

Background: The role of the outermost M4 transmembrane α-helix in prokaryotic pentameric ligand-gated ion channel (pLGIC) function is unknown.
Results: Interactions between the M4 C terminus and both the adjacent M3 α-helix and the β6-β7 loop are essential in one, but not another prokaryotic pLGIC.
Conclusion: M4 contributes differently to maturation/function.
Significance: Variations in M4 may contribute to subunit-specific functional differences.
Keywords: lipid-protein interaction, membrane protein, nicotinic acetylcholine receptors (nAChR), receptor regulation, receptor structure-function, ELIC, GLIC, M4, intramembrane aromatic interactions, pentameric ligand-gated ion channels
Abstract
The role of the outermost transmembrane α-helix in both the maturation and function of the prokaryotic pentameric ligand-gated ion channels, GLIC and ELIC, was examined by Ala scanning mutagenesis, deletion mutations, and mutant cycle analyses. Ala mutations at the M4-M1/M3 interface lead to loss-of-function phenotypes in GLIC, with the largest negative effects occurring near the M4 C terminus. In particular, two aromatic residues at the M4 C terminus form a network of π-π and/or cation-π interactions with residues on M3 and the β6-β7 loop that is essential for both maturation and function. M4-M1/M3 interactions appear to be optimized in GLIC with even subtle structural changes at this interface leading to detrimental effects. In contrast, mutations along the M4-M1/M3 interface of ELIC typically lead to gain-of-function phenotypes, suggesting that these interactions in ELIC are not optimized for channel function. In addition, no cluster of interacting residues involving the M4 C terminus, M3, and the β6-β7 loop was found, suggesting that the M4 C terminus plays little role in ELIC maturation or function. This study shows that M4 makes distinct contributions to the maturation and gating of these two closely related homologs, suggesting that GLIC and ELIC exhibit divergent features of channel function.
Introduction
Pentameric ligand-gated ion channels (pLGICs)2 are the sites of action for a variety of both endogenous and exogenous compounds that alter synaptic communication by either potentiating or inhibiting channel function. Increasing interest has focused on allosteric modulators that interact with the transmembrane domain (TMD) (1–4). The TMD of the prototypic pLGIC, the muscle-type nicotinic acetylcholine receptor (nAChR), is sensitive to a variety of compounds, including lipids (5, 6). Both cholesterol and anionic lipids are important for nAChR function (7–13). In the absence of these activating lipids, the nAChR adopts an uncoupled conformation that binds agonist, but in most membranes does not undergo agonist-induced conformational transitions (14–16).
The outermost lipid-exposed transmembrane α-helix, M4 (one in each nAChR subunit), is likely a site for lipid sensing (17–20). Mutations along the lipid-facing surface of M4 alter channel gating, suggesting that altered M4-lipid interactions influence coupling between the agonist site and channel gate (21–25). One model proposes that lipids potentiate gating by enhancing M4 interactions with the adjacent α-helices, M1 and M3, to promote effective interactions between the M4 C terminus and the Cys-loop (14), a structure at the interface between the extracellular domain (ECD) and TMD that plays a key role in coupling agonist binding to channel gating (26–29). Interactions between the M4 C terminus and Cys-loop may be essential for the Cys-loop to adopt a conformation that promotes channel function (14, 30). In the absence of effective M4-M1/M3 interactions, the M4 lipid-sensor model proposes that the Cys-loop adopts a non-functional conformation leading to the uncoupled state (14).
As the prokaryotic pLGICs, GLIC and ELIC, are expressed in sufficient quantities for biophysical and crystallographic studies, both are excellent models for studying the mechanisms underlying pLGIC lipid sensing. Furthermore, the M4 α-helix in crystal structures of GLIC interacts extensively with M1/M3, with lipids bound at both the M4-M1 and M4-M3 interfaces (31–33). In the ELIC crystal structure, the C-terminal 5 residues of M4 are not resolved, possibly due to differential crystal packing, the absence of bound lipids, and/or detergent perturbation of weaker M4 C-terminal interactions with M1/M3 (34–37). Interestingly, differences in pore diameter are observed when GLIC is crystallized in the presence versus absence of agonist (38). A locally closed pore was also detected with several disulfide cross-linked GLIC mutants (39). These results suggest that crystalline GLIC with tight M4-M1/M3 interactions undergoes agonist-induced conformational transitions. In contrast, ELIC has not yet been crystallized in open and closed structures, even though structures of ELIC have been solved in the presence and absence of bound agonist (36, 40). This raises the possibility that crystallized ELIC, with weakened or no interactions between the M4 C terminus and adjacent structures at the ECD/TMD interface, adopts a conformation that does not undergo agonist-induced conformational transitions (37). Single amino acid substitutions engineered to weaken M4-M1/M3 interactions in GLIC reduce, whereas substitutions engineered to enhance M4-M1/M3 interactions in ELIC promote channel function (41). The intrinsic strengths of M4-M1/M3 interactions also influence the functional sensitivities of GLIC and ELIC to lipids (42, 43). Modulatable M4-M1/M3 interactions may play a role in lipid sensing.
Here, we examine the role of M4 in the maturation and gating of GLIC and ELIC. Through Ala scanning mutagenesis, M4 deletion mutations, and mutant cycle analyses, we show that the M4-M1/M3 interactions are optimized in GLIC for channel function, whereas in ELIC they are not. We identify a cluster of interacting residues on the M4 C terminus, M3, and the β6-β7 loop that is essential for GLIC maturation and function, with no analogous cluster located in ELIC. Our study shows that M4 contributes differently to the maturation and gating of these two closely related homologs, and suggests that GLIC and ELIC exhibit divergent features of channel function/modulation.
Experimental Procedures
The GLIC or ELIC coding sequences following an α7 nAChR signal sequence in the plasmids pSP64 and pTLN, respectively, were linearized by EcoRI and MluI, respectively, and used to produce capped cRNA by in vitro transcription using the mMESSAGE mMACHINE® SP6 kit (Ambion). Stage V-VI oocytes were injected with the appropriate cRNA, and allowed to incubate 1 to 4 days at 16 °C, as described elsewhere (41). Whole cell currents were measured in response to either pH or cysteamine concentration jumps using a two-electrode voltage clamp apparatus (OC-725C oocyte clamp; Harvard Apparatus, Holliston, MA). Whole cell currents were recorded for GLIC in MES buffer (140 mm NaCl, 2.8 mm KCl, 2 mm MgCl2, and 10 mm MES) and for ELIC in HEPES buffer (150 mm NaCl, 0.5 mm BaCl2, and 10 mm HEPES, pH 7.0, 1 mm DTT), with the transmembrane voltage clamped at voltages between −10 and −60 mV, depending on the levels of protein expression. Each individual dose-response was fit with a variable slope sigmoidal dose-response curve, and the individual EC50 and Hill coefficients were averaged to give the presented values ± S.D. For the presented dose-response curves, the individual dose-responses were normalized, and then each data point averaged. Curve fits of the averaged data are presented, with the error bars representing the standard error. To directly compare the magnitude of the changes in EC50 between the various mutants, ELIC results were converted to log values. The ΔpH50/ΔEC50 values presented in Figs. 2 and 4 represent the differences between mutant and WT pH50/log(EC50) values. Statistical significance was tested using a one-way ANOVA, followed by Dunnett's post hoc test.
FIGURE 2.

GLIC and ELIC M4 Ala scan heat map. The changes in the EC50 values resulting from the mutation of each M4 residue to Ala are heat-mapped onto M4 for both (A) GLIC (Protein Data Bank code 4HFI) and (B) homology model of ELIC (based on the GLIC structure (41)). The magnitude of the shift in EC50 (log scale) is depicted via color intensity, with no change in EC50 in white, gain-of-function in red, and loss-of-function in blue. A non-functional mutation, GLIC P300A (NC), is shown in black. The EC50 of the corresponding P304A mutant (colored gray) in ELIC is not presented, due to altered desensitization kinetics. C, graphical representation of EC50 changes (log scale, ΔEC50 = EC50Mut − EC50WT), colored as gain/loss-of-function. Residues are grouped by helical turn (gray boxes), and vertically aligned with their position on the adjacent M4 structure. Wild type Ala residues are shown in gray. Changes in pH50/EC50 were statistically significant by one-way ANOVA followed by Dunnett's post hoc test for all Ala mutants in GLIC, except for G316A, I309A, I308A, L304A, F299A, I297A, S295A and P285A, and for all Ala mutants in ELIC, except I319A, V313A, I310A, and G306A (see Table 1 for complete list of data). Error bars represent S.D. (GLIC) and log S.D. (ELIC).
FIGURE 4.
