

Background: Switch Regions of G protein Gα subunits activate downstream cell signaling events.
Results: Gα13 Switch Region 2 forms a calcium-dependent bi-molecular complex with the head domain of talin.
Conclusion: Binding of the Gα13 to the talin head domain promotes αIIbβ3 integrin activation.
Significance: These results provide a new paradigm for inside-out signaling and αIIbβ3 integrin activation in platelets.
Keywords: G protein-coupled receptor (GPCR), integrin, signal transduction, talin, thrombosis
Abstract
Even though GPCR signaling in human platelets is directly involved in hemostasis and thrombus formation, the sequence of events by which G protein activation leads to αIIbβ3 integrin activation (inside-out signaling) is not clearly defined. We previously demonstrated that a conformationally sensitive domain of one G protein, i.e. Gα13 switch region 1 (Gα13SR1), can directly participate in the platelet inside-out signaling process. Interestingly however, the dependence on Gα13SR1 signaling was limited to PAR1 receptors, and did not involve signaling through other important platelet GPCRs. Based on the limited scope of this involvement, and the known importance of G13 in hemostasis and thrombosis, the present study examined whether signaling through another switch region of G13, i.e. Gα13 switch region 2 (Gα13SR2) may represent a more global mechanism of platelet activation. Using multiple experimental approaches, our results demonstrate that Gα13SR2 forms a bi-molecular complex with the head domain of talin and thereby promotes β3 integrin activation. Moreover, additional studies provided evidence that Gα13SR2 is not constitutively associated with talin in unactivated platelets, but becomes bound to talin in response to elevated intraplatelet calcium levels. Collectively, these findings provide evidence for a novel paradigm of inside-out signaling in platelets, whereby β3 integrin activation involves the direct binding of the talin head domain to the switch region 2 sequence of the Gα13 subunit.
Introduction
Cells possess multiple G protein signaling pathways that contribute to the different cellular responses involved in development, function, survival, and disease (1, 2). Consequently, G protein signaling remains an area of intense investigation. It is well established that classic heterotrimeric G protein signaling first requires ligand binding to a G-protein-coupled receptor. The G-protein subunits (Gα, Gβ, and Gγ) then become activated and interact with downstream effectors to initiate signal transduction events (3, 4). In this regard, the conformationally sensitive switch regions 1 and 2 of the Gα subunit are known to be directly involved in G protein activation and participate in its downstream signaling mechanisms (5–7). G13 is one such G protein that is ubiquitously expressed and abundant in all cell types. It is known to be essential for numerous in vivo processes including embryogenesis, angiogenesis, chemokinesis, hemostasis, and thrombosis (2–4). In this connection, we previously demonstrated that human platelet shape change, aggregation, and secretion can be dependent on Gα13 switch region 1 (Gα13SR1)3 signaling (8). However, these studies also provided evidence that the critical importance of this Gα13SR1 signaling pathway is limited to PAR1-mediated platelet activation. Based on this consideration, the present study examined whether a separate Gα13 switch region signaling mechanism, i.e. Gα13SR2 may explain the global importance of G13 for in vivo platelet function. Using peptide affinity chromatography of native platelet proteins and immunoaffinity purification of native platelet Gα13-protein interactions, our results demonstrate that the amino acid sequence of Gα13SR2 (but not Gα13SR1) directly binds to the talin-αIIbβ3 integrin-kindlin-3 complex in human platelets. Furthermore, dissociation of this complex revealed that the binding partner for Gα13SR2 is the head domain (also designated as FERM domain) of talin (and not αIIb, β3 integrin or kindlin-3. Importantly, this Gα13SR2-talin binding interaction was promoted by increased intraplatelet calcium levels and prevented by calcium chelation. The ability of talin to form a specific complex with Gα13SR2 was further confirmed by bi-molecular binding measurements using recombinant talin head domain, Gα13SR2 peptides and GST-G13SR2 fusion proteins. Lastly, studies measuring fibronectin adhesion of NIH3T3 fibroblasts suggest that the binding interaction between talin and Gα13SR2 is not limited to platelet signaling, but may represent a more universal mechanism of integrin activation.
Experimental Procedures
Reagents
Human platelet concentrates (PRP) were purchased from Life Source Blood Services (Glenview, IL). The G13SR2pep (Myr-VGGQRSERKRWFECFDS), the G13SR2227 mutant pep (Myr-VGGQASERK RWFECFDS), the G13SR2232 mutant pep (Myr-VGGQRSERKAWFECFDS), the G13SR2random pep (Myr-GFDEWEVSFKGCQRRSR), the G13SR1pep (Myr-LLARRPTAGIHEY), the G13SR1random pep (Myr-LIRPTLHRATLEG), the TRAP1-peptide (SFLLR NPNDKYEPF), the TRAP4-peptide (AYPGKF) and all biotinylated peptide derivatives were synthesized and HPLC purified (>95% pure) by the Research Resource Center, University of Illinois, Chicago. Reagents were from the following sources: ADP and dimethyl-BAPTA-AM (Invitrogen); U46619 (Cayman Chemical); polyclonal rabbit anti-kindlin-3 and the monoclonal anti-αIIb, anti-β3, whole talin antibodies, and fibronectin (Abcam); HRP-conjugated goat anti-rabbit antibody (Cell Signaling); BCA protein assay kit and nitrocellulose membranes (Bio-Rad), Pierce Supersignal kit, TMB and ECL chemiluminescent substrates (Pierce Biochemicals); Streptavidin-HRP (Life Technologies); nitrocellulose blotting membranes, pGEX6p2, and glutathione-Sepharose 4B resin (GE Life Sciences); IPTG and nickel metal affinity chromatography (GoldBio); Src ELISA activation assay kit (Millipore); RhoA G-LISATM activation assay kit and cell lysis buffer (Cytoskeleton); SulfoLink immoblization kit for peptides and the FITC-PAC1 antibody (Thermo Fisher Scientific); PAC1 monoclonal antibody (Biolegend); protein A-Sepharose beads (Sigma-Aldrich); trypsin EDTA (Corning); Rap1 antibody (Bethyl Laboratories); Gα13, His-probe, and GST antibody (Santa Cruz Biotechnology); GFP-C1 plasmid (Clontech Laboratories, Inc); Immulon 2 Removawells (Dynatech Laboratories, Inc). The TA205 antibody was a generous gift from Dr. Stephen Lam (University of Illinois). GFP tagged Full length Talin plasmid was a generous gift from Dr. Jun Qin (Cleveland Clinic, Lerner Research Institute). All reagents used were of analytical grade.
Human Platelet Functional Studies
The platelet count in the freshly drawn PRP was adjusted to 3 × 108 platelets/ml with calcium-free Tyrode's buffer (pH 7.4). Platelets were pre-incubated with peptides or vehicle for 1 min prior to incubation with U46619, TRAP1, TRAP4, ADP, or A23187. Aggregation was measured using the turbidimetric method (9), with a model 400 Chrono-Log aggregometer.
Solubilized Platelet Membrane Preparation
Solubilized platelet membranes were prepared as previously described (10). Briefly, platelets were sonicated and the membranes were sedimented by ultracentrifugation (100,000 × g) for 45 min at 4 °C. The membrane pellet was then solubilized in cold buffer (25 mm Tris-HCl, 5 mm MgCl2, pH 7.4, plus 10 mm CHAPS and 5 mm DTT).
Ca2+ Mobilization Assay
Human PRP was diluted to 2.5 × 106 platelets/ml in Tyrode's buffer containing 0.1% BSA. Cells were loaded for 1 h at 37 °C with FLIPR calcium-sensitive dye, according to the manufacturer's protocol. The samples were measured using a FlexStation plate reader (Molecular Devices). Cells were excited at 485 nm, and Ca2+ fluorescence was detected at an emission wavelength of 525 nm (11).
