

Background: Sulfide quinone oxidoreductase is a flavoprotein that catalyzes the first step in H2S oxidation.
Results: Transient kinetic studies reveal the presence of an equally intense charge-transfer (CT) band in the presence of sulfite as with sulfide.
Conclusion: Only the CT intermediate formed in the presence of sulfide is kinetically competent.
Significance: An off-pathway reaction could be promoted under pathologically high sulfite concentrations.
Keywords: electrochemistry, enzyme kinetics, flavin, flavoprotein, hydrogen sulfide, oxidation-reduction (redox)
Abstract
The first step in the mitochondrial sulfide oxidation pathway is catalyzed by sulfide quinone oxidoreductase (SQR), which belongs to the family of flavoprotein disulfide oxidoreductases. During the catalytic cycle, the flavin cofactor is intermittently reduced by sulfide and oxidized by ubiquinone, linking H2S oxidation to the electron transfer chain and to energy metabolism. Human SQR can use multiple thiophilic acceptors, including sulfide, sulfite, and glutathione, to form as products, hydrodisulfide, thiosulfate, and glutathione persulfide, respectively. In this study, we have used transient kinetics to examine the mechanism of the flavin reductive half-reaction and have determined the redox potential of the bound flavin to be −123 ± 7 mV. We observe formation of an unusually intense charge-transfer (CT) complex when the enzyme is exposed to sulfide and unexpectedly, when it is exposed to sulfite. In the canonical reaction, sulfide serves as the sulfur donor and sulfite serves as the acceptor, forming thiosulfate. We show that thiosulfate is also formed when sulfide is added to the sulfite-induced CT intermediate, representing a new mechanism for thiosulfate formation. The CT complex is formed at a kinetically competent rate by reaction with sulfide but not with sulfite. Our study indicates that sulfide addition to the active site disulfide is preferred under normal turnover conditions. However, under pathological conditions when sulfite concentrations are high, sulfite could compete with sulfide for addition to the active site disulfide, leading to attenuation of SQR activity and to an alternate route for thiosulfate formation.
Introduction
Coupling of hydrogen sulfide (H2S) oxidation to oxidative phosphorylation was first reported almost 30 years ago in the bivalve, Solemya reidi (1). Twenty years later, H2S was described as the first inorganic substrate for human cells (2). Toxic when elevated, H2S is a signaling molecule that mediates a variety of physiological effects impacting major organ systems including the cardiovascular, central nervous, and digestive systems (3–5). H2S is biosynthesized from the sulfur-containing amino acids, cysteine and homocysteine, in reactions catalyzed by two enzymes in the transsulfuration pathway, cystathionine β-synthase and γ-cystathionase (6–9). A third enzyme, mercaptopyruvate sulfurtransferase, catalyzes H2S production from a cysteine catabolite, mercaptopyruvate (10, 11). Tissues such as liver maintain a high metabolic flux of sulfur through H2S with the balance between biogenesis and oxidation rates being critical for maintaining low steady-state levels of H2S (12–14).
In mammals, the sulfide oxidation pathway resides in the mitochondrion and produces thiosulfate and sulfate for excretion. The potential role of this pathway in generating reactive sulfur species for H2S-based signaling has been raised (15). An alternative to this pathway for sulfide oxidation exists in red blood cells, which lack mitochondria and utilize methemoglobin instead to generate thiosulfate from sulfide (16).
The first step in the mitochondrial sulfide oxidation pathway is catalyzed by sulfide quinone oxidoreductase (SQR),3 a member of the flavin disulfide reductase family, which includes such well studied enzymes as thioredoxin reductase, glutathione reductase, and dihydrolipoamide dehydrogenase (17). In general, these enzymes catalyze pyridine nucleotide-dependent reductions of various substrates. A characteristic of this family is the presence of two redox centers: an active site disulfide (or cysteine sulfenic acid or a mixed disulfide between cysteine and CoA) and an FAD, which are utilized for relaying electron equivalents to or from the substrate. In mammals, SQR is anchored to the inner mitochondrial membrane. Although a structure for human SQR is not available, it has been modeled using available SQR structures (see Fig. 1, A and B). The FAD is noncovalently bound (18), and the active site disulfide in the resting enzyme is presumed to form between Cys-201 and Cys-379.
FIGURE 1.

Structure and mechanism of SQR. A, the structure of a single subunit of human SQR modeled using I-TASSER. FAD (pink) and the active site cysteine residues, Cys-201 and Cys-379, are shown in sphere representation. B, close-up of the active site showing the redox-active cofactors, flavin, and a pair of cysteine residues. C, postulated reaction mechanism of SQR in which sulfide adds into the disulfide bond in the active site, forming a persulfide intermediate and a CT complex between the liberated thiolate and oxidized FAD (FADox). The addition of sulfide or sulfite to the persulfide produces hydrodisulfide or thiosulfate and is accompanied by electron transfer to FAD and reformation of the disulfide bond. The electrons from reduced FAD (FADred) are passed to CoQ to complete the catalytic cycle. D, elution profile of SQR and gel filtration standards in 50 mm Tris buffer (pH 8) containing 0.3% DHPC, and 200 mm NaCl. The standards used were: 1, thyroglobulin (670 kDa); 2, γ-globulin (158 kDa); 3, ovalbumin (44 kDa); 4, myoglobin (17 kDa); and 5, vitamin B12 (1.35 kDa).
Because SQR is critical for maintaining low intracellular H2S levels, insights into its reaction mechanism are important for therapeutic targeting. SQR couples the oxidation of sulfide to the reduction of quinone via the intermediacy of an active site persulfide (18, 19) (see Fig. 1C). The reaction is postulated to involve attack of the sulfide anion on an active site disulfide, forming cysteine persulfide, presumably on Cys-379 as it releases the thiolate of Cys-201 that is proximal to and on the re face of the FAD (see Fig. 1B). Attack of an acceptor, e.g. sulfite or a second mole of sulfide, leads to the transfer of sulfane sulfur, forming thiosulfate or hydrodisulfide, respectively. Concomitantly, the active site disulfide is re-formed and the flavin is reduced. In the second half-reaction, FADH2 transfers its electrons to coenzyme Q (CoQ), which binds on the si face of flavin and in a pocket facing the membrane (20).