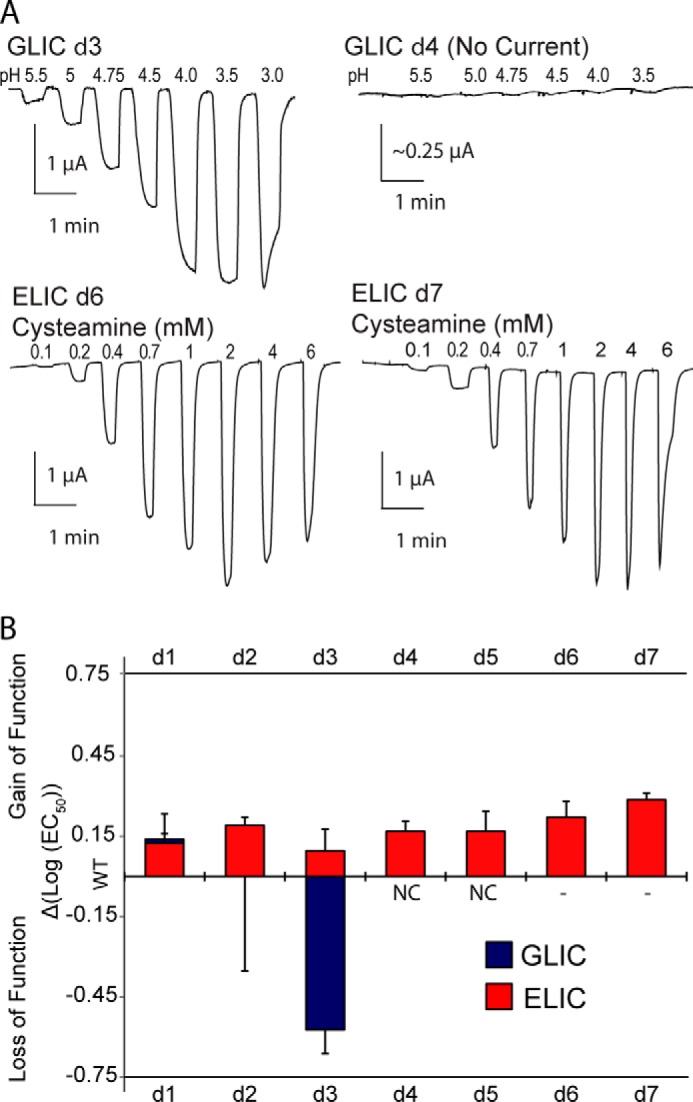
Effects of M4 C-terminal deletions on the function of GLIC and ELIC. A, whole cell electrophysiological traces recorded from oocytes expressing the key C-terminal deletion mutants in GLIC and ELIC (log scale, ΔEC50 = EC50Mut − EC50WT). dn corresponds to the number (n) of C-terminal residues removed. B, effects of the C-terminal deletions on pH50/EC50. Error bars represent S.D. (GLIC) and log S.D. (ELIC). NC, no current. EC50 changes in all ELIC mutants and GLIC d1 and d3 were statistically significant as determined by one-way ANOVA followed by Dunnett's post hoc test. See Table 2 for EC50 values.
GLIC Surface Expression
To detect surface expression of the GLIC M4 deletion mutants, the Loop C (β9-β10) sequence KPANFALEDRLESK (GLIC extracellular domain) was replaced by the sequence SERFYECCKEPYPD from the α7 nAChR, as described by Wang et al. (45). Oocytes expressing the various GLIC constructs were incubated with 2.5 nm 125I-α-Bungarotoxin (α-BTX) (143.8 Ci/mmol; PerkinElmer Life Sciences) and 1 mg/ml of bovine serum albumin (Sigma) in MOR2 (82 mm NaCl, 2.5 mm KCl, 5 mm MgCl2, 1 mm NaH2PO4, 5 mm HEPES, 0.2 mm CaCl2, pH 7.4) at room temperature for 2 h, and then washed 5× with 2 ml of MOR2 (42). 125I-α-BTX binding was quantified by γ counting. Nonspecific binding was determined by the amount of toxin bound to mock-injected oocytes under the same conditions.
Results
GLIC M4 Ala Scan
To probe the functional role of M4 in GLIC, we first performed an Ala scan of the entire α-helix, with the effects of each Ala mutation on channel function assessed using the two-electrode voltage clamp apparatus. The injection of wild type GLIC cRNA into Xenopus oocytes led to the expression of proton-activated channels on the oocyte surface that responded to protons in a dose-dependent manner (Fig. 1), with a pH50 value of 5.03 ± 0.08 (n = 38), consistent with published data (46). Twenty-five of the 26 M4 Ala mutants gave robust proton-activated currents, with the mutations displaying either no (ΔpH50 ≤ 0.09 for 8/26 mutants) or statistically significant changes in pH50 (0.09 < ΔpH50 ≤ 0.6 for 17/26 mutants, p < 0.05) relative to wild type. Five mutations led to shifts in pH50 >0.5 pH units, which correspond to a 5-fold increase in the concentration of protons required to elicit half-maximal gating. The lack of dramatic effects on pH50 is not surprising given that the introduced changes in side chain chemistry are predominantly modest and that they occur at positions peripheral to the primary structures implicated in coupling the agonist site to the channel gate (26–29). The magnitudes of the individual changes in pH50 for each Ala mutation are heat-mapped onto the GLIC M4 α-helix in Fig. 2 (see also Table 1). Note that the proton binding sites for GLIC activation are located primarily in the ECD (47), although an intramembranous His residue on M2 is essential (45, 48). Given the long distance between the sites of mutation and the agonist (proton) binding sites, the shifts in ligand sensitivity likely reflect changes in the ability to couple agonist binding to channel gating. This assertion is consistent with the previous observations that mutations in M4 of the muscle-type nAChR have no effect on agonist affinity (25, 49, 50).
FIGURE 1.
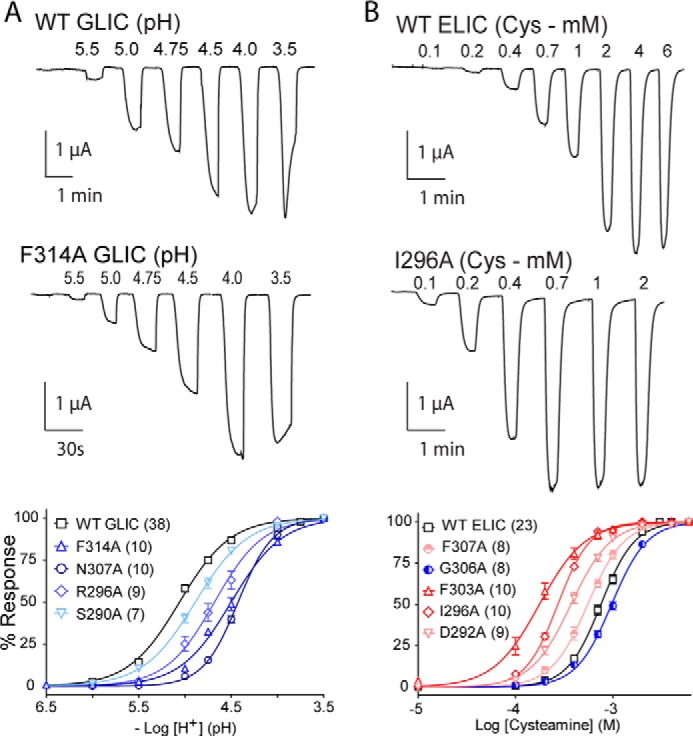
Functional characterization of GLIC and ELIC mutants. Whole cell electrophysiological traces recorded using the two-electrode voltage clamp apparatus. Currents were recorded from Xenopus oocytes expressing either GLIC (A) or ELIC (B) in response to either protons or cysteamine, respectively. The lower panel presents dose response curves (normalized current (I/Imax) versus ligand concentration) for select Ala mutants, with n = number of averaged traces. Error bars represent S.E.
TABLE 1.
