

Background: There are regulatory interactions between ENaC and extracellular factors.
Results: Mutations of multiple α subunit knuckle residues activate ENaC by suppressing the inhibitory effect of Na+. Channels lacking the β or γ subunit knuckle have processing defects.
Conclusion: Interactions between the α subunit knuckle and palm/finger domains regulate ENaC.
Significance: Intrasubunit domain-domain interactions have important regulatory roles.
Keywords: acid-sensing ion channel (ASIC), allosteric regulation, epithelial sodium channel (ENaC), ion channel, sodium channel
Abstract
The extracellular regions of epithelial Na+ channel subunits are highly ordered structures composed of domains formed by α helices and β strands. Deletion of the peripheral knuckle domain of the α subunit in the αβγ trimer results in channel activation, reflecting an increase in channel open probability due to a loss of the inhibitory effect of external Na+ (Na+ self-inhibition). In contrast, deletion of either the β or γ subunit knuckle domain within the αβγ trimer dramatically reduces epithelial Na+ channel function and surface expression, and impairs subunit maturation. We systematically mutated individual α subunit knuckle domain residues and assessed functional properties of these mutants. Cysteine substitutions at 14 of 28 residues significantly suppressed Na+ self-inhibition. The side chains of a cluster of these residues are non-polar and are predicted to be directed toward the palm domain, whereas a group of polar residues are predicted to orient their side chains toward the space between the knuckle and finger domains. Among the mutants causing the greatest suppression of Na+ self-inhibition were αP521C, αI529C, and αS534C. The introduction of Cys residues at homologous sites within either the β or γ subunit knuckle domain resulted in little or no change in Na+ self-inhibition. Our results suggest that multiple residues in the α subunit knuckle domain contribute to the mechanism of Na+ self-inhibition by interacting with palm and finger domain residues via two separate and chemically distinct motifs.
Introduction
Epithelial Na+ channels (ENaCs)3 have important roles in the regulation of extracellular fluid volume and blood pressure (1, 2). A variety of factors fine tune ENaC activity in response to varying physiological conditions (3). Among these regulatory factors is extracellular Na+, which inhibits ENaC activity by a well documented process referred to as Na+ self-inhibition (3–5). Recent studies using heterologously expressed channels suggest that Na+ self-inhibition reflects a reduction in channel open probability and functions as an important mechanism that modulates ENaC gating (2, 6–14). Given the role of ENaC in blood pressure regulation, it has been suggested that variants that alter Na+ self-inhibition may contribute to the development of human hypertension (15). Despite the identification of numerous ENaC residues where substitutions alter Na+ self-inhibition (7, 10, 16–23) and the recent identification of an extracellular Na+ binding site mediating the Na+ self-inhibition response (24), a mechanistic understanding of how external Na+ reduces channel activity is lacking.
Clues regarding allosteric changes that occur in association with extracellular Na+ binding and Na+ self-inhibition were provided by the resolved structure of acid-sensing ion channel 1 (ASIC1), a related ENaC/degenerin family member (25, 26). The resolved homotrimeric ASIC1 structures reveal a highly organized extracellular region linked to transmembrane domains (Fig. 1). The extracellular region of each subunit is composed of distinct domains. The more peripheral thumb, finger, and knuckle domains are formed by α helices, and proximal domains (palm and β ball) are formed by β strands. The knuckle domain includes two short helices (α6 and α7) arranged in a helix-loop-helix conformation that is linked to the palm domain (25). Based on comparisons of conducting and non-conducting ASIC1 structures, Baconguis and Gouaux (26) suggested that the knuckle domain, together with the upper portions of the palm domain, forms a rigid structural scaffold, in contrast to the more flexible lower palm domain. A human ENaC polymorphism (αW493R), located at the junction of the palm and knuckle domains, activates human ENaC in association with diminished Na+ self-inhibition (27) (Fig. 1). We also described a human ENaC variant (γL511Q) at the junction of the upper and lower palm domain that exhibited a phenotype similar to αW493R (23). These observations, taken together, are consistent with a role of this region in the allosteric regulation of ENaC.
FIGURE 1.

Knuckle domain location and sequence alignments. A, chicken ASIC1 structural model showing knuckle domain locations in the channel complex. The trimeric cASIC1 model was built from structural coordinates of the minimal functional cASIC1 (PDB entry 4NYK superseding 3HGC (48)). The three cASIC1 subunits are rendered as solid ribbons in dark, intermediate, and light gray using PyMOL (49). The knuckle domain residues of three subunits are rendered as space-filled spheres in red, blue, or green. B, top view of the same model in A. C, sequence alignments of human, mouse, and rat ENaC subunits and cASIC1. Alignments were done using Vector NTI 11 (Invitrogen). Identical residues are in red, and conserved residues are in blue. Mouse ENaC residues that were deleted in this study are underlined. Chicken ASIC1 residues within the α6 and α7 helices are indicated by two orange bars.
We recently reported that in-frame deletion of exon 11 of the human α subunit, which encodes the helix-turn-helix component of the knuckle domain (Fig. 1C), dramatically increased whole cell ENaC currents and channel open probability, in association with a loss of the Na+ self-inhibition response (28). In this study, we investigated the functional roles of individual residues in the α subunit knuckle domain by performing cysteine-scanning mutagenesis and examining the effects of these substitutions on Na+ self-inhibition. We discovered that substitutions within a cluster of hydrophobic residues in the proximal half of the knuckle domain and within a stretch of polar residues in the distal half of this domain greatly dampen Na+ self-inhibition. Interestingly, the functional roles of α subunit knuckle domain residues are not shared by the homologous residues in the β or γ subunits, suggesting that specific sites in the α subunit knuckle domain modulate ENaC gating. Moreover, unlike the α subunit, β and γ subunits lacking the knuckle domain are not processed to mature forms, suggesting that the β and γ subunit knuckle domains are required for proper ENaC biogenesis.
Experimental Procedures
Site-directed Mutagenesis
Point mutations and deletions were introduced into mouse α, β, and γ ENaC cDNAs using the QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). Target mutations were verified by DNA sequencing. Wild type (WT) and mutant ENaC cRNAs were synthesized by in vitro transcription using T3 RNA polymerase (Ambion, Inc.), purified by an RNA purification kit (Qiagen), and quantified by spectrophotometry.