The details of the reductive half-reactions involving the active site cysteines and FAD are not well understood and appear to be distinct in SQRs from lower and higher organisms. In mammals, an acceptor is needed in each turnover cycle to move the persulfide species out of the active site. Under in vitro conditions, a number of small molecules can serve as acceptors, including cyanide, sulfite, cysteine, and homocysteine (18, 19, 21), whereas under physiological conditions, GSH is expected to be the primary persulfide acceptor for human SQR (19, 21). SQRs from lower organisms convert sulfide to elemental sulfur or hydropolysulfides whose length is presumably limited by the steric constraints within the active site. A polysulfide bridge with three sulfur atoms between the active site cysteines was seen in the structure of Acidianus ambivalens SQR (22), whereas density interpreted as a cyclooctane sulfur bound to an active site cysteine was observed in the Aquifex aeolicus SQR (20).
In this study, we have used transient enzyme-monitored kinetics to examine the flavin reduction half-reaction catalyzed by human SQR. Formation of a charge-transfer (CT) complex between oxidized flavin and the free thiolate is predicted to accompany persulfide formation in the SQR reaction (see Fig. 1C). Indeed, a CT species was observed when human SQR was mixed with 1–2 equivalents of sulfide. The CT absorbance band extended out to 900 nm and exhibited a maximum at 673 nm with an intensity ratio for 390:673 nm of ∼1:0.26 (18). In this study, we report formation of intense CT bands (intensity ratios of 390:675 nm ranging from 1:0.51 to 1:0.65) centered at 675 nm when human SQR is mixed with the nucleophiles, sulfite or sulfide. We evaluated the kinetic competence of the CT intermediate formed in the presence of sulfide or sulfite. To further characterize the unusual CT band, we also determined the redox potential of the SQR-bound FAD. Our studies reveal an off-pathway reaction catalyzed by SQR that might be important under pathological conditions in which sulfite concentrations are elevated.
Experimental Procedures
Materials
All chemicals were reagent-grade and used without further purification. The following reagents were purchased from Sigma-Aldrich: sodium salts of sulfide, sulfite, indigo carmine, and xanthine; 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB, or Ellman's reagent); and coenzyme Q1 (CoQ1). Monobromobimane (FluoroPure grade) was obtained from Thermo Fisher Scientific. 1,2-Diheptanoyl-sn-glycero-3-phosphocholine (DHPC) was from Avanti Polar Lipids. Xanthine oxidase used for flavin redox titrations was a generous gift from Dr. Bruce Palfey (University of Michigan).
Purification of SQR and Determination of Its Oligomeric Structure
Human SQR was purified as reported previously (19). For determination of its oligomeric structure, SQR was further purified using a 25-ml Superdex 200 (GE Healthcare) column with a flow rate of 0.5 ml min−1. The protein was eluted with 50 mm Tris buffer, pH 8, containing 0.3% DHPC and 200 mm NaCl. The native molecular mass was determined by comparison with gel filtration standards (Bio-Rad) with known molecular masses.
Redox Potential Determination of SQR-bound FAD
The redox potential of SQR-bound flavin was determined using the method originally developed by Massey (23), with indigo carmine (Em = −125 mV versus normal hydrogen electrode) as a reference dye (where Em is midpoint redox potential). A solution of SQR (A450 ∼0.2) containing indigo carmine (A611 ∼0.2), 100 μm xanthine, and 5 μm methyl viologen (mediator) in 50 mm Tris-Cl, pH 8.0, with 0.03% DHPC was deoxygenated in an anaerobic cuvette by cycles of equilibration with argon and evacuation. The solution was kept on ice during this process to minimize protein precipitation. The redox titration was then initiated by the addition of a catalytic amount of xanthine oxidase and carried out to completion over a period of 2–3 h at 6 °C. Scans were recorded on a UV-2501 PC Shimadzu spectrophotometer at 10-min intervals to monitor the reduction of the flavin and the reference dye. During the titration of indigo carmine alone, an isosbestic point at 465 nm was observed. Therefore, reduction of SQR-bound FAD in the presence of the dye was followed at this wavelength. The ratio of the oxidized and reduced species over the course of titration was calculated based on the absorbance at 640 and 465 nm, for the dye and SQR-bound FAD, respectively. The standard reduction potential of the solution was determined using the known mid-point potential of indigo carmine (Em,dye = −125 mV) and the Nernst equation (Eq. 1),
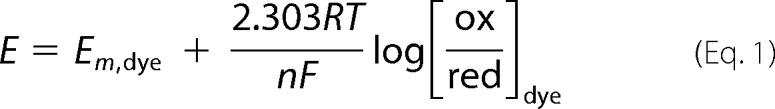 |
where R is the universal gas constant, T is the absolute temperature (298 K where the Em,dye was determined), n is the number of electrons in the reduction, and F is Faraday's constant. The solution potential versus log[ox/red] data (where ox and red are oxidized and reduced) for SQR from three independent titrations were combined and fitted to a linear function to yield the mid-point potential for the SQR-bound FAD.
SQR Activity Assays
SQR activity was measured by following the reduction of CoQ1 at 278 nm (ϵ = 12,000 m−1 cm−1) (24). The reactions were performed aerobically at 4 °C in 50 mm potassium phosphate buffer, pH 7.4, with 0.03% (w/v) DHPC. The reactions contained the following: 60 μm CoQ1, 0.1 mg/ml bovine serum albumin, 200 μm sodium sulfide (except when sulfide was the sole substrate, in which case it was present at 600 μm), 600 μm sodium sulfite, and 0.3 μg of SQR. All reagents, excluding sulfide and SQR, were preincubated at 4 °C for 3 min, after which the reactions were initiated by the addition of sulfide and SQR. A kcat value of 52 s−1 was measured for the reaction containing sulfide and sulfite, and a kcat value of 6 s−1 was measured for the reaction with sulfide only.
Stopped-flow Spectroscopy
All rapid-mixing spectroscopic experiments were carried out aerobically at 4 °C, on an SF-61 DX2 double mixing stopped-flow system from Hi-Tech Scientific equipped with a diode-array detector (300–700 nm range). The concentration of SQR for the studies was determined by the 450 nm absorbance of bound FAD (ϵ = 11,500 m−1 cm−1). Typically, a 20 μm solution of SQR in the reaction buffer (either 50 mm Tris-Cl, pH 8.0, with 0.03% DHPC, or 50 mm potassium phosphate, pH 7.4, with 0.03% DHPC) was mixed at 1:1 (v/v) ratio with solutions of sulfide or sulfite at various concentrations. The syringe containing SQR was surrounded by an ice bath throughout the experiment to stabilize the protein. Changes in the spectrum of SQR-bound flavin were then monitored between 1.5 ms and 150 s. The kinetic traces at selected wavelengths were fitted using the KinetAsyst and/or Kinetic Studio software packages. SQR from at least two independent protein preparations was employed in stopped-flow studies. In the experiments with CoQ1, the quinone was added to the syringe containing the sulfur substrate.