The effects of M4 Ala mutations on the function of GLIC and ELIC
| Dose-responsea | |||||||
|---|---|---|---|---|---|---|---|
| GLIC |
ELIC |
||||||
| Mutant | pH50 | Hill slope | n | Mutant | EC50 | Hill slope | n |
| mm | |||||||
| WT | 5.03 ± 0.08 | 1.42 ± 0.49 | 38 | WT | 0.93 ± 0.13 | 2.13 ± 0.36 | 23 |
| F317A | 5.41 ± 0.08b | 1.31 ± 0.30 | 8 | L321A | 0.58 ± 0.08b | 2.99 ± 0.31 | 10 |
| G316A | 5.01 ± 0.08 | 1.74 ± 0.48 | 8 | T320A | 0.59 ± 0.06b | 2.53 ± 0.73 | 8 |
| F315A | 4.69 ± 0.01b,c | 1.71 ± 0.07 | 8 | I319A | 0.86 ± 0.16 | 2.20 ± 0.38 | 11 |
| F314A | 4.48 ± 0.02b,c | 1.61 ± 0.11 | 10 | G318A | 0.76 ± 0.13b | 2.13 ± 0.16 | 10 |
| L313A | 4.78 ± 0.05b | 1.14 ± 0.13 | 6 | R317A | 0.55 ± 0.11b | 2.41 ± 0.32 | 10 |
| F312A | 4.50 ± 0.01b | 1.84 ± 0.04 | 6 | I316A | 0.63 ± 0.10b | 2.52 ± 0.27 | 10 |
| Ala-311 | — | — | — | V315A | 0.60 ± 0.08b | 2.54 ± 0.14 | 9 |
| L310A | 4.47 ± 0.02b | 1.57 ± 0.11 | 6 | L314A | 0.42 ± 0.05b | 2.60 ± 0.36 | 8 |
| I309A | 5.05 ± 0.05 | 1.59 ± 0.24 | 7 | V313A | 0.95 ± 0.12 | 2.58 ± 0.41 | 10 |
| I308A | 5.08 ± 0.02 | 1.55 ± 0.09 | 6 | C312A | 0.55 ± 0.08b | 2.11 ± 0.26 | 10 |
| N307A | 4.42 ± 0.01b | 2.43 ± 0.13 | 10 | G311A | 0.51 ± 0.10b | 2.02 ± 0.20 | 11 |
| Ala-306 | — | — | — | I310A | 0.88 ± 0.09 | 1.99 ± 0.11 | 10 |
| L305A | 5.11 ± 0.09d | 1.53 ± 0.41 | 9 | Ala-309 | — | — | — |
| L304A | 5.08 ± 0.13 | 1.57 ± 0.74 | 7 | L308A | 0.52 ± 0.08b | 2.76 ± 0.53 | 8 |
| F303A | 4.48 ± 0.09b | 1.85 ± 0.65 | 8 | F307A | 0.57 ± 0.09b | 2.48 ± 0.24 | 8 |
| V302A | 4.81 ± 0.04b | 1.64 ± 0.27 | 7 | G306A | 0.97 ± 0.10 | 2.23 ± 0.23 | 8 |
| V301A | 4.92 ± 0.06e | 1.51 ± 0.35 | 8 | L305A | 0.57 ± 0.04b | 2.36 ± 0.40 | 8 |
| P300A | NCf | NC | 8 | P304A | —g | —g | 10 |
| F299A | 5.04 ± 0.09 | 1.44 ± 0.38 | 6 | F303A | 0.19 ± 0.07b | 2.03 ± 0.40 | 10 |
| Ala-298 | — | — | — | Ala-302 | — | — | — |
| I297A | 4.99 ± 0.08 | 1.20 ± 0.26 | 6 | L301A | 0.40 ± 0.07b | 2.03 ± 0.34 | 9 |
| R296A | 4.65 ± 0.03b | 1.61 ± 0.13 | 9 | R300A | 0.40 ± 0.05b | 2.54 ± 0.37 | 11 |
| S295A | 5.12 ± 0.1 | 1.36 ± 0.39 | 6 | C299A | 0.56 ± 0.09b | 2.50 ± 0.18 | 9 |
| Ala-294 | — | — | — | R298A | 0.58 ± 0.21b | 2.09 ± 0.22 | 8 |
| R293A | 4.90 ± 0.12b | 1.32 ± 0.49 | 8 | Q297A | 0.56 ± 0.09b | 2.90 ± 0.43 | 7 |
| T292A | 4.86 ± 0.05b | 1.69 ± 0.32 | 6 | I296A | 0.27 ± 0.03b | 2.80 ± 0.45 | 10 |
| I291A | 4.78 ± 0.05b | 1.15 ± 0.15 | 7 | L295A | 0.50 ± 0.11b | 2.26 ± 0.28 | 8 |
| S290A | 4.89 ± 0.06b | 1.53 ± 0.30 | 7 | L294A | 0.69 ± 0.17b | 2.20 ± 0.13 | 8 |
| Ala-289 | — | — | — | D293A | 0.46 ± 0.21b | 2.13 ± 0.30 | 8 |
| Ala-288 | — | — | — | D292A | 0.38 ± 0.07b | 2.18 ± 0.20 | 9 |
| R287A | 4.93 ± 0.03d | 1.50 ± 0.14 | 6 | E291A | 0.41 ± 0.06b | 2.66 ± 0.48 | 11 |
| Ala-286 | — | — | — | V290A | 0.53 ± 0.10b | 2.00 ± 0.13 | 6 |
| P285A | 4.96 ± 0.07 | 1.47 ± 0.31 | 6 | G289A | 0.57 ± 0.03b | 2.15 ± 0.11 | 6 |
a Measurements performed 1–4 days after cRNA injection (Vhold ranging from −10 to −60 mV). Error values represent standard deviation.
b p < 0.001 relative to WT ELIC/GLIC via one-way ANOVA followed by Dunnet's post hoc test.
c Data from Ref. 39.
d p < 0.05 relative to WT GLIC via one-way ANOVA followed by Dunnet's post hoc test.
e p < 0.01 relative to WT GLIC via one-way ANOVA followed by Dunnet's post hoc test.
f No significant current observed up to 4 days after cRNA injection.
g Functionally distinct, traditional EC50 values were not obtained.
Several important trends are observed in the GLIC Ala scan data set. First, 15 of the 17 function-altering mutations (ΔpH50 >0.09) led to an increase in the concentration of protons required to elicit channel gating and thus a decrease in channel function. The only mutation that led to a gain-of-function, F317A, is located at the C terminus of M4, where it projects away from the core of the protein to interact with adjacent phospholipids. Second, the deleterious effects of the Ala mutations are greater near the C terminus of M4, proximal to structures at the interface between the ECD and TMD (the β1-β2 and β6-β67 loops of the ECD and the M2-M3 linker of the TMD) that play a role coupling agonist binding to channel gating (Fig. 3). Third, the deleterious effects are greater for residues located on the inner face of M4, oriented toward M1/M3, suggesting that interactions between the M4 C terminus and either the adjacent transmembrane α-helices or the loops at the ECD/TMD interface are important for function. Finally, the largest negative effects on channel function came from Ala substitution of aromatic residues on the M1/M3-facing surface of M4, consistent with the demonstrated essential role of aromatic interactions driving M4 binding to M1/M3 during folding (51). Two or more simultaneous aromatic to Ala substitutions at the M4-M1/M3 interface in GLIC lead to a complete loss of folding and/or function (41).
FIGURE 3.
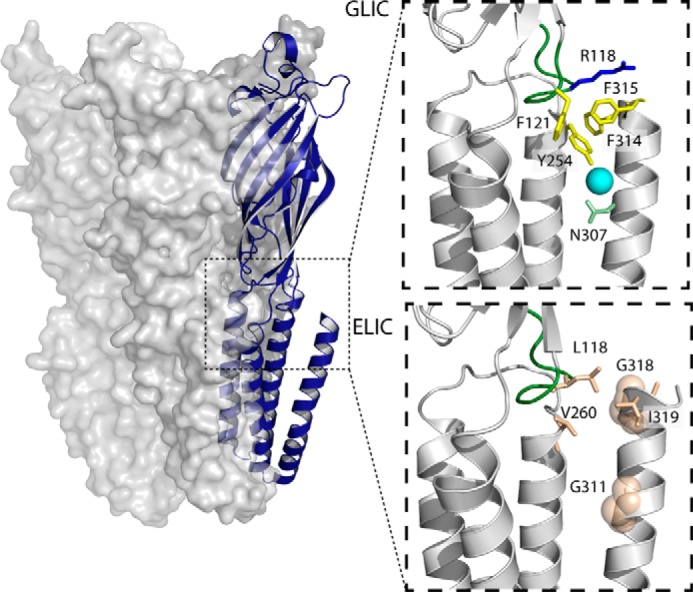
Interactions at the ECD/TMD interface of GLIC and ELIC involving M4. The structure of GLIC, with a single subunit shown in a blue schematic representation is shown on the left, with the dashed box highlighting the approximate area zoomed in on the right. The top right panel shows interactions between M4 and the ECD/TMD interface in GLIC (Protein Data Bank code 4HFI). The bottom right shows interactions between M4 and the ECD/TMD interface in a homology model of ELIC (based on the GLIC structure). Interacting residues are colored according to their physicochemical properties, with aliphatic residues in tan, aromatic residues in yellow, charged residues (Arg-118) in blue, and polar residues (Asn-307) in green. A water molecule is shown as a cyan sphere. For visibility, the glycine residues are depicted as transparent spheres, and the β6-β7 loop is highlighted in green.