ENaC Expression and Two-electrode Voltage Clamp
ENaC expression in Xenopus oocytes and current measurements by two-electrode voltage clamp were performed as reported previously (29). Stage V and VI oocytes with the follicle cell layer removed were injected with 0.5 ng of each subunit (α, β, and γ) cRNA in a volume of 50 nl and incubated at 18 °C in modified Barth's solution (MBS; 88 mm NaCl, 1 mm KCl, 2.4 mm NaHCO3, 15 mm HEPES, 0.3 mm Ca(NO3)2, 0.41 mm CaCl2, 0.82 mm MgSO4, 10 μg/ml streptomycin sulfate, 100 μg/ml gentamycin sulfate, pH 7.4). All experiments were performed at room temperature (20–24 °C) 20–52 h following injection. Oocytes were placed in a recording chamber from Warner Instruments (Hamden, CT) and perfused with a constant flow rate of 12–15 ml/min. The perfusion solution contained 110 mm NaCl, 2 mm KCl, 2 mm CaCl2, and 10 mm HEPES and had the pH adjusted at 7.4. Voltage clamp was performed using an Axoclamp 900A computer-controlled microelectrode amplifier and DigiData 1440A interface controlled by pClamp version 9.2 (Molecular Devices Corp., Sunnyvale, CA). The protocol for harvesting oocytes from Xenopus laevis was approved by the University of Pittsburgh institutional animal care and use committee.
Surface Expression
Surface expression of αβγ ENaC in oocytes was determined using a mouse β or γ subunit with an extracellular FLAG epitope tag, as reported previously (30). Briefly, oocytes were injected with 2 ng/subunit of the three ENaC cRNAs with either the β or γ subunit containing an extracellular FLAG epitope tag (DYKDDDDK). The FLAG tag was placed immediately following residue Thr137 in the β subunit, as described previously (31). The γ subunit tag was generated by replacing residues Val131–Thr138 with a FLAG sequence. Robust amiloride-sensitive Na+ currents confirmed that ENaCs containing a FLAG-tagged β or γ subunit were functional. Two days after cRNA injection, a surface expression assay was performed on ice, except for the last step, which was at room temperature (20–24 °C). Following a 30-min incubation in antibiotic-free MBS supplemented with 1% bovine serum albumin (MBS/BSA), oocytes were incubated for 1 h with MBS/BSA supplemented with 1 μg/ml of a mouse monoclonal anti-FLAG antibody (M2, Sigma). Oocytes were then washed in MBS/BSA and incubated with MBS/BSA supplemented with 1 μg/ml of a horseradish peroxidase coupled to a secondary antibody (peroxidase-conjugated AffiniPure F(ab′)2 fragment goat anti-mouse IgG, Jackson ImmunoResearch, West Grove, PA) for 1 h. Cells were washed and transferred to MBS without BSA. Individual oocytes were placed in a 96-well plate, and 100 μl of SuperSignal ELISA Femto maximum sensitivity substrates (Thermo Scientific, Rockford, IL) was added to each well. Cells were then incubated at room temperature for 1 min, and chemiluminescence in relative light units was quantified in a GloMax-Multi+ detection system (Promega, Madison, WI).
Immunoprecipitation of ENaC Expressed in Xenopus Oocytes
Oocytes were injected with mouse ENaC cRNAs where either the β or γ subunit had both an N-terminal HA and C-terminal V5 tag. Epitope-tagged subunits were either WT or a knuckle domain deletion mutant, as indicated. 24 h after injection, 20 oocytes for each condition were transferred to a 1.5-ml conical tube with a snap cap, and all liquid was removed. Detergent solution (400 μl of 100 mm NaCl, 40 mm KCl, 1 mm EDTA, 10% glycerol, 1% Nonidet P-40, 0.4% deoxycholate, 20 mm HEPES, pH 7.4, with Calbiochem Protease Inhibitor Mixture III (EDTA-free)) was added to each tube before homogenization by pipetting up and down with a Pipetteman (Gilson) and then with a BD insulin syringe. The homogenate was mixed on a vortex for 30 s and centrifuged for 10 min (200 × g) in an Eppendorf 5417C centrifuge at 4 °C. The supernatant was centrifuged a second time for 10 min (200 × g) at 4 °C, and that resulting supernatant was centrifuged again for 10 min (20,800 × g) at 4 °C (20-μl aliquots were retained as “total extract”). The final supernatant was mixed with 400 μl of detergent solution and incubated overnight with end-over-end mixing with 40 μl of a 50% slurry of anti-V5 antibodies conjugated to beads (Bethyl). The beads were (i) washed twice with HEPES-buffered saline (10 mm HEPES, pH 7.4, 150 mm NaCl) containing 1% Triton X-100, (ii) washed once with HEPES-buffered saline containing 0.01% SDS, and (iii) washed once with HEPES-buffered saline. The washed beads were heated for 5 min at 95 °C with 30 μl of Bio-Rad 2× Laemmli sample buffer containing 0.14 m β-mercaptoethanol and spun, and the supernatant was loaded onto an SDS gel. Immunoblots were probed with anti-V5 antibodies as described previously (32). Total extract (whole cell lysate) was also subjected to SDS-PAGE and immunoblotting with an anti-β-actin antibody (Sigma), as described previously (33, 34).
Na+ Self-inhibition
Na+ self-inhibition was examined as reported previously (7). Briefly, a low [Na+] bath solution (NaCl-1; 1 mm NaCl, 109 mm N-methyl-d-glucamine, 2 mm KCl, 2 mm CaCl2, 10 mm HEPES, pH 7.4) was rapidly replaced with a high [Na+] bath solution (NaCl-110; 110 mm NaCl, 2 mm KCl, 2 mm CaCl2, 10 mm HEPES, pH 7.4), whereas the oocytes expressing ENaCs were continuously clamped to a membrane potential of −60 or −100 mV. Bath solution exchange was done with a Teflon valve computer-controlled perfusion system (AutoMate Scientific Inc., Berkeley, CA). At the end of each experiment, 10 μm amiloride was added to the bath solution to determine the amiloride-insensitive portion of the whole-cell current. The magnitude of Na+ self-inhibition was represented by the ratio of the steady-state current (Iss) to peak current (Ipeak). Iss was determined as the current at 40 s following the Ipeak. Both Iss and Ipeak were amiloride-sensitive currents. The time constants (τ) for Na+ self-inhibition were estimated by fitting the first 40-s current decay from Ipeak with a standard exponential function in the Clampfit software. To minimize complications due to variability in the Na+ self-inhibition response between batches of oocytes, the responses of WT channels were always tested in an alternating manner with mutants in the same batch of oocytes.