SQR Titration with Sulfite
The formation of an SQR CT complex was monitored in a quartz cuvette at 6 °C in a UV-visible range with a 2501 PC Shimadzu spectrophotometer. Stock solutions of sulfite (4–400 mm) were prepared in deoxygenated water immediately prior to titrations. Aliquots (1 μl) of the stock solution were added to 200 μl of aerobic 19 μm SQR in 50 mm Tris-Cl buffer, pH 8.0, containing 0.03% (w/v) DHPC, starting with the least concentrated stock, and the resulting flavin spectrum was recorded after mixing each aliquot with SQR solution. The absorbance of the flavin CT complex (675 nm) versus the concentration of sulfite corrected for dilution was fitted to a hyperbolic function in Origin 7.0 to obtain the apparent Kd. Formation of the CT complex followed in these experiments requires not only initial sulfite binding to SQR, but also its subsequent nucleophilic attack on the active site disulfide. Hence, we refer to the Kd determined as “apparent” because it includes contributions from both the binding and the chemical steps.
Characterization of the SQR-Sulfite CT Complex
The SQR-sulfite CT complex was generated by treating 200 μl of 12 μm SQR in 50 mm Tris-Cl buffer, pH 8.0, containing 0.03% DHPC, with sodium sulfite at a final concentration of 400 μm. The resulting CT complex was repeatedly washed with ice-cold buffer by ultrafiltration to remove excess sulfite, to minimize its reaction with the DTNB used to determine the total thiol content. A 2-μl aliquot of 33.3 mm DTNB stock (in ethanol) was added to the washed SQR-sulfite CT complex in 200 μl of total volume and allowed to react for 20 min at room temperature in the dark. The mixture was passed through a 10-kDa molecular mass cut-off Amicon filter, and the 5-thio-2-nitrobenzoate (TNB) anion product in the flow-through was quantified by its absorbance at 412 nm (ϵ = 14,500 m−1 cm−1). SQR retained above the filter was repeatedly washed with buffer to remove any unbound DTNB and TNB anion. The flavin spectrum of the washed enzyme was similar to that of as-purified SQR. The amount of recovered enzyme was assessed based on the absorbance at 450 nm (ϵ = 11,500 m−1 cm−1). Reactions of DTNB with untreated SQR and controls lacking SQR were run in parallel with sulfite-treated SQR samples. The experiments were repeated twice.
The formation of thiosulfate from the reaction between the SQR-sulfite CT complex and sulfide was assessed by HPLC. Briefly, 200 μl of 19 μm SQR in 50 mm Tris-Cl buffer, pH 8.0, containing 0.03% DHPC was mixed with 750 μm sodium sulfite (final concentration) in a cuvette. The accumulation of the CT complex was monitored, and the protein was thoroughly washed by ultrafiltration using ice-cold buffer to remove unbound sulfite. Sodium sulfide (100 μm) was added to the protein, and the mixture was derivatized with monobromobimane and analyzed by HPLC as described previously (16). A control lacking SQR was included in the analysis to correct for non-enzymatic solution chemistry between sulfide and sulfite. The reactions were repeated three times, with consistent results.
Generation of a Structural Model for Human SQR
The I-TASSER method (25) was used to generate a structural model for human SQR. The program identified six structural templates from the Protein Data Bank (PDB) (3hyx, 3hyw, 3pkg, 1fcd, 3vrd, and 3t2z), which it then used to construct an atomic model of human SQR. The structural templates included other SQRs (3hyx, 3hyw, 3pkg, and 3t2z), a flavocytochrome c sulfide dehydrogenase (1fcd), and a flavocytochrome c (3vrd).
Results
Oligomeric Organization of SQR and the Redox Potential of SQR-bound FAD
As isolated in the presence of 0.03% DHPC-containing buffer, SQR elutes in the void volume of a Superdex 200 gel filtration column, suggesting protein aggregation (not shown). However, in the presence of 0.3% DHPC, which does not affect SQR activity, the protein elutes as a symmetric peak with an estimated molecular mass of ∼90 kDa (Fig. 1D). Because the subunit molecular mass of human SQR is 47 kDa, the gel filtration data reveal that the protein exists as a dimer in solution, like other members of the flavin disulfide reductase superfamily (17).
Next, we determined the two-electron redox potential of SQR-bound FAD using a spectrochemical method as described under “Experimental Procedures.” Nernst analysis of the data pooled from three independent titrations yielded a value of −123 ± 7 mV versus the normal hydrogen electrode (Fig. 2).
FIGURE 2.
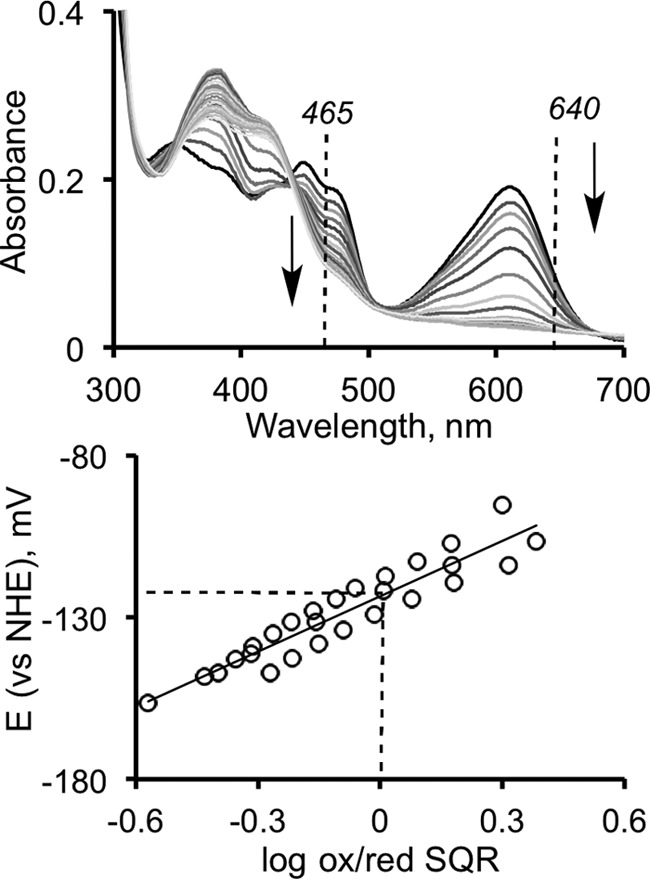
Redox titration of SQR-bound FAD. The titration was performed as described under “Experimental Procedures” in the presence of indigo carmine as a reference dye. Top panel, FAD reduction was monitored at 465 nm (the isosbestic point for the reference dye), and the reduction of the dye was followed at 640 nm. Bottom panel, Nernst-plot analysis of the estimated standard reduction potential of the solution versus the fraction of oxidized SQR-bound FAD. The spectrum (top) is representative of three independent experiments, whereas the lower panel shows data from all three experiments. NHE, normal hydrogen electrode; ox/red, oxidized/reduced.