Note that the mutation of a polar residue, N307A, led to a relatively large drop in the pH50 (∼0.6 pH units). Asn-307 is located in the C-terminal half of M4 where it forms a hydrogen bond via a bridging water molecule with Tyr-254 on M3 (see Fig. 3) (33). Finally, no pH-activated currents were observed with the P300A mutant, suggesting that this mutation abrogates channel function and/or the folding, oligomerization, and thus trafficking of the mutant to the cell surface. P300 is located near the middle of M4, where it kinks the α-helix such that the M4 C terminus interacts directly with both the N terminus of M3 and the β6-β7 loop (Fig. 3). This kink may be important for positioning the M4 C terminus for effective interactions with M3 and the β6-β7 loop (see below), and might also position the C terminus of M4 so that it helps mask a pre-M1 sequence, which in muscle-type nAChRs acts as an ER-retention signal (see “Discussion”) (52).
GLIC M4 C-terminal Deletions
We further explored the role of M4 in channel function by sequentially deleting residues located at the M4 C terminus (Fig. 4; Table 2). Eliminating the terminal one (Phe-317; dF) or two (Gly-316 and Phe-317; dGF) residues, which project away from the remainder of the TMD into the lipid bilayer, had little effect on channel function, consistent with the minimal effects of the Ala mutations at these two sites. In contrast, the additional deletion of Phe-315 (dFGF) led to a relatively large drop in ligand sensitivity (ΔpH50 = 0.7). Phe-315 projects toward the TMD, forming π-π and/or cation-π interactions with Tyr-254 on M3 and Phe-121 and Arg-118 on the β6-β7 loop (Fig. 3). Further deletion of Phe-314 (dFFGF) and then Leu-313 (dLFFGF) led to a loss of detectible proton-activated current, suggesting a loss of function and/or impaired folding/trafficking of the mutants to the cell surface.
TABLE 2.
The effects of M4 C-terminal deletions on the function of GLIC and ELIC
| Dose-response | |||||||
|---|---|---|---|---|---|---|---|
| GLIC |
ELIC |
||||||
| Mutant | pH50 | Hill slope | n | Mutant | EC50 | Hill slope | n |
| mm | |||||||
| WT | 5.03 ± 0.08 | 1.42 ± 0.49 | 38 | WT | 0.93 ± 0.13 | 2.13 ± 0.36 | 23 |
| 1. dF | 5.15 ± 0.13a | 1.65 ± 0.26 | 9 | 1 - dL | 0.62 ± 0.04b | 2.30 ± 0.26 | 5 |
| 2. dGF | 5.03 ± 0.20 | 1.42 ± 0.20 | 8 | 2 - dTL | 0.56 ± 0.05b | 2.67 ± 0.42 | 11 |
| 3. dFGF | 4.42 ± 0.18b | 1.64 ± 0.46 | 10 | 3 - dITL | 0.81 ± 0.08a | 2.62 ± 0.36 | 8 |
| 4. dFFGF | NCc | NC | 8 | 4 - dGITL | 0.68 ± 0.07b | 2.66 ± 0.43 | 9 |
| 5. dLFFGF | NC | NC | 8 | 5 - dRGITL | 0.68 ± 0.09b | 2.63 ± 0.55 | 9 |
| 6 - dIRGITL | 0.54 ± 0.08b | 2.40 ± 0.55 | 10 | ||||
| 7 - dVIRGITL | 0.50 ± 0.06b | 2.45 ± 0.28 | 12 | ||||
a p < 0.05 relative to WT ELIC/GLIC via one-way ANOVA followed by Dunnet's post hoc test.
b p < 0.001 relative to WT ELIC/GLIC via one-way ANOVA followed by Dunnet's post hoc test.
c NC, no current.
To test whether the loss of proton-activated current reflected reduced coupling of binding to gating or reduced expression on the oocyte surface, we engineered an α-BTX binding site into the wild type GLIC background (referred to as α-BTX-WT), as well as into each of the 5 C-terminal M4 deletion mutants (see Ref. 45). We expressed each α-BTX deletion mutant in oocytes, characterizing each pH50 value. We also correlated the maximal current elicited by each mutant at pH 4.0 with the level of surface expression, as measured by radiolabeled α-BTX binding to that same oocyte (Fig. 5). No α-BTX binding was observed with the α-BTX-dFFGF and α-BTX-dLFFGF mutants, suggesting that the lack of activity is due to a loss of surface expression. Interestingly, the α-BTX-dFGF mutant expressed on the cell surface, but the maximal response normalized to the level of expression is greatly reduced relative to the α-BTX-WT, α-BTX-dF, and α-BTX-dGF mutants. This result confirms that Phe-315 plays a key role in the coupling of proton binding to channel gating.
FIGURE 5.

Surface expression of GLIC and ELIC C-terminal deletions. WT and deletion mutant αBTX-GLIC surface expression in oocytes over time, measured via binding of 125I-labeled α-BTX binding assays. Error bars represent S.D. Inset graph: maximum current in each oocyte at pH 4.0 plotted versus the level of surface expression in the same oocyte.
Interactions between the C Terminus of M4 and Both M3 and the β6-β7 Loop
Residues at the C terminus of M4 in GLIC interact with residues on both the adjacent transmembrane α-helix, M3, and the β6-β7 loop (Fig. 3). As noted, Phe-315 is within roughly 4 Å of three residues, Tyr-254 on M3 and both Phe-121 and Arg-118 on the β6-β7 loop, thus potentially forming a network of both π-π and cation-π interactions that serve to enhance channel function. Phe-314 is relatively close to both Tyr-254 on M3 and Met-252 on the M2-M3 linker. Each of the noted aromatic and charged residues was mutated to Ala, and each led to a loss-of-function (Table 3). Double Ala mutations of potentially interacting pairs of residues were also generated, but in all but one case these double mutants did not lead to functional channels on the oocyte surface, suggesting that these interactions are essential for folding/trafficking and/or function. Only the R118A/F315A double mutant gave proton-activated currents. The pH50 of the R118A/F315A double mutant is essentially identical to that of each single mutation (R118A and F315A) suggesting that the two residues are energetically coupled, and that this coupling plays a role in channel function. Taken together, the data show that interactions between the M4 C terminus and both M3 and the β6-β7 loop are essential for folding and/or channel function, as previously suggested for the Torpedo nAChR (14).
TABLE 3.
Effects of mutations at the ECD/TMD interface on the function of GLIC and ELIC
| pH50 | n | |
|---|---|---|
| GLIC mutant | ||
| R118A | 4.41 ± 0.22a | 7 |
| F121A | 4.66 ± 0.20a | 8 |
| F315A | 4.69 ± 0.01a | 8 |
| R118A/F315A | 4.54 ± 0.31a | 8 |
| F121A/F315A | NC | |
| R118A/F121A | NC | |
| R118A/F121A/F315A | NC | |
| Y254A | 4.58 ± 0.03a | 8 |
| N307A/Y254A | 4.39 ± 0.10a | 9 |
| ELIC mutant | EC50 (mm) | n |
| L118R | 0.35 ± 0.11a | 8 |
| I319D | 0.89 ± 0.13 | 8 |
| I319F | 0.62 ± 0.18a | 6 |
| I319F/L118R | 0.56 ± 0.18a | 9 |
| L118R/I319D | 0.51 ± 0.19a | 6 |
| G311N | 0.32 ± 0.07a | 9 |
| V260Y | 0.47 ± 0.21a | 4 |
| G311N/V260Y | 0.41 ± 0.10a | 7 |
| E3: V260Y/G318F/I319F | 0.18 ± 0.02a | 9 |
| E3 + G311N | 0.64 ± 0.15a | 8 |
| E3 + L118R | 0.08 ± 0.03a | 6 |
a p < 0.001 relative to WT ELIC/GLIC via one-way ANOVA followed by Dunnet's post hoc test.
ELIC M4 Ala Scan
An equivalent M4 Ala scan was performed to explore the role of the M4 α-helix in ELIC function. Wild type ELIC cRNA injected into oocytes led to robust cysteamine-activated currents on the oocyte surface (Fig. 1), with EC50 values of 0.94 ± 0.16 mm consistent with published data (36, 53). Similar to the Ala mutations in GLIC, Ala mutations of M4 residues in ELIC led to relatively small changes in function (<10-fold), but the results were strikingly different from a qualitative perspective. Whereas M4 Ala mutations in GLIC typically had detrimental effects on channel function, the majority of the M4 Ala mutations in ELIC led to modest but statistically significant (p < 0.001) gain-of-function phenotypes (ΔEC50 ≥ 0.09 log units) (27/31 mutants), with no significant loss-of-function mutations observed (Fig. 3 and Table 1). In addition, the Ala mutations eliciting the greatest change in EC50 were located in the N-terminal half, as opposed to the C-terminal half of M4. Consistent with the Ala scan of GLIC, the strongest effects were again observed with the mutation of residues lining the inner face of M4 that orients toward M1/M3. Thus, in both pLGICs, interactions at the M4-M1/M3 interface most strongly influence channel function.