ENaC Degradation in Yeast
Yeast Saccharomyces cerevisiae strains were grown at 26 °C using standard methods, and medium preparation and transformation were performed as described (35). The WT yeast strain used was BY4742 from Open Biosystems (Thermo Scientific). Construction of pRS426GPD-αENaC-HA, pRS426GPD-βENaC-HA, and pRS426GPD-γENaC-HA was described previously (36). The same cloning strategy and restriction enzymes were used to construct pRS426GPD-αENaCΔknuckle-HA, pRS426GPD-βENaCΔknuckle-HA, and pRS426GPD-γENaCΔknuckle-HA from the knuckle deletion constructs. Cycloheximide chase analyses of the ENaC subunits were performed as published (36), and cell lysates from chase samples were generated using alkaline lysis, followed by trichloroacetic acid precipitation (37). Proteins were resolved by SDS-PAGE before Western blot analysis. The ENaC subunits were detected using horseradish peroxidase (HRP)-conjugated anti-HA antibodies (clone 3F10; Roche Applied Science) at a dilution of 1:5000. Western blots were also probed with anti-glucose-6-phosphate dehydrogenase (Sigma) rabbit antisera, which served as a loading control. The glucose-6-phosphate dehydrogenase primary antibody was detected with a donkey HRP-conjugated anti-rabbit IgG secondary antibody (GE Healthcare). The Western blot signals were imaged using enhanced chemiluminescence (Pierce) and visualized on a Bio-Rad ChemiDoc XRS+ system. Quantitation was carried out using ImageJ (version 1.44; National Institutes of Health).
ENaC Degradation in FRT Cells
Fisher rat thyroid (FRT) cells were kindly provided by Peter Snyder (University of Iowa). FRT cells were cultured in DMEM/F-12 medium supplemented with 8% fetal bovine serum. Cells were plated in plastic wells (12-well size from Costar, Corning, NY) and the next day (18 h) transfected using Lipofectamine 2000 with plasmids encoding α, β, and γ mouse ENaC subunits as described in the figure legends. In each case one subunit had both N-terminal HA and C-terminal V5 tags, whereas the other two had no epitope tags. Knuckle domains were deleted where indicated in epitope-tagged subunits. Cycloheximide (1000× stock in DMSO) was added to cells in fresh medium at a final concentration of 100 μg/ml (Calbiochem EMD Millipore, Billerica, MA). At 0, 1, 2, 4, or 6 h following the addition of cycloheximide, the cells were washed with Dulbecco's phosphate-buffered saline with Mg2+ and Ca2+ (Corning Cellgro, Manassas, VA). Cells were solubilized at 4 °C in detergent solution (0.4% sodium deoxycholate, 1% Nonidet P-40, 63 mm EDTA, and 50 mm Tris-HCl, pH 8) with 1% protease inhibitor mixture III (EMD Millipore, Billerica, MA; pH 7.4). ENaC was immunoprecipitated with anti-V5 antibodies conjugated to beads, as described previously (32), before SDS-PAGE and immunoblotting with anti-V5 antibodies. An aliquot of the initial cell extract (5%) was retained for SDS-PAGE and immunoblotting for β-actin.
Statistical Analyses
Data are presented as mean ± S.E. Significance comparisons between WT and mutant groups expressed in the same batches of oocytes were performed with Student's t test. Data obtained in Xenopus oocytes were analyzed using Microsoft Excel 2013 and Origin Pro version 8.5 (OriginLab Corp., Northampton, MA). Data obtained in yeast and FRT cells were analyzed by two-way analysis of variance with Tukey post hoc test using SigmaPlot version 12 (Systat Software Inc., San Jose, CA).
Results
We recently reported that deletion of the human α subunit residues encoded by exon 11 (αGlu500–αLys517; Fig. 1C) increased ENaC activity, reflecting a loss of Na+ self-inhibition (28). These residues form the double helix bundle of the knuckle domain (Fig. 1). We asked whether mouse ENaCs with a similar α subunit knuckle domain deletion (residues Gln526–Val548; underlined in Fig. 1C) also exhibit increased ENaC activity and a loss of Na+ self-inhibition. Mouse ENaCs with an α subunit Gln526–Val548 deletion (denoted as αΔK (αΔKnuckle)) had significantly larger ENaC currents (Fig. 2A) with no significant change in cell surface expression when expressed in oocytes, as compared with WT (Fig. 2B). The αΔK mutant exhibited a significant blunting of the Na+ self-inhibition response following transition from a low (1 mm) to a high (110 mm) Na+ bath (Fig. 3). The absence of Na+ self-inhibition is reflected by a significant increase in the steady state/peak current ratio (Iss/Ipeak) and a reduction in the rate of current decay following the peak current.
FIGURE 2.