Sulfide-induced CT Complex Formation
Sulfide can serve as both an electron donor and a sulfane sulfur acceptor in the SQR-catalyzed reaction (Fig. 1C) (18, 19). A low intensity CT species was reported to form when sulfide was mixed with SQR under anaerobic conditions (absorbance intensity ratio of 390:673 nm ≈ 1:0.26) (18). To assess the kinetic competence of the CT species, we used stopped-flow spectroscopy to monitor its rate of formation and decay. Formation of a broad CT complex with an absorbance maximum at 675 nm was readily observed under these conditions (Fig. 3A). CT complex formation was accompanied by a decrease in FAD absorption at 450 and 473 nm (shoulder). The spectral changes exhibited isosbestic points at 423 and 500 nm (Fig. 3A). Another spectral change was observed in the 385 nm region associated with oxidized flavin. It involved broadening and a red shift to 390 nm along with a slight increase in intensity at this wavelength (Fig. 3B). This feature will be discussed later. The amplitude of the CT species was directly proportional to the sulfide concentration (not shown). At high sulfide concentrations (1 mm), >60% of the starting oxidized flavin absorbance was associated with the CT species, giving a 390:675 nm absorbance ratio of 1:0.65 (Fig. 3B).
FIGURE 3.
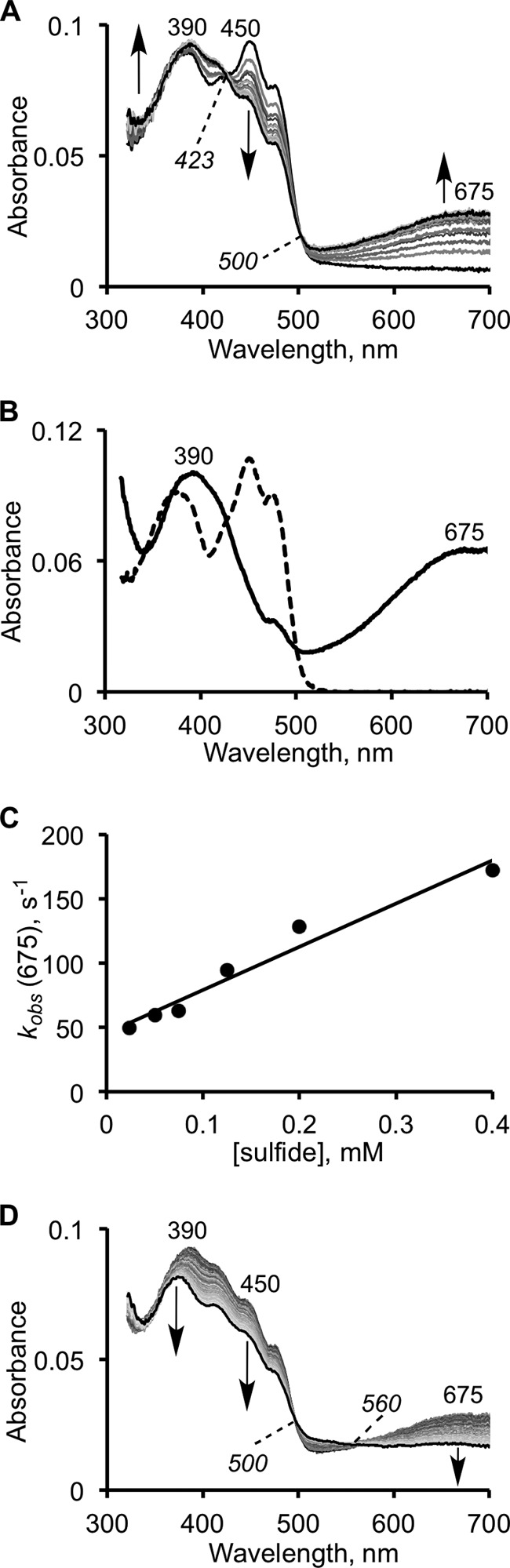
Kinetics of CT complex formation on SQR in the presence of sulfide. A, reaction between 10 μm SQR and 125 μm Na2S in 50 mm Tris-Cl buffer, pH 8.0, with 0.03% DHPC, was monitored over a period of 15 s. The first phase of the reaction is associated with the formation of an SQR-sulfide CT complex. B, representative spectrum of the fully developed CT complex (solid line) in the presence of an excess of sulfide (1 mm) and oxidized SQR (dashed line). C, dependence of the observed rate constant for formation of the CT complex on sulfide concentration, in 50 mm potassium phosphate buffer, pH 7.4, with 0.03% DHPC. The kinetics of CT complex decay (0.2 s−1) are unaffected by the changes in sulfide concentration. D, decay of the CT complex is accompanied by flavin reduction at 450 nm.
The observed rate constant for CT formation showed a dependence on sulfide concentration and was ∼5-fold higher at pH 7.4 (in 50 mm phosphate buffer) than at pH 8.0 (in 50 mm Tris-Cl), at all sulfide concentrations tested. The rate constant for accumulation of the CT species at 600 μm sulfide was 200 s−1 at pH 7.4 and 4 °C (Fig. 3C), which is well above the kcat of 6 s−1 measured under the same conditions by monitoring the rate of CoQ1 reduction (sulfide served as both the sulfur donor and the acceptor under these conditions). Hence, formation of the CT complex in the presence of sulfide represents a kinetically competent step when sulfide is the sulfur donor. The bimolecular rate constant for formation of the CT complex is calculated to be 3.36 × 105 m−1 s−1, whereas the koff estimated from the y-intercept is 45.5 s−1. Decomposition of the CT complex in the binary SQR-sulfide complex is slow (0.2 s−1), independent of sulfide concentration, and a second isosbestic point at 560 nm was observed in this phase of the reaction (Fig. 3D).