The Ala scan also revealed several unexpected effects. Contrasting the results obtained from GLIC, mutation of either of the two M1/M3-facing aromatic residues in the cytoplasmic half of M4 led to a gain, rather than a loss-of-function, with F303A leading to the largest observed increase in ligand sensitivity (∼0.7 log units). As neither of these M4 aromatic residues (Phe-307 or Phe-303) is within range of an aromatic binding partner on M1/M3, their uncompensated-for bulk may impede, rather than enhance, the interaction of M4 with M1/M3. Second, the Ala-substitution of an inward-facing Ile (I296A) in the N-terminal half of M4 led to the second highest gain-of-function. It is possible that, as with aromatics, the mutation to a less bulky hydrophobic residue leads to more effective van der Waals contact with the adjacent Leu-278 and Ala-282 located on M3. Finally, a substantial effect was observed with the Ala substitution of a proline residue, which is conserved in the M4 of many pLGICs. Unlike the analogous P300A mutation in GLIC, P304A in ELIC remained functional, but exhibited more rapid desensitization kinetics. The effects of the P304A mutation on ELIC desensitization are explored in more detail elsewhere.
ELIC M4 C-terminal Deletions
The consistent gain-of-function phenotypes observed with the Ala mutations suggest that unlike GLIC, M4-M1/M3 interactions in ELIC are not optimized for effective channel function. We next explored the functional role of the M4 C terminus by performing C-terminal deletions. In stark contrast to the C-terminal deletions in GLIC, deletions of up to 7 residues (dL → dVIRGITL), i.e. two full turns at the C terminus of M4, led to no change, or even modest gain-of-function phenotypes (Fig. 4 and Table 2). Thus, interactions between residues located at the M4 C terminus and residues on either M3 or the β6-β7 loop in ELIC are not essential, or even important, for channel function. Although this is perhaps not surprising given that the M4 C terminus in ELIC has primarily aliphatic residues, with Gly and Ile facing the M1/M3 interface, even deletion of the charged Arg-317 had no detrimental effect. As noted, the final 5 residues in the C-terminal half of M4 α-helix are not resolved in the ELIC crystal structure (34, 35). The crystal structure suggests weak interactions between the M4 C terminus and either M1/M3 or the β6-β7 loop. One explanation of our data is that removal of the loosely bound and thus likely dynamic M4 C terminus allows the remainder of M4 to interact more effectively with M1/M3 to promote channel function.
ELIC C-terminal M4 Interactions with M3 and the β6-β7 Loop
As we had previously observed that enhancing M4-M3 interactions via aliphatic-to-aromatic substitutions had a positive effect on ELIC channel function (41), we next tested whether introducing additional M4-M3 interactions would further improve the coupling of binding to gating. In GLIC, M4 Asn-307 (M4) is linked to Tyr-254 (M3) via a bridging water molecule (Fig. 3). We inserted these residues at corresponding positions in ELIC, giving the mutant G311N/V260Y. Although individually each mutation induced a mild gain-of-function (EC50 = 0.32 ± 0.07 and 0.47 ± 0.21 mm, respectively) relative to the WT EC50 (0.94 ± 0.16 mm), no further enhancement was observed with the G311N/V260Y double mutant (EC50 = 0.41 ± 0.1 mm). The G311N mutation was also superimposed onto an ELIC mutant containing the equivalent C-terminal aromatic cluster to that found in GLIC (I319F/G318F/V260Y), but in this case the G311N mutation actually reduced channel function relative to the mutant containing only the aromatic cluster (EC50 = 0.64 ± 0.15 for G311N/I319F/G318F/V260Y versus EC50 = 0.18 ± 0.02 for I319F/G318F/V260Y).
We also tested whether enhanced interactions between M4 and the β6-β7 loop could improve coupling between the agonist site and channel gate. In GLIC, an interaction between Phe-315 on M4 and Arg-118 in the β6-β7 loop promotes channel function. Although the corresponding individual mutations I319F (EC50 = 0.62 ± 0.18 mm) and L118R (EC50 = 0.35 ± 0.11 mm) in ELIC led to functional gains, the double L118R/I319F mutant gave no further enhancement (EC50 = 0.56 ± 0.18 mm). A double L118R/I319D mutant was also generated to replace the engineered cation-π interaction with an engineered salt bridge, but although I319D gave a WT-like EC50 value (0.89 ± 0.12), the double mutant had little additional effect (EC50 = 0.51 ± 0.19 mm). It may be, however, that these residues are too distant/improperly oriented in the actual protein to promote M4-β6-β7 interactions. With that in mind, we superimposed the β6-β7 loop L118R mutation onto a “M4-M1/M3 stabilized” ELIC mutant, which possesses the full C-terminal M4 aromatic cluster that is found in GLIC (44). Introduction of the L118R mutation into M4-M1/M3-stabilized ELIC did lead to a slight enhancement (EC50 goes from 0.18 ± 0.02 with the aromatic cluster (V260Y/G318F/I319F) to 0.08 ± 0.03 with both the aromatic cluster and the L118R mutation). Thus it appears that in WT ELIC, there are no interactions between the C terminus of M4 and β6-β7 loop that impact on channel function. With the addition of aromatics to stabilize C-terminal M4-M3 interactions, however, enhanced M4 interactions with the β6-β7 loop slightly improve gating. In other words, as ELIC becomes more like GLIC in terms of its M4-M1/M3 interactions, its function becomes more easily modulated through mutation of M4 and/or the β6-β7 loop.
Discussion
This work sheds light on the mechanism(s) by which the outermost transmembrane M4 α-helix influences pLGIC function, and suggests further that M4 makes distinct contributions to the maturation and function of the two closely related prokaryotic pLGICs, GLIC and ELIC. The latter conclusion was unexpected, but is based on several observations.
First, an Ala scan of GLIC M4 shows that almost any disruption of the M4-M1/M3 interface has a detrimental effect on function, suggesting that M4-M1/M3 interactions are optimized in GLIC. In contrast, Ala mutations of residues at the M4-M1/M3 interface in ELIC typically lead to gain-of-function phenotypes, suggesting that M4-M1/M3 interactions are less than optimal. In fact, the replacement of large bulky hydrophobic side chains at the M4-M1/M3 interface in ELIC with a smaller methyl group of alanine seemingly leads to more effective M4-M1/M3 interactions that promote channel function. Even greater improvements in ELIC function are observed with the introduction of interacting aromatic residues at the M4-M1/M3 interface (44).
Second, a cluster of interacting residues on M3, M4, and the β6-β7 loop (i.e. at the ECD/TMD interface) in GLIC is essential to the maturation and function of GLIC, whereas no such counterpart is found in ELIC. Specifically, the aromatic residues, Phe-314 and Phe-315, on the M4 C terminus in GLIC form π-π and/or cation-π interactions with Tyr-254 on M3 and both Phe-121 and Arg-118 on the β6-β7 loop. Ala mutations of any of these interacting residues leads to a loss-of-function, whereas double Ala mutations of every interacting pair, except Phe-315 and Arg-118, completely eliminates folding and/or function (see also (44)). Furthermore, deletion of the final three M4 residues to eliminate Phe-315 (i.e. deletion of Phe-317, Gly-316, and Phe-315) leads to a substantial loss of both folding/cell surface expression and function, with the additional deletion of Phe-314 completely abrogating cell surface expression of GLIC. In contrast, Ala mutations of either of the residues in ELIC that are analogous to the residues Phe-314 and Phe-315 in GLIC (i.e. ELIC Gly-317 and Ile-318) actually leads to a slight potentiation of channel function. In addition, deletion of two full turns at the M4 C terminus (7 residues) has no noticeable effect on expression and actually leads to a left shift in EC50 corresponding to a slight gain-of-function. We also engineered the interactions involving the M4 C terminus and residues on both M3 and the β6-β7 loop that are found in GLIC into ELIC, with limited benefits to channel function. It appears that ELIC has evolved to fold, traffic to the cell surface, and function in the absence of effective interactions between the M4 C terminus and residues at the ECD/TMD coupling interface.
Finally, the contrasting contributions of the M4 C terminus to the maturation and function of GLIC and ELIC were highlighted by the Ala mutation of a lipid-facing Pro residue located midway along M4 in both prokaryotic pLGICs. In the GLIC structure, this proline (Pro-300) leads to a kink in M4 that positions the M4 C terminus so that it forms the above noted π-π and cation-π interactions with other residues at the ECD/TMD coupling interface. The P300A mutation should remove this kink and thus straighten the M4 α-helix, leading to the elimination or weakening of these essential interactions between the M4 C terminus and both M3 and the β6-β7 loop. Consistent with this hypothesis, the P300A mutation completely eliminates GLIC folding/trafficking and/or function. Intriguingly, the analogous Pro residue in ELIC (Pro-304) also leads to a kink in M4, which should position the M4 C terminus to interact with M3 and possibly the β6-β7 loop, yet the P304A mutant of ELIC still forms functional channels that traffic to the oocyte surface. The ability of P304A to gate open is consistent with our contention that interactions between the M4 C terminus and other residues at the ECD/TMD coupling interface are not essential for folding/trafficking or function. The observation that the P304A mutation leads to rapid desensitization kinetics, however, demonstrates that the C-terminal half of M4 still interacts with the adjacent M1 and M3 α-helices to shape the agonist-induced response. This finding is consistent with a recent study, which detected movement of M4 upon desensitization of GLIC (Ref. 54, and see also Refs. 55–58).