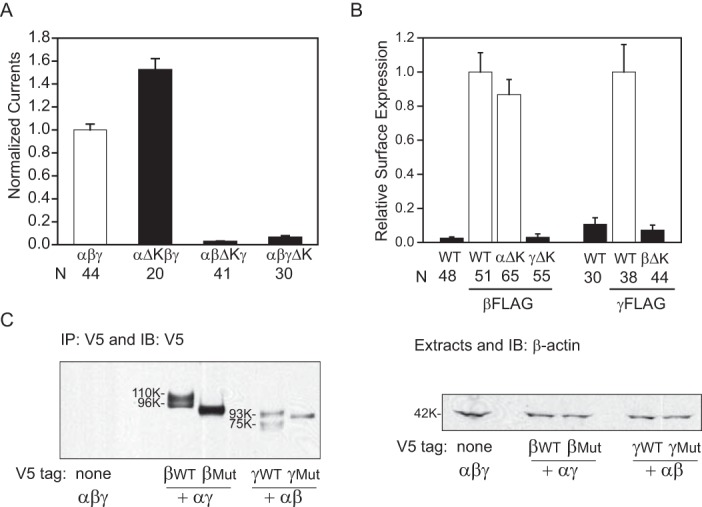
Knuckle domain deletions result in subunit-specific changes in ENaC activity and surface expression. A, normalized currents in oocytes expressing mouse αβγ, αΔKβγ, αβΔKγ, or αβγΔK ENaCs. Normalized currents represent amiloride-sensitive currents measured at −100 mV from individual oocytes, divided by the average of the amiloride-sensitive current of oocytes expressing WT channels from the same batch of oocytes. Data were pooled from two batches of oocytes. The amiloride-sensitive currents from the two batches of oocytes expressing WT ENaC were −7.5 ± 0.6 μA (n = 27) and −12.7 ± 0.7 μA (n = 17). Black columns indicate values that were significantly different from WT controls (p < 0.01). B, relative levels of surface expression. Oocytes were injected with three ENaC subunit cRNAs for αβγ (WT), αΔKβγ (αΔK), αβΔKγ (βΔK), and αβγΔK (γΔK). For α and γ mutants, a β subunit with a FLAG epitope was used, whereas a γ subunit with a FLAG epitope was used with the β subunit mutant. Relative light units measured from individual oocytes were normalized to the average relative light units of the same batch of oocytes expressing the FLAG-tagged WT control. Data were from four batches of oocytes. Black columns, values that were significantly different from the FLAG WT control group (p < 0.01). C, deletion of the knuckle domain affects processing of ENaC expressed in Xenopus oocytes. Oocytes (20 oocytes/group) were injected with αβγ cRNAs, where either the β or γ subunit had both an N-terminal HA and C-terminal V5 tag. Epitope-tagged subunits were either WT or had a knuckle domain deletion mutant (Mut), as indicated. αβγ lacking an epitope tag was used as a negative control. The following day, ENaC was immunoprecipitated (IP) from oocyte extracts with anti-V5 antibodies conjugated to beads and immunoblotted with anti-V5 antibodies. Molecular weights of the immature β (96K) and γ (93K) subunits and the mature β (110K) and γ (75K) subunits are indicated beside the immunoblots (IB) for WT subunits. Mobility of mutant subunits in each case was slightly faster than wild-type subunits, as expected for the knuckle deletions (∼3 kDa). No mature forms of the β and γ mutant subunits were observed, even upon longer exposures. An aliquot of the oocyte extract (5%) for each group was immunoblotted with anti-β-actin antibodies (42 kDa). Error bars, S.E.
FIGURE 3.
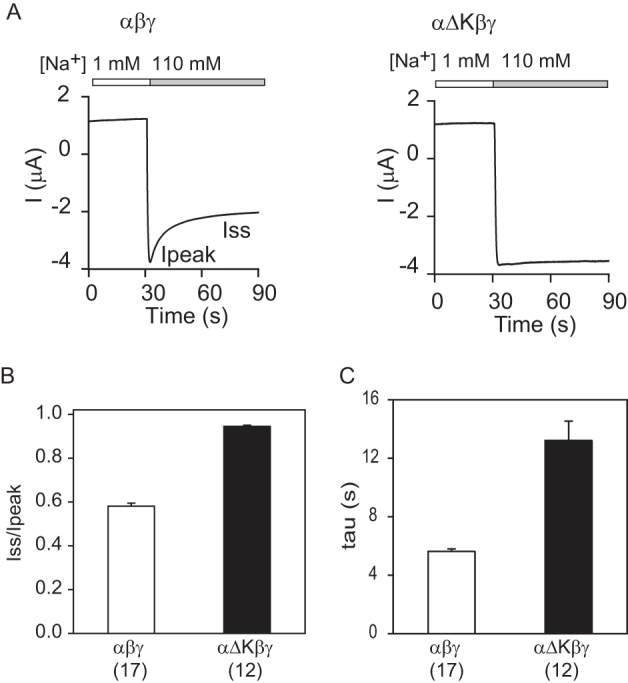
α subunit knuckle domain deletion results in a loss of Na+ self-inhibition. A, representative traces for Na+ self-inhibition responses in oocytes expressing αβγ and αΔKβγ. Oocytes were clamped at −60 or −100 mV, and whole-cell currents were continuously recorded while bath [Na+] was rapidly increased from 1 mm (open bar) to 110 mm (gray bar). Inward currents are shown as negative values by convention. The current decline following the increase in bath [Na+] from a peak current (Ipeak) to a steady-state current (Iss) represents Na+ self-inhibition. B, Na+ self-inhibition magnitudes (Iss/Ipeak) of αβγ and αΔKβγ. The Iss/Ipeak values were obtained as described under “Experimental Procedures.” Data were collected in three batches of oocytes with the total oocyte numbers shown in parentheses. C, Na+ self-inhibition time constants of αβγ and αΔKβγ. The Iss/Ipeak ratio and τ of αΔKβγ were significantly greater than that of WT (black columns, p < 0.001). Data were collected in three batches of oocytes. Total oocyte numbers are shown in parentheses below. Error bars, S.E.
As we noted with human ENaC (28), deletions of the β or γ subunit knuckle domain (βΔK, ΔGlu466–Ile490; γΔK, ΔGlu483–Leu507, underlined in Fig. 1C) exhibited markedly reduced amiloride-sensitive currents and surface expression (Fig. 2). To demonstrate that β and γ subunits with knuckle domain deletions were expressed in oocytes, we examined whole cell expression of ENaC subunits in oocytes injected with cRNAs for αβ*ΔKγ or αβγ*ΔK (where the asterisk represents a V5 tag at the C terminus). WT αβ*γ or αβγ* expression was also examined in the same batches of oocytes for comparison (Fig. 2C). β*ΔK expression was slightly lower than that of WT β*, but the difference was not statistically significant (mutant/WT, 0.79 ± 0.18, n = 3, p > 0.05). In contrast, γ*ΔK expression was significantly less than that of WT γ* (mutant/WT, 0.46 ± 0.10, n = 3, p < 0.05). However, the reduced whole cell expression does not account for reduction of channel activity or surface expression. These results suggest that a reduction in protein expression was not primarily responsible for reduced channel activity and surface expression as a result of a β or γ knuckle domain deletion. We also found that mature forms of the β and γ subunit were seen with WT ENaC. These mature forms are a slower migrating (110-kDa) β subunit, reflecting N-glycan maturation, as well as a faster migrating (75-kDa) γ subunit, reflecting N-glycan maturation and proteolytic processing (32, 38, 39). Only immature forms of the β or γ subunit were observed when the knuckle domain was deleted.