BAPTA Loading of Platelets
Platelets in PRP were loaded with diethel-BAPTA-AM (15 μm) for 30 min at 37 °C.
RhoA G-LISA Activation
RhoA activation was measured with a G-LISA™ assay. The platelets were treated with agonists alone or were pre-incubated with 250 μm peptide prior to stimulation with the agonists. After 5 min, platelet lysates were prepared, and the bound active Rho-family protein was detected at 490 nm.
Src ELISA Activation
Src activation was measured with a Src ELISA activation assay. The platelets were treated with agonists alone or were pre-incubated with 250 μm of peptide prior to stimulation with the agonists. After 15 min, platelet lysates were prepared, and the bound phosphorylated Src was detected at 450 nm.
SulfoLink Peptide Affinity Columns
All the peptides used for affinity chromatography were synthesized with a N-terminal cystine and coupled to the SulfoLink agarose beads according to the manufacturer's protocol.
Antibody Affinity Columns
Antibody against Gα13 was raised in rabbits immunized with N-terminal peptide (40–48 amino acid sequence) of Gα13 (12). This antibody was used to generate an immunoaffinity protein-A Sepharose column to purify Gα13 and its associated proteins from solubilized human platelets. The specific column-bound proteins were eluted using a 2.5 pH glycine buffer into neutralization buffer (pH 8.0).
Western Blotting
Solubilized platelet membranes or eluates from the peptide affinity columns were normalized for protein concentration using the BCA protein assay. Samples containing 25–50 μg of protein and molecular weight markers were resolved on 10% SDS/polyacrylamide gels. Primary antibodies (13–15) were used at the following dilutions: anti-talin-monoclonal TA205 (13) (1:1,000), anti-α2b-abcam11027 (14) (1:250), anti-β3-abcam7167 (14) (1:250) and anti-kindlin-3-abcam77050 (14) (1:200), p115RhoGEF (15) (1:500). The blots were washed and incubated with the appropriate HRP-conjugated secondary antibodies (1:2,000) for 1 h at room temperature. Bands were visualized using the Pierce Supersignal kit.
Flow Cytometry Analysis of Platelets using FITC-labeled PAC1 Antibody
The platelet count in human PRP was adjusted to 1 × 106 platelets/ml using Tyrode's buffer (pH 7.4). The platelets were stimulated with 0.5 μm U46619 or 25 μm TRAP1 for 5 min and then labeled for 15 min at room temperature with FITC-conjugated PAC1 antibody against activated αllbβ3. The samples were then analyzed by flow cytometry in a Beckman Coulter CyAn II Flow Cytometer.
Recombinant Proteins
Talin head domain (TalinH, aa1–400) in pET15b (Novagen) plasmid was a generous gift from Dr. Ed Plow (Cleveland Clinic, Lerner Research Institute). TalinH and β3 integrin cytoplasmic domain (aa715–761) were cloned into pGEX6p2. Full Length p115RhoGEF, a generous gift from Dr. Tohru Kozasa (University of Tokyo), was cloned into pET15b expression plasmid. G13SR2 and G13SR2random were cloned into pGEX6p2 using overlapping primer PCR using the following sequences: G13SR2 Fwd 5′-CGGGGATCCGTAGGTGGTCAGCGTTCGGAACGCAAGCGTTGGTT-3′; G13SR2 Rev 5′-CGGGTCGACTCATGAGTCGAAGCATTCGAACCAACGCTTGCGTTCCG-3′; G13SR2random Fwd 5′-CGGGGATCCGGTTGCCGTAAGGAAGTCTTTAGCGATCGCCAGTG-3′; and G13SR2random Rev 5′-CGGGTCGACTCACTCGCGGCTGCCGAACCACTGGCGATCGCTAAAGA-3′. RALGDS plasmid was a generous gift from Cheryl Arrowsmith (Addgene plasmid # 25331). Each plasmid was transformed into BL21-DE3 strain Escherichia coli and induced with 0.2 mm IPTG at an optimal density of 0.6–1.0 for 3 h at 37 °C. The pET15b constructs were purified using nickel metal affinity chromatography while pGEX6p2 constructs were purified using Glutathione Sepharose 4B resin according to manufacturer's specifications. Eluted proteins were dialyzed overnight in a PBS (pH 7.4) and 5% glycerol solution.
Dot Blot Assay
Recombinant proteins were pipetted directly onto nitrocellulose blotting membranes, rinsed briefly in PBS (pH 7.4) and 0.1% Tween 20 (PBST), and incubated with 5% Milk-PBST for 1 h at room temperature. The membrane was rinsed briefly and incubated with 5% BSA-PBST with 0.5 μm biotinylated peptide for 1 h at room temperature. Subsequently, each blot was incubated with 5% BSA-PBST with streptavidin-HRP for 30 min at room temperature. Each blot was developed using ECL chemiluminescent substrates.
GST Pull-down Assay
Recombinant GST-G13SR2 and GST-G13SR2random proteins (10 μg each) were incubated with glutathione-Sepharose 4B resin in Binding Buffer (50 mm Tris-HCl, pH 8, 150 mm NaCl, 2 mm EDTA, 1 mm MgCl2, 0.1% Nonidet P-40, 5% BSA) for 1 h at room temperature. Beads were washed in the binding buffer twice and incubated with 40 μg of His-TalinH for 1 h at room temperature, followed by two washes using the wash buffer (50 mm Tris-HCl, pH 8, 150 mm NaCl, 2 mm EDTA, 1 mm MgCl2, 0.1% Nonidet P-40). Samples were eluted off beads by boiling in 3× SDS sample buffer. Samples were resolved using 10% SDS-PAGE Gels, transferred to nitrocellulose membranes, and probed with an anti-talin head domain antibody.
Construction of Mouse Gα13 Plasmids
A 2.8 kb cDNA encoding wild-type mouse Gα13 (GeneBankTM ID 057665) subcloned into the pCMV-SPORT6 vector (clone ID 4918717; Open Biosystems). Three new plasmids were then created based on this vector by selectively mutating either the Gα13SR2 Arg227 to Ala or the Gα13SR2 Arg232 to Ala using the Stratagene II site-directed mutagenesis kit (Agilent Technologies) following the manufacturer's instructions. The primers used for these point mutation reactions (R227A, 5′-GTTGATGTAGGTGGCCAGGCATCAGAACGGAAACGCTG-3′ and R232A, 5′-GCCAGAGATCAGAACGGAAAGCCTGGTTTGAATGCTTTGACA-3′ were made by Integrated DNA Technologies. All plasmids used in experiments were amplified from α5-competent E. coli cultures (NEB) and then purified using endotoxin-free maxiprep kits (Qiagen) and sequenced by the DNA Services Facility at the UIC Research Resources Center.
Cell Culture and Plasmid Transfection
Mouse embryonic fibroblasts (NIH3T3) were purchased from ATCC and grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% calf bovine serum (CBS), 100 units/ml of penicillin and 0.1 mg/ml streptomycin, under 10% CO2 at 37 °C. When the cells reached 30–40% confluence, the plasmid transient transfection was carried out using Fugene HD (Promega) according to manufacturer's instructions. Forty-eight hours after the transfection, the cells were either trypsinized with 0.25% Trypsin EDTA for the cell adhesion assay or harvested for Western blot using cell lysis buffer).