The distinct contributions of M4 to the folding and function of GLIC and ELIC mirrors the distinct conformations of M4 observed in the GLIC and ELIC crystal structures. GLIC has an abundance of aromatic, polar, and charged residues that contribute to tight M4-M1/M3 interactions along the entire length of M4. Although many interactions are likely important facilitators of M4-M1/M3 interactions, aromatic interactions play a dominant role in M4 binding to M1/M3 during pLGIC folding (51). The abundant aromatic interactions at the M4-M1/M3 interface in GLIC are sufficiently strong to maintain tight M4 “binding” to M1/M3, even in the detergent-solubilized state. This tight binding may facilitate channel function, as GLIC crystallized in the presence and absence of agonist exhibits differences in pore diameter that likely reflect channel gating (31, 32, 38, 39). In ELIC, however, the M4-M1/M3 interface is richer in aliphatic residues, and there are fewer aromatic, polar, and charged residues, particularly involving the M4 C terminus. Significantly, the five C-terminal M4 residues in the ELIC crystal structure are unresolved. Furthermore, although ELIC has been crystallized in both the presence and absence of bound agonist, no changes in pore diameter were observed (36, 40). Although further studies may yet lead to the crystallization of both open and closed ELIC conformations, one possible interpretation of the crystal structures is that the absence of effective interactions between the M4 C terminus and other residues at the ECD/TMD interface leads to an ELIC conformation that does not undergo conformational transitions (37). Although the latter result appears to conflict with our M4 deletions mutations, which suggest that the M4 C terminus has no role in channel function, detergent solubilization followed by crystallization may lead to a more detrimental conformational perturbation than that observed with the M4 C-terminal deletion mutants of ELIC expressed in oocyte membranes.
The distinct contributions of M4 to the folding and function of GLIC and ELIC may also reflect different pathways for agonist-induced channel gating. GLIC is proton activated. A GLIC chimera formed between the GLIC ECD and the glycine receptor TMD is responsive to protons, showing that ECD protonation sites act to gate open the channel (47). An intramembrane proton binding site (His) also plays an important role in activation (45, 48). Conversely, ELIC is activated by primary amines, which bind to the canonical agonist binding site found at the interface between subunits in the ECDs of eukaryotic pLGICs. One possible interpretation is that distinct roles for M4 in gating arise due to distinct mechanisms of activation, with GLIC activation intimately associated with protonation of the TMD His residue, whereas ELIC activation occurs through an allosteric pathway similar to that of eukaryotic pLGICs. This interpretation, however, is paradoxical with the observation that GLIC requires extensive interactions between the M4 C terminus and residues at the ECD/TMD interface for folding and function, whereas ELIC does not. Regardless, the data suggest that there may be distinct features in the mechanisms of activation in GLIC and ELIC, a finding supported by the observation that ELIC exhibits atypical gating and cation-conduction properties relative to other pLGICs (59). Double electron-electron resonance experiments also suggest that the x-ray structures of ELIC are not appropriate models for the resting state (60). Taken together, these findings caution against the utility of comparing different conformations of GLIC and ELIC to probe the mechanisms underlying agonist-induced gating.
The effects of M4 C-terminal deletions on the folding and cell surface expression of GLIC are intriguing given that a mutation leading to truncation at a Cys residue located distal to the M4 α-helix in the muscle-type ϵ-subunit alters nAChR trafficking to the cell surface leading to one form of congenital myasthenic syndrome (61). The M4 C terminus also plays an essential role in the maturation and function of the Torpedo nAChR, an α7–5HT3A chimeric homopentamer, and the full-length 5HT3A homopentamer (30, 62, 63). One possibility is that the truncation mutations influence the masking of an endoplasmic retention signal located at the N terminus of the M1 (pre-M1) transmembrane α-helix (52). In the muscle-type nAChR, this sequence (PL(F/Y)(F/Y)XXN) is masked through both inter-subunit and M4-M1/M3 interactions. Although the corresponding pre-M1 sequence in GLIC (RQYFSYI) differs from that of the nAChR consensus, it may be sufficient to influence the trafficking of GLIC in oocytes. Specifically, an unmasking of this sequence by M4 C-terminal deletions could lead to a loss of cell surface expression. Interestingly, the same M4 deletions in ELIC have no effect on folding/trafficking. The analogous pre-M1 sequence in ELIC (RNPSYYL), however, exhibits greater variability relative to the muscle-type nAChR pre-M1 sequence, thus perhaps allowing ELIC to traffic in the absence of the M4 C terminus.
Previous studies addressing the role of M4 in pLGIC function have tended to focus on lipid-facing mutations in the muscle-type nAChR, including detailed kinetic analyses of a lipid facing αC418W mutation that leads to one form of congenital myasthenic syndrome (22, 50, 64, 65). M4 Trp-scanning mutations have also been performed on the M4 α-helix of the α, β, γ, and δ subunits of the nAChR, with some mutations leading to loss-, and others to gain-of-function phenotypes (66–69). In fact, identical mutations of analogous M4 residues in different subunits can have opposite effects on channel function (24, 25, 70). The two αM4 α-helices both move as a single unit roughly halfway along the reaction coordinate leading from the resting to the channel open state, with the movements of both βM4 and ϵM4 following that of αM4 (49). These along with our study highlight the importance of M4 in pLGIC function.
Our data show that M4 contributes differently to the maturation and function of the two closely related prokaryotic homologs, GLIC and ELIC. Specifically, the M4 C terminus plays an essential role in GLIC, whereas the role of the M4 C terminus in ELIC is less defined. An important extension of this work will be to assess how variable M4 sequences in different pLGICs influence both their response to agonist and their lipid sensitivities. This work builds on a previous study, which showed that sequence variations in M4 contribute to the functional differences between fetal and adult forms of the muscle-type nAChR (44). Sequence variations in the M4 transmembrane α-helices of different nAChR subunits may contribute to distinct subtype-specific channel properties and thus organismal health.
Author Contributions
C. M. H., P. F. J., and J. E. B. designed the research project. C. M. H. and P. F. J. generated the mutants and acquired/processed the data. C. M. H. and J. E. B. wrote the paper, and C. M. H. prepared all figures.
This work was supported by Canadian Institutes of Health Research Grant 111243 (to J. E. B.). The authors declare that they have no conflicts of interest with the contents of this article.
- pLGIC
- pentameric ligand-gated ion channel
- α-BTX
- α-bungarotoxin
- ECD
- extracellular domain
- ELIC
- Erwinia ligand-gated ion channel
- GLIC
- Gloebacter ligand-gated ion channel
- nAChR
- nicotinic acetylcholine receptor
- TMD
- transmembrane domain
- ANOVA
- analysis of variance.