The reduced levels of activity and surface expression observed with channels bearing the βΔK or γΔK mutant could reflect aberrant subunit folding associated with enhanced endoplasmic reticulum-associated degradation. We have shown previously that individual ENaC subunits expressed in yeast undergo fairly rapid endoplasmic reticulum-associated degradation (36). Therefore, we performed cycloheximide chase experiments of individual ENaC subunits in yeast to determine whether deletion of the knuckle domain accelerated the rate of subunit degradation. The logic underlying the use of the yeast system is that quantitative data can be acquired in a rapid and facile manner, and gross alterations in the folding of an individual channel subunit are readily apparent. Positive results would then be followed-up by more laborious studies in higher cells. However, we found that subunits with a deleted knuckle domain (αΔK, βΔK, or γΔK) exhibited rates of degradation similar to their WT counterparts (Fig. 4).
FIGURE 4.
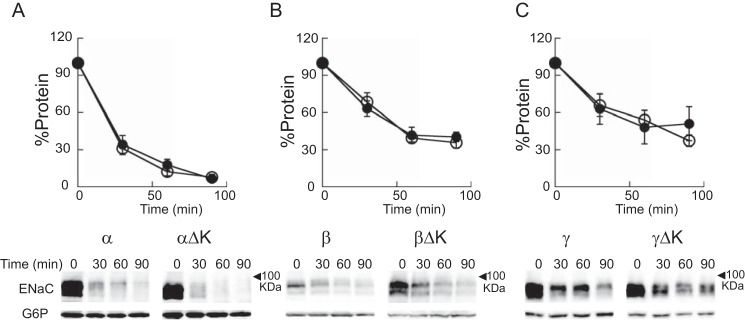
Deletion of a knuckle domain does not change the degradation rates of individual ENaC subunits. Cycloheximide chase experiments were performed as described under “Experimental Procedures” in WT yeast strains expressing a mouse ENaC subunit with a C-terminal HA epitope tag. A, α (open circles) or αΔK (closed circles); B, β (open circles) or βΔK (closed circles); C, γ (open circles) or γΔK (closed circles). Representative blots are shown below each graph. Chase reactions were performed at 37 °C, and lysates were immunoblotted with anti-HA antisera (ENaC) and with anti-glucose-6-phosphate dehydrogenase (G6P) as a loading control. Data represent the means of 3–7 experiments ± S.E. (error bars). No significant difference at each time point between the WT and mutant subunits was found (all p > 0.05, from two-way analysis of variance).
We next examined whether knuckle domain deletions affect ENaC subunit degradation when all three subunits are co-expressed in FRT cells. We previously showed that processing of ENaC subunits in the biosynthetic pathway includes furin-dependent cleavage of the α and γ subunits as well as maturation of N-glycans (32, 40) and that only the cleaved forms of the α and γ subunits had mature N-glycans (32). β subunit maturation was associated with the appearance of a slower migrating, full-length β subunit (32). We performed cycloheximide chase experiments in FRT cells expressing all three ENaC subunits, comparing WT channels with channels where one subunit lacked a knuckle domain (Fig. 5). Both full-length (95-kDa) and cleaved (65-kDa) forms of the α subunit were readily detected in WT and αΔKβγ channels (knuckle domain deletion reduces the corresponding molecular mass of the full-length and cleaved forms by ∼3 kDa). In contrast, we found only the forms corresponding to an immature β subunit (96 kDa, but not the slower migrating mature 110-kDa β subunit) and immature γ subunit (93 kDa, but not the 75-kDa cleaved γ subunit) when channels lacked the β or γ subunit knuckle domain, respectively. Deletion of the α subunit knuckle domain did not significantly affect degradation of the corresponding full-length (95-kDa) or cleaved (65-kDa) forms of the α subunit, although a significant difference was observed at one time point (2 h) for the mature 65-kDa form (Fig. 5B). Similarly, deletion of the β or γ subunit knuckle domain did not alter the rate of degradation of the β or γ subunit, although a significant difference was observed at one time point (1 h) for the immature 93-kDa γ subunit (Fig. 5G).
FIGURE 5.

Deletion of the knuckle domain of the β or γ subunit, but not the α subunit, slows ENaC maturation. FRT cells were transfected with mouse αβγ ENaC where either the α (A–C), β (D–F), or γ (G–I) subunit had both an N-terminal HA and C-terminal V5 tag. Epitope-tagged subunits were either wild-type (WT) or mutant with knuckle domain deletions as indicated. Cycloheximide was added to cells growing on plastic (12-well size) at 100 μg/ml, and cells were recovered at varying times (t = 0, 1, 2, 4, or 6 h) and extracted in the presence of protease inhibitors. ENaC was immunoprecipitated with anti-V5 antibodies conjugated to beads and immunoblotted with anti-V5 antibodies. Aliquots of the initial cell extracts were subjected to immunoblotting with anti-β-actin antibodies. The results from three experiments are presented as the means ± S.E. (error bars) (A, B, D, E, G, and H), and a representative immunoblot is shown in each case (C, F, and I). Molecular weights of the immature α (95K), β (96K), and γ (93K) subunits and the mature α (65K), β (110K), and γ (75K) subunits are indicated beside the immunoblots for the WT subunits. The mobilities of the mutant subunits in each case were slightly faster than for the WT subunits, as expected for the knuckle deletions (∼3 kDa). The mature form of the mutant α subunit was observed (* in C), but mature forms of the β and γ mutant subunits were not observed, even upon longer exposures (data not shown). Rates of degradation analyzed with two-way analysis of variance were similar between WT and mutant subunits with no difference for the α subunit (p > 0.05 for both immature (A) and mature (B) forms) and for the β subunit (p > 0.05 in D). There was a small but significant difference in the degradation rates of the WT and mutant γ subunit (p < 0.05 for immature γ subunit in G). Significant differences were also observed between WT mature (65 kDa) and the corresponding mutant mature α subunits at t = 2 h and between WT immature (93 kDa) and the corresponding mutant mature γ subunits at t = 1 h (*, p < 0.05). J, whole cell, steady-state levels of WT and mutant subunits were assessed at t = 0 and normalized to β-actin expression (n = 3). Whereas WT and αΔKβγ (66.8 ± 27.1%) channels had similar levels of α subunit expression (p > 0.05, open bar), αβΔKγ (38.8 ± 13.5%) and αβγΔK (39.8 ± 10.9%) channels had significantly reduced whole cell levels of β and γ subunit expression, respectively, when compared with WT (p < 0.05, black bars). For WT subunits, both mature and immature forms were included in the quantification.
