., Lafuste P., Ruiz-Bonilla V., Jardí M., Gherardi R., Christov C., Dierssen M., Carmeliet P., Degen J. L., Dewerchin M., Muñoz-Cánoves P. (2008) Fibrinogen drives dystrophic muscle fibrosis via a TGFβ/alternative macrophage activation pathway. Genes Dev. 22, 1747–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hartel J. V., Granchelli J. A., Hudecki M. S., Pollina C. M., Gosselin L. E. (2001) Impact of prednisone on TGF-β1 and collagen in diaphragm muscle from mdx mice. Muscle Nerve 24, 428–432 [DOI] [PubMed] [Google Scholar]
- 51. Bernasconi P., Torchiana E., Confalonieri P., Brugnoni R., Barresi R., Mora M., Cornelio F., Morandi L., Mantegazza R. (1995) Expression of transforming growth factor-β1 in dystrophic patient muscles correlates with fibrosis: pathogenetic role of a fibrogenic cytokine. J. Clin. Invest. 96, 1137–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cohn R. D., van Erp C., Habashi J. P., Soleimani A. A., Klein E. C., Lisi M. T., Gamradt M., ap Rhys C. M., Holm T. M., Loeys B. L., Ramirez F., Judge D. P., Ward C. W., Dietz H. C. (2007) Angiotensin II type 1 receptor blockade attenuates TGF-β-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 13, 204–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mehra A., Wrana J. L. (2002) TGF-beta and the Smad signal transduction pathway. Biochem. Cell Biol. 80, 605–622 [DOI] [PubMed] [Google Scholar]
- 54. Kumar S., Kim C. W., Simmons R. D., Jo H. (2014) Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler. Thromb. Vasc. Biol. 34, 2206–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mann C. J., Perdiguero E., Kharraz Y., Aguilar S., Pessina P., Serrano A. L., Muñoz-Cánoves P. (2011) Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 1, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Goyenvalle A., Seto J. T., Davies K. E., Chamberlain J. (2011) Therapeutic approaches to muscular dystrophy. Hum. Mol. Genet. 20, R69–R78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Davies K. E., Grounds M. D. (2007) Modified patient stem cells as prelude to autologous treatment of muscular dystrophy. Cell Stem Cell 1, 595–596 [DOI] [PubMed] [Google Scholar]
- 58. MacDonald E. M., Cohn R. D. (2012) TGFβ signaling: its role in fibrosis formation and myopathies. Curr. Opin. Rheumatol. 24, 628–634 [DOI] [PubMed] [Google Scholar]
- 59. Schessl J., Zou Y., Bönnemann C. G. (2006) Congenital muscular dystrophies and the extracellular matrix. Semin. Pediatr. Neurol. 13, 80–89 [DOI] [PubMed] [Google Scholar]
- 60. Stedman H. H., Sweeney H. L., Shrager J. B., Maguire H. C., Panettieri R. A., Petrof B., Narusawa M., Leferovich J. M., Sladky J. T., Kelly A. M. (1991) The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352, 536–539 [DOI] [PubMed] [Google Scholar]
- 61. Desguerre I., Mayer M., Leturcq F., Barbet J. P., Gherardi R. K., Christov C. (2009) Endomysial fibrosis in Duchenne muscular dystrophy: a marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 68, 762–773 [DOI] [PubMed] [Google Scholar]
- 62. Yamazaki M., Minota S., Sakurai H., Miyazono K., Yamada A., Kanazawa I., Kawai M. (1994) Expression of transforming growth factor-β1 and its relation to endomysial fibrosis in progressive muscular dystrophy. Am. J. Pathol. 144, 221–226 [PMC free article] [PubMed] [Google Scholar]
- 63. Sun G., Haginoya K., Wu Y., Chiba Y., Nakanishi T., Onuma A., Sato Y., Takigawa M., Iinuma K., Tsuchiya S. (2008) Connective tissue growth factor is overexpressed in muscles of human muscular dystrophy. J. Neurol. Sci. 267, 48–56 [DOI] [PubMed] [Google Scholar]
- 64. Gosselin L. E., Williams J. E., Deering M., Brazeau D., Koury S., Martinez D. A. (2004) Localization and early time course of TGF-β1 mRNA expression in dystrophic muscle. Muscle Nerve 30, 645–653 [DOI] [PubMed] [Google Scholar]
- 65. Andreetta F., Bernasconi P., Baggi F., Ferro P., Oliva L., Arnoldi E., Cornelio F., Mantegazza R., Confalonieri P. (2006) Immunomodulation of TGF-β1 in mdx mouse inhibits connective tissue proliferation in diaphragm but increases inflammatory response: implications for antifibrotic therapy. J. Neuroimmunol. 175, 77–86 [DOI] [PubMed] [Google Scholar]
- 66. Schmierer B., Hill C. S. (2007) TGF β-SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 8, 970–982 [DOI] [PubMed] [Google Scholar]
- 67. Goldstein J. A., Bogdanovich S., Beiriger A., Wren L. M., Rossi A. E., Gao Q. Q., Gardner B. B., Earley J. U., Molkentin J. D., McNally E. M. (2014) Excess SMAD signaling contributes to heart and muscle dysfunction in muscular dystrophy. Hum. Mol. Genet. 23, 6722–6731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Reinhart B. J., Slack F. J., Basson M., Pasquinelli A. E., Bettinger J. C., Rougvie A. E., Horvitz H. R., Ruvkun G. (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403, 901–906 [DOI] [PubMed] [Google Scholar]
- 69. Lagos-Quintana M., Rauhut R., Meyer J., Borkhardt A., Tuschl T. (2003) New microRNAs from mouse and human. RNA 9, 175–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Roush S., Slack F. J. (2008) The let-7 family of microRNAs. Trends Cell Biol. 18, 505–516 [DOI] [PubMed] [Google Scholar]
- 71. Drummond M. J., McCarthy J. J., Sinha M., Spratt H. M., Volpi E., Esser K. A., Rasmussen B. B. (2011) Aging and microRNA expression in human skeletal muscle: a microarray and bioinformatics analysis. Physiol. Genomics 43, 595–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hurschler B. A., Harris D. T., Grosshans H. (2011) The type II poly(A)-binding protein PABP-2 genetically interacts with the let-7 miRNA and elicits heterochronic phenotypes in Caenorhabditis elegans. Nucleic Acids Res. 39, 5647–5657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Brais B., Bouchard J. P., Xie Y. G., Rochefort D. L., Chrétien N., Tomé F. M., Lafrenière R. G., Rommens J. M., Uyama E., Nohira O., Blumen S., Korczyn A. D., Heutink P., Mathieu J., Duranceau A., Codère F., Fardeau M., Rouleau G. A. (1998) Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 18, 164–167 [DOI] [PubMed] [Google Scholar]
- 74. Carlson M. E., Suetta C., Conboy M. J., Aagaard P., Mackey A., Kjaer M., Conboy I. (2009) Molecular aging and rejuvenation of human muscle stem cells. EMBO Mol. Med. 1, 381–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kropp J., Degerny C., Morozova N., Pontis J., Harel-Bellan A., Polesskaya A. (2015) miR-98 delays skeletal muscle differentiation by down-regulating E2F5. Biochem. J. 466, 85–93 [DOI] [PubMed] [Google Scholar]
- 76. Boriek A. M., Rodarte J. R. (1994) Inferences on passive diaphragm mechanics from gross anatomy. J. Appl. Physiol. 77, 2065–2070 [DOI] [PubMed] [Google Scholar]