

Background: Csd6 is one of the cell shape-determining proteins in H. pylori.
Results: The active site of Csd6 is tailored to function as an l,d-carboxypeptidase in the peptidoglycan-trimming process.
Conclusion: Csd6 constitutes a new family of l,d-carboxypeptidase.
Significance: The substrate limitation of Csd6 is a strategy that H. pylori uses to regulate its helical cell shape and motility.
Keywords: cell motility; Helicobacter pylori; peptidoglycan; protein structure; structure-function; Csd6; HP0518; L,D-carboxypeptidase; cell shape; flagellin
Abstract
Helicobacter pylori causes gastrointestinal diseases, including gastric cancer. Its high motility in the viscous gastric mucosa facilitates colonization of the human stomach and depends on the helical cell shape and the flagella. In H. pylori, Csd6 is one of the cell shape-determining proteins that play key roles in alteration of cross-linking or by trimming of peptidoglycan muropeptides. Csd6 is also involved in deglycosylation of the flagellar protein FlaA. To better understand its function, biochemical, biophysical, and structural characterizations were carried out. We show that Csd6 has a three-domain architecture and exists as a dimer in solution. The N-terminal domain plays a key role in dimerization. The middle catalytic domain resembles those of l,d-transpeptidases, but its pocket-shaped active site is uniquely defined by the four loops I to IV, among which loops I and III show the most distinct variations from the known l,d-transpeptidases. Mass analyses confirm that Csd6 functions only as an l,d-carboxypeptidase and not as an l,d-transpeptidase. The d-Ala-complexed structure suggests possible binding modes of both the substrate and product to the catalytic domain. The C-terminal nuclear transport factor 2-like domain possesses a deep pocket for possible binding of pseudaminic acid, and in silico docking supports its role in deglycosylation of flagellin. On the basis of these findings, it is proposed that H. pylori Csd6 and its homologs constitute a new family of l,d-carboxypeptidase. This work provides insights into the function of Csd6 in regulating the helical cell shape and motility of H. pylori.
Introduction
Helicobacter pylori is a spiral-shaped Gram-negative bacterium that colonizes the upper gastrointestinal tract in approximately half of the world's human population. Its infection of the gastric mucosa is associated with various gastric diseases, including chronic gastritis, peptic ulcer, mucosa-associated lymphoid tissue lymphoma, and gastric adenocarcinoma (1, 2). H. pylori is regarded as a primary factor for gastric cancer development (1) and was classified as a group I carcinogen by the International Agency for Research on Cancer of the World Health Organization. In recent years, H. pylori infection has also been implicated with some extra-digestive diseases (3). Typical treatment regimens for eradicating H. pylori consist of a proton pump inhibitor (for example, omeprazole) and the antibiotics such as clarithromycin and amoxicillin (or metronidazole). However, increasing drug resistance in H. pylori requires the discovery of new antibiotics (4).
High motility of H. pylori is important for its colonization of the human stomach and its survival in the viscous gastric mucosa (5–7). The helical cell shape of H. pylori is believed to facilitate penetration of the viscous epithelial mucous layer via a cork-screwing mechanism (8–10). Several H. pylori mutants with altered cell shapes exhibited attenuated colonization (11). The peptidoglycan layer, the major component of the bacterial cell wall, plays an essential role in withstanding the turgor pressure and in determining the cell shape (12, 13). It is made of linear polysaccharide chains that consist of alternating β-1,4-linked N-acetylglucosamine-N-acetylmuramic acid disaccharide units, with a pentapeptide linked to N-acetylmuramic acid (14). In H. pylori, the pentapeptide sequence is l-Ala1-γ-d-Glu2-mDAP3-d-Ala4-d-Ala5 (or -Gly5), where mDAP5 refers to meso-2,6-diaminopimelate, and the neighboring peptides are cross-linked exclusively by the 4→3 linkage between the main chain of d-Ala4 from one strand and the side chain of mDAP3 from another strand (15) to form a mesh-like structure termed peptidoglycan sacculus (16, 17). Colonization and infection by H. pylori also rely on the ability of the bacterium to move toward the part of gastric mucosa with more neutral pH. Besides the helical morphology, powerful flagella of H. pylori are responsible for its high motility through the viscous gastric mucous layer (5–7). The flagella provide a propulsive torque as well as a rotary movement of the cell body; a helical cell shape of H. pylori generates a corkscrew-like rotation (7–9).
In many bacteria, including H. pylori, the peptidoglycan layer is remodeled by a number of cell-wall hydrolases as well as synthetases for the peptidoglycan maturation, regulation of cell-wall growth, cell division, peptidoglycan turnover and recycling, cell lysis, and the release of peptidoglycan fragments for host-pathogen interactions (18). In H. pylori, an amidase AmiA (19), peptidases Csd1–4, potential regulators Csd5 and CcmA (20–22), and another peptidase Csd6 (23) are required to tailor the peptidoglycan layer to generate the helical cell shape. We have recently reported the crystal structures of both Csd4 (HP1075 in strain 26695) (24) and Csd3 (HP0506 in strain 26695) (25). Csd4 is a Zn2+-dependent d,l-carboxypeptidase (d,l-CPase) of the M14 metallopeptidase family and cleaves the γ-d-Glu2-mDAP3 bond of the muramyl tripeptide to produce the muramyl dipeptide (21, 24). Csd3 (also known as HdpA) belongs to the M23 metallopeptidase family and has both d,d-endopeptidase and d,d-carboxypeptidase (d,d-CPase) activities (22). Csd6 (encoded by the hp0518 gene in H. pylori 26695 strain) was identified as another member of the peptidoglycan trimming pathway and also a cell-shape determinant of H. pylori (23). The transposon mutant H. pylori with the disruption of the csd6 gene or the csd6 deletion mutant displayed a straight rod shape and an increase in tetrapeptide-containing muropeptides (23). Incubation of the recombinant hexahistidine-tagged Csd6 with tetrapeptide-rich sacculi from the Δcsd1csd6 mutant H. pylori resulted in complete conversion of the monomeric tetrapeptides to tripeptides (23). In Campylobacter jejuni, another helically shaped gastrointestinal pathogen, a homolog of Csd6 has been characterized as an l,d-CPase and was named as Pgp2 (26). Loss of pgp2 resulted in the morphology defect, changes in the peptidoglycan muropeptide profile, reduced motility, and moreover, a decreased interaction with the host (26). Between Csd6 and Pgp2, an overall amino acid sequence identity of 36% and similarity of 58% exist. Given the significance of mDAP-containing muramyl tripeptide as an agonist for the cytosolic innate immune receptor Nod1 (27, 28), an increased amount of muramyl tripeptide as a result of l,d-CPase reaction by Pgp2 (or Csd6 from H. pylori) was suggested to effect the Nod1 activation and ultimately the NF-κB transcriptional activity (26). The complete absence of tripeptide-containing muropeptides in Δpgp2 peptidoglycan did indeed reflect the reduced Nod1 activation, despite no effect in either intracellular survival or IL-8 secretion (26).
A bioinformatics analysis on the basis of the amino acid sequence predicts that residues 67–200 of H. pylori Csd6 form a YkuD domain (formerly called ErfK/YbiS/YcfS/YnhG; “l,d-transpeptidase” catalytic domain; Pfam 03734) that can possibly catalyze nonclassical 3→3 cross-linking of peptidoglycan. The YkuD domain-containing l,d-transpeptidases (l,d-TPases) have been identified in a range of bacteria, including Enterococcus faecium, Bacillus subtilis, and Mycobacterium tuberculosis (7, 26, 29, 30). They generate 3→3 cross-linkages of peptidoglycan, instead of 4→3 cross-linking catalyzed by classical d,d-transpeptidases, resulting in high level resistance to β-lactam antibiotics (31) or in peptidoglycan remodeling for dormancy in M. tuberculosis (32). However, the Helicobacter peptidoglycan layer is cross-linked exclusively by 4→3 linkages (15, 20, 33). Consistent with the absence of 3→3 cross-linked muropeptides in the H. pylori peptidoglycan sacculus, Csd6 was shown to exhibit the l,d-CPase activity only with no “transpeptidase” cross-linking activity against the muropeptides (23). However, it still remains unanswered why Csd6 has no “transpeptidase” cross-linking activity, despite the presence of a putative “l,d-TPase” domain and conservation of the catalytic Cys/His residues.
In many bacterial pathogens, flagella are best known for conferring motility and virulence, as well as serving as an export apparatus for virulence factors (34) and sensing the viscosity of a medium (35). Flagellar filaments of H. pylori are composed of two copolymerized flagellins (FlaA and FlaB) (36), which are heavily O-glycosylated with pseudaminic acid (Pse5Ac7Ac; Pse), a sialic acid resembling sugar (37, 38). The regulation of such post-translational modification is critical for the assembly of functional flagella (37, 39). Interestingly, a previous work highlighted the unique role of H. pylori G27 Csd6 that involves deglycosylation of FlaA; the Δcsd6 mutant exhibited altered motility to facilitate host-cell interaction and superior colonization (40). It suggests that Csd6 binds Pse molecules or deglycosylates O-glycosylated FlaA proteins.
To better understand the molecular function of H. pylori Csd6, its biochemical, biophysical, and structural characterizations have been performed in this study. Mass analyses using the synthetic muramyl peptides indicate that Csd6 functions only as the l,d-carboxypeptidase (l,d-CPase) and not as the l,d-transpeptidase (l,d-TPase). Analytical ultracentrifugation and single molecule fluorescence resonance energy transfer (FRET) analyses indicate that Csd6 exists as a dimer in solution. A Csd6 monomer has a three-domain architecture consisting of the N-terminal domain (NTD; Val-13–Asn-56), the middle l,d-CPase domain (Lys-67–Glu-202), and the C-terminal nuclear transport factor 2-like domain (NTF2-like domain; Thr-209–Lys-330). The NTD shows very remote structural similarity to other known protein structures and plays a key role in dimerization. The middle catalytic domain has an overall fold of the l,d-TPase domain, validating the bioinformatics prediction. However, here this catalytic domain is referred to as the “l,d-CPase” domain, because we as well as others show that Csd6 functions only as an l,d-CPase. The Csd6 l,d-CPase domain resembles those of well characterized l,d-TPases, but its pocket-shaped active site is uniquely defined by the four loops I–IV, among which loops I and III show the most distinct variations in sequence length and conformation from known l,d-TPases. The d-Ala-complexed structure suggests possible binding modes of both the substrate and product to the l,d-CPase domain. The NTF2-like domain possesses a deep pocket for possible binding of a hydrophobic ligand such as Pse, and an in silico docking study supports its role in the control of the glycosylation level of flagellin. This work provides further insights into the strategy of H. pylori for regulating its helical cell shape and motility, which are crucial for its virulence. The reported structural information would serve as the foundation in a search for new drug targets to fight infections by H. pylori.