Cell Adhesion Assay
Non-treated, Corning® 96 Well Black Flat Bottom Polystyrene Microplates were coated with 200 μl of mouse fibronectin (10 μg/ml), at 37 °C for 1 h. The plates were rinsed with 200 μl of PBS twice, and the nonspecific binding sites were blocked with 200 μl heat-treated 1% BSA/PBS solution 37 °C for 1 h. Cultured NIH3T3 cells were harvested with 0.25% trypsin EDTA, and the cell suspension was centrifuged (80 × g) for 2 min. The pelleted cells were resuspended in colorless DMEM (Mediatech, Inc) at a concentration of 5 × 106 cells/ml. 5 μl/ml of calcein AM (Life Technologies) was then added, and the cell suspension was incubated at 37 °C for 30 min. The cells were then separated from the suspension medium by centrifugation (80 × g) for 2 min and resuspended (5 × 106 cells/ml) in adhesion buffer (Hank's balanced salt solution plus 1% BSA). Following resuspension, 100 μl of the cells were added to each well, and the plates were incubated for 2 h at 37 °C. Finally, the wells were washed 4× with PBS buffer read for fluorescence at 517 nm using a fluorescein filter.
Quantification of Western Blots/Dot Blots and Statistical Analysis
NIH software, ImageJ, was used for densitometric analysis of blots. Unless otherwise specified, significance was determined between samples using unpaired Student's t test (p < 0.05). All results are representative of data obtained using at least three separate experiments/and or 3 separate units of human PRP.
Rap1 Activity Assay
Rap1 activity was determined as previously described (16). Human PRP was adjusted to 2 × 108 platelets/ml in Tyrode's buffer. Platelets were pre-treated with either 150 μm Myr-G13SR2pep, 150 μm Myr-G13SR2Random pep, DMSO, or 10 ng/ml PGI2 for 2 min prior to stimulation with 1.0 μm A23187 for 5 min on ice with gentle agitation. Platelets were immediately lysed using 2× RIPA buffer with Complete Protease Inhibitor Mixture (Roche) for 15 min with gentle agitation. Clarified lysate was obtained through centrifugation at 14,000 × g at 4 °C for 10 min. The supernatant was incubated with 5 μg of RalGDS bound to nickel bead resin (GoldBio) for 90 min at 4 °C with gentle agitation. Resin was washed with 1× RIPA buffer and eluted by boiling in SDS loading buffer. Bound Rap1 was detected via Western blotting using anti-Rap1 antibody.
TalinH ELISA
Recombinant 6x-His tagged Talin head domain (talinH) was added to Immulon2 Removawells (Dynatech Laboratories, Inc.) in 50 mm sodium carbonate/bicarbonate buffer (pH 9.4) and incubated overnight at 4 °C to generate a standard curve. Samples were blocked with 5% BSA-PBST for 1 h at room temperature, washed thoroughly, and incubated with an anti-His antibody (Santa Cruz Biotechnology). TalinH concentration was determined using TMB substrate at 450 nm. Subsequently, GST-G13SR2, GST- G13SR2Random pep, and GST were added to Immulon 2 wells at concentrations ranging from 1.0 μg/ml to 1.0 ng/ml and probed with an anti-GST antibody (Santa Cruz Biotechnology). Equal loading concentrations were normalized by absorbance values. To determine talinH binding, GST fusion proteins were added to Immulon2 wells in equal concentrations overnight at 4 °C. Each well was blocked in 5% BSA-PBST, incubated with talinH, and washed thoroughly. TalinH binding was measured by probing with an anti-His antibody and developed using TMB substrate at 450 nm. Each value was converted to picogram of talinH using the linear range of the standard curve. Each analysis was performed in triplicate and statistical significance was determined using unpaired Student's t test (p < 0.05).
PAC1 Integrin Activation Assay
CHO cells stably expressing αIIbβ3 integrins (CHOA5 cells) were a generous gift from Dr. Mark Ginsberg. CHOA5 cells were transfected with either Gα13 wild type (WT), or Gα13 R227A or R232A mutants. After 24 h integrin activation was determined using a PAC1 monoclonal antibody (Biolegend) and an Alexa555 conjugated secondary antibody (Life Technologies). Cells were fixed in 0.5% PFA for 15 min at room temperature and washed thoroughly in PBS. Cells were blocked and permeabilized for 30 min on ice using a PBS buffer containing 4% BSA and 0.1% Saponin. After thorough washing in PBS, Gα13 was probed using an anti-Gα13 antibody (Santa Cruz) and an Alexa 405 conjugated secondary antibody (Life Technologies). Cells were analyzed using a BD LSRii flow cytometer and PAC1 activation was measured for all cells and Gα13-positive cells. To evaluate the effect of Talin and Gα13 together, CHOA5 cells were co transfected with Gα13 WT, R227A, or R232A and either GFP (Control) or GFP Talin (Experimental). Integrin activation was determined by gating for Gα13-, GFP-, and PAC1-positive cells. The overall change in percent integrin activation was determined as follows: ΔActivation = (% Experimental/% Control). Each analysis was performed in triplicate and statistical significance was determined using unpaired Student's t test (p < 0.05).
Results
Agonist Activation of αIIbβ3 Is Dependent on Gα13 Switch Region 2 Signaling
To investigate Gα13SR2 signaling in human platelets, a myristolyated peptide comprising the complete amino acid sequence of Gα13SR2 was synthesized. This peptide was employed as both a competitive inhibitor of Gα13SR2 signaling in intact platelets and as an immobilized ligand to enable the purification of Gα13SR2-associated proteins. In the intact platelet studies, it was necessary to demonstrate that the myristolyated Gα13SR2 peptide gained access to the platelet cytosolic compartment. To this end, human PRP was treated with myristoylated G13SR2pep, non-myristoylated G13SR2pep, or Vehicle (DMSO). After incubation, the PRP samples were centrifuged and the supernatant was removed to obtain the unbound fraction as shown in Fig. 1a (left panel). The pellet was then lysed and centrifuged to separate the membrane and cytosolic fractions as shown in the middle and right panel of Fig. 1a, respectively. Western blotting data clearly show that the non-myristoylated peptide is primarily located in the unbound fraction, while the myristoylated peptide is primarily taken up into the cytosolic fraction. These results demonstrate that myristolyated G13SR2pep permeates the platelet plasma membrane and has access to intraplatelet signaling processes. The next series of experiments examined whether this Gα13SR2 peptide (G13SR2pep) interfered with human platelet aggregation in PRP. It was found (Fig. 1b) that G13SR2pep blocked platelet aggregation (trace c) stimulated by all the agonists examined, including U46619, TRAP1, ADP, TRAP4, and A23187. Clearly, this inhibitory profile is quite different from that associated with a peptide representing Gα13SR1 (G13SR1pep), which only blocked aggregation mediated by TRAP1 (trace b). Furthermore, a single change in the amino acid sequence of G13SR2pep at amino acid 227 (from Arg to Ala; G13SR2227 mutant pep) significantly reduced its inhibitory effects (trace d), demonstrating the high degree of specificity of G13SR2pep. To investigate the mechanism by which G13SR2pep blocks platelet aggregation, we first examined its role in primary platelet aggregation stimulated by ADP (3 μm) in human PRP. Specifically, aspirin (ASA) treatment was used to block the secondary aggregation process that derives from the secretion of platelet dense granules. The results (aggregation upper panel and secretion lower panel) are presented in Fig. 1c. Trace e and bar e illustrate the ADP-mediated aggregation and secretion response in PRP pretreated with 75 μm G13SR2Random pep alone. It can be seen that pretreatment of the PRP with 1.0 mm ASA resulted in partial inhibition of this aggregation (upper panel, trace f) and a complete inhibition of dense granule secretion (lower panel, bar f), relative to no ASA treatment (trace e and bar e). Thus, in the presence of 1.0 mm ASA, the residual aggregation (trace f) represents the primary aggregation response. Furthermore, the results illustrate that G13SR2pep (75 μm) caused almost complete inhibition of the residual, ADP-induced, primary aggregation response (upper panel, trace g), even though it only produced partial inhibition of secretion (lower panel, bar g). Finally, when platelets were treated with both 1.0 mm ASA and 75 μm G13SR2pep both aggregation (trace h) and secretion (bar h) were completely blocked. Similar results were obtained using A23187 under the same experimental conditions (data not shown). These data demonstrate that G13SR2pep peptide is capable of blocking primary human platelet aggregation.