We also examined whole cell, steady-state expression levels of ENaCs in FRT cells expressing WT channels or channels where one subunit lacked a knuckle domain. Whereas WT and αΔKβγ channels had similar levels of the α subunit (normalized to β-actin expression at t = 0), the αβΔKγ and αβγΔK channels had significantly reduced steady-state levels of the β and γ subunit, respectively, when compared with the WT channel (Fig. 5J; p < 0.05).
Because a loss of the α subunit knuckle domain (αΔK) led to channel activation and a loss of Na+ self-inhibition, we examined whether there were specific residues in this domain that contributed to the observed phenotype. We systematically mutated individual residues within the α subunit knuckle domain (Gln520–Gly547) to Cys and examined the functional effects of these mutations. All of the mutants were functional when expressed in oocytes (Figs. 6 and 7). Half of the mutations (14 of 28) significantly reduced the Na+ self-inhibition response, as reflected by an increased Iss/Ipeak, compared with WT. Of these 14 mutants that had an increased Iss/Ipeak, 11 also showed greater time constants, indicating a slowed Na+ self-inhibition response. Two mutants (P521C and T540C) had greater time constants than WT, but the differences were not significant. One mutant (K544C) showed a time constant indistinguishable from that of WT, suggestive of a selective effect of the mutation on the magnitude of Na+ self-inhibition.
FIGURE 6.

Mutation of specific α subunit knuckle domain residues diminished the Na+ self-inhibition response. Representative current traces for Na+ self-inhibition responses were from oocytes expressing either WT αβγ or mutant α subunits with WT β and γ subunits. The mutated residues are shown above each panel. Oocytes were clamped at −60 or −100 mV, and whole-cell currents were continuously recorded while bath [Na+] was rapidly increased from 1 mm (open bar) to 110 mm (gray bar).
FIGURE 7.
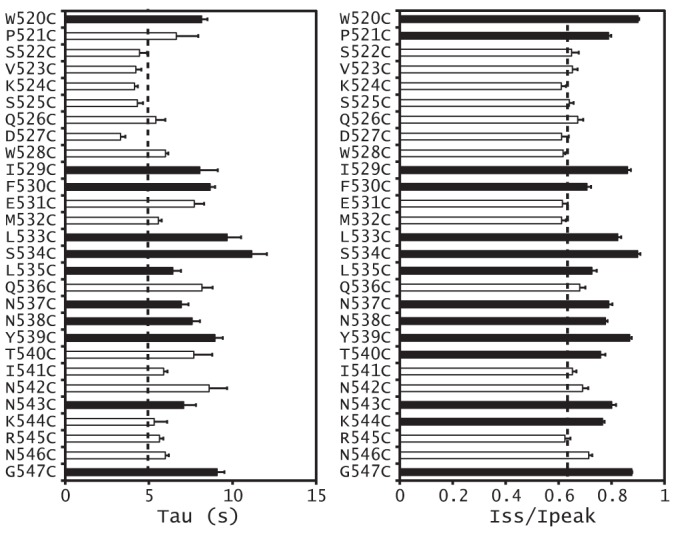
Magnitude and time constant of the Na+ self-inhibition response of α subunit knuckle domain mutants. The Iss/Ipeak (right) and τ (left) values were obtained as described under “Experimental Procedures.” Values that are significantly different from that of WT in the same batch of oocytes are shown as black bars (p < 0.01, n = 4–7). Dashed lines, averaged WT values from all tested oocytes. Error bars, S.E.
We have previously observed subunit specificity regarding the effects of mutations on Na+ self-inhibition (7, 10, 19). Consequently, we examined whether knuckle domain mutations at specific sites exhibited subunit specificity. We chose three sites where α subunit knuckle domain Cys substitutions significantly reduced Na+ self-inhibition (αPro521, αIle529, and αSer534; Figs. 6 and 7). When we expressed channels with Cys mutations in the corresponding sites in the β or γ subunit, Na+ self-inhibition was either similar to WT (mutations at the sites corresponding to αIle529 and αSer534) or was moderately reduced (mutations at the sites corresponding to αPro521; Fig. 8). Combined with the data presented above, these results suggest that α knuckle domain residues have an important role in the Na+ self-inhibition response when compared with their corresponding β and γ subunit residues.
FIGURE 8.
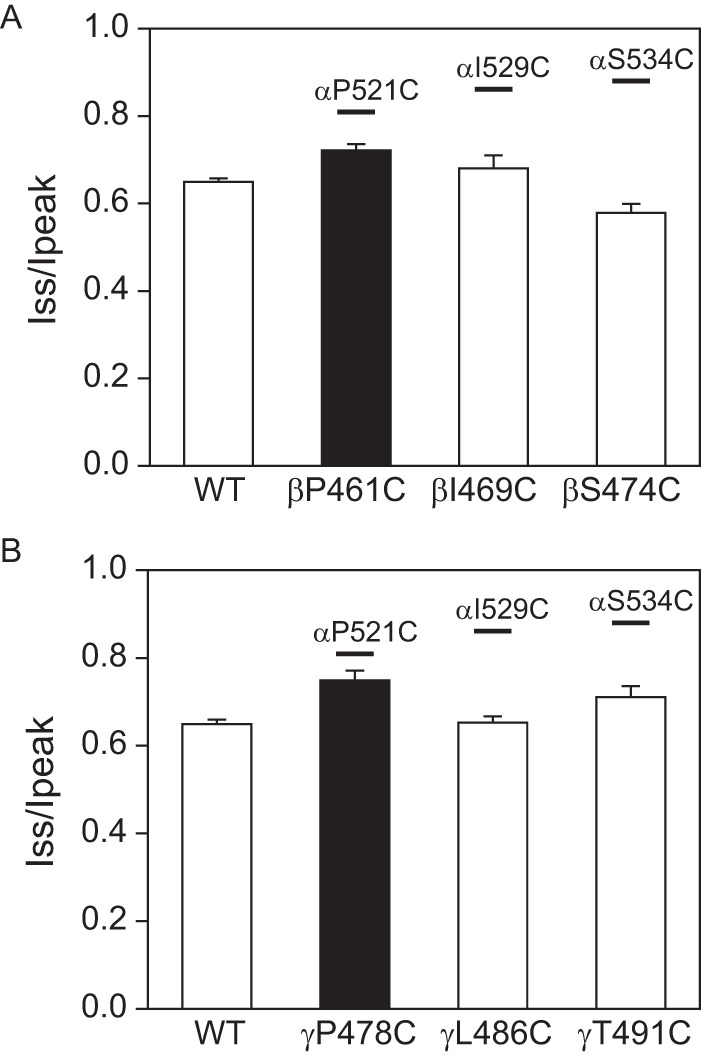
Effects of β and γ knuckle domain mutations on Na+ self-inhibition. A, Iss/Ipeak values obtained in oocytes expressing αβγ (WT), αβP461Cγ, αβI469Cγ, or αβS474Cγ (n = 6–7). The black bar indicates that the Iss/Ipeak for αβP521Cγ was significantly greater than that of WT (p < 0.01). Boldface lines above the three bars show the mean Iss/Ipeak values for channels with the corresponding α subunit mutation (from Fig. 6). B, Iss/Ipeak values in oocytes expressing αβγ (WT), αβγP478Cγ, αβγL486C, or αβγT491C (n = 6–9). The black bar indicates that the Iss/Ipeak for αβγP478C was significantly greater than that of WT (p < 0.01). Lines above the three bars are as described in A.