Experimental Procedures
Protein Expression and Purification
The PCR-amplified csd6 gene from H. pylori 26695 strain, encompassing residues Val-13–Lys-330 of the gene product (HP0518), was cloned into the expression vector pET-28b(+) (Novagen) to express the recombinant Csd6 protein fused with a His6-containing tag at its N terminus. It was overexpressed in Escherichia coli Rosetta2(DE3)pLysS cells using a Luria Broth culture medium. Protein expression was induced by 0.5 mm isopropyl β-d-thiogalactopyranoside, and the cells were incubated for an additional 20 h at 18 °C following growth to mid-log phase at 37 °C. The cells were lysed by sonication in buffer A (50 mm Tris-HCl, pH 7.9, 500 mm sodium chloride, and 50 mm imidazole) containing 10% (v/v) glycerol and 1 mm phenylmethylsulfonyl fluoride. The crude lysate was centrifuged at 36,000 × g for 1 h. The recombinant Csd6 protein showed a tendency to aggregate at 4 °C; therefore, it was purified at room temperature in two column chromatography steps. The supernatant was applied to an affinity chromatography column of HiTrap Chelating HP (GE Healthcare), which was previously equilibrated with buffer A. Upon eluting with a gradient of imidazole in the same buffer, the Csd6 protein was eluted at 120–150 mm imidazole concentration. The eluted protein was applied to a HiLoad XK-16 Superdex 200TM column (GE Healthcare), which was previously equilibrated with 20 mm Tris-HCl, pH 7.9, and 150 mm sodium chloride. Fractions containing the Csd6 protein were pooled and concentrated to 5.8 mg ml−1 for crystallization using a YM10 ultrafiltration membrane (Amicon). This construct gave the best crystals in crystallization experiments. Other constructs were also tried for expression and crystallization. The Val-13–Lys-330 construct fused with a C-terminal His6-containing tag was not expressed in E. coli Rosetta2(DE3)pLysS cells. The constructs covering residues 4–330 or 17–330 were expressed at very low levels and were insoluble. The construct covering residues 1–330 (full-length) was expressed in a soluble form, but its crystals diffracted poorly to 7 Å only.
As a positive control in the l,d-TPase assay, LdtMt2, an l,d-TPase encoded by the rv2518c gene of M. tuberculosis H37Rv strain, was used. The N-terminal region (Leu-20–Ala-42) of LdtMt2 is predicted to form a putative transmembrane helix; the gene covering residues Ala-55–Ala-408, fused with a His6-containing tag at its C terminus, was cloned into pET-21a(+) (Novagen). The recombinant LdtMt2 was overexpressed and purified essentially as above.
Crystallization, X-ray Data Collection, and Structural Determination
To solve the phase problem by anomalous diffraction, ethyl mercury thiosalicylate (EMTS)-derivative crystals of the recombinant Csd6 were obtained at 23 °C by co-crystallization in the presence of EMTS by the sitting-drop vapor diffusion method. Sitting drops were prepared by mixing 3 μl of the protein solution and 2 μl of the reservoir solution (250 mm potassium nitrate, 18% (w/v) polyethylene glycol 3,350, and 10 mm EMTS). EMTS-derivatized crystals grew to approximate dimensions of 0.1 × 0.1 × 0.05 mm within a few days. Single-wavelength anomalous diffraction data were collected at 100 K from an EMTS-derivatized crystal of Csd6 using a cryoprotectant solution containing 25% (v/v) glycerol added to the reservoir solution (Table 1). The raw data were processed and scaled using the program suite HKL2000 (41). Two mercury sites in two monomers of Csd6 in the crystallographic asymmetric unit were located using the AutoSol program of the PHENIX software package (42). The initial phases were further improved by density modification using the automatic model building program Resolve (43). The initial model was improved through iterative cycles of model building with Coot (44) and refinement with Refmac5 of the CCP4 program suite (45, 46).
TABLE 1.
Data collection and refinement statistics
| A. Data collection | |||
| Data set | EMTS peak | Csd6-unbound | Csd6-Ala |
| Beamline sourcea | PLS BL-4A | PF BL-1A | PLS BL-5C |
| Space group | P212121 | P212121 | P212121 |
| a, b, c (Å) | 61.8, 89.6, 128.7 | 63.0, 91.0, 127.8 | 63.4, 90.6, 127.9 |
| X-ray wavelength (Å) | 1.0048 | 1.0000 | 0.9796 |
| Resolution rangeb (Å) | 50.0–2.90 (2.95–2.90) | 50.0–2.03 (2.07–2.03) | 50.0–2.04 (2.08–2.04) |
| No. of total reflectionsb | 373,689 (18,354) | 229,193 (11,715) | 227,623 (11,074) |
| No. of unique reflectionsb | 30,311 (1,515)c | 47,555 (2,343) | 47,575 (2,307) |
| Completeness (%)b | 99.9 (100)c | 98.3 (98.1) | 99.9 (100) |
| 〈I〉/〈σI〉b | 35.0 (4.7)c | 25.0 (3.0) | 29.3 (3.4) |
| Wilson B factor (Å2) | 53.1 | 34.2 | 30.5 |
| Rmergeb,d (%) | 11.0 (73.1)c | 9.5 (47.0) | 8.6 (53.7) |
| B. SAD phasing | Figure of merit (before/after density modification) 0.34/0.66 | ||
| C. Model refinement | |||
| PDB ID code | 4XZZ | 4Y4V | |
| Resolution range (Å) | 30.0–2.03 | 20.0–2.04 | |
| Rwork/Rfreee (%) | 19.6/24.8 | 19.1/24.2 | |
| No. of non-hydrogen atoms/average B-factor (Å2) | |||
| Total | 5,589/40.6 | 5,606/37.8 | |
| Protein | 5,248/40.4 | 5,249/38.7 | |
| Water oxygen | 323/41.5 | 315/40.5 | |
| Glycerol | 18/63.4 | 24/58.9 | |
| d-Ala | −/− | 18/44.6 | |
| R.m.s. deviations from ideal geometry | |||
| Bond lengths (Å)/angles (°) | 0.009/1.33 | 0.010/1.39 | |
| R.m.s. Z-scores | |||
| Bond lengths (Å)/angles (°) | 0.46/0.61 | 0.49/0.65 | |
| Ramachandran (%)f | |||
| Favored/outliers | 98.6/0.00 | 98.4/0.00 | |
| Poor rotamers (%)f | 0.00 | 0.70 | |
a PF and PLS stand for Photon Factory, Japan, and Pohang Light Source, Korea, respectively.
b Values in parentheses refer to the highest resolution shell.
c Friedel pairs were treated as separate observations.
d Rmerge = ΣhΣi|I(h)i − 〈I(h)〉|/ΣhΣiI(h)i, where I(h) is the intensity of reflection h; Σh is the sum over all reflections, and Σi is the sum over i measurements of reflection h.
e Rwork = Σ‖Fobs| − |Fcalc‖/Σ|Fobs|, where Rfree is calculated for a randomly chosen 5% of reflections, which were not used for structure refinement, and Rwork is calculated for the remaining reflections.
f Values were obtained using MolProbity.
Native crystals of ligand-free Csd6 were grown at 23 °C by the sitting-drop vapor diffusion method. Sitting drops were prepared by mixing 0.5 μl of the protein solution, 0.1 μl of a 10-fold diluted microseed crystal solution, and 0.4 μl of the reservoir solution (1.5% (v/v) Tacsimate, pH 7.0, 22% (w/v) PEG 3,350, and 100 mm sodium-HEPES, pH 7.5). The microseed crystals were originally obtained with ligand-free Csd6 using the above reservoir solution. The crystals grew to approximate dimensions of 0.2 × 0.1 × 0.05 mm within a few days. X-ray diffraction data for the ligand-free Csd6 (Csd6-unbound) were collected at 100 K using a cryoprotectant solution containing 20% (v/v) glycerol added to the reservoir solution (Table 1). X-ray diffraction data for the d-Ala-bound Csd6 (Csd6-Ala) were collected at 100 K after the native crystal was preincubated for 2 min in a cryoprotectant solution containing both 20% (v/v) glycerol and 96 mm d-Ala, which were added to the reservoir solution (Table 1). The structures of both Csd6-unbound and Csd6-Ala were determined by molecular replacement with the program MolRep (47) using the refined model of EMTS-derivatized Csd6. Stereochemistry of the refined models was evaluated using MolProbity (48). Data collection and refinement statistics are given in Table 1.
In Silico Docking
Induced fit docking (IFD) (Schrödinger Suite Induced Fit Docking protocol; Glide version 5.7, Prime version 3.0, Schrödinger, LLC, New York) (49, 50) was employed to predict the binding mode of Pse with consideration of protein flexibility. The Csd6 structure (monomer A model) was prepared for docking calculations using the Protein Preparation Wizard implemented in Maestro (version 9.2, Schrödinger, LLC, New York). The structure of Pse was built in Maestro 9.2, and the possible conformations of the ligand were generated using LigPrep (version 2.5, Schrödinger, LLC, New York). Pse was docked onto Csd6 using the following steps. (i) The receptor grid of Csd6 was defined as an enclosing box at the centroid of the key amino acid residues (i.e. Trp-227, Lys-258, Tyr-297, Lys-313, and Glu-329) in the binding pocket. (ii) In the initial Glide docking stage, a potential docking with van der Waals radius scaling of 0.5 for the protein and ligand was performed retaining a maximum number of 20 poses per ligand. (iii) Residues within 5.0 Å of ligand poses were kept free to move in the Prime refinement step, and the side chains were further optimized. (iv) Poses within 30 kcal mol−1 of the energy cutoff in the previous step were re-docked using Glide XP. (v) The binding energy (IFDScore) for each output pose was computed as implemented in the IFD protocol. The best docked poses were also reproduced by a different docking program, Surflex-Dock of Sybyl X 2.0 (Tripos Int., St. Louis, MO). All the computations were undertaken on an Intel® XeonTM Quad-core 2.5 GHz workstation with Linux Cent OS release 5.5.
As a positive control of the docking method, IFD of sialic acid (N-acetylneuraminic acid) and Pse onto Micromonospora viridifaciens sialidase (PDB code 1EUS) (51) and Pseudomonas aeruginosa pseudaminidase (PDB code 2W38) (52) was performed. The docked conformation of sialic acid showed a root-mean-square (r.m.s.) deviation of 1.5 Å from the co-crystallized 2-deoxy-2,3-dideoxy-N-acetylneuraminic acid (a sialic acid mimic) in M. viridifaciens sialidase (51). The binding mode of Pse in P. aeruginosa pseudaminidase predicted by the present docking calculation was very similar to that of the reported docking result (52). These results validate the present docking method.