Effect of CoQ1 on the Kinetics of Sulfide-induced CT Complex Formation
In the presence of sulfide alone, the catalytic cycle cannot proceed beyond flavin reduction. So next, we examined the kinetics of sulfide-induced CT complex formation in the presence of the final electron acceptor, CoQ1. The spectral changes for SQR-bound flavin were similar but not identical in the presence and absence of CoQ1 (Fig. 4, A and B). In the presence of CoQ1, the second isosbestic point at 560 nm was not observed and the amplitude of the absorption changes at 390 nm was smaller. The presence of CoQ1 increased the rate of CT complex appearance, suggesting that the reaction occurred more efficiently in a ternary SQR-sulfide-CoQ1 complex. The rate constant for CT complex formation in the presence of 400 μm sulfide and 100 μm CoQ1 was 470 s−1 versus 165 s−1 in the absence of CoQ1. The rate of CT decomposition was difficult to compare because of turnover occurring with both substrates, which were in excess of SQR. Additionally, in the presence of CoQ1, sulfide serves as both an electron donor and a sulfane sulfur acceptor forming the hydrodisulfide product under these conditions (Fig. 1C) (18). Hence, the CT complex remained in pseudo-steady-state until CoQ1 was consumed.
FIGURE 4.
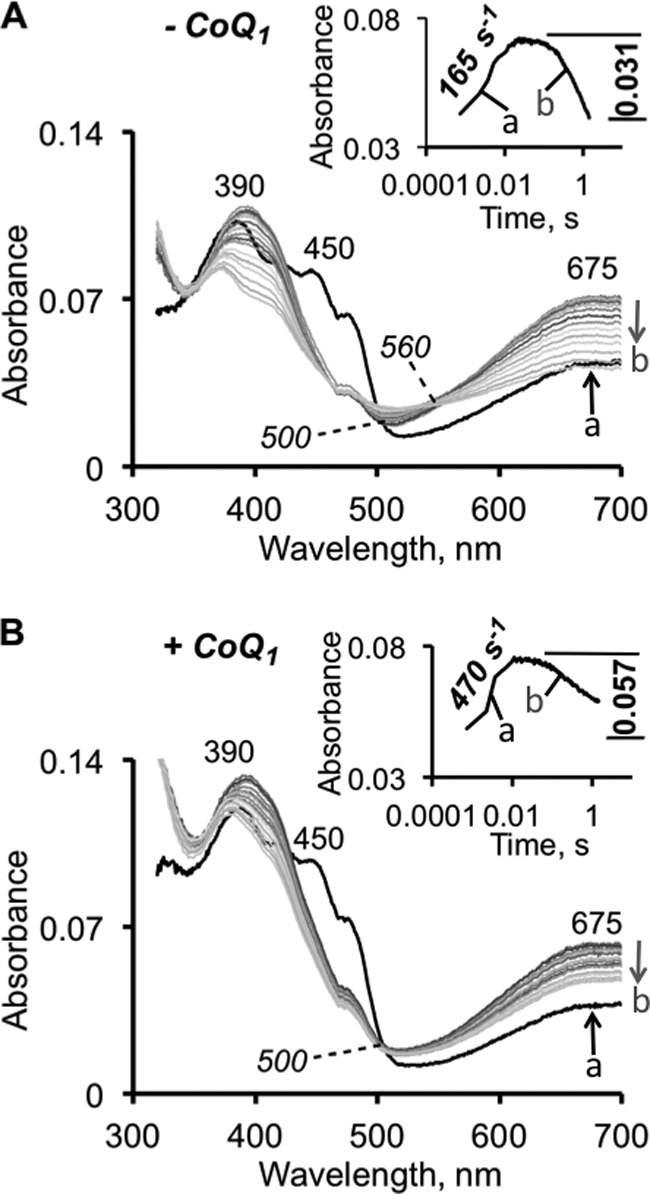
Kinetics of sulfide-dependent CT complex formation in the presence of CoQ1. The kinetics of CT formation (indicated by the up arrow a) and decay (indicated by the down arrow b) were monitored after mixing 10 μm SQR with 400 μm Na2S ± 100 μm CoQ1 (final concentrations; CoQ1 was included in the sulfide syringe) in 50 mm Tris-Cl buffer, pH 8.0, containing 0.03% DHPC. Due to fast CT formation at this higher sulfide concentration, the first recorded spectrum (shown in black in both panels) already possesses CT features. Although spectrally similar, the reactions without (A) and with (B) CoQ1 differ kinetically. The rate constant for the formation of the CT complex is enhanced in the presence of CoQ1. The rate constants and amplitudes for the kinetics measured at 675 nm are shown in the insets.
Formation of a CT Complex in the Presence of Sulfite
Sulfite is an efficient sulfane sulfur acceptor in the SQR-catalyzed reaction (18, 19), i.e. it functions to resolve the protein-bound persulfide intermediate formed in the presence of sulfide, thereby forming thiosulfate (Fig. 1C). However, an alternative mechanism in which sulfite adds first into the disulfide bond forming a sulfocysteine derivative can also be considered. The second step in this mechanism would involve resolution of the sulfocysteine intermediate by sulfide to give thiosulfate, disulfide, and reduced flavin. To test this postulate, we monitored the spectral changes upon the addition of sulfite to SQR and observed the robust formation of a stable CT complex (Fig. 5A). The absorbance features of the SQR-sulfite CT complex are very similar to the CT complex formed in the presence of sulfide, although it exhibits a slightly lower absorbance ratio of 390:675 nm of 1:0.51. The intensity of the CT absorbance was dependent on sulfite concentration, and its relative stability allowed estimation of a KD,app for sulfite of 160 ± 11 μm (Fig. 5, B and C).
FIGURE 5.

Characterization of the SQR-sulfite CT complex. A, a CT complex is formed upon the addition of 400 μm sulfite to 20 μm SQR and is resolved in the presence of 600 μm sulfide. The final spectrum is that of the reduced flavin (because sulfide is in excess) as observed previously (18). B, titration of 19 μm SQR with sulfite (0–3 mm) in 50 mm Tris-Cl buffer, pH 8.0, containing 0.03% DHPC (n = 3). The starting and final spectra are shown in black. C, the dependence of the CT absorbance change (average of three experiments) on sulfite concentration. D, DTNB (0.33 mm) treatment of the CT complex formed in A, after washing to remove excess sulfite, leads to the disappearance of CT complex, suggesting that a thiol in SQR participates in the CT complex. The decrease in absorbance of the “washed” CT complex likely resulted from a partial reversal to the oxidized protein during the handling step. Note that spectrophotometer was blanked with a DTNB solution, which itself absorbs in the visible region (400–500 nm).
The addition of sulfide led to the disappearance of the sulfite-induced CT complex (Fig. 5A) and a general bleaching of the flavin absorption spectrum. Similarly, the addition of DTNB led to loss of the sulfite-induced CT complex (Fig. 5D). The DTNB sensitivity of the spectrum is consistent with the involvement of a thiolate group in the CT interaction. Analysis of the SQR-sulfite mixture that had been treated with DTNB under native conditions indicated the presence of three thiols per monomer versus two in a sample of untreated SQR. Native SQR contains a total of four cysteines per monomer, of which two exist as the active site disulfide in the resting enzyme. When the CT complex obtained in the presence of sulfite was washed thoroughly to remove unbound sulfite and then reacted with sulfide, thiosulfate was detected in the mixture (not shown).