FIGURE 1.

Effects of G13SR1pep and G13SR2pep on human platelet function. a, myristoylated or non-myristoylated peptide were incubated with platelet PRP, which was then separated into unbound, membrane bound, or cytosolic fractions. An antibody raised against G13SR2 sequence was used for immunoblotting. ImageJ densiometric analysis indicates that roughly 5% of the myristoylated peptide remains unbound in the supernatant and that 95% partitions to the cytosolic fraction. b, traces denoted as a are representative of PRP pretreated with either 500 μm G13SR1random pep or G13SR2random pep. Unless otherwise specified in the figure; traces b, c, and d are representative of PRP pre-treated with 500 μm G13SR1pep, 150 μm G13SR2pep, or 150 μm G13SR2mutant pep, respectively. From left to right, the agonists used to induce platelet aggregation are U46619, TRAP1, ADP, TRAP4, and A23187. c, shows the effects of G13SR2pep and aspirin (ASA) on ADP-induced (3 μm) platelet aggregation (top panel) and secretion (bottom panel) in human PRP. Traces and bars e–h represent PRP preincubated with 75 μm G13SR2random pep, 1.0 mm ASA, 75 μm G13SR2pep, and 75 μm G13SR2pep plus 1.0 mm ASA, respectively. d, illustrates the effects of 500 μm G13SR1pep and 500 μm G13SR2pep on calcium, RhoA, and Src activation induced by 15 μm TRAP1 or 0.5 μm U46619. The agonist-mediated calcium, RhoA and Src responses in the absence of peptide have been standardized to 100%. e shows the activation state of Rap1 when treated with 1.0 μm A23187, vehicle (DMSO), or 1.0 μm A23187 with either 10 ng/ml PGI2, 150 μm G13SR2pep, or 150 μm G13SR2random pep. The top panel depicts Rap1 (GTP) pulled down by recombinant RalGDS from human platelet PRP, while the lower panel depicts total Rap1 present in the platelet lysate, each probed with an anti-Rap1 antibody. f and g show the effects of G13SR2pep on human platelet αllbβ3 integrin activation as measured by FITC-PAC-1 antibody labeling. Panel f trace 1 depicts resting platelets. Trace 2 represents platelets stimulated with 15 μm TRAP1. Trace 3 represents platelets pretreated with 500 μm G13SR2random pep, followed by 15 μm TRAP1 stimulation. Trace 4 represents platelets pretreated with 150 μm of G13SR2pep, followed by 15 μm TRAP1 stimulation. In panel g, trace 1 depicts resting platelets. Trace 2 represents platelets stimulated with 0.5 μm U46619. Trace 3 represents platelets pretreated with 500 μm G13SR2random pep, followed by 0.5 μm U46619 stimulation. Trace 4 represents platelets pretreated with 150 μm of G13SR2pep, followed by 0.5 μm U46619 stimulation. All results are representative of data obtained using at least three separate units of human PRP, and representative data were chosen for each figure.
The next experiments investigated the fundamental differences between G13SR1 and G13SR2 signaling pathways by comparing the abilities of G13SR1pep and G13SR2pep to modulate downstream platelet effectors. It was found that G13SR1pep substantially blocked RhoA, calcium, and Src activation induced by TRAP1 but not by U46619. Surprisingly however, G13SR2pep did not block these signaling pathways induced by either TRAP1 or U46619 (Fig. 1d). These findings indicate that G13SR1 and G13SR2 have completely different signaling profiles in human platelets, and that unlike G13SR1 signaling, the signaling events mediated by Gα13SR2 lie further downstream from RhoA, calcium mobilization, or Src.
Based on these findings, experiments were undertaken to investigate whether G13SR2pep interferes with a separate and well-known mediator of platelet aggregation, i.e. the Rap1/RIAM signaling pathway. To examine this possibility, human PRP was activated with 1.0 μm A23187 to mobilize calcium, and treated with either PGI2 to inhibit Rap1 activation, or with G13SR2pep or G13SR2random pep. After treatment, the platelets were lysed and incubated with RalGDS to harvest active GTP-bound form of Rap1 that was in turn detected using immunoblotting with a Rap1 antibody (Fig. 1, panel e) The lower panel of blots show total Rap1 in PRP, while the upper panel of blots show the active form of Rap1 upon each treatment (Fig. 1). It is clear that A23187 increases the amount of active Rap1 as compared with platelets treated with vehicle (DMSO), while PGI2 treatment abrogates Rap1 activation in agreement with previous findings (16). Finally, when pre-treated with G13SR2pep or G13SR2random pep, the activity of Rap1 remained unchanged. Together, these data indicate that G13SR2pep acts downstream of the calcium mobilization and Rap1 pathways.
On this basis, we next examined the possibility that Gα13SR2 modulates a later step in the platelet activation process, i.e. αIIbβ3 integrin activation (17). These experiments employed FITC-PAC1 antibody and flow cytometry analysis to measure agonist-mediated activation of platelet αIIbβ3 (18). It was found that G13SR2pep did indeed inhibit αIIbβ3 activation in response to both TRAP1 (Fig. 1f) and U46619 (Fig. 1g). Thus, while Gα13SR1 signaling induces calcium mobilization, RhoA activation and Src activation specifically through PAR1 (19) receptor stimulation, Gα13SR2 signaling is recruited by multiple platelet receptors and acts at a further downstream target involving αIIbβ3 activation.
Gα13 Switch Region 2 Interacts with the Talin-αllbβ3-kindlin-3 Complex by Binding to the Talin Head Domain
To identify the mechanism by which Gα13SR2 modulates αllbβ3 function, we performed experiments to determine whether the amino acid sequence of Gα13SR2 interacts with αllbβ3 integrin or an αllbβ3-associated protein such as talin (20). To this end, peptide-affinity chromatography was employed. Briefly, G13SR2pep was coupled to a SulfoLink agarose column to purify Gα13SR2-associated proteins from solubilized human platelets. Western blot analysis of the affinity eluates from the G13SR2pep column demonstrated the purification of intact talin, αllbβ3, and kindlin-3 (Fig. 2a). The specificity of this affinity purification was demonstrated by the finding that an affinity column using G13SR2random pep did not purify any of these platelet proteins (Fig. 2a). These findings provide evidence that the amino acid sequence comprising Gα13SR2 directly binds to a protein complex (13, 21–26) containing talin, αllbβ3, and kindlin-3. This notion was further confirmed by measuring the association of endogenous platelet Gα13 with this protein complex. Specifically, an antibody generated against the N-terminal peptide sequence of Gα13 (12) was coupled to an immunoaffinity column (Gα13-Ab) and used to purify Gα13-associated proteins from solubilized platelets. Western blot analysis of the eluates from the Gα13-Ab immunoaffinity column again demonstrated the purification of talin, αllbβ3, and kindlin-3 (Fig. 2b), whereas an immunoaffinity column using the pre-immune IgG did not result in the purification of any of these proteins (Fig. 2b).
FIGURE 2.