Peptidase Assay by Mass Analysis
As potential substrates, synthetic muramyl tetrapeptide (β-methyl N-acetylmuramic acid-l-Ala1-γ-d-Glu2-mDAP3-d-Ala4) and muramyl pentapeptide (β-methyl N-acetylmuramic acid-l-Ala1-γ-d-Glu2-mDAP3-d-Ala4-d-Ala5) were prepared as described previously (Fig. 1, A and B) (53). The analyte (muramyl tetrapeptide or muramyl pentapeptide) at 5 mm was incubated for 2 h at 37 °C with the recombinant Csd6 protein (5 μm), which was dissolved in 20 mm sodium phosphate, pH 6.0, and 150 mm sodium chloride. A sample solution (1 μl) was mixed on the target with a fresh saturated matrix solution of 2,5-dihydroxy benzoic acid dissolved in 0.1% (v/v) trifluoroacetic acid and 50% (v/v) acetonitrile. For sample deposition, a 384-position stainless steel sample plate was used. Mass spectra were acquired on a matrix-assisted laser desorption/ionization quadrupole ion trap time-of-flight mass spectrometer (MALDI-QIT-TOF MS, AXIMA QIT; Shimadzu/Kratos, Manchester, UK) equipped with a nitrogen laser (337 nm, 3-ns pulse width, maximum pulse rate of 10 Hz). Mass spectra were obtained in a positive ion mode. Helium was used for trapping and cooling ions in the ion source. The pressure in the trap was held at 4 × 10−3 torr. Each spectrum constituted an average of 200 profiles. All spectra were externally calibrated with bradykinin (757.3992 Da), angiotensin II (1046.5418 Da), angiotensin I (1296.6848 Da), Glu-fibrinopeptide B (1570.6768 Da), and N-acetyl renin substrate (1800.9432 Da) in TOFMixTM (Shimadzu, Japan). Acquisition and data processing were controlled by the LanchpadTM software. The m/z values of multiple sodium adduct ions from the substrates and products are shown in Fig. 1, A–E. As a positive control, the peaks were detected corresponding to the dimeric cross-linked species between the muramyl tetrapeptide and muramyl tripeptide (Fig. 1C), a product of the l,d-TPase reaction catalyzed by the recombinant LdtMt2 (Ala-55–Ala-408). The l,d-TPase domain (residues Asp-251–Val-378) of M. tuberculosis LdtMt2 shows sequence identity and similarity of 22 and 38%, respectively, with the Csd6 l,d-CPase domain (residues Lys-67–Glu-202).
FIGURE 1.

l,d-CPase activity of Csd6. A and B, mass spectra and structural formulas of the synthesized muramyl tetrapeptide (A) and muramyl pentapeptide (B). The observed peaks correspond to a series of sodium adduct ions of the peptides. C–E, mass spectra of the peptide samples upon incubation with LdtMt2 and Csd6. In the control reaction catalyzed by LdtMt2, the dimeric cross-linked tetra-tripeptide species is produced from the muramyl tetrapeptide (C). In contrast, in the reaction catalyzed by Csd6, the muramyl tetrapeptide is converted to the muramyl tripeptide but not to dimeric cross-linked tetra-tripeptide species (D). Upon incubation with Csd6, the muramyl pentapeptide undergoes no change (E). The observed m/z values for the charged species with bound sodium ions (up to four: [M + Na]+1, [M + 2Na-H]+1, [M + 3Na-2H]+1, and [M + 4Na-3H]+1) agree with the calculated m/z values for muramyl tri-, muramyl tetra-, muramyl penta-, or dimeric cross-linked tetra-tripeptides (average relative mass of neutral species = 679.67, 750.75, 821.83, or 1412.41, respectively).
To evaluate the importance of specific residues in the l,d-CPase activity of Csd6, each of seven residues in the active site was mutated into alanine as follows: E110A, Y132A, Y133A, H155A, W158A, H160A, and C176A. The mutations were verified by DNA sequencing. The wild type and these single mutants of Csd6 were incubated with the substrate muramyl tetrapeptide for 30 min at 23 °C. Mass spectra were acquired as described above.
Analytical Ultracentrifugation
To determine the oligomeric state of the recombinant Csd6 in solution, equilibrium sedimentation and sedimentation velocity experiments were carried out using a Beckman ProteomeLab XL-A analytical ultracentrifuge in 20 mm Tris-HCl buffer, pH 7.9, containing 150 mm sodium chloride and 1.5 mm tris(2-carboxyethyl)phosphine at 20 °C. For the equilibrium sedimentation experiment, the absorbance from the Csd6 samples was measured at 235 and 280 nm using a six-sector cell at two speeds (16,000 and 20,000 rpm) and at three different Csd6 monomer concentrations (2.74, 3.65, and 4.56 μm) with a loading volume of 135 μl. For the sedimentation velocity experiment, the Csd6 samples at two different monomer concentrations (0.40 and 5.00 μm) were measured in double-sector cells at 30,000 rpm, and the collected data were analyzed by SEDFIT and SEDPHAT programs available on line. The concentration of the recombinant Csd6 protein was calculated using ϵ280 nm = 54,780 m−1 cm−1.
Single Molecule Fluorescence Resonance Energy Transfer (FRET)
To analyze the dimeric form of Csd6 in solution, a single molecule FRET technique incorporating alternating laser excitation (ALEX) was employed (54, 55). Briefly, in this method the fluorescence signal of a dimeric molecule was analyzed one-by-one, and the result is presented in a two-dimensional E-S graph (Fig. 3A), where E is the FRET efficiency and S denotes the Cy3/Cy5 molar ratio in a dimer. The Csd6 sample was divided into 2 aliquots, each of which was labeled on the sole cysteine residue (Cys-176) with the maleimide-reactive Cy3 dye (donor) or the Cy5 dye (acceptor). Subsequently, Cy3-labeled Csd6 (Cy3-Csd6) and Cy5-labeled Csd6 (Cy5-Csd6) were mixed at 2 μm final monomer concentration in a buffer (20 mm Tris-HCl, pH 7.9, and 150 mm sodium chloride) for 3 h at 37 °C. The protein sample was diluted further to 100 pm using a single molecule buffer (20 mm Tris-HCl, pH 7.9, 150 mm sodium chloride, 5% (v/v) glycerol, 1 mm 2-mercaptoethylamine, and 0.01% (v/v) bovine serum albumin). The single molecule data were acquired from a 10-min measurement.
FIGURE 3.
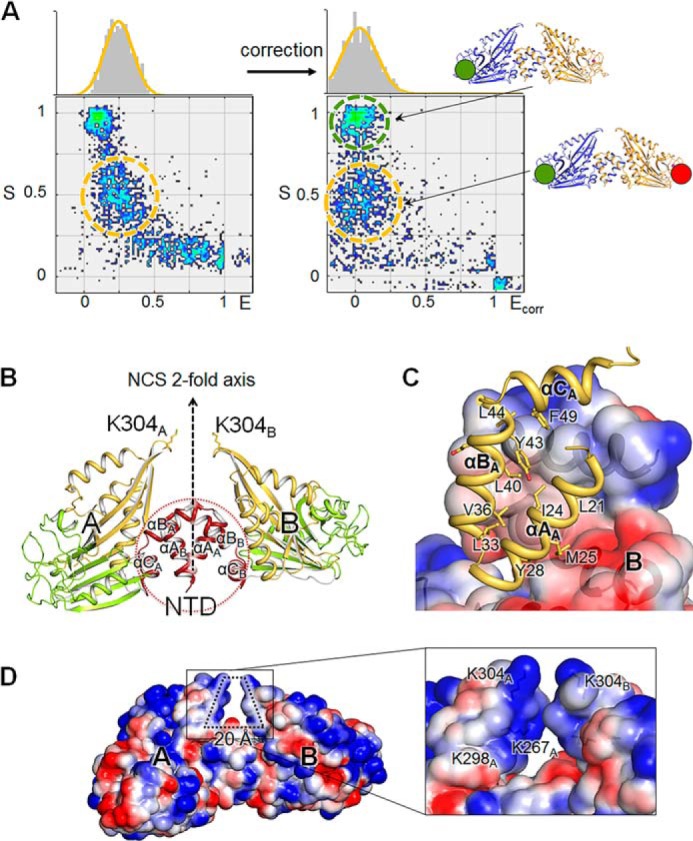
Dimeric structure of H. pylori Csd6. A, determination of the dimeric model of Csd6 by single molecule FRET technique. Single molecule FRET data are presented in a two-dimensional E-S graph, where E is the FRET efficiency and S denotes the Cy3 (donor)/Cy5 (acceptor) molar ratio in a dimer (54). Three clusters correspond to the dimers formed as follows: (i) between two Cy3-Csd6 monomers; (ii) between two Cy5-Csd6 monomers; and (iii) between a Cy3-Csd6 monomer and a Cy5-Csd6 monomer. The cluster appearing at S ∼ 1 (green dotted ellipse) with E ≈ 0 corresponds to Cy3-labeled Csd6 dimers. Csd6 dimers labeled with both Cy3 and Cy5 appear at S ∼ 0.5 (orange dotted ellipse) (55). B, ribbon diagram of the Csd6 homodimer (model I) colored as in Fig. 2A. In this dimer, A and B monomers are related by a noncrystallographic pseudo 2-fold symmetry. C, hydrophobic interactions between the NTDs of A and B monomers, which are shown in the ribbon diagram and the electrostatic surface diagram, respectively. D, electrostatic potential surface diagrams of the Csd6 dimer molecule. A detailed view of the specific hole formed by the dimerization is shown in the black lined box.
Surface Plasmon Resonance Experiment
The kinetics and affinity of Csd6 with the synthetic muramyl tetrapeptide and muramyl tripeptide (β-methyl N-acetylmuramic acid-l-Ala1-γ-d-Glu2-mDAP3) as a reaction product were assessed using a Reichert SR7500 surface plasmon resonance (SPR) dual channel instrument (Reichert, Depew, NY). Purified Csd6 in 20 mm sodium acetate, pH 5.5, was immobilized using the standard amino coupling at 20 μl min−1 on a carboxymethyl dextran hydrogel surface sensor chip (Reichert, Depew, NY) until saturation was achieved. The running buffer B used in all SPR experiments was 20 mm HEPES, pH 7.9, and 150 mm sodium chloride. SPR experiments were performed at 25 °C. The muramyl tetrapeptide or muramyl tripeptide at concentrations of 15.6, 31.3, 62.5, 125, 250, and 500 μm was injected over the Csd6-chip at 30 μl min−1 for 5 min for association analyses. Subsequently, the running buffer was flowed over the chip for an additional 6 min (30 μl min−1) for dissociation analyses. Regeneration of the chip was carried out using 20 mm sodium hydroxide. Binding was detected as a change in the refractive index at the surface of the chip as measured by the response unit. A reference flow cell was used to record the response by bovine serum albumin (BSA) as a positive control, and the response by BSA was subtracted from each sample. SPR data were fit using the Scrubber2 software.
Results
Csd6 Functions as an l,d-CPase but Not as an l,d-TPase
To verify the molecular function of H. pylori Csd6 in peptidoglycan modification, the peptidase activity of Csd6 was measured in vitro with the synthetic muramyl tetrapeptide and muramyl pentapeptide (Fig. 1, A and B) as potential substrates using mass analyses. When the muramyl pentapeptide was incubated with the recombinant Csd6(13–330), no reaction product could be detected (Fig. 1C). This suggests that Csd6 does not have “endo”-, “d,d-carboxy”-, or “l,d-trans”-peptidase activities with the muramyl pentapeptide. The l,d-CPase domain of Csd6 shows limited but significant sequence identity (22%) with the l,d-TPase domain of M. tuberculosis LdtMt2. M. tuberculosis LdtMt2 is a functional l,d-TPase, catalyzing the formation of 3→3 cross-links between the muramyl tetrapeptide and the muramyl tripeptide (56). Therefore, as a positive control for measuring the l,d-TPase activity, the muramyl tetrapeptide was incubated with the recombinant M. tuberculosis LdtMt2. In this control reaction, not only the muramyl tripeptide (Tri in Fig. 1C) but also the dimeric cross-linked species (Tetra-Tri in Fig. 1C) were detected as the reaction products. In comparison, when the muramyl tetrapeptide was incubated with the recombinant Csd6, the muramyl tripeptide was detected as the sole reaction product of trimming without any dimeric cross-linked species (Fig. 1D). This result implies that Csd6 would not be active for cross-linking, if the muramyl tripeptide were used as a substrate. The present data, together with the previous report that Csd6 converts monomeric tetrapeptides of the peptidoglycan sacculus into tripeptides (23), clearly establish that H. pylori Csd6 functions as an l,d-“carboxypeptidase” devoid of a cross-linking l,d-TPase, despite the presence of the conserved Cys/His residues, which are characteristics of the l,d-TPase domains. The active-site features of Csd6, as discussed in more detail below, explain why Csd6 is inactive as l,d-TPase.