To further explore the possibility that the efficient turnover (monitored by CoQ1 reduction) observed in the presence of sulfite and sulfide is due to sulfite acting as a sulfur donor, the kinetic competence of the SQR-sulfite CT intermediate was assessed (Fig. 6). CT complex formation kinetics was biphasic, and the data were fitted by a double exponential equation. The amplitude of the first phase was independent of sulfite concentration, whereas the amplitude of the second phase was dependent on sulfite concentration (not shown). The rate constants for the two phases (1.0 and 0.1 s−1 at 600 μm sulfite) were significantly smaller than the kcat value of 52 s−1 determined under the same conditions for the SQR reaction. From the dependence of the kobs for CT formation on sulfite concentration, the kon for sulfite was estimated to be 103 m−1 s−1 and the koff was estimated to be 0.017 s−1, yielding a KD,app for sulfite of 165 μm, which is very similar to that obtained by spectral titration (Fig. 5C). Unlike sulfide, the kinetics of sulfite-induced CT complex formation were largely unaffected by the presence of CoQ1. The rate constants for the two phases at 200 μm sulfite were 0.47 and 0.11 s−1 (without CoQ1), and 0.50 and 0.16 s−1 (with CoQ1) (Fig. 6B).
FIGURE 6.
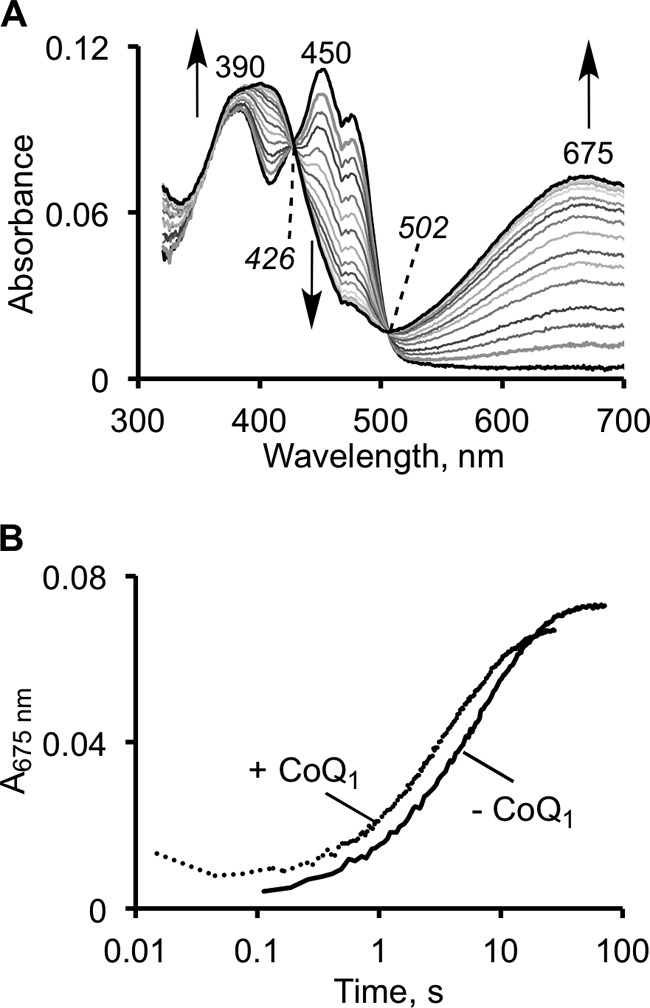
Stopped-flow kinetics of the SQR reaction with sulfite. A, reaction between 10 μm SQR and 200 μm sulfite in 50 mm potassium phosphate buffer, pH 7.4, with 0.03% DHPC, monitored over a period of 75 s. B, kinetics of CT formation in the absence and presence of 30 μm CoQ1. The kinetic traces could be fitted to a two-exponential model as described under “Results.” The addition of CoQ1 did not significantly alter the kinetics of CT complex formation, and similar kinetics were observed in 50 mm Tris-Cl buffer, pH 8.0.
Discussion
Despite the critical role played by SQR in H2S homeostasis, mechanistic insights into its catalytic cycle have been limited. Under Vmax conditions, using CoQ1 as electron acceptor, SQR exhibits a 7-fold higher catalytic efficiency at pH 7.4 in the presence of sulfide and sulfite versus when sulfide is used as both the sulfur donor and the acceptor (Fig. 7A, path I versus path II) (19). This difference in catalytic efficiency is even greater (140-fold) at pH 8.5 (18). However, although it has been assumed that the reaction is initiated by the addition of sulfide into the active site disulfide bond (Fig. 7A), the chemically feasible alternative, i.e. the addition of sulfite to the disulfide bond (Fig. 7B), has not been considered previously. In both reactions, thiosulfate is the product. In contrast, when sulfide is the only substrate, it serves as both the sulfur donor and the acceptor in the flavin reductive half-reaction that generates hydrodisulfide (Fig. 7A, path I). In this study, we have examined the transient kinetics of the flavin reductive half-reaction in the presence of sulfide or sulfite.
FIGURE 7.

Mechanistic alternatives for the flavin reductive half-reaction catalyzed by human SQR. A, reductive half-reaction in which sulfide is the first substrate to add to the disulfide bond forming a CT complex. The reaction branches at this point into paths I (sulfide as acceptor) and II (sulfite as acceptor), leading to the formation of hydrodisulfide and thiosulfate, respectively, and to reduced flavin (FADH2). Under Vmax conditions, the kcat for path II is 5-fold greater than for path I at pH 7.4 (19). B, reductive half-reaction of SQR in which sulfite is the first substrate and adds to the disulfide bond to give a sulfocysteine intermediate. This reaction is slow and is followed by sulfide addition to sulfocysteine to give thiosulfate and reduced flavin.
SQR catalyzes the oxidation of H2S to persulfide (E°′ = 280 mV (15, 26)) with concomitant reduction of ubiquinone to ubiquinol (E°′ = 65 mV, (27)). The two-electron oxidation of H2S proceeds via reduction of SQR-bound FAD, whose redox potential has been determined in this study to be −123 ± 7 mV, which is significantly more positive than that for FAD free in solution (−208 mV) (28). Thus, the two half-reactions are both predicted to be thermodynamically favorable with ΔG°′ estimates of −7.2 kcal mol−1 for the reduction of flavin by H2S, and −2.7 kcal mol−1 for reduction of ubiquinone by FADH2. The ΔG°′ for the overall reduction of ubiquinone by H2S is −9.9 kcal mol−1.