Discussion
The goal of this study was to explore the functional role of the extracellular knuckle domain and to identify specific residues involved in Na+ self-inhibition. As with the human α subunit (28), deletion of the mouse α subunit knuckle domain led to channel activation. The lack of change in ENaC surface expression when the mouse α subunit knuckle domain is deleted, in association with the markedly impaired Na+ self-inhibition response, suggests that channel activation reflects an increase in its open probability. In fact, we found that human ENaCs lacking the α subunit knuckle domain had an open probability that was significantly greater than WT (28). Our results suggest that the α subunit knuckle domain functions as a suppressor of ENaC activity in the presence of extracellular Na+.
In contrast to channels lacking an α subunit knuckle domain, deletions of mouse (Figs. 1 and 2) or human (28) β or γ subunit knuckle domains resulted in a dramatic reduction in ENaC activity. This phenomenon primarily reflects reduced channel expression at the cell surface (Fig. 2) (28), which does not appear to be due to an enhanced rate of channel degradation as assessed by cycloheximide chase assays. We did not observe enhanced degradation of individual subunits lacking their knuckle domain when expressed in the yeast system (Fig. 4) or when all three subunits (αβγ with an individual subunit lacking a knuckle domain) were expressed in FRT cells. Interestingly, the β or γ knuckle domain deletion prevented subunit maturation in both oocytes and FRT cells. Moreover, levels of both mutant subunits were significantly lower than WT in FRT cells, reflecting reduced protein synthesis and/or increased degradation. Together, these results suggest that channels with β and γ subunits lacking knuckle domains do not efficiently transit the Golgi complex or trans-Golgi network, where processing events associated with subunit maturation occur. Our findings suggest that the mutant channels are retained in the endoplasmic reticulum and undergo endoplasmic reticulum-associated degradation.
It is not clear why channels with an α subunit knuckle domain deletion do not exhibit a reduction in channel expression or maturation. One possible explanation is that during biogenesis, subunit-specific interactions with molecular chaperones, as we previously reported (41), make this distinction. These differences are probably not due to a deletion of an N-glycosylation site because all three knuckle domains have a putative N-glycosylation site although at slightly different locations.
The knuckle domain is formed by two α helices connected by a short loop. The sites where Cys substitutions alter Na+ self-inhibition, mapped onto the resolved ASIC1 structure, are present in both helices as well as in a loop preceding the helices and the loop connecting the helices (Fig. 9). Two residues precede and three are within the proximal α helix (Trp520, Pro521, Ile529, Phe530, and Leu533). These are all non-polar amino acids, and the side chains of corresponding residues within the ASIC1 crystal structure are directed toward a hydrophobic pocket between the knuckle and palm domains (Fig. 9, green side chains). Trp520 is conserved in the three ENaC subunits (Fig. 1), and substitutions at this site in the human (Trp493) or rat (Trp520) α subunit similarly stimulate channel activity (27). We speculate that this hydrophobic contact plays a role in the mechanism of Na+ self-inhibition.
FIGURE 9.

α subunit knuckle domain residues mapped on the resolved ASIC1 structure. A, structural model for the extracellular region of an ASIC1 subunit (right) and the enlarged knuckle and upper palm domains (left). Sticks illustrate ASIC1 residues that are homologous to those of α subunit, where Cys substitutions greatly suppressed Na+ self-inhibition. Nonpolar residues are shown in green and polar residues in red. B, knuckle domain sequence alignments of the mouse α subunit and chicken ASIC1. As in A, sites in the α subunit where Cys substitutions greatly suppressed Na+ self-inhibition are shown in green (non-polar) or red (polar). A Gly residue is shown in gray. Tyrosine has both nonpolar (aromatic ring) and polar (hydroxyl group) features. Tyr393 of cASIC1 is shown in green because its nonpolar ring contacts other nonpolar residues. Tyr399 is shown in red because its polar hydroxyl group contacts polar side chains of other residues. C, α helical wheel projections of the α subunit knuckle domain α helices, indicated in B. Non-polar and polar residues are shown in green and red, respectively. The Gly residue is in gray. Stars identify residues where mutations significantly reduced Na+ self-inhibition.
How might alterations at these knuckle domain sites impair the allosteric changes associated with Na+ self-inhibition? In ASIC1, the knuckle domain and upper palm β-strands have been suggested to function as a structurally conserved scaffold (26). We suggest that the hydrophobic contacts between the knuckle and palm domains may function as an anchor to convey conformational changes initiated by Na+ binding to the channel gate. Mutation-induced disruption of this anchor may prevent the synchronized movements of the lower palm domain and thus sever the conformational wave. In agreement with this notion, we found that Na+ self-inhibition was disrupted by Cys substitutions at specific sites that are expected to disrupt hydrophobic packing with the upper palm domain.