References
- 1. Baenziger J. E., Corringer P. J. (2011) 3D structure and allosteric modulation of the transmembrane domain of pentameric ligand-gated ion channels. Neuropharmacology 60, 116–125 [DOI] [PubMed] [Google Scholar]
- 2. Nury H., Delarue M., Corringer P. J. (2011) X-ray structures of general anesthetics bound to their molecular targets. Med. Sci. (Paris) 27, 1056–1057 [DOI] [PubMed] [Google Scholar]
- 3. Nury H., Van Renterghem C., Weng Y., Tran A., Baaden M., Dufresne V., Changeux J. P., Sonner J. M., Delarue M., Corringer P. J. (2011) X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature 469, 428–431 [DOI] [PubMed] [Google Scholar]
- 4. Forman S. A., Chiara D. C., Miller K. W. (2015) Anesthetics target interfacial transmembrane sites in nicotinic acetylcholine receptors. Neuropharmacology 96, 169–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barrantes F. J. (2015) Phylogenetic conservation of protein-lipid motifs in pentameric ligand-gated ion channels. Biochim. Biophys. Acta 1848, 1796–1805 [DOI] [PubMed] [Google Scholar]
- 6. Baenziger J. E., Hénault C. M., Therien J. P., Sun J. (2015) Nicotinic acetylcholine receptor-lipid interactions: mechanistic insight and biological function. Biochim. Biophys. Acta 1848, 1806–1817 [DOI] [PubMed] [Google Scholar]
- 7. Baenziger J. E., Morris M. L., Darsaut T. E., Ryan S. E. (2000) Effect of membrane lipid composition on the conformational equilibria of the nicotinic acetylcholine receptor. J. Biol. Chem. 275, 777–784 [DOI] [PubMed] [Google Scholar]
- 8. daCosta C. J., Ogrel A. A., McCardy E. A., Blanton M. P., Baenziger J. E. (2002) Lipid-protein interactions at the nicotinic acetylcholine receptor: a functional coupling between nicotinic receptors and phosphatidic acid-containing lipid bilayers. J. Biol. Chem. 277, 201–208 [DOI] [PubMed] [Google Scholar]
- 9. Hamouda A. K., Sanghvi M., Sauls D., Machu T. K., Blanton M. P. (2006) Assessing the lipid requirements of the Torpedo californica nicotinic acetylcholine receptor. Biochemistry 45, 4327–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. daCosta C. J., Medaglia S. A., Lavigne N., Wang S., Carswell C. L., Baenziger J. E. (2009) Anionic lipids allosterically modulate multiple nicotinic acetylcholine receptor conformational equilibria. J. Biol. Chem. 284, 33841–33849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Criado M., Eibl H., Barrantes F. J. (1984) Functional properties of the acetylcholine receptor incorporated in model lipid membranes: differential effects of chain length and head group of phospholipids on receptor affinity states and receptor-mediated ion translocation. J. Biol. Chem. 259, 9188–9198 [PubMed] [Google Scholar]
- 12. Criado M., Eibl H., Barrantes F. J. (1982) Effects of lipids on acetylcholine receptor: essential need of cholesterol for maintenance of agonist-induced state transitions in lipid vesicles. Biochemistry 21, 3622–3629 [DOI] [PubMed] [Google Scholar]
- 13. Fong T. M., McNamee M. G. (1986) Correlation between acetylcholine receptor function and structural properties of membranes. Biochemistry 25, 830–840 [DOI] [PubMed] [Google Scholar]
- 14. daCosta C. J., Baenziger J. E. (2009) A lipid-dependent uncoupled conformation of the acetylcholine receptor. J. Biol. Chem. 284, 17819–17825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baenziger J. E., Ryan S. E., Goodreid M. M., Vuong N. Q., Sturgeon R. M., daCosta C. J. (2008) Lipid composition alters drug action at the nicotinic acetylcholine receptor. Mol. Pharmacol. 73, 880–890 [DOI] [PubMed] [Google Scholar]
- 16. daCosta C. J., Dey L., Therien J. P., Baenziger J. E. (2013) A distinct mechanism for activating uncoupled nicotinic acetylcholine receptors. Nat. Chem. Biol. 9, 701–707 [DOI] [PubMed] [Google Scholar]
- 17. Xu Y., Barrantes F. J., Luo X., Chen K., Shen J., Jiang H. (2005) Conformational dynamics of the nicotinic acetylcholine receptor channel: a 35-ns molecular dynamics simulation study. J. Am. Chem. Soc. 127, 1291–1299 [DOI] [PubMed] [Google Scholar]
- 18. Williamson P. T., Zandomeneghi G., Barrantes F. J., Watts A., Meier B. H. (2005) Structural and dynamic studies of the g-M4 trans-membrane domain of the nicotinic acetylcholine receptor. Mol. Membr. Biol. 22, 485–496 [DOI] [PubMed] [Google Scholar]
- 19. Antollini S. S., Xu Y., Jiang H., Barrantes F. J. (2005) Fluorescence and molecular dynamics studies of the acetylcholine receptor gM4 transmembrane peptide in reconstituted systems. Mol. Membr. Biol. 22, 471–483 [DOI] [PubMed] [Google Scholar]
- 20. Hénault C. M., Sun J., Therien J. P., daCosta C. J., Carswell C. L., Labriola J. M., Juranka P. F., Baenziger J. E. (2015) The role of the M4 lipid-sensor in the folding, trafficking, and allosteric modulation of nicotinic acetylcholine receptors. Neuropharmacology 96, 157–168 [DOI] [PubMed] [Google Scholar]
- 21. Li L., Schuchard M., Palma A., Pradier L., McNamee M. G. (1990) Functional role of the cysteine 451 thiol group in the M4 helix of the γ subunit of Torpedo californica acetylcholine receptor. Biochemistry 29, 5428–5436 [DOI] [PubMed] [Google Scholar]
- 22. Lee Y. H., Li L., Lasalde J., Rojas L., McNamee M., Ortiz-Miranda S. I., Pappone P. (1994) Mutations in the M4 domain of Torpedo californica acetylcholine receptor dramatically alter ion channel function. Biophys. J. 66, 646–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lasalde J. A., Tamamizu S., Butler D. H., Vibat C. R., Hung B., McNamee M. G. (1996) Tryptophan substitutions at the lipid-exposed transmembrane segment M4 of Torpedo californica acetylcholine receptor govern channel gating. Biochemistry 35, 14139–14148 [DOI] [PubMed] [Google Scholar]
- 24. Bouzat C., Roccamo A. M., Garbus I., Barrantes F. J. (1998) Mutations at lipid-exposed residues of the acetylcholine receptor affect its gating kinetics. Mol. Pharmacol. 54, 146–153 [DOI] [PubMed] [Google Scholar]
- 25. Bouzat C., Barrantes F., Sine S. (2000) Nicotinic receptor fourth transmembrane domain: hydrogen bonding by conserved threonine contributes to channel gating kinetics. J. Gen. Physiol. 115, 663–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee W. Y., Sine S. M. (2005) Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature 438, 243–247 [DOI] [PubMed] [Google Scholar]
- 27. Grutter T., de Carvalho L. P., Dufresne V., Taly A., Edelstein S. J., Changeux J. P. (2005) Molecular tuning of fast gating in pentameric ligand-gated ion channels. Proc. Natl. Acad. Sci. U.S.A. 102, 18207–18212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lummis S. C., Beene D. L., Lee L. W., Lester H. A., Broadhurst R. W., Dougherty D. A. (2005) Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature 438, 248–252 [DOI] [PubMed] [Google Scholar]
- 29. Jha A., Cadugan D. J., Purohit P., Auerbach A. (2007) Acetylcholine receptor gating at extracellular transmembrane domain interface: the Cys-loop and M2-M3 linker. J. Gen. Physiol. 130, 547–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pons S., Sallette J., Bourgeois J. P., Taly A., Changeux J. P., Devillers-Thiéry A. (2004) Critical role of the C-terminal segment in the maturation and export to the cell surface of the homopentameric α7-5HT3A receptor. Eur. J. Neurosci. 20, 2022–2030 [DOI] [PubMed] [Google Scholar]
- 31. Bocquet N., Nury H., Baaden M., Le Poupon C., Changeux J. P., Delarue M., Corringer P. J. (2009) X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457, 111–114 [DOI] [PubMed] [Google Scholar]
- 32. Hilf R. J., Dutzler R. (2009) Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 457, 115–118 [DOI] [PubMed] [Google Scholar]
- 33. Sauguet L., Poitevin F., Murail S., Van Renterghem C., Moraga-Cid G., Malherbe L., Thompson A. W., Koehl P., Corringer P. J., Baaden M., Delarue M. (2013) Structural basis for ion permeation mechanism in pentameric ligand-gated ion channels. EMBO J. 32, 728–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hilf R. J., Dutzler R. (2008) X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 452, 375–379 [DOI] [PubMed] [Google Scholar]
- 35. Pan J., Chen Q., Willenbring D., Yoshida K., Tillman T., Kashlan O. B., Cohen A., Kong X. P., Xu Y., Tang P. (2012) Structure of the pentameric ligand-gated ion channel ELIC cocrystallized with its competitive antagonist acetylcholine. Nat. Commun. 3, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zimmermann I., Dutzler R. (2011) Ligand activation of the prokaryotic pentameric ligand-gated ion channel ELIC. PLos Biol. 9, e1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. daCosta C. J., Baenziger J. E. (2013) Gating of pentameric ligand-gated ion channels: structural insights and ambiguities. Structure 21, 1271–1283 [DOI] [PubMed] [Google Scholar]