In the presence of potential sulfur donors, an unusually intense long-wavelength CT band centered at 675 nm is observed (Figs. 3B and 5B). In SQR, the CT complex is predicted to form following the addition of the sulfur donor substrate to the disulfide liberating a cysteine thiolate that charge-transfers to the oxidized flavin (Fig. 7). In principle, CT complexes can be formed by the interaction between a substrate/ligand that is electron-rich with the C(4a)-N(5) position of oxidized flavin or between reduced flavin and an electron-deficient ligand. When the electronic distribution involves reduced flavin and an oxidized ligand, a bluish CT band centered between 550 and 650 nm is often seen, whereas in the opposite situation, i.e. interaction between oxidized flavin and reduced ligand, a greenish CT band at longer wavelengths can be observed (29). The energy of the charge-transfer band, νCT, is governed by the electron affinity of the acceptor and the electron-donating capacity of the donor. Although the correlation is more appropriately made between νCT and the one-electron flavin redox potential (28, 30), correlations between the two-electron flavin redox potential and νCT have also been observed (28, 31).
A CT band that is similar to that observed with human SQR is seen in acyl-CoA dehydrogenases and has been ascribed to a tightly bound CoA-persulfide that donates into oxidized flavin (28). However, the spectrum of the fully developed SQR CT complex is remarkable in its intensity, and to our knowledge, unlike any other flavin CT intermediate reported previously. We ascribe the intensity of the CT band seen with human SQR to electron donation from the Cys-201 thiolate to FAD along with contributions from the nearby electron-rich persulfide (Cys-S-S−) or sulfocysteine (Cys-S-SO32−) formed on Cys-379 in the presence of sulfide or sulfite, respectively (Fig. 7, A and B).
A nearly symmetric band with a maximum at 390 nm accompanies formation of the 675 nm CT band in the presence of small molecule sulfur donors (Figs. 3B and 5B). As shown in Fig. 7, the Cys-201 thiolate nucleophile is postulated to attack the oxidized FAD to form a C4 adduct. The 390 nm absorption feature that forms at the same rate as the CT band in SQR bears some resemblance to the C4a-sulfur adduct centered at 384 nm in the thiol/disulfide oxidoreductase, lipoamide dehydrogenase (32). It also resembles the thiol-flavin C4a adduct in mercuric reductase, another member of this oxidoreductase family, which exhibits an absorbance maximum between 390 and 395 nm (33). Under different conditions, an enhanced 380 nm feature was observed when CoQ1 was used to reoxidize sulfide-reduced SQR (18). Despite the resemblance to C4a adducts of flavin, the 390 nm flavin band in SQR is not fully characteristic of one. We know of no C4a-flavin adduct that exhibits significant CT interactions. The maximal absorbances of C4a-flavin adducts have been observed between 370 and 390 nm. It is known that the dielectric constant of the flavin binding site, the planarity of the bound flavin, and interactions between the cofactor and active site residues can tune the spectral characteristics of flavin adducts significantly (34). The identity of the 390 nm species in SQR and the kinetics of its formation and decay are under investigation.
Sulfite is a very efficient sulfane sulfur acceptor in the presence of sulfide (as the sulfur donor) and exhibits a kcat/Km,sulfite of 1.7 × 106 m−1 s−1 at pH 7.4 (19). This value is 7-fold higher than the reaction in which sulfide acts as both donor and acceptor (19). Unexpectedly, we found that sulfite can also add into the disulfide bond generating a CT band with spectral properties similar to that observed with sulfide (Figs. 3 and 5). The chemistry of sulfite addition to cysteine disulfide has precedence in the formation of sulfocysteine, a biomarker for sulfite oxidase deficiency (35). Sulfite can also add to the N5 and C4a positions of flavin, with the latter reaction being favored when the C6 position has a bulky and charged substituent (36).
The rate constant for the addition of sulfite to SQR, however, is too slow for this step to be kinetically competent for thiosulfate formation and rules out the mechanism in Fig. 7B under turnover conditions. Hence, in the presence of both sulfite and sulfide, SQR favors the use of sulfide as the donor and sulfite, the acceptor. The kon for sulfite is significantly lower (103 m−1 s−1) than compared with that for sulfide (3.36 × 105 m−1 s−1). However, under pathological conditions when sulfite levels are elevated, it is possible that SQR can be subverted to form thiosulfate by the mechanism shown in Fig. 7B. This could, in turn, inhibit H2S oxidation by tying up SQR in a slow conversion of sulfite and sulfide to thiosulfate. Although thiosulfate levels are elevated in sulfite oxidase deficiency (35), changes, if any, in H2S levels associated with this disease are not known.
In summary, we have demonstrated the formation of an unusually intense CT intermediate that is formed at a kinetically competent rate in the presence of sulfide. We also report an alternate reaction in which sulfite can add to the disulfide bond in SQR, which might be relevant under pathological conditions of sulfite oxidase deficiency. Based on the flavin midpoint potential determined in this study, both flavin reduction (by sulfide) and oxidation (by ubiquinone) are predicted to be thermodynamically favorable.
Author Contributions
T. V. M., P. K. Y., and D. P. B. designed, performed and analyzed the experiments. T. V. M. and P. K. Y. wrote parts of the manuscript. R. B. helped conceive the experiments, analyzed the data, and co-wrote the manuscript. All authors edited and approved the final version of the manuscript.
This work was supported by National Institutes of Health Grant GM112455 (to R. B.) and by a postdoctoral fellowship from the American Heart Association (14POST18760003) (to P. K. Y.). The authors declare that they have no conflicts of interest with the contents of this article.
- SQR
- sulfide quinone oxidoreductase
- CT
- charge-transfer
- CoQ1
- coenzyme Q1
- DHPC
- 1,2-diheptanoyl-sn-glycero-3-phosphocholine
- DTNB
- 5,5′-dithiobis(2-nitrobenzoic acid).