Binding of G13SR2pep or native platelet Gα13 protein to talin in solubilized human platelet membranes. Because the preparation of platelet membranes involves physical manipulation, the results from these studies reflect partially activated platelets. In panel a the solubilized platelet membrane eluates from the G13SR2pep or the G13SR2random pep affinity columns were immunoblotted for whole talin (220 kDa) αllbβ3 (130 kDa, 115 kDa) and kindlin-3 (72 kDa). In panel b the solubilized platelet membrane eluates from the Gα13-Ab immunoaffinity column or the pre-immune IgG column were immunoblotted for Gα13 (43 kDa), talin, αllbβ3, and kindlin-3. In panels c and d the components of the talin-αllbβ3-kindlin-3 complex were dissociated by heating the solubilized platelet membrane samples at 100 °C for 45 min prior to affinity chromatography. The resulting eluates from the G13SR2pep (or G13SR2random pep) columns (panel c) and the Gα13Ab immunoaffinity (or control pre-immune IgG) columns (panel d) were immunoblotted for talin, αllb, β3, and kindlin-3. In panel e the solubilized platelet membrane eluates from the Gα13-Ab immunoaffinity column or the control pre-immune IgG column were immunoblotted for the talin head domain. In panel f competition binding was performed by incubating solubilized platelet membranes with 250 μm of G13SR2pep (or G13SR2random pep) as the competing agent prior to Gα13-Ab immunoaffinity column chromatography. In panel g the solubilized platelet membrane eluates from an affinity columns using G13SR2pep or a mutant peptide with a single amino acid substitution (R227A; G13SR2227 mutant pep) were immunoblotted for the talin head domain. In panel h the solubilized platelet membrane eluate from a peptide affinity columns using G13SR1pep was immunoblotted for whole talin, αllb, β3, kindlin-3, and p115-RhoGEF. In cases where the gel was probed with more than one antibody, the panel represents a composite of separate blots where each lane was run using its own molecular weight standards. All results are representative of data obtained using at least three separate units of human PRP, and representative data were chosen for each panel.
The next series of experiments were designed to identify the binding partner of Gα13 within the protein complex. Our data demonstrate that Gα13 binds to talin, but not to αllb, β3 integrin or kindlin-3. Specifically, when the talin-αllbβ3-kindlin-3 complex was dissociated by heat treatment of solubilized platelets, both the G13SR2pep affinity column and the Gα13-Ab immunoaffinity column purified only talin and not αllb, β3 integrin, or kindlin-3 (Fig. 2, c and d). In addition, since the talin 8D4 antibody is known to detect both whole talin as well as the talin rod domain (21), the absence of the talin rod domain (Fig. 2, a and b) suggests that the head domain of talin serves as the binding region for Gα13. This finding was confirmed by blotting the Gα13-Ab immunoaffinity column eluate with the TA205 antibody that specifically recognizes the talin head domain (13) (Fig. 2e). Evidence that the binding of Gα13 to the talin head domain is mediated through the Gα13SR2 sequence was provided by binding competition studies. In these experiments, solubilized platelet membranes were pre-incubated with G13SR2pep before protein purification by the Gα13-Ab immunoaffinity column. The results demonstrated that competition with G13SR2pep inhibited native Gα13 binding to the talin head domain (Fig. 2f). Taken together, these findings suggest that Gα13 specifically binds to the talin head domain through the G13SR2 sequence, and does not directly associate with either αllb, β3 integrin, or kindlin-3. Furthermore, the aggregation data shown in Fig. 1a suggest that Arg-227 within the G13SR2 sequence may be important for talin binding. This notion was confirmed by the finding that an affinity column using the G13SR2227 mutant pep (R227A) was not effective in purifying the talin head domain (Fig. 2g).
Our previous results demonstrated that G13 switch region 1 (G13SR1) binds to and signals through p115-RhoGEF in human platelets. In order to examine whether G13SR1 also interacts with the talin-αllbβ3-kindlin-3 complex, we again employed peptide-affinity chromatography as described above. In this case, the column was prepared using G13SR1pep coupled to SulfoLink agarose beads. However, Western blot analysis of the affinity eluates from the G13SR1pep column demonstrated only the purification p115-RhoGEF and not the purification of talin, αllb, β3 integrin, or kindlin-3 (Fig. 2h).
Additional experiments provided independent confirmation of the direct binding interaction between Gα13SR2 and the talin head domain. To this end, we employed a bi-molecular in vitro binding reaction outside of the platelet milieu. Using a dot blot assay, it was found that recombinant talin head domain directly binds to G13SR2pep but not to the G13SR2random pep (Fig. 3, a and b). Moreover, the specificity of G13SR2pep-talin binding complex was demonstrated by using two different G13SR2 mutant peptides, each with a single R to A substitution at positions 227 or 232 (G13SR2227 mutant pep and G13SR2232 mutant pep). The results demonstrated (Fig. 3, a and b) that each mutant exhibited a significantly reduced binding affinity for talin head domain. To further demonstrate the specificity of this binding interaction between G13SR2pep and talin head domain, we utilized an alternate experimental approach. Specifically, recombinant GST-G13SR2 and GST-G13SR2random fusion proteins were generated and incubated with recombinant talin head domain to quantify their binary interactions. The results show that while GST-G13SR2 bound to talin, GST-G13SR2random did not (Fig. 3c). Taken together, these results add additional support to the notion that G13SR2 can form a bi-molecular complex with the talin head domain.
FIGURE 3.

Bi-molecular binding between G13SR2 and talin head domain (talinH). Panel a depicts a dot blot of talin (2 μg, 1 μg, 0.5 μg) probed with biotinylated G13SR2 peptides (G13SR2pep, G13SR2227 mutant pep, G13SR2232 mutant pep, and G13SR2random pep). Panel b depicts quantification of the bottom row of panel a. The symbol (*) represent significance (p < 0.05) relative to G13SR2pep binding to His-talinH. Panel c depicts a GST pull-down assay where His-talinH is bound to either GST-G13SR2 or GST-G13SR2random fusion proteins. GST-G13SR2 and GST-G13SR2random inputs are shown by Ponceau stain (bottom panel), His-talinH input is shown using Coomassie staining (middle panel), and bound His-talinH (top panel) is shown using an anti-talin antibody. Panel d depicts a dot blot where GST-talinH (top row), GST-β3 cytoplasmic tail (middle row), and GST (bottom row) are probed with G13SR1pep (first column), G13SR1random pep (second column), G13SR2pep (third column), or G13SR2random pep (fourth column) biotinylated peptides. Panel e depicts quantification of panel d. The symbol (*) represents significance (p < 0.05) between GST-talinH binding to G13SR2pep and G13SR2pep random. Panel f depicts a dot blot where recombinant His-p115-RhoGEF full length is probed with G13SR1pep or G13SR1random pep biotinylated peptides. To the left is a quantification of this dot blot. The symbol (*) represents significance (p < 0.05) between G13SR1pep and G13SR1random pep. Panel g depicts a dot blot where GST-β3 cytoplasmic tail and GST are overlaid with recombinant His-talinH, and probed using anti-talin antibody. The bar illustrates quantification of this dot blot. The symbol (*) represents significance (p < 0.05) between GST-β3 cytoplasmic tail and GST. Panel h depicts a talin standard curve ELISA. Panel i depicts the binding of talin to GST fusion proteins in an ELISA. Absorbance values (450 nm) were recorded for talin binding to GST-G13SR2, GST-G13SR2Random, and GST. Absorbance values were converted to picograms using the talin standard curve in panel h, and significance was established using the Student's unpaired t test. Each experiment was repeated three times (n = 3) and representative data were chosen for each figure.