Within the intervening loop and distal α helix, Cys substitution at nine sites (Ser534, Leu535, Asn537, Asn538, Tyr539, Thr540, Asn543, Lys544, and Gly547) in the α subunit knuckle domain significantly reduced Na+ self-inhibition (Figs. 6 and 7). The first three sites (Ser534, Leu535, and Asn537) lack corresponding residues in ASIC1 and cannot be mapped to the ASIC1 crystal structure. The next five are sites where Cys substitutions impair Na+ self-inhibition correspond to ASIC1 residues with polar side chains that project toward the finger domain within the same subunit (Fig. 9). There is a low degree of homology between the finger domains in ASIC1 and ENaC subunits, and the α subunit has an additional 73 residues in its finger domain that are absent in ASIC1 (42). Therefore, it is difficult to infer specific knuckle and finger domain interactions based on the resolved ASIC1 structure. However, numerous studies have highlighted the importance of the finger domain in modulating ENaC activity, particularly in response to proteases and extracellular Na+ (14, 24, 40, 43–47). For example, an extracellular Na+ binding site that facilitates Na+ self-inhibition, consisting of the β6-β7 loop where it approaches the finger domain α1 helix, was recently identified (24).
In summary, we identified multiple residues within the α subunit knuckle domain where Cys substitutions alter Na+ self-inhibition. We propose that the α subunit knuckle domain facilitates allosteric structural conformational changes that occur in response to the binding of extracellular Na+. Our results suggest that functionally important residues are organized into two distinct motifs that interact with the palm and finger domains via hydrophobic and polar interactions, respectively.
Author Contributions
S. S. and T. R. K. conceived and coordinated the study and wrote the paper. J. C., E. C. R., and K. L. W. performed and/or analyzed the experiments shown in Figs. 2, 3, 6, 7, and 8. M. E. Y., T. M. B., and J. L. B. designed, performed, and/or analyzed the experiments shown in Fig. 4. C. L. K. and R. P. H. designed, performed, and/or analyzed the experiments shown in Figs. 2C and 5. All authors reviewed the results and approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 ES014701, R37 DK051391, R01 DK098204, R01 GM075061, T32 DK061296, P30 DK079307, and K01 DK90195. The authors declare that they have no conflicts of interest with the contents of this article.
- ENaC
- epithelial Na+ channel
- ASIC
- acid-sensing ion channel
- MBS
- modified Barth's solution.
References
- 1. Soundararajan R., Pearce D., Hughey R. P., Kleyman T. R. (2010) Role of epithelial sodium channels and their regulators in hypertension. J. Biol. Chem. 285, 30363–30369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sheng S., Hallows K. R., Kleyman T. R. (2012) in Seldin and Giebisch's The Kidney: Physiology & Pathophysiology, 5th Ed (Alpern R. J., Caplan M. J., Moe O. W., eds) pp. 983–1017, Academic Press, Inc., New York [Google Scholar]
- 3. Kashlan O. B., Kleyman T. R. (2012) Epithelial Na+ channel regulation by cytoplasmic and extracellular factors. Exp. Cell Res. 318, 1011–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Driessche W., Zeiske W. (1985) Ionic channels in epithelial cell membranes. Physiol. Rev. 65, 833–903 [DOI] [PubMed] [Google Scholar]
- 5. Garty H., Palmer L. G. (1997) Epithelial sodium channels: function, structure, and regulation. Physiol. Rev. 77, 359–396 [DOI] [PubMed] [Google Scholar]
- 6. Chraïbi A., Horisberger J. D. (2002) Na self inhibition of human epithelial Na channel: temperature dependence and effect of extracellular proteases. J. Gen. Physiol. 120, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sheng S., Bruns J. B., Kleyman T. R. (2004) Extracellular histidine residues crucial for Na+ self-inhibition of epithelial Na+ channels. J. Biol. Chem. 279, 9743–9749 [DOI] [PubMed] [Google Scholar]
- 8. Sheng S., Perry C. J., Kleyman T. R. (2004) Extracellular Zn2+ activates epithelial Na+ channels by eliminating Na+ self-inhibition. J. Biol. Chem. 279, 31687–31696 [DOI] [PubMed] [Google Scholar]
- 9. Sheng S., Carattino M. D., Bruns J. B., Hughey R. P., Kleyman T. R. (2006) Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am. J. Physiol. Renal Physiol. 290, F1488–F1496 [DOI] [PubMed] [Google Scholar]
- 10. Maarouf A. B., Sheng N., Chen J., Winarski K. L., Okumura S., Carattino M. D., Boyd C. R., Kleyman T. R., Sheng S. (2009) Novel determinants of epithelial sodium channel gating within extracellular thumb domains. J. Biol. Chem. 284, 7756–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Collier D. M., Snyder P. M. (2009) Extracellular protons regulate human ENaC by modulating Na+ self-inhibition. J. Biol. Chem. 284, 792–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Collier D. M., Snyder P. M. (2009) Extracellular chloride regulates the epithelial sodium channel. J. Biol. Chem. 284, 29320–29325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han D. Y., Nie H. G., Su X. F., Shi X. M., Bhattarai D., Zhao M., Zhao R. Z., Landers K., Tang H., Zhang L., Ji H. L. (2011) 8-(4-Chlorophenylthio)-guanosine-3′,5′-cyclic monophosphate-Na stimulates human alveolar fluid clearance by releasing external Na+ self-inhibition of epithelial Na+ channels. Am. J. Respir. Cell Mol. Biol. 45, 1007–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kashlan O. B., Kleyman T. R. (2011) ENaC structure and function in the wake of a resolved structure of a family member. Am. J. Physiol. Renal Physiol. 301, F684–F696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Warnock D. G., Kusche-Vihrog K., Tarjus A., Sheng S., Oberleithner H., Kleyman T. R., Jaisser F. (2014) Blood pressure and amiloride-sensitive sodium channels in vascular and renal cells. Nat. Rev. Nephrol. 10, 146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sheng S., Maarouf A. B., Bruns J. B., Hughey R. P., Kleyman T. R. (2007) Functional role of extracellular loop cysteine residues of the epithelial Na+ channel in Na+ self-inhibition. J. Biol. Chem. 282, 20180–20190 [DOI] [PubMed] [Google Scholar]
- 17. Winarski K. L., Sheng N., Chen J., Kleyman T. R., Sheng S. (2010) Extracellular allosteric regulatory subdomain within the γ subunit of