References
- 1. Powell M. A., Somero G. N. (1986) Hydrogen sulfide oxidation is coupled to oxidative phosphorylation in mitochondria of Solemya reidi. Science 233, 563–566 [DOI] [PubMed] [Google Scholar]
- 2. Goubern M., Andriamihaja M., Nübel T., Blachier F., Bouillaud F. (2007) Sulfide, the first inorganic substrate for human cells. FASEB J. 21, 1699–1706 [DOI] [PubMed] [Google Scholar]
- 3. Kimura H. (2010) Hydrogen sulfide: from brain to gut. Antioxid. Redox Signal. 12, 1111–1123 [DOI] [PubMed] [Google Scholar]
- 4. Polhemus D. J., Lefer D. J. (2014) Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ. Res. 114, 730–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kabil O., Motl N., Banerjee R. (2014) H2S and its role in redox signaling. Biochim. Biophys. Acta 1844, 1355–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chiku T., Padovani D., Zhu W., Singh S., Vitvitsky V., Banerjee R. (2009) H2S biogenesis by cystathionine γ-lyase leads to the novel sulfur metabolites, lanthionine and homolanthionine, and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 284, 11601–11612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singh S., Padovani D., Leslie R. A., Chiku T., Banerjee R. (2009) Relative contributions of cystathionine β-synthase and γ-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 284, 22457–22466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stipanuk M. H., Beck P. W. (1982) Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 206, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kabil O., Vitvitsky V., Xie P., Banerjee R. (2011) The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid. Redox Signal. 15, 363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yadav P. K., Yamada K., Chiku T., Koutmos M., Banerjee R. (2013) Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 288, 20002–20013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shibuya N., Tanaka M., Yoshida M., Ogasawara Y., Togawa T., Ishii K., Kimura H. (2009) 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 11, 703–714 [DOI] [PubMed] [Google Scholar]
- 12. Kabil O., Banerjee R. (2010) The redox biochemistry of hydrogen sulfide. J. Biol. Chem. 285, 21903–21907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vitvitsky V., Kabil O., Banerjee R. (2012) High turnover rates for hydrogen sulfide allow for rapid regulation of its tissue concentrations. Antioxid. Redox Signal. 17, 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kabil O., Vitvitsky V., Banerjee R. (2014) Sulfur as a signaling nutrient through hydrogen sulfide. Annu. Rev. Nutr. 34, 171–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mishanina T. V., Libiad M., Banerjee R. (2015) Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 11, 457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vitvitsky V., Yadav P. K., Kurthen A., Banerjee R. (2015) Sulfide oxidation by a noncanonical pathway in red blood cells generates thiosulfate and polysulfides. J. Biol. Chem. 290, 8310–8320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Argyrou A., Blanchard J. S. (2004) Flavoprotein disulfide reductases: advances in chemistry and function. Prog. Nucleic Acid Res. Mol. Biol. 78, 89–142 [DOI] [PubMed] [Google Scholar]
- 18. Jackson M. R., Melideo S. L., Jorns M. S. (2012) Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51, 6804–6815 [DOI] [PubMed] [Google Scholar]
- 19. Libiad M., Yadav P. K., Vitvitsky V., Martinov M., Banerjee R. (2014) Organization of the human mitochondrial H2S oxidation pathway. J. Biol. Chem. 289, 30901–30910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marcia M., Ermler U., Peng G., Michel H. (2009) The structure of Aquifex aeolicus sulfide:quinone oxidoreductase, a basis to understand sulfide detoxification and respiration. Proc. Natl. Acad. Sci. U.S.A. 106, 9625–9630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hildebrandt T. M., Grieshaber M. K. (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 275, 3352–3361 [DOI] [PubMed] [Google Scholar]
- 22. Brito J. A., Sousa F. L., Stelter M., Bandeiras T. M., Vonrhein C., Teixeira M., Pereira M. M., Archer M. (2009) Structural and functional insights into sulfide:quinone oxidoreductase. Biochemistry 48, 5613–5622 [DOI] [PubMed] [Google Scholar]
- 23. Massey V. A. (1991) A simple method for the determination of redox potentials. in Flavins and Flavoproteins (Curti B., Rochi S., Zanetti G., eds), pp. 59–66, Water DeGruyter & Co., Berlin, Germany [Google Scholar]
- 24. Banerjee R., Chiku T., Kabil O., Libiad M., Motl N., Yadav P. K. (2015) Assay methods for H2S biogenesis and catabolism enzymes. Methods Enzymol. 554, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y. (2015) The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Giles G. I., Jacob C. (2002) Reactive sulfur species: an emerging concept in oxidative stress. Biol. Chem. 383, 375–388 [DOI] [PubMed] [Google Scholar]
- 27. Urban P. F., Klingenberg M. (1969) On the redox potentials of ubiquinone and cytochrome b in the respiratory chain. Eur. J. Biochem. 9, 519–525 [DOI] [PubMed] [Google Scholar]
- 28. Williamson G., Engel P. C., Mizzer J. P., Thorpe C., Massey V. (1982) Evidence that the greening ligand in native butyryl-CoA dehydrogenase is a CoA persulfide. J. Biol. Chem. 257, 4314–4320 [PubMed] [Google Scholar]
- 29. Ghisla S., Thorpe C. (2004) Acyl-CoA dehydrogenases: a mechanistic overview. Eur. J. Biochem. 271, 494–508 [DOI] [PubMed] [Google Scholar]
- 30. Wagner M. A., Trickey P., Chen Z. W., Mathews F. S., Jorns M. S. (2000) Monomeric sarcosine oxidase: 1. Flavin reactivity and active site binding determinants. Biochemistry 39, 8813–8824 [DOI] [PubMed] [Google Scholar]
- 31. Abramovitz A. S., Massey V. (1976) Interaction of phenols with old yellow enzyme: physical evidence for charge-transfer complexes. J. Biol. Chem. 251, 5327–5336 [PubMed] [Google Scholar]
- 32. Thorpe C., Williams C. H., Jr. (1976) Spectral evidence for a flavin adduct in a monoalkylated derivative of pig heart lipoamide dehydrogenase. J. Biol. Chem. 251, 7726–7728 [PubMed] [Google Scholar]
- 33. Sahlman L., Lambeir A. M., Lindskog S. (1986) Rapid-scan stopped-flow studies of the pH dependence of the reaction between mercuric reductase and NADPH. Eur. J. Biochem. 156, 479–488 [DOI] [PubMed] [Google Scholar]
- 34. Ghisla S., Massey V., Lhoste J. M., Mayhew S. G. (1974) Fluorescence and optical characteristics of reduced flavines and flavoproteins. Biochemistry 13, 589–597 [DOI] [PubMed] [Google Scholar]
- 35. Shih V. E., Abroms I. F., Johnson J. L., Carney M., Mandell R., Robb R. M., Cloherty J. P., Rajagopalan K. V. (1977) Sulfite oxidase deficiency: biochemical and clinical investigations of a hereditary metabolic disorder in sulfur metabolism. N. Engl. J. Med. 297, 1022–1028 [DOI] [PubMed] [Google Scholar]
- 36. Hevesi L., Bruice T. C. (1973) Reaction of sulfite with isoalloxazines. Biochemistry 12, 290–297 [DOI] [PubMed] [Google Scholar]