Gα13 Switch Region 2 Directly Binds to Talin but Not to β3 Integrin
Our previous experiments (Fig. 2d) indicated that Gα13 is capable of forming a ternary complex with talin but not with β3 integrin. This notion was further investigated by additional dot blot binding assays measuring the ability of biotinylated G13SR2pep, (or G13SR2random pep) to bind with recombinant GST-talin head domain, GST-β3 cytoplasmic tail, or GST control. It was found that G13SR2pep bound only to GST-talin head domain, but not to the GST-β3 cytoplasmic tail (Fig. 3, d and e). Furthermore, the specificity of the binding interaction between Gα13SR2 and talin head domain was again demonstrated by the finding that biotinylated G13SR1pep bound to neither GST-talin head domain nor GST-β3 cytoplasmic tail (Fig. 3, d and e), but did effectively bind to recombinant full-length p115-RhoGEF (Fig. 3f). This preference of Gα13SR1 for binding to p115-RhoGEF is consistent with our findings in solubilized platelet membranes (Fig. 2h) and our previously published results (8). To demonstrate that the recombinant GST-β3 integrin cytoplasmic tail employed in the previous experiment (Fig. 3, d and e) was indeed functional, we tested the binding activity of GST-β3 integrin cytoplasmic tail with talin head domain (22). As expected, talin head domain significantly bound GST-β3 integrin (Fig. 3g) under the same experimental conditions as employed in Fig. 3, d and e. Finally, an ELISA was used to examine Gα13SR2 interaction with talin. Using a talinH standard curve (Fig. 3h), the picograms of talinH bound to GST, G13SR2pep, or GST- G13SR2random pep is illustrated in Fig. 3i. Collectively, the above findings provide strong evidence that Gα13SR2 mediates integrin activation by directly binding to the talin head domain and not to the β3 integrin cytoplasmic tail.
Intraplatelet Calcium Stimulates the Binding of Gα13 Switch Region 2 to Talin
We next examined a possible mechanistic basis for the binding interaction between Gα13SR2 and talin head domain. Since intraplatelet calcium is known to be a common mediator of β3 integrin activation, we first tested the effects of the calcium ionophore A23187 on Gα13-talin binding. Specifically, intact human platelets (PRP) were treated with 10 μm A23187, solubilized, and then subjected to Gα13-Ab immunoaffinity chromatography as described above. The results demonstrated (Fig. 4a) that elevation of intraplatelet calcium by A23187 caused a notable increase in Gα13-talin head domain binding. This observation was further confirmed by chelating available calcium with EGTA and BAPTA. Under these conditions, the binding of Gα13 to talin was completely abolished, even in the presence of A23187 (Fig. 4a). These results indicate that Gα13 is not constitutively associated with talin in truly un-activated platelets, but only becomes bound to talin in the presence of elevated calcium.
FIGURE 4.

Effect of Gα13 on integrin activation. Fig. 4 panel a shows that increased intraplatelet calcium levels promote native Gα13-talin binding. Platelets from untreated human PRP or A23187 (10 μm)-treated PRP were solubilized and subjected to Gα13-Ab immunoaffinity column chromatography. In separate experiments, the PRP was supplemented with EGTA (3 mm) and the platelets were loaded with BAPTA prior to A23187 (10 μm) stimulation. The platelets were then solubilized and subjected to Gα13-Ab immunoaffinity column chromatography. Gα13 blotting, shown in the left two lanes, was used as a loading control. All results are representative of data obtained using at least 3 separate units of human PRP, and representative data were chosen. In panel b cultured mouse embryonic fibroblasts (NIH3T3) were transfected with plasmids containing Gα13 wild-type, Gα13 R227A, or Gα13 R232A. Forty-eight hours after transfection, the cells were harvested for Western blot analysis of Gα13 or allowed to adhere to fibronectin-coated plates. The Western blots illustrated at the top of the figure illustrate comparable levels of Gα13 expression in each case. In panel c a Gα13 plasmid or vector alone was transfected into CHO cells stably expressing platelet αIIbβ3 integrins (CHOA5). Percent integrin activation was determined using a monoclonal PAC1 antibody. In panel d the Gα13 wild-type, Gα13 R227A, or Gα13 R232A plasmids were transfected into CHOA5 cells, gated for Gα13 expression and the percent of cells with active integrins was quantified. In panel e each Gα13 construct was co-transfected with either GFP or GFP tagged full-length talin and the change in integrin activation between Gα13 co-expression with GFP and GFP-talin. Significance (*) (p < 0.05) was determined by ANOVA using GraphPad PRISM 5.04 statistical software (San Diego, CA). All results are representative of at least three separate experiments (n = 3).
Gα13 Switch Region 2 Signaling Is Essential for Adhesion of NIH3T3 Cells
To determine whether Gα13SR2-talin signaling is limited to human platelets or whether it may serve as a more global mechanism of integrin activation, cell adhesion experiments were performed using fibroblasts. Specifically, we expressed both wild-type and mutant forms of the Gα13 protein in cultured NIH3T3 cells and measured the effects of these Gα13SR2 mutations on cell adhesion to fibronectin. The rationale for these experiments derives from our previous observations indicating that arginine 227 and arginine 232 contained within Gα13SR2 each play a critical role in Gα13SR2-talin binding and integrin activation. Thus, if Gα13SR2 signaling is in fact essential for the process of cell adhesion, overexpression of the mutant forms of Gα13 should interfere with this process, due to the loss of important arginine residues within the Gα13SR2 sequence. The results from these experiments are consistent with this notion. Specifically, Western blot analysis (Fig. 4b) of transfected NIH3T3 cells demonstrated that the expression levels of the wild-type Gα13 were comparable to the expression levels of both the Gα13 R227A and the Gα13 R232A mutant proteins. However, despite these equal expression levels, the Gα13SR2 mutant (R227A and R232A) cells were significantly less capable of adhering to fibronectin compared with the wild-type Gα13SR2 cells (Fig. 4b). Collectively, these findings suggest that in addition to human platelet inside-out signaling, Gα13SR2 may play a fundamental role in the process of integrin-mediated cell adhesion in other cell types.
To further examine the effect of Gα13 in integrin activation, we transfected wild type Gα13 and Gα13 SR2 mutants into CHO cells stably expressing αIIbβ3, (CHOA5 cells). After a 24 h transfection, each sample was gated for Gα13 expression and integrin activation using a PAC1 antibody. The results demonstrate that Gα13 increases the total percent of cells with active integrins when compared with vector alone (Fig. 4c). Similarly, the Gα13 or Gα13 SR2 mutants were transfected into CHOA5 cells (Fig. 4d). After gating for Gα13 expression and PAC1 activation, the data show that SR2 mutations significantly reduced the percent of cells with active integrins. To determine if Gα13 SR2 is mediating integrin activation through talin, we co-transfected the Gα13 constructs with either GFP or GFP tagged full-length talin. CHOA5 cells were gated for Gα13, GFP, or GFP-talin, and PAC1 signal, and the change in integrin activation between GFP and GFP-talin cells was calculated. The results demonstrate that WT Gα13 causes substantially greater integrin activation than either of the Gα13 SR2 mutants (Fig. 4e).
Discussion
In the present study, the underlying basis for platelet inside-out signaling has been further investigated by examining a direct mechanistic link between Gα13 signaling and αllbβ3 integrin activation. The results demonstrate evidence for such a link involving switch region 2 of the G13 α subunit. Our past (8) and present findings suggest that Gα13 actually plays two important and distinct roles in the platelet activation process, depending on whether signaling occurs through its Switch Region 1 or through its Switch Region 2 (see model in Fig. 5). In this regard, we found that signaling through Gα13SR1 is a classical GPCR-linked event that derives from agonist interaction with a G13-coupled receptor. The activated Gα13SR1 then causes stimulation of multiple downstream effectors, including RhoA, calcium, and Src, among others. The specificity of this signaling pathway is consistent with the notion that inhibition of Gα13SR1 signaling has very selective and limited effects on platelet function. Thus, Gα13SR1 inhibitors only block signaling by GPCRs that are primarily coupled to Gα13, i.e. PAR1 (Fig. 1b, trace b) (8), and do not block platelet aggregation in response to other platelet agonists, e.g. ADP, TRAP4, U46619, A23187, etc., because these agonists do not heavily rely upon G13 signal transduction (8). Our present and previous demonstration that Gα13SR1 signaling is specific for PAR1-mediated activation is inconsistent with recent work (27, 28) describing a binding interaction between Gα13SR1 and platelet β3 integrin, as a basis for αllbβ3 integrin activation. If this binding interaction were indeed the mechanism of Gα13-mediated integrin activation in platelets, one would expect inhibition of G13SR1 signaling to not only block platelet stimulation caused by PAR1, but platelet stimulation caused by all activating agents. However, this was not found to be the case (8). Moreover, the present studies have been unable to demonstrate the direct binding of either native platelet Gα13 or G13SR1pep to β3 integrin in a bi-molecular reaction. The basis for our inability to demonstrate this direct binding interaction is presently unclear.