Csd6 Monomer Is Organized into a Three-domain Architecture
To provide a structural basis of understanding the molecular function, the crystal structures of Csd6 have been determined, both in the ligand-free state (“Csd6-unbound”) (Fig. 2A) and in the d-Ala-bound state (“Csd6-Ala”) (Table 1). Refined models of both structures are essentially identical to each other with an r.m.s. deviation of 0.17 Å for 632 eq Cα positions. They account for residues Met-15–Lys-330 in each of two Csd6 monomers in an asymmetric unit (ASU). In the case of Csd6-unbound structure, two monomers in the ASU were similar to each other with an r.m.s. deviation of 0.64 Å for 316 Cα atom pairs, except for the N-terminal residues (Leu-15–Asp-41) showing r.m.s. deviations greater than 1.0 Å with a maximum Cα deviation (7.6 Å) at Leu-15. For the Csd6-Ala model, two monomers in the ASU agree with an r.m.s. deviation of 0.61 Å for 316 Cα atom pairs, except for the N-terminal residues (Leu-15–Asp-41) showing r.m.s. deviations greater than 1.0 Å with a maximum Cα deviation (8.1 Å) at Leu-15. The amino acid residues with the largest deviation encompass the entire helix αA and the N-terminal half of αB.
FIGURE 2.

Overall structure and the oligomeric state of H. pylori Csd6. A, ribbon diagram (upper panel) and topology diagram (bottom panel) of the Csd6-unbound structure. The NTD, l,d-CPase domain, and NTF2-like domain are shown in red, green, and yellow, respectively. The secondary structure elements have been defined by the DSSP program (77). The nucleophile Cys-176 of the l,d-CPase domain is shown in a cyan stick model. B, equilibrium sedimentation data for Csd6 at an ultracentrifugal speed of 16,000 rpm using 3.65 μm protein at 20 °C. The circles are experimental data, and the solid line is a fitting line for an ideal monomer model. The two dotted lines are fitting lines for ideal monomer and trimer models. Distributions of the residuals for monomer, dimer, and trimer models are shown in the inset panel. These data indicate that Csd6 exists as homogeneous dimers in solution. C, two putative models of the Csd6 dimer in the crystal. Dimer model I (upper panel) and model II (bottom panel) are shown in ribbon diagrams. The side chain sulfur atoms of Cys-176 are shown as purple spheres. Model I is favored by the single molecule ALEX-FRET data (shown in Fig. 3A). D, plots of the residuals for dimer species model of Csd6 at 0.40 μm (upper) and 5.00 μm (lower), and the distribution of sedimentation coefficient (c(s) versus s, where s is in Svedberg unit, S) from the sedimentation velocity experiments.
The Csd6 monomer consists of three domains of unequal sizes as follows: the NTD (Val-13–Asn-56), the middle l,d-CPase domain (Lys-67–Glu-202), and the C-terminal NTF2-like domain (Thr-209–Lys-330) (Fig. 2A). The NTD consists of three α-helices (αA–αC), and the helix αC is positioned at one end of the antiparallel coiled-coil formed by helices αA and αB. The l,d-CPase domain consists of two curved β-sheets (β3↓-β2↑-β1↓-β8↓-β4↑ and β4↑-β5↓-β6↑-β7↑), which share the β4-strand, as well as two α-helices (αD and αE). The NTF2-like domain forms an antiparallel four-stranded β-sheet (β9↓-β10↑-β11↓-β12↑), whose concave side is packed by three α-helices (αF–αH). DALI structural similarity searches (57) revealed no significant match with either the entire Csd6 with three domains or any combination of two connected domains in Csd6, suggesting that the three-domain architecture of Csd6 is unique. This is in line with our observation that the organization of the l,d-CPase domain and the NTF2-like domain in Csd6 and its homologs is unique among the l,d-TPase and l,d-CPase families according to the conserved domain searches using the InterPro database (58).
In the Csd6 monomer structure, three domains are arranged in a tripartite leaf-like fashion with a single chain connecting two adjacent domains (Fig. 2A). Virtually no interaction exists between the NTD and the l,d-CPase domain; the buried surface area at the interface is only 36 Å2, as calculated by the PISA server (59). Much more extensive interactions exist between the l,d-CPase domain and the NTF2-like domain, with a buried surface area of 718 Å2. The latter interface is mainly formed between strands β4 and β8 of the l,d-CPase domain and strands β9–β11 of the NTF2-like domain. It is primarily lined with hydrophobic residues of both domains (Val-119, Tyr-120, Tyr-146, Phe-197, and Ile-199 of the l,d-CPase domain and Ile-278, Tyr-280, Tyr-290, Val-292, and Tyr-316 of the NTF2-like domain). The NTD (helix αC) and the NTF2-like domain (helix αF) interact with each other, burying a surface area of 410 Å2 at the interface.
NTD of Csd6 Plays a Dominant Role in Homodimerization
Equilibrium sedimentation measurements indicate that the Csd6 protein exists as a homogeneous dimer in solution (at the tested Csd6 monomer concentration range of 2.74–4.56 μm). The data fit well to a dimer model, and a representative result measured at 16,000 rpm using the 3.65 μm monomer concentration is presented in Fig. 2B. Further sedimentation velocity experiments at 0.40 and 5.00 μm Csd6 monomer concentrations also indicate the oligomeric state of Csd6 as a dimer, without any discernable dissociation of a dimer into monomers, down to 0.40 μm (Fig. 2D). In the crystal of Csd6, two possible models of the dimer could be identified. The dimer model I has a buried surface area of 1,080 Å2 per monomer (6.7% of the monomer surface area) at the interface, as analyzed by the PISA server (Fig. 2C, upper panel) (59). In the dimer model I, the two monomers are related by noncrystallographic “pseudo” 2-fold symmetry. The 2-fold symmetry is broken primarily by the significant difference in the orientation of N-terminal helices αA and αB between the two monomers. The dimer model II has a buried surface area of 726 Å2 (4.5% of the monomer surface area) (Fig. 2C, lower panel). In the dimer model II, the two monomers are related by an exact crystallographic 2-fold symmetry. The ΔiG values (solvation free energy gain upon formation of the interface) are −15.8 and −2.1 kcal mol−1 for models I and II, with the complexation significance scores of 1.000 and 0.000, respectively. These complexation significance score values imply that the interface in model I, but not in model II, plays an essential role in complex formation. Therefore, we conclude that the dimer model I in the crystal represents the biologically relevant Csd6 dimers in solution. This conclusion is supported experimentally by the distance between two Cys-176 residues in the dimer as estimated using the single molecule ALEX-FRET technique (Fig. 3A). The single molecule ALEX-FRET experiment was performed at 100 pm monomer concentration, which is much lower than that used for analytical ultracentrifugation, to avoid ensemble averaging (i.e. to avoid the case of having two noninteracting species) (60). After direct excitation leakage and buffer background corrections, the distance between Cy3 and Cy5 dipoles in the dye-labeled Csd6 dimer is estimated to be 107 Å from the corrected FRET efficiency (Ecorr ≈0.03), using Ro (Förster distance) of 60 Å for the Cy3 and Cy5 pair. In the crystal structure of Csd6-unbound, the distance between sulfur atoms of two Cys-176 residues is 96 and 66 Å in dimer models I and II, respectively. This result unambiguously favors model I, supporting the assignment of dimer model I in the crystal as the dimer in solution (Fig. 2C, upper panel).
The Csd6 dimer (i.e. dimer model I) is elongated with approximate dimensions of 100 × 80 × 50 Å (Fig. 3B). In this dimer, two l,d-CPase domains are well separated from each other, and their active sites are open toward the bulk solvent (Fig. 3B). The bulk of the buried surface area at the dimerization interface is contributed by the NTD (82.7%), with the linker between the NTD and the l,d-CPase domain contributing 5.7% and the NTF2-like domain contributing 11.6%. The N-terminal helix αA of the NTD takes slightly different conformations in the two monomers, with a maximum Cα deviation of 8.1 Å at Leu-15. The two NTDs pack against each other through their antiparallel coiled-coils (helices αA and αB), at an angle of about 60°, to form a four-helix bundle (Fig. 3B); numerous hydrophobic side chains are present at the interface. The αC-helices from two NTDs cover the sides of the four-helix bundle. Seven hydrogen bonds (involving Arg-26, Tyr-28, Gln-29, Gly-37, Asp-41, Glu-53, Tyr-63, Gln-65, and Phe-272) and a salt bridge (involving Arg-26 and Glu-53) exist at the interface (Fig. 3C). Interestingly, the Csd6 dimer has a deep crevice of an approximate size of 20 × 20 Å, with a widened bottom (indicated by dotted lines in Fig. 3D). The NTDs provide the bottom of the crevice, and the C-terminal NTF2-like domains form the two sides of the crevice. The sides of the crevice are lined with positively charged residues (Lys-267, Lys-298, and Lys-304) but no negatively charged residues (Fig. 3D).
A DALI search with the Csd6 NTD (Val-13–Asn-56) alone showed very remote structural similarities to other known protein structures. The highest Z-score was obtained with human phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α isoform (PDB code 4L2Y-A; an r.m.s. deviation of 2.1 Å for 41 equivalent Cα positions in residues 16–56, a Z-score of 4.6, and a sequence identity of 5%). The Csd6 NTD is perhaps functionally more related to the L27N domain of Mus musculus PALS1-associated tight junction protein (PDB code 1VF6-D; an r.m.s. deviation of 4.2 Å for 41 eq Cα positions in residues 16-56, a Z-score of 4.3, and a sequence identity of 12%) (61). L27 domains, such as PALS1-L27N, have been established as a protein-binding module that brings multiple proteins into complexes for signaling, cell polarity, and epithelial morphogenesis (62, 63). Like the Csd6 NTD, the L27 domain is composed of three α-helices. However, unlike the Csd6 NTD that plays a key role in homodimerization, two different L27 domains (e.g. PALS1-L27N domain and PATJ-L27) heterodimerize by building a compact four-helix bundle structure through the first two helices from each L27 domain (61).