- 38. Sauguet L., Shahsavar A., Poitevin F., Huon C., Menny A., Nemecz À., Haouz A., Changeux J. P., Corringer P. J., Delarue M. (2014) Crystal structures of a pentameric ligand-gated ion channel provide a mechanism for activation. Proc. Natl. Acad. Sci. U.S.A. 111, 966–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prevost M. S., Sauguet L., Nury H., Van Renterghem C., Huon C., Poitevin F., Baaden M., Delarue M., Corringer P. J. (2012) A locally closed conformation of a bacterial pentameric proton-gated ion channel. Nat. Struct. Mol. Biol. 19, 642–649 [DOI] [PubMed] [Google Scholar]
- 40. Gonzalez-Gutierrez G., Lukk T., Agarwal V., Papke D., Nair S. K., Grosman C. (2012) Mutations that stabilize the open state of the Erwinia chrisanthemi ligand-gated ion channel fail to change the conformation of the pore domain in crystals. Proc. Natl. Acad. Sci. U.S.A. 109, 6331–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carswell C. L., Henault C. M., Murlidaran S., Therien J. P., Juranka P. F., Surujballi J. A., Brannigan G., Baenziger J. E. (2015) Role of the fourth transmembrane α-helix in the allosteric modulation of pentameric ligand-gated ion channels. Structure 23, 1655–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Labriola J. M., Pandhare A., Jansen M., Blanton M. P., Corringer P. J., Baenziger J. E. (2013) Structural sensitivity of a prokaryotic pentameric ligand-gated ion channel to its membrane environment. J. Biol. Chem. 288, 11294–11303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carswell C. L., Sun J., Baenziger J. E. (2015) Intramembrane aromatic interactions influence the lipid sensitivities of pentameric ligand-gated ion channels. J. Biol. Chem. 290, 2496–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bouzat C., Bren N., Sine S. M. (1994) Structural basis of the different gating kinetics of fetal and adult acetylcholine receptors. Neuron 13, 1395–1402 [DOI] [PubMed] [Google Scholar]
- 45. Wang H. L., Cheng X., Sine S. M. (2012) Intramembrane proton binding site linked to activation of bacterial pentameric ion channel. J. Biol. Chem. 287, 6482–6489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bocquet N., Prado de Carvalho L., Cartaud J., Neyton J., Le Poupon C., Taly A., Grutter T., Changeux J. P., Corringer P. J. (2007) A prokaryotic proton-gated ion channel from the nicotinic acetylcholine receptor family. Nature 445, 116–119 [DOI] [PubMed] [Google Scholar]
- 47. Duret G., Van Renterghem C., Weng Y., Prevost M., Moraga-Cid G., Huon C., Sonner J. M., Corringer P. J. (2011) Functional prokaryotic-eukaryotic chimera from the pentameric ligand-gated ion channel family. Proc. Natl. Acad. Sci. U.S.A. 108, 12143–12148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rienzo M., Lummis S. C., Dougherty D. A. (2014) Structural requirements in the transmembrane domain of GLIC revealed by incorporation of noncanonical histidine analogs. Chem. Biol. 21, 1700–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mitra A., Bailey T. D., Auerbach A. L. (2004) Structural dynamics of the M4 transmembrane segment during acetylcholine receptor gating. Structure 12, 1909–1918 [DOI] [PubMed] [Google Scholar]
- 50. Shen X. M., Deymeer F., Sine S. M., Engel A. G. (2006) Slow-channel mutation in acetylcholine receptor αM4 domain and its efficient knockdown. Ann. Neurol. 60, 128–136 [DOI] [PubMed] [Google Scholar]
- 51. Haeger S., Kuzmin D., Detro-Dassen S., Lang N., Kilb M., Tsetlin V., Betz H., Laube B., Schmalzing G. (2010) An intramembrane aromatic network determines pentameric assembly of Cys-loop receptors. Nat. Struct. Mol. Biol. 17, 90–98 [DOI] [PubMed] [Google Scholar]
- 52. Wang J. M., Zhang L., Yao Y., Viroonchatapan N., Rothe E., Wang Z. Z. (2002) A transmembrane motif governs the surface trafficking of nicotinic acetylcholine receptors. Nat. Neurosci. 5, 963–970 [DOI] [PubMed] [Google Scholar]
- 53. Zimmermann I., Marabelli A., Bertozzi C., Sivilotti L. G., Dutzler R. (2012) Inhibition of the prokaryotic pentameric ligand-gated ion channel ELIC by divalent cations. PLos Biol. 10, e1001429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Velisetty P., Chalamalasetti S. V., Chakrapani S. (2014) Structural basis for allosteric coupling at the membrane-protein interface in Gloeobacter violaceus ligand-gated ion channel (GLIC). J. Biol. Chem. 289, 3013–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kinde M. N., Chen Q., Lawless M. J., Mowrey D. D., Xu J., Saxena S., Xu Y., Tang P. (2015) Conformational changes underlying desensitization of the pentameric ligand-gated ion channel ELIC. Structure 23, 995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Velisetty P., Chakrapani S. (2012) Desensitization mechanism in prokaryotic ligand-gated ion channel. J. Biol. Chem. 287, 18467–18477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Velisetty P., Chalamalasetti S. V., Chakrapani S. (2012) Conformational transitions underlying pore opening and desensitization in membrane-embedded Gloeobacter violaceus ligand-gated ion channel (GLIC). J. Biol. Chem. 287, 36864–36872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tillman T. S., Seyoum E., Mowrey D. D., Xu Y., Tang P. (2014) ELIC-α7 nicotinic acetylcholine receptor (α7nAChR) chimeras reveal a prominent role of the extracellular-transmembrane domain interface in allosteric modulation. J. Biol. Chem. 289, 13851–13857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gonzalez-Gutierrez G., Grosman C. (2015) The atypical cation-conduction and gating properties of ELIC underscore the marked functional versatility of the pentameric ligand-gated ion-channel fold. J. Gen. Physiol. 146, 15–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dellisanti C. D., Ghosh B., Hanson S. M., Raspanti J. M., Grant V. A., Diarra G. M., Schuh A. M., Satyshur K., Klug C. S., Czajkowski C. (2013) Site-directed spin labeling reveals pentameric ligand-gated ion channel gating motions. PLos Biol. 11, e1001714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ealing J., Webster R., Brownlow S., Abdelgany A., Oosterhuis H., Muntoni F., Vaux D. J., Vincent A., Beeson D. (2002) Mutations in congenital myasthenic syndromes reveal an epsilon subunit C-terminal cysteine, C470, crucial for maturation and surface expression of adult AChR. Hum. Mol. Genet. 11, 3087–3096 [DOI] [PubMed] [Google Scholar]
- 62. Tobimatsu T., Fujita Y., Fukuda K., Tanaka K., Mori Y., Konno T., Mishina M., Numa S. (1987) Effects of substitution of putative transmembrane segments on nicotinic acetylcholine receptor function. FEBS Lett. 222, 56–62 [DOI] [PubMed] [Google Scholar]
- 63. Butler A. S., Lindesay S. A., Dover T. J., Kennedy M. D., Patchell V. B., Levine B. A., Hope A. G., Barnes N. M. (2009) Importance of the C-terminus of the human 5-HT3A receptor subunit. Neuropharmacology 56, 292–302 [DOI] [PubMed] [Google Scholar]
- 64. Ortiz-Miranda S. I., Lasalde J. A., Pappone P. A., McNamee M. G. (1997) Mutations in the M4 domain of the Torpedo californica nicotinic acetylcholine receptor alter channel opening and closing. J. Membr. Biol. 158, 17–30 [DOI] [PubMed] [Google Scholar]
- 65. Tamamizu S., Lee Y., Hung B., McNamee M. G., Lasalde-Dominicci J. A. (1999) Alteration in ion channel function of mouse nicotinic acetylcholine receptor by mutations in the M4 transmembrane domain. J. Membr. Biol. 170, 157–164 [DOI] [PubMed] [Google Scholar]
- 66. Caballero-Rivera D., Cruz-Nieves O. A., Oyola-Cintron J., Torres-Nunez D. A., Otero-Cruz J. D., Lasalde-Dominicci J. A. (2012) Tryptophan scanning mutagenesis reveals distortions in the helical structure of the θM4 transmembrane domain of the Torpedo californica nicotinic acetylcholine receptor. Channels (Austin) 6, 111–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Diaz-De Leon R., Otero-Cruz J. D., Torres-Nunez D. A., Casiano A., Lasalde-Dominicci J. A. (2008) Tryptophan scanning of the acetylcholine receptor's βM4 transmembrane domain: decoding allosteric linkage at the lipid-protein interface with ion-channel gating. Channels (Austin) 2, 439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ortiz-Acevedo A., Melendez M., Asseo A. M., Biaggi N., Rojas L. V., Lasalde-Dominicci J. A. (2004) Tryptophan scanning mutagenesis of the γM4 transmembrane domain of the acetylcholine receptor from Torpedo californica. J. Biol. Chem. 279, 42250–42257 [DOI] [PubMed] [Google Scholar]
- 69. Tamamizu S., Guzmán G. R., Santiago J., Rojas L. V., McNamee M. G., Lasalde-Dominicci J. A. (2000) Functional effects of periodic tryptophan substitutions in the α M4 transmembrane domain of the Torpedo californica nicotinic acetylcholine receptor. Biochemistry 39, 4666–4673 [DOI] [PubMed] [Google Scholar]
- 70. Bouzat C., Gumilar F., del Carmen Esandi M., Sine S. M. (2002) Subunit-selective contribution to channel gating of the M4 domain of the nicotinic receptor. Biophys. J. 82, 1920–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]