FIGURE 5.
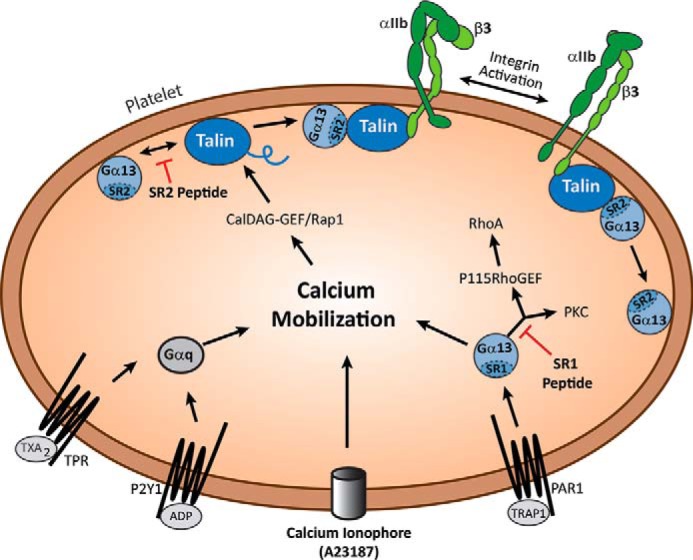
A model for two separate Gα13 Switch Region signaling pathways. G13SR1 is a classical receptor-coupled signal transduction pathway where agonist interaction with a surface membrane GPCR leads to G13SR1 activation and subsequent downstream signaling events, e.g. calcium mobilization, RhoA and Src activation, etc. In contrast, signaling through G13SR2 is not directly GPCR-dependent, but rather calcium-dependent. In this case, elevation of intraplatelet calcium promotes the recruitment of talin by the CalDAG-GEF/Rap1/RIAM pathway, binding of G13SR2 to whole talin, cleavage of the talin rod domain, binding of the Gα13-talin head complex to the cytoplasmic tail of β3 integrin, and dissociation of Gα13 from the talin head complex. Because of these significant differences in signaling mechanisms, selective inhibition of G13SR1 and G13SR2 signaling produces markedly different effects on human platelet function. Thus, inhibition of G13SR1 signaling only blocks PAR1-mediated platelet aggregation, whereas inhibition of G13SR2 signaling blocks aggregation in response to all platelet agonists.
On the other hand, it is apparent that the signaling mechanism of Gα13SR2 is quite different from that of Gα13SR1 in the sense that it is not directly coupled to GPCR activation. Rather, it appears to occur as a consequence of GPCR-mediated increases in intraplatelet calcium, and to be directly linked to talin activation of β3 integrin. Furthermore, since integrin activation serves as the final common pathway for platelet-platelet adhesion, inhibition of Gα13SR2 signaling produces global inhibition of platelet aggregation in response to all agonists (Fig. 1b, trace c).
Because of these two divergent signaling pathways, the sequence of events that ultimately leads to integrin activation may be quite different depending on the GPCR that is stimulated. Thus, for GPCRs that are extensively coupled to G13, such as PAR1, Gα13SR1, and Gα13SR2 signaling events are sequentially induced, where PAR1-mediated activation of Gα13SR1 leads to intraplatelet calcium mobilization (8). This increase in intraplatelet calcium levels then promotes multiple platelet signaling events (29–31) including binding of Gα13SR2 to whole talin and subsequent cleavage of the talin rod domain, presumably by calpain (32–34). Once formed, the Gα13SR2-talin head domain complex binds to the β3 cytoplasmic tail leading to integrin activation.
In contrast, other platelet receptors that do not extensively couple to G13 (e.g. P2Y1, TPR, PAR4, etc.) are capable of increasing intraplatelet calcium levels by separate signaling mechanisms, such as Gq-mediated PLC activation and consequent IP3 formation. Therefore, in such instances, elevated calcium is itself sufficient to initiate Gα13SR2-mediated integrin activation, and GPCR-mediated Gα13SR1 signaling is not required. Taken together, our findings therefore provide a new paradigm for inside-out platelet signaling by identifying Gα13SR2 as an essential link between membrane receptor stimulation, increased intracellular calcium levels, and β3 integrin activation.
Author Contributions
S. S. participated in writing the manuscript as well as in the design of experiments and analysis of the data in Fig. 1 (b, d, f, and g), Fig. 2 (a–h), Fig. 4a, and preparation of the model in Fig. 5. J. S. participated in writing the manuscript as well as in the design of experiments and analysis of the data in Fig. 1 (a and e), Fig. 3 (a–i), Fig. 4 (c–e), and preparation of the model in Fig. 5. X. Z. participated in writing the manuscript as well as in the design of experiments and analysis of the data in Fig. 4b. A. H. C. participated in writing the manuscript, as well as overseeing, coordinating, and conceiving the experiments performed at Tufts University. G. C. L. conceived the study, participated in writing the manuscript, as well as overseeing, coordinating and conceiving the experiments performed at the University of Illinois at Chicago.
Acknowledgments
We thank Dr. Jin-Sheng Huang for generating the random sequences for the switch region peptides, G13SR1pep and G13SR2pep. We also wish to thank Lan Lan Dong for technical assistance, Dr. Fozia Mir for assistance in preparation of affinity columns, and Dr. Jun Qin and Ashley Holly for their technical expertise in flow cytometry and CHO cell experiments. We thank Donna-Marie Mironchuk for help with the graphic design and preparation of figures.
This work was supported in part by National Institutes of Health Grant HL24530-29 (to G. C. L.), HL089517 (to A. H. C.), and grant-in-aid from the American Heart Association (to A. H. C.). The authors declare that they have no conflicts of interest with the contents of this article.
- SR
- switch region
- talinH
- talin head domain
- PRP
- platelet-rich plasma
- ASA
- aspirin.