Active-site of Csd6 l,d-CPase Domain Is Tailored for the l,d-CPase Activity
A bioinformatics analysis on the basis of amino acid sequence predicts H. pylori Csd6 to have an overall fold of the l,d-TPase catalytic domain over residues 67–200. The structure reported here validates this prediction (Figs. 2A and 4A). However, this catalytic domain is referred to as the l,d-CPase domain, because Csd6 is shown to function only as an l,d-CPase and not as an l,d-TPase against the muramyl tetrapeptide. According to the DALI search, the overall fold of the Csd6 l,d-CPase domain (Lys-67–Glu-202) (Fig. 4A) resembles those of well characterized l,d-TPases such as B. subtilis LdtBs (Fig. 4B) (PDB code 1Y7M; a Z-score of 14.8, an r.m.s. deviation of 1.8 Å, and a sequence identity of 13% for 109 equivalent Cα positions) (64), E. faecium Ldtfm (Fig. 4C) (PDB code 1ZAT; a Z-score of 12.1, an r.m.s. deviation of 2.7 Å, and a sequence identity of 14% for 111 eq Cα positions) (29), and M. tuberculosis LdtMt2 (Fig. 4D) (PDB code 4GSU; a Z-score of 11.7, an r.m.s. deviation of 2.1 Å, and a sequence identity of 14% for 107 eq Cα positions) (30). Unlike Csd6, all of these l,d-TPases are monomeric enzymes (29, 30, 64). Furthermore, the l,d-CPase domain of Csd6 contains the characteristic sequence motif H(S/G/D)X14–19C (Fig. 4E), where X stands for any amino acid and the strictly conserved residues are in boldface. This motif is conserved among proteins containing an l,d-TPase catalytic domain and provides the catalytic triad residues, which form a hydrogen bond network in a wide range of Cys/Ser-based proteolytic enzymes (65). The catalytic triad of Csd6 consists of His-160, Gly-161, and Cys-176 (marked by red dots in Fig. 4E), corresponding to His-336, Ser-337, and Cys-354 of LdtMt2 (30). However, H. pylori Csd6 is unrelated in its sequence and structure to previously characterized l,d-CPases such as l,d-carboxypeptidases A (LdcA) from P. aeruginosa (66) and Novosphingobium aromaticivorans DSM 12444 (67), and l,d-carboxypeptidases B (LdcB) from Streptococcus pneumoniae, Bacillus anthracis, and Bacillus subtilis (68). LdcA is a Ser-based l,d-CPase. The latter three LdcB proteins belong to the LAS (lysostaphin, d-Ala-d-Ala metallopeptidases, sonic hedgehog) family of Zn2+-dependent peptidases (68). This raises an intriguing question why H. pylori Csd6 functions only as an l,d-CPase, and not as an l,d-TPase, despite the overall l,d-TPase fold and the presence of the conserved catalytic triad.
FIGURE 4.

Comparison of Csd6 and three l,d-TPases. A–D, the ribbon diagrams (left panels) and surface diagrams (right panels) of the l,d-CPase domain of H. pylori Csd6 (A) and the l,d-TPase domains of B. subtilis LdtBs (B), E. faecium Ldtfm (C), and M. tuberculosis LdtMt2 (D). The four loops I–IV are colored in yellow, green, purple, and light blue, respectively. One or two paths that allow access to the catalytic triad are indicated by red arrows. In the meropenem-complexed LdtMt2 structure (PDB code 4GSU), the paths A and B correspond to left and right arrows, respectively (30). Meropenem was modeled into each domain structure by structural superimposition of the corresponding domains using the meropenem-complexed LdtMt2 (PDB code 4GSU). E, structure-based sequence alignment was performed by PROMALS3D (80), and the alignment was presented by GeneDoc. Structures of the l,d-CPase domain of H. pylori Csd6 (Swiss-Prot accession code O25255; PDB code 4XZZ) and l,d-TPase catalytic domains of B. subtilis LdtBs (O34816; 1Y7M), E. faecium Ldtfm (Q3Y185; 1ZAT), and M. tuberculosis LdtMt2 (O53223; 4GSU) were used in the alignment. In the case of C. jejuni Pgp2 (A1VZP0), no structural information was available, and thus only the sequence is used. Red dots indicate the catalytic triad (His-160, Gly-161, and Cys-176 in H. pylori Csd6). The four loops I–IV are shown in colored boxes as in A.
A structural comparison of Csd6 l,d-CPase domain with l,d-TPase domains reveals that the shape of the active site in the Csd6 l,d-CPase domain seems to be tailored for the l,d-carboxypeptidation, i.e. the active site is a deep pocket and the catalytic triad positioned at the bottom of this pocket is accessible via a single narrow path (Fig. 4A), as in typical carboxypeptidases (69). In contrast, the active site in the corresponding domains of the above three l,d-TPases is an elongated groove, and the catalytic triad is accessible via two paths (Fig. 4, B–D) (30). The different shapes of the active sites are primarily dictated by the length and conformation of the loops around the entrance(s) to the active sites. Structural comparison and sequence alignment of the Csd6 l,d-CPase domain with the corresponding domains of three l,d-TPases (Fig. 4) show that unique features of Csd6 that distinguish it from other l,d-TPases reside primarily in the following four loop regions: loop I (β3-β4 loop; Ala-98–Ile-117); loop II (β4-β5 loop; Leu-126–Leu-136); loop III (β5-αD-β6 loop; Asn-141–Ile-157); and loop IV (β6-β7 loop; Met-162–Cys-176).
The reaction catalyzed by l,d-TPases such as LdtMt2 occurs in two sequential steps as follows: acylation of the donor substrate (“carboxypeptidation”) and deacylation by the acceptor substrate for cross-linking (“transpeptidation”) (70). As a consequence, l,d-TPases such as LdtMt2 have two accessible paths for donor and acceptor substrates (indicated by red arrows in Fig. 4, B–D), thereby allowing an approach of the acceptor substrate upon acylation of the donor substrate by the catalytic Cys residue for transpeptidation. For a better understanding of the substrate-binding site (Fig. 4, A–D), meropenem (mimicking the substrate) was modeled into Csd6 by superimposing its l,d-CPase domain and the l,d-TPase domain of the meropenem-complexed LdtMt2 (30). The position of the covalently bound meropenem represents the path for the donor substrate (“path A”). The active-site clefts in three l,d-TPases reveal a long groove for binding two substrates, which is contributed by the four loops I–IV (Fig. 4, B–D). In LdtMt2 and Ldtfm, the extended loop II (green colored in Fig. 4, C and D) forms a lid to cover the active-site cleft, while exposing two discrete paths for the access of two substrates. However, the loop II of LdtBs is much shorter, and the active-site cleft is not covered with a lid (green colored in Fig. 4B). Like LdtBs, the loop II is much shorter in Csd6, and the active-site cleft is not covered with a lid. Moreover, in the Csd6 structure, the corresponding paths to the active-site cleft for access of two substrates in three l,d-TPases are blocked due to the distinct length and conformations of the loops I–IV, and the active-site cleft opens vertically toward the solvent (indicated by a red arrow in Fig. 4A), allowing access of only one muramyl peptide for carboxypeptidation. This difference in the shape of the active-site cleft is the likely reason for the enzyme serving as a carboxypeptidase, to the exclusion of the transpeptidase activity.
Among the four loops I–IV, the loops I and III show the most distinct variations in sequence length and conformation. They are considerably longer (8 and 12 residues, respectively) in H. pylori Csd6 than in the l,d-TPases (Fig. 4E). Loop III contains the helix αD, which protrudes from the active site. Together with loop I, it provides one side of the active-site cleft to recognize a substrate. The C. jejuni Pgp2, a Csd6 homolog, is predicted to contain an l,d-TPase domain on the basis of its amino acid sequence (Fig. 4E), but it was shown to function as an l,d-CPase (26). We notice that the four loops of Pgp2 show a highly similar pattern with those of Csd6 (Fig. 4E). Therefore, it is anticipated that the observed unique structural features of these four loops around the active-site cleft are conserved among Csd6 homologs in other ϵ-proteobacteria.
d-Ala-complexed Structure Reveals Binding Modes of Both the Substrate and Product
By analogy with the first carboxypeptidation step of LdtMt2, His-160 of Csd6 serves as a proton acceptor to assist a nucleophilic attack by Cys-176. In the present Csd6 structure, the side chains of His-160 and Cys-176 are suitably predisposed for a cooperative action with the main-chain carbonyl of Gly-161 (Fig. 5A). His-160Nϵ2 interacts with Cys-176Sγ with a distance of 3.96 Å and His-160Nδ1 interacts with the main-chain oxygen atom of Gly-161 with a distance of 2.75 Å (Fig. 5A). In addition, the l,d-CPase domain of Csd6 shows a well defined oxyanion hole to facilitate catalysis. The backbone amide nitrogen atoms of Lys-174, Gly-175, and Cys-176 (asterisks in Fig. 5A) could stabilize the transient oxyanion species of the polarized carbonyl bond, as in LdtMt2 (70).
FIGURE 5.

d-Ala-complexed Csd6 and key residues for the l,d-CPase activity. A, ribbon diagram and the accessible inner surface of the active site in the Csd4-Ala structure. The bound d-Ala molecules (d-Ala′ and d-Ala″) (upper panel) and the mDAP (bottom panel) modeled on the basis of d-Ala′ and Wat1 are shown in stick models. The omit mFo − DFc maps for d-Ala′ and d-Ala″ (contoured at 2.0σ) and the side chain oxygen atom of Cys-176 oxidized as the sulfenic acid (contoured at 2.5σ) are colored in blue. The oxyanion hole is asterisked and also shown as blue dotted lines in the bottom panel. Red dotted lines indicate the interactions among residues of the catalytic triad. B, l,d-carboxypeptidation activities of the wild-type Csd6 (with the treatment of EDTA or not) and the mutants (E110A, Y132A, Y133A, H155A, W158A, H160A, and C176A) with the muramyl tetrapeptide. C, SPR experiments with immobilized Csd6 and the muramyl tetrapeptide (or muramyl tripeptide) as an analyte at different concentrations (15.6, 31.3, 62.5, 125, 250, and 500 μm) are shown in traces colored as orange, light blue, purple, green, red, and blue, respectively.
To understand the structural determinants for ligand binding, the structure of Csd6 bound with the reaction product d-Ala (Csd6-Ala in Table 1) was determined. In the Csd6-Ala model, two d-Ala molecules are well ensconced within the active site of monomer A with clear electron density (marked as d-Ala′ and d-Ala″ in Fig. 5A), whereas only one d-Ala molecule is present in monomer B at the corresponding position as d-Ala′ in monomer A. The different binding states of monomers A and B in the crystal seem to be correlated with the oxidation state of Cys-176. Its side chain is oxidized to sulfenic acid only in monomer A, as indicated by the extra electron density (C176OH in Fig. 5A). An oxygen atom of sulfenic acid possibly stabilizes the weak binding of the product d-Ala″N in monomer A. d-Ala′ is located between loops I and III, whereas d-Ala″ is trapped by loops II–IV, right in front of the nucleophile Cys-176. d-Ala′ is too far from the predicted site of the scissile bond for Cys-176 to act, but it interacts with Lys-107Nζ, Glu-110Oϵ2, His-155N,O,Nδ1, and Gly-111N, as well as Lys-174N, and the main-chain oxygen atom of Leu-113 via a water molecule. Interestingly, the side-chain portion of mDAP, part of the Csd6 substrate muramyl-l-Ala1-γ-d-Glu2-mDAP3-d-Ala4, is structurally identical to a d-Ala molecule. Therefore, we suggest that d-Ala′ and d-Ala″ represent the portion of the substrate and the end product, d-Ala, respectively, for the link between mDAP3 and d-Ala4 (the scissile peptide bond) of the muramyl tetrapeptide. mDAP was modeled into the Csd6 l,d-CPase domain on the basis of the positions of d-Ala′ and Wat1 bound in the oxyanion hole. The modeled mDAP fits well into the active-site cleft (bottom panel of Fig. 5A). It makes proper interactions with the oxyanion hole and is suitably positioned for a nucleophilic attack by Cys-176, thus providing the substrate-binding mode. Dissociation constants (KD) of Csd6 for its complex with d-Ala (or mDAP) have also been measured to be 224 (or 94) μm using SPR measurements. The measured association and dissociation rate constants (ka and kd) are 1.05 m−1 s−1 and 2.30 × 10−4 s−1 for d-Ala and 1.49 m−1 s−1 and 1.40 × 10−4 s−1 for mDAP, respectively.