References
- 1. Brass L. F., Manning D. R., Cichowski K., Abrams C. S. (1997) Signaling through G proteins in platelets: to the integrins and beyond. Thromb. Haemost. 78, 581–589 [PubMed] [Google Scholar]
- 2. Wettschureck N., Offermanns S. (2005) Mammalian G proteins and their cell type specific functions. Physiol. Rev. 85, 1159–1204 [DOI] [PubMed] [Google Scholar]
- 3. Offermanns S., Mancino V., Revel J. P., Simon M. I. (1997) Vascular System Defects and Impaired Cell Chemokinesis as a Result of Gα13 Deficiency. Science 275, 533–536 [DOI] [PubMed] [Google Scholar]
- 4. Moers A., Nieswandt B., Massberg S., Wettschureck N., Grüner S., Konrad I., Schulte V., Aktas B., Gratacap M-P., Simon M. I., Gawaz M., Offermanns S. (2003) G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat. Med. 9, 1418–1422 [DOI] [PubMed] [Google Scholar]
- 5. Milburn M. V., Tong L., deVos A. M., Brünger A., Yamaizumi Z., Nishimura S., Kim S. H. (1990) Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science 247, 939–945 [DOI] [PubMed] [Google Scholar]
- 6. Sonde J., Lambright D. G., Noel J. P., Hamm H.E., Sigler P.B. (1994) GTPase mechanism of G proteins from the 1.7-A crystal structure of transducin α-GDP-AIF-4. Nature 217, 503–516 [DOI] [PubMed] [Google Scholar]
- 7. Lambright D. G., Noel J. P., Hamm H. E., Sigler P. B. (1994) Structural determinants for activation of the α-subunit of a heterotrimeric G protein. Nature 369, 621–628 [DOI] [PubMed] [Google Scholar]
- 8. Huang J-S., Dong L., Kozasa T., Le Breton G. C. (2007) Signaling through G(α)13 switch region I is essential for protease-activated receptor 1-mediated human platelet shape change, aggregation, and secretion. J. Biol. Chem. 282, 10210–10222 [DOI] [PubMed] [Google Scholar]
- 9. Michal F., Born G. V. (1971) Effect of the rapid shape change of platelets on the transmission and scattering of light through plasma. Nat. New Biol. 24, 220–222 [DOI] [PubMed] [Google Scholar]
- 10. Kim S., Lim C. T., Lam S., Hall C-T., Komiotis D., Venton D. L., Le Breton G. C. (1992) Purification of the human blood platelet thromboxane A2/prostaglandin H2 receptor protein. Biochem. Pharmacol 43, 313–322 [DOI] [PubMed] [Google Scholar]
- 11. Southgate E. L., He R. L., Gao J.-L., Murphy P. M., Nanamori M., Ye R. D. (2008) Identification of formyl peptides from Listeria monocytogenes and Staphylococcus aureus as potent chemoattractants for mouse neutrophils. J. Immunol. 181, 1429–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Djellas Y., Manganello J. M., Antonakis K., Le Breton G. C. (1999) Identification of Gα13 as one of the G-proteins that couple to human platelet thromboxane A2 receptors. J. Biol. Chem. 274, 14325–14330 [DOI] [PubMed] [Google Scholar]
- 13. Patil S., Jedsadayanmata A., Wencel-Drake J. D., Wang W., Knezevic I., Lam S.C.-T. (1999) Identification of a Talin-binding Site in the Integrin β3 Subunit Distinct from the NPLY Regulatory Motif of Post-ligand Binding Functions: The talin N-terminal head domain interacts with the membrane-proximal region of the β3 cytoplasmic tail. J. Biol. Chem. 274, 28575–28583 [DOI] [PubMed] [Google Scholar]
- 14. Bai Y., Durbin H., Hogg N. (1984) Monoclonal antibodies specific for platelet glycoproteins react with human monocytes. Blood 64, 139–146 [PubMed] [Google Scholar]
- 15. Galvagni F., Pennacchini S., Salameh A., Rocchigiani M., Neri F., Orlandini M., Petraglia F., Gotta S., Sardone G. L., Matteucci G., Terstappen G. C., Oliviero S. (2010) Endothelial cell adhesion to the extracellular matrix induces c-Src-dependent VEGFR-3 phosphorylation without the activation of the receptor intrinsic kinase activity. Circ. Res. 106, 1839–1848 [DOI] [PubMed] [Google Scholar]
- 16. Franke B., Akkerman J., Bos J. L. (1997) Rapid Ca2+ mediated activation of Rap1 in human platelets. EMBO 16, 252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Phillips D. R., Fitzgerald L. A., Charo I. F., Parise L. (1987) The platelet membrane glycoprotein IIb/IIIa complex. Structure, function, and relationship to adhesive protein receptors in nucleated cells. Ann. N.Y. Acad. Sci. 509, 177–187 [DOI] [PubMed] [Google Scholar]
- 18. Shattil S. J., Cunningham M., Hoxie J. A. (1987) Detection of activated platelets in whole blood using activation-dependent monoclonal antibodies and flow cytometry. Blood 70, 307–315 [PubMed] [Google Scholar]
- 19. Kahn M. L., Nakanishi-Matsui M., Shapiro M. J., Ishihara H., Coughlin S. R. (1999) Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Invest. 103, 879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burridge K., Connell L. (1983) A new protein of adhesion plaques and ruffling membranes. J. Cell Biol. 97, 359–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Calderwood D. A., Zent R., Grant R., Rees D. J. G., Hynes R. O., Ginsberg M. H. (1999) The talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274, 28071–28074 [DOI] [PubMed] [Google Scholar]
- 22. Tadokoro S., Shattil S. J., Eto K., Tai V., Liddington R. C., de Pereda J. M., Ginsberg M. H., Calderwood D. A. (2003) Talin binding to integrin β tails: a final common step in integrin activation. Science 302, 103–106 [DOI] [PubMed] [Google Scholar]
- 23. Shattil S. J., Newman P. J. (2004) Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood 104, 1606–1615 [DOI] [PubMed] [Google Scholar]
- 24. Moser M., Legate K. R., Zent R., Fässler R. (2009) The tail of integrins, talin, and kindlins. Science 324, 895–899 [DOI] [PubMed] [Google Scholar]
- 25. Ma Y. Q., Yang J., Pesho M. M., Vinogradova O., Qin J., Plow E. F. (2006) Regulation of integrin αIIbβ3 activation by distinct regions of its cytoplasmic tails. Biochemistry 45, 6656–6662 [DOI] [PubMed] [Google Scholar]
- 26. Bledzka K., Bialkowska K., Nie H., Qin J., Byzova T., Wu C., Plow E. F., Ma Y-Q. (2010) Tyrosine phosphorylation of integrin β3 regulates kindlin-2 binding and integrin activation. J. Biol. Chem. 285, 30370–30374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gong H., Shen B., Flevaris P., Chow C., Lam S. C.-T., Voyno-Yasenetskaya T. A., Kozasa T., Du X. (2010) G Protein Subunit Gα13 Binds to Integrin αIIbβ3 and Mediates Integrin “Outside-In” Signaling. Science 327, 340–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shen B., Zhao X., O'Brien K. A., Stojanovic-Terpo A., Delaney M. K., Kim K., Cho J., Lam S. C. T., Du X. (2013) A directional switch of integrin signalling and a new anti-thrombotic strategy. Nature 503, 131–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. White J. G., Rao G. H., Gerrard J. M. (1974) Effects of the Ionophore A23187 on blood platelets I. Influence on aggregation and secretion. Am. J. Pathol. 77, 151–156 [PMC free article] [PubMed] [Google Scholar]
- 30. Brace L. D., Venton D. L., Le Breton G. C. (1985) Thromboxane A /prostaglandin H mobilizes calcium in human blood platelets. Am. J. Physiol. 249, H1-H7 [DOI] [PubMed] [Google Scholar]
- 31. Jin J., Daniel J. L., Kunapuli S. P. (1998) Molecular basis for ADP-induced platelet activation. II. The P2Y1 receptor mediates ADP-induced intracellular calcium mobilization and shape change in platelets. J. Biol. Chem. 273, 2030–2034 [DOI] [PubMed] [Google Scholar]
- 32. Fox J. E., Phillips D. R. (1983) Stimulus-induced activation of the calcium-dependent protease within platelets. Cell Motility 3, 579–588 [DOI] [PubMed] [Google Scholar]
- 33. Yan B., Calderwood D. A., Yaspan B., Ginsberg M. H. (2001) Calpain cleavage promotes talin binding to the β3 integrin cytoplasmic domain. J. Biol. Chem. 276, 28164–28170 [DOI] [PubMed] [Google Scholar]
- 34. Kuchay S. M., Chishti A. H. (2007) Calpain-mediated regulation of platelet signaling pathways. Curr. Opin. Hematol. 14, 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]