To further confirm the role of potential key residues in the active-site cleft, seven single amino acid mutant proteins (E110A, Y132A, Y133A, H155A, W158A, H160A, and C176A) were prepared. When the l,d-carboxypeptidation activities of the wild-type Csd6 and the mutants against the muramyl tetrapeptide for a limited time (30 min) were compared, mutation of Glu-110, Tyr-133, His-155, and Trp-158, as well as His-160 and Cys-176 that belong to the catalytic triad, resulted in a complete loss of the l,d-carboxypeptidation activity (Fig. 5B). Glu-110, Tyr-133, and Trp-158 are strictly conserved in C. jejuni Pgp2 as well (Fig. 4E). Lack of l,d-carboxypeptidation activity in E110A and H155A mutants supports that Glu-110 and His-155 recognize the side chain of mDAP, as shown in d-Ala′ bound to Csd6 and mDAP modeled into the position of d-Ala′ (Fig. 5A). Trp-158 holds the aliphatic side chain of d-Ala″, whereas Tyr-133 likely interacts with the main chain of mDAP, possibly via a water molecule such as Wat2 in Fig. 5A. Meanwhile, the retained activity of the Y132A mutant indicates that Tyr-132 is not important for the l,d-carboxypeptidation activity of Csd6 (Fig. 5B). The l,d-carboxypeptidation activity of Csd6 is independent of metal ions, as there is no change upon EDTA treatment (Fig. 5B).
To estimate the kinetics and affinity of Csd6 with the muramyl tetrapeptide (as a substrate) and muramyl tripeptide (as a product), SPR experiments were conducted. The association rate constant (ka) and the dissociation rate constant (kd) for the muramyl tripeptide are roughly 3 and 50× larger than those for the muramyl tetrapeptide, respectively (Fig. 5C). As a consequence, the KD values for the muramyl tetrapeptide and the muramyl tripeptide are 27.0 and 510 μm, respectively, indicating that the muramyl tripeptide dissociates readily from Csd6 as the product of Csd6-catalyzed l,d-CPase reaction. This result is also consistent with the muramyl tripeptide not being used as the substrate for the possible l,d-TPase cross-linking reaction in the subsequent step.
C-terminal NTF2-like Domain Has a Putative Binding Pocket for Pseudaminic Acid
H. pylori Csd6 is distinct from the known l,d-TPases by having a C-terminal NTF2-like domain (residues Thr-209–Lys-330), following its l,d-CPase domain. The combination of these two domains is conserved in the so-called ErfK/YbiS/YcfS/YnhG family proteins from both Helicobacter and Campylobacter species (Fig. 6). It was reported that csd6 gene-deficient transposon mutants of H. pylori G27 exhibited altered motility in comparison with its parental strain, and the altered pattern of motility resulted from elevated levels of O-linked FlaA Pse-glycosylation of flagellin (40). H. pylori flagellin filaments are post-translationally modified by glycosylation with Pse, a nine carbon sugar derivative that resembles sialic acid, which is typically found on mammalian cell surfaces (Fig. 7B) (37, 38). The transposon insertions causing the altered motility were located at either positions +765 or +803 bp from the 5′ terminus of the csd6 gene (40), corresponding to the region encoding the NTF2-like domain. Furthermore, the recombinant H. pylori G27 Csd6 protein expressed in E. coli was capable of deglycosylating the purified FlaA protein (40). These results suggest a possible function of the Csd6 NTF2-like domain as a deglycosylase, i.e. pseudaminidase.
FIGURE 6.

Sequence alignment of Csd6 homologs. Sequence alignment of Csd6 homologs from H. pylori strain 26695 (HP0518; Swiss-Prot accession code O25255), from Wolinella succinogenes (Q7MRJ6), from Arcobacter nitrofigilis (D5V196), from Campylobacter concisus strain 13826 (A7ZCK9), and from Thermovibrio ammonificans (E8T4Z0) was performed and presented by ClustalX (78) and ESPript (79). The secondary structure of Csd6 is presented above the aligned sequences. The four loops I–IV around the active site of the Csd6 l,d-CPase domain are indicated with boxes colored as in Fig. 4E.
FIGURE 7.
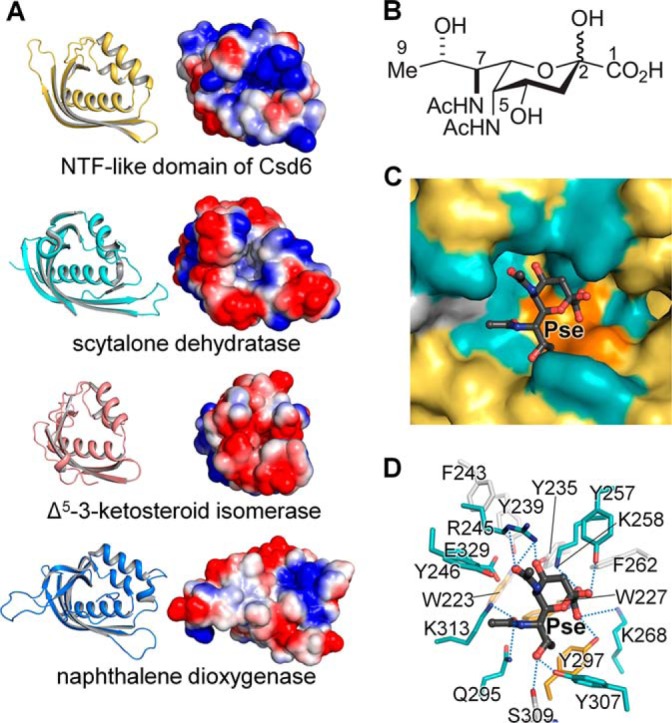
NTF-like domain of Csd6. A, ribbon diagrams and electrostatic potential surface diagrams of the NTF-like domains of Csd6, scytalone dehydratase, Δ5-3-ketosteroid isomerase, and naphthalene dioxygenase. B, chemical structure of Pse. C and D, in silico docking of Pse onto the pocket in the NTF-like domain of Csd6. The potential binding pocket is shown in surface representation (C), and the residues around the docked ligand are shown as stick models (D).
The present structure shows that the overall fold of the Csd6 NTF2-like domain differs from other nonviral sialidase or pseudaminidase of the six-bladed β-propeller fold (52, 71). Instead, it is shared with other NTF2-like superfamily members such as the Magnaporthe grisea scytalone dehydratase (72) (PDB code 1IDP; a Z-score of 14.3 and a sequence identity of 8% for 118 eq Cα positions), Pseudomonas putida Δ5-3-ketosteroid isomerase (73) (PDB code 1OPY; a Z-score of 12.0 and a sequence identity of 8% for 103 eq Cα positions), and Rhodococcus sp. naphthalene dioxygenase (74) (PDB code 2B1X; a Z-score of 14.1 and a sequence identity of 7% for 116 eq Cα positions) (Fig. 7A). None of these enzymes bears any significant sequence or functional homology to each other, except that they share common hydrophobic substrates. This suggests that the fold of NTF2-like proteins may be a suitable scaffold for building a pocket to bind hydrophobic ligands. Although the Csd6 NTF2-like domain differs considerably from these enzymes in the sequence and the potential function, it shares a deep, largely hydrophobic pocket that is open to the solvent (Fig. 7, A and C). This potential ligand-binding pocket is lined with numerous hydrophobic residues (Trp-223, Trp-227, Tyr-235, Tyr-239, Phe-243, Tyr-246, Tyr-257, Tyr-297, and Tyr-307). In addition, four positively charged residues (Arg-245, Lys-258, Lys-268, and Lys-313), and a negatively charged residue (Glu-329) are located within the pocket (Fig. 7D). The bold-face residues are strictly conserved in Csd6 homologs in ϵ-proteobacteria (Fig. 6). The pocket is somewhat elongated in its shape, unlike hemispherical pockets in scytalone dehydratase and Δ5-3-ketosteroid isomerase (Fig. 7, A and C). The Csd6 NTF2-like domain is highly basic with a calculated pI of 9.4; the electrostatic potential surface shows a highly positively charged surface around its deep pocket (Fig. 7A), compared with the above three NTF2-like superfamily enzymes.
In light of the previous report that H. pylori Csd6 is involved in the deglycosylation of flagellin (40) and the presence of a deep pocket in the NTF2-like domain of Csd6 for possible binding of a hydrophobic ligand with an acidic group(s) such as Pse, an in silico docking study has been performed. It shows that Pse can fit well into the pocket of the NTF-like domain (Fig. 7C). However, it remains to be determined whether Csd6 has pseudaminidase activity.
Discussion
We have determined the first crystal structure of Csd6, one of the cell shape-determining proteins in H. pylori. Each of its three domains is associated with a distinct function. The NTD plays a dominant role in homodimerization. The C-terminal NTF2-like domain has a putative binding pocket for Pse. The central l,d-CPase domain has the l,d-TPase fold but the shape of its active-site pocket explains why Csd6 possesses only the l,d-carboxypeptidation activity, despite the presence of the conserved catalytic residues for the potential transpeptidase reaction.
H. pylori Csd6, as well as its close homolog Pgp2 from C. jejuni, contributes to the helical morphology of the cells. Interestingly, H. pylori Csd6 and C. jejuni Pgp2 are unrelated in their sequence and structure to previously characterized l,d-CPases such as LdcA and LdcB from other microorganisms. They are thus presumed to form a third family of l,d-CPases. However, it is currently impossible to assign and distinguish genuine l,d-TPases from the functional l,d-CPases via bioinformatics analyses of amino acid sequences alone. Our finding that Csd6-specific active-site loops I and III are much longer than the corresponding loops in known l,d-TPases (Fig. 4E) enables us to distinguish l,d-CPases possessing the l,d-TPase fold from bona fide l,d-TPases. Indeed, when amino acid sequences of Csd6 homologs in ϵ-proteobacteria are aligned, Csd6-specific loops I and III display high levels of sequence conservation (Fig. 6). In light of this observation, the current annotation of H. pylori Csd6, as well as its homologs in ϵ-proteobacteria, as belonging to the protein family containing an l,d-TPase catalytic domain needs to be revised. We propose that they should be classified as a third distinct family of l,d-CPase, as initially noted by others (75).
Enhanced cell attachment of H. pylori leads to an increased immune response of the host (76). Too much attachment may thus be detrimental to the bacterium, suggesting a role for motility in the proper balance of bacterial levels during infection. This is intriguing, given that H. pylori Csd6 is known to be involved in regulating motility through trimming of the peptidoglycan-derived peptides as an l,d-CPase or through affecting the Pse glycosylation level of flagellin. This study shows that the active-site of the Csd6 l,d-TPase domain is tailored to function as the l,d-CPase, which converts muramyl tetrapeptides into muramyl tripeptides. As Csd6 is nonfunctional as l,d-TPase, muramyl tripeptides in the peptidoglycan cannot be 3→3 cross-linked. This is consistent with the previous reports that the Helicobacter peptidoglycan layer lacks 3→3 cross-links (15, 20, 33). An in silico docking shows that the Csd6 NTF-like domain presents a potential binding site for Pse, supporting its role in the control of the glycosylation level of flagellin. Relaxation of peptidoglycan 4→3 cross-linking was reported to promote H. pylori's helical shape and stomach colonization (20). Csd6 plays a critical role in determining the helical cell shape of H. pylori, because it can limit the supply of substrates for peptidoglycan 4→3 cross-linking. In conclusion, our study on the multidomain, multifunctional Csd6 provides further insights into the strategy of H. pylori for regulating its helical cell shape and motility, which are crucial for its virulence.
Author Contributions
H. S. K., H. N. I., S. M., S. C., N. K. L., S. J. K., J. Y. K., B. W. H., B. I. L., and S. W. S. designed the study and wrote the paper. H. N. I., D. R. A., J. Y. Y., J. Y. J., and H. J. Y. purified and crystallized Csd6 protein and determined its x-ray structure. S. M., D. H., and M. L. synthesized the muramyl peptides. J. Y., M. C., and S. C. conducted docking calculations. C. K. and N. K. L. performed the ALEX-FRET experiment. S. J. K. performed the equilibrium sedimentation experiment. J. Y. K. and G. B. performed mass spectrometric assays of enzyme activities. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank beamline staffs for assistance during x-ray diffraction experiments at Pohang Light Source (beamlines BL-5C and BL-7A), SPring-8 (beamlines BL-26B1 and BL44XU), and Photon Factory (beamlines BL-1A, BL-5A, BL-17A, NE3A, and NW12A). We also thank Dr. Sunghwan Kim and Young-Jin Son of the New Drug Development Center, Daegu-Gyeongbuk Medical Innovation Foundation, Korea, for performing sedimentation velocity experiments.
This work was supported by the Korea Ministry of Science, ICT and Future Planning, National Research Foundation of Korea Grant 2013R1A2A1A05067303, Innovative Drug Research Center for Metabolic and Inflammatory Disease (2007-0056817), the Korea Ministry of Health, Welfare and Family Affairs, Korea Healthcare Technology R&D Project A092006 (to S. W. S.), and by the Ministry of Science, ICT and Future Planning, the National Research Foundation of Korea, National Leading Research Lab Program, Grant 2011-0028885 (to the S. C. laboratory). This work was supported in part by National Institutes of Health Grant AI090348 (to S. M.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 4XZZ and 4Y4V) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- mDAP
- meso-2,6-diaminopimelate
- d,l-CPase
- d,l-carboxypeptidase
- d,d-CPase
- d,d-carboxypeptidase
- l,d-TPases
- l,d-transpeptidases
- l,d-CPase
- l,d-carboxypeptidase
- Pse
- pseudaminic acid
- NTD
- N-terminal domain
- NTF2
- nuclear transport factor 2
- EMTS
- ethyl mercury thiosalicylate
- IFD
- induced fit docking
- Neu5Ac2en
- 2-deoxy-2,3-dideoxy-N-acetylneuraminic acid
- r.m.s.
- root-mean-square
- MALDI-QIT-TOF MS
- matrix-assisted laser desorption/ionization quadrupole ion trap time-of-flight mass spectrometer
- SPR
- surface plasmon resonance
- ASU
- asymmetric unit
- ALEX
- alternating-laser excitation
- PDB
- Protein Data Bank.
References
- 1. Roesler B. M., Rabelo-Gonçalves E. M., Zeitune J. M. (2014) Virulence factors of Helicobacter pylori: a review. Clin. Med. Insights Gastroenterol. 7, 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kusters J. G., van Vliet A. H., Kuipers E. J. (2006) Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19, 449–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roubaud Baudron C., Franceschi F., Salles N., Gasbarrini A. (2013) Extragastric diseases and Helicobacter pylori. Helicobacter 18, 44–51 [DOI] [PubMed] [Google Scholar]
- 4. Malfertheiner P., Selgrad M., Bornschein J. (2012) Helicobacter pylori: clinical management. Curr. Opin. Gastroenterol. 28, 608–614 [DOI] [PubMed] [Google Scholar]
- 5. Ottemann K. M., Lowenthal A. C. (2002) Helicobacter pylori uses motility for initial colonization and to attain robust infection. Infect. Immun. 70, 1984–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schreiber S., Konradt M., Groll C., Scheid P., Hanauer G., Werling H. O., Josenhans C., Suerbaum S. (2004) The spatial orientation of Helicobacter pylori in the gastric mucus. Proc. Natl. Acad. Sci. U.S.A. 101, 5024–5029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lertsethtakarn P., Ottemann K. M., Hendrixson D. R. (2011) Motility and chemotaxis in Campylobacter and Helicobacter. Annu. Rev. Microbiol. 65, 389–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berg H. C., Turner L. (1979) Movement of microorganisms in viscous environments. Nature 278, 349–351 [DOI] [PubMed] [Google Scholar]
- 9. Hazell S. L., Lee A., Brady L., Hennessy W. (1986) Campylobacter pyloridis and gastritis: association with intercellular spaces and adaptation to an environment of mucus as important factors in colonization of the gastric epithelium. J. Infect. Dis. 153, 658–663 [DOI] [PubMed] [Google Scholar]
- 10. Worku M. L., Sidebotham R. L., Walker M. M., Keshavarz T., Karim Q. N. (1999) The relationship between Helicobacter pylori motility, morphology and phase of growth: implications for gastric colonization and pathology. Microbiology 145, 2803–2811 [DOI] [PubMed] [Google Scholar]
- 11. Wyckoff T. J., Taylor J. A., Salama N. R. (2012) Beyond growth: novel functions for bacterial cell wall hydrolases. Trends Microbiol. 20, 540–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scheffers D. J., Pinho M. G. (2005) Bacterial cell wall synthesis: new insights from localization studies. Microbiol. Mol. Biol. Rev. 69, 585–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vollmer W., Bertsche U. (2008) Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim. Biophys. Acta 1778, 1714–1734 [DOI] [PubMed] [Google Scholar]
- 14. Vollmer W., Blanot D., de Pedro M. A. (2008) Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32, 149–167 [DOI] [PubMed] [Google Scholar]
- 15. Costa K., Bacher G., Allmaier G., Dominguez-Bello M. G., Engstrand L., Falk P., de Pedro M. A., García-del Portillo F. (1999) The morphological transition of Helicobacter pylori cells from spiral to coccoid is preceded by a substantial modification of the cell wall. J. Bacteriol. 181, 3710–3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meroueh S. O., Bencze K. Z., Hesek D., Lee M., Fisher J. F., Stemmler T. L., Mobashery S. (2006) Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl. Acad. Sci. U.S.A. 103, 4404–4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sauvage E., Kerff F., Terrak M., Ayala J. A., Charlier P. (2008) The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32, 234–258 [DOI] [PubMed] [Google Scholar]
- 18. Vollmer W., Joris B., Charlier P., Foster S. (2008) Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 32, 259–286 [DOI] [PubMed] [Google Scholar]
- 19. Chaput C., Ecobichon C., Cayet N., Girardin S. E., Werts C., Guadagnini S., Prévost M. C., Mengin-Lecreulx D., Labigne A., Boneca I. G. (2006) Role of AmiA in the morphological transition of Helicobacter pylori and in immune escape. PLoS Pathog. 2, e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sycuro L. K., Pincus Z., Gutierrez K. D., Biboy J., Stern C. A., Vollmer W., Salama N. R. (2010) Peptidoglycan cross-linking relaxation promotes Helicobacter pylori's helical shape and stomach colonization. Cell 141, 822–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sycuro L. K., Wyckoff T. J., Biboy J., Born P., Pincus Z., Vollmer W., Salama N. R. (2012) Multiple peptidoglycan modification networks modulate Helicobacter pylori's cell shape, motility, and colonization potential. PLoS Pathog. 8, e1002603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bonis M., Ecobichon C., Guadagnini S., Prévost M. C., Boneca I. G. (2010) A M23B family metallopeptidase of Helicobacter pylori required for cell shape, pole formation and virulence. Mol. Microbiol. 78, 809–819 [DOI] [PubMed] [Google Scholar]
- 23. Sycuro L. K., Rule C. S., Petersen T. W., Wyckoff T. J., Sessler T., Nagarkar D. B., Khalid F., Pincus Z., Biboy J., Vollmer W., Salama N. R. (2013) Flow cytometry-based enrichment for cell shape mutants identifies multiple genes that influence Helicobacter pylori morphology. Mol. Microbiol. 90, 869–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim H. S., Kim J., Im H. N., An D. R., Lee M., Hesek D., Mobashery S., Kim J. Y., Cho K., Yoon H. J., Han B. W., Lee B. I., Suh S. W. (2014) Structural basis for the recognition of muramyltripeptide by Helicobacter pylori Csd4, a dl-carboxypeptidase controlling the helical cell shape. Acta Crystallogr. D Biol. Crystallogr. 70, 2800–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. An D. R., Kim H. S., Kim J., Im H. N., Yoon H. J., Yoon J. Y., Jang J. Y., Hesek D., Lee M., Mobashery S., Kim S. J., Lee B. I., Suh S. W. (2015) Structure of Csd3 from Helicobacter pylori, a cell shape-determining metallopeptidase. Acta Crystallogr. D Biol. Crystallogr. 71, 675–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frirdich E., Vermeulen J., Biboy J., Soares F., Taveirne M. E., Johnson J. G., DiRita V. J., Girardin S. E., Vollmer W., Gaynor E. C. (2014) Peptidoglycan l,d-carboxypeptidase Pgp2 influences Campylobacter jejuni helical cell shape and pathogenic properties and provides the substrate for the dl-carboxypeptidase Pgp1. J. Biol. Chem. 289, 8007–8018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Girardin S. E., Boneca I. G., Carneiro L. A., Antignac A., Jéhanno M., Viala J., Tedin K., Taha M. K., Labigne A., Zähringer U., Coyle A. J., DiStefano P. S., Bertin J., Sansonetti P. J., Philpott D. J. (2003) Nod1 detects a unique muropeptide from Gram-negative bacterial peptidoglycan. Science 300, 1584–1587 [DOI] [PubMed] [Google Scholar]
- 28. Chamaillard M., Hashimoto M., Horie Y., Masumoto J., Qiu S., Saab L., Ogura Y., Kawasaki A., Fukase K., Kusumoto S., Valvano M. A., Foster S. J., Mak T. W., Nuñez G., Inohara N. (2003) An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4, 702–707 [DOI] [PubMed] [Google Scholar]
- 29. Biarrotte-Sorin S., Hugonnet J. E., Delfosse V., Mainardi J. L., Gutmann L., Arthur M., Mayer C. (2006) Crystal structure of a novel β-lactam-insensitive peptidoglycan transpeptidase. J. Mol. Biol. 359, 533–538 [DOI] [PubMed] [Google Scholar]
- 30. Kim H. S., Kim J., Im H. N., Yoon J. Y., An D. R., Yoon H. J., Kim J. Y., Min H. K., Kim S. J., Lee J. Y., Han B. W., Suh S. W. (2013) Structural basis for the inhibition of Mycobacterium tuberculosis l,d-transpeptidase by meropenem, a drug effective against extensively drug-resistant strains. Acta Crystallogr. D Biol. Crystallogr. 69, 420–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mainardi J. L., Legrand R., Arthur M., Schoot B., van Heijenoort J., Gutmann L. (2000) Novel mechanism of β-lactam resistance due to bypass of d,d-transpeptidation in Enterococcus faecium. J. Biol. Chem. 275, 16490–16496 [DOI] [PubMed] [Google Scholar]
- 32. Lavollay M., Arthur M., Fourgeaud M., Dubost L., Marie A., Veziris N., Blanot D., Gutmann L., Mainardi J. L. (2008) The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by l,d-transpeptidation. J. Bacteriol. 190, 4360–4366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chaput C., Labigne A., Boneca I. G. (2007) Characterization of Helicobacter pylori lytic transglycosylases Slt and MltD. J. Bacteriol. 189, 422–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Young G. M., Schmiel D. H., Miller V. L. (1999) A new pathway for the secretion of virulence factors by bacteria: the flagellar export apparatus functions as a protein-secretion system. Proc. Natl. Acad. Sci. U.S.A. 96, 6456–6461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCarter L., Silverman M. (1990) Surface-induced swarmer cell differentiation of Vibrio parahaemolyticus. Mol. Microbiol. 4, 1057–1062 [DOI] [PubMed] [Google Scholar]
- 36. Kostrzynska M., Betts J. D., Austin J. W., Trust T. J. (1991) Identification, characterization, and spatial localization of two flagellin species in Helicobacter pylori flagella. J. Bacteriol. 173, 937–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schirm M., Soo E. C., Aubry A. J., Austin J., Thibault P., Logan S. M. (2003) Structural, genetic and functional characterization of the flagellin glycosylation process in Helicobacter pylori. Mol. Microbiol. 48, 1579–1592 [DOI] [PubMed] [Google Scholar]
- 38. Logan S. M. (2006) Flagellar glycosylation–a new component of the motility repertoire? Microbiology 152, 1249–1262 [DOI] [PubMed] [Google Scholar]
- 39. Josenhans C., Vossebein L., Friedrich S., Suerbaum S. (2002) The neuA/flmD gene cluster of Helicobacter pylori is involved in flagellar biosynthesis and flagellin glycosylation. FEMS Microbiol. Lett. 210, 165–172 [DOI] [PubMed] [Google Scholar]
- 40. Asakura H., Churin Y., Bauer B., Boettcher J. P., Bartfeld S., Hashii N., Kawasaki N., Mollenkopf H. J., Jungblut P. R., Brinkmann V., Meyer T. F. (2010) Helicobacter pylori HP0518 affects flagellin glycosylation to alter bacterial motility. Mol. Microbiol. 78, 1130–1144 [DOI] [PubMed] [Google Scholar]
- 41. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 42. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Terwilliger T. C. (2003) Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr. D Biol. Crystallogr. 59, 38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 46. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 48. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yoo J., Choi S., Medina-Franco J. L. (2013) Molecular modeling studies of the novel inhibitors of DNA methyltransferases SGI-1027 and CBC12: implications for the mechanism of inhibition of DNMTs. PLoS ONE 8, e62152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Medina-Franco J. L., Méndez-Lucio O., Yoo J. (2014) Rationalization of activity cliffs of a sulfonamide inhibitor of DNA methyltransferases with induced-fit docking. Int. J. Mol. Sci. 15, 3253–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gaskell A., Crennell S., Taylor G. (1995) The three domains of a bacterial sialidase: a β-propeller, an immunoglobulin module and a galactose-binding jelly-roll. Structure 3, 1197–1205 [DOI] [PubMed] [Google Scholar]
- 52. Xu G., Ryan C., Kiefel M. J., Wilson J. C., Taylor G. L. (2009) Structural studies on the Pseudomonas aeruginosa sialidase-like enzyme PA2794 suggest substrate and mechanistic variations. J. Mol. Biol. 386, 828–840 [DOI] [PubMed] [Google Scholar]
- 53. Lee M., Zhang W., Hesek D., Noll B. C., Boggess B., Mobashery S. (2009) Bacterial AmpD at the crossroads of peptidoglycan recycling and manifestation of antibiotic resistance. J. Am. Chem. Soc. 131, 8742–8743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim C., Kim J. Y., Kim S. H., Lee B. I., Lee N. K. (2012) Direct characterization of protein oligomers and their quaternary structures by single-molecule FRET. Chem. Commun. 48, 1138–1140 [DOI] [PubMed] [Google Scholar]
- 55. Lee N. K., Kapanidis A. N., Wang Y., Michalet X., Mukhopadhyay J., Ebright R. H., Weiss S. (2005) Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys. J. 88, 2939–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cordillot M., Dubée V., Triboulet S., Dubost L., Marie A., Hugonnet J. E., Arthur M., Mainardi J. L. (2013) In vitro cross-linking of Mycobacterium tuberculosis peptidoglycan by l,d-transpeptidases and inactivation of these enzymes by carbapenems. Antimicrob. Agents Chemother. 57, 5940–5945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mitchell A., Chang H. Y., Daugherty L., Fraser M., Hunter S., Lopez R., McAnulla C., McMenamin C., Nuka G., Pesseat S., Sangrador-Vegas A., Scheremetjew M., Rato C., Yong S. Y., Bateman A., et al. (2015) The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 60. Hohlbein J., Craggs T. D., Cordes T. (2014) Alternating-laser excitation: single-molecule FRET and beyond. Chem. Soc. Rev. 43, 1156–1171 [DOI] [PubMed] [Google Scholar]
- 61. Li Y., Karnak D., Demeler B., Margolis B., Lavie A. (2004) Structural basis for L27 domain-mediated assembly of signaling and cell polarity complexes. EMBO J. 23, 2723–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Doerks T., Bork P., Kamberov E., Makarova O., Muecke S., Margolis B. (2000) L27, a novel heterodimerization domain in receptor targeting proteins Lin-2 and Lin-7. Trends Biochem. Sci. 25, 317–318 [DOI] [PubMed] [Google Scholar]
- 63. Harris B. Z., Venkatasubrahmanyam S., Lim W. A. (2002) Coordinated folding and association of the LIN-2, -7 (L27) domain. An obligate heterodimerization involved in assembly of signaling and cell polarity complexes. J. Biol. Chem. 277, 34902–34908 [DOI] [PubMed] [Google Scholar]
- 64. Bielnicki J., Devedjiev Y., Derewenda U., Dauter Z., Joachimiak A., Derewenda Z. S. (2006) B. subtilis ykuD protein at 2.0 Å resolution: insights into the structure and function of a novel, ubiquitous family of bacterial enzymes. Proteins 62, 144–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dodson G., Wlodawer A. (1998) Catalytic triads and their relatives. Trends Biochem. Sci. 23, 347–352 [DOI] [PubMed] [Google Scholar]
- 66. Korza H. J., Bochtler M. (2005) Pseudomonas aeruginosa l,d-carboxypeptidase, a serine peptidase with a Ser-His-Glu triad and a nucleophilic elbow. J. Biol. Chem. 280, 40802–40812 [DOI] [PubMed] [Google Scholar]
- 67. Das D., Hervé M., Elsliger M. A., Kadam R. U., Grant J. C., Chiu H. J., Knuth M. W., Klock H. E., Miller M. D., Godzik A., Lesley S. A., Deacon A. M., Mengin-Lecreulx D., Wilson I. A. (2013) Structure and function of a novel l,d-carboxypeptidase a involved in peptidoglycan recycling. J. Bacteriol. 195, 5555–5566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hoyland C. N., Aldridge C., Cleverley R. M., Duchêne M. C., Minasov G., Onopriyenko O., Sidiq K., Stogios P. J., Anderson W. F., Daniel R. A., Savchenko A., Vollmer W., Lewis R. J. (2014) Structure of the LdcB l,d-carboxypeptidase reveals the molecular basis of peptidoglycan recognition. Structure 22, 949–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Covaleda G., del Rivero M. A., Chávez M. A., Avilés F. X., Reverter D. (2012) Crystal structure of novel metallocarboxypeptidase inhibitor from marine mollusk Nerita versicolor in complex with human carboxypeptidase A4. J. Biol. Chem. 287, 9250–9258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Silva J. R., Roitberg A. E., Alves C. N. (2014) Catalytic mechanism of l,d-transpeptidase 2 from Mycobacterium tuberculosis described by a computational approach: insights for the design of new antibiotics drugs. J. Chem. Inf. Model. 54, 2402–2410 [DOI] [PubMed] [Google Scholar]
- 71. Kim S., Oh D. B., Kang H. A., Kwon O. (2011) Features and applications of bacterial sialidases. Appl. Microbiol. Biotechnol. 91, 1–15 [DOI] [PubMed] [Google Scholar]
- 72. Motoyama T., Nakasako M., Yamaguchi I. (2002) Crystallization of scytalone dehydratase F162A mutant in the unligated state and a preliminary x-ray diffraction study at 37 K. Acta Crystallogr. D Biol. Crystallogr. 58, 148–150 [DOI] [PubMed] [Google Scholar]
- 73. Kim S. W., Cha S. S., Cho H. S., Kim J. S., Ha N. C., Cho M. J., Joo S., Kim K. K., Choi K. Y., Oh B. H. (1997) High-resolution crystal structures of Δ5-3-ketosteroid isomerase with and without a reaction intermediate analogue. Biochemistry 36, 14030–14036 [DOI] [PubMed] [Google Scholar]
- 74. Gakhar L., Malik Z. A., Allen C. C., Lipscomb D. A., Larkin M. J., Ramaswamy S. (2005) Structure and increased thermostability of Rhodococcus sp. naphthalene 1,2-dioxygenase. J. Bacteriol. 187, 7222–7231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cadby I. T., Lovering A. L. (2014) Life in the “old bag” yet: structure of peptidoglycan l,d-carboxypeptidases. Structure 22, 932–934 [DOI] [PubMed] [Google Scholar]
- 76. Guruge J. L., Falk P. G., Lorenz R. G., Dans M., Wirth H. P., Blaser M. J., Berg D. E., Gordon J. I. (1998) Epithelial attachment alters the outcome of Helicobacter pylori infection. Proc. Natl. Acad. Sci. U.S.A. 95, 3925–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 78. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 79. Gouet P., Robert X., Courcelle E. (2003) ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 31, 3320–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pei J., Kim B. H., Grishin N. V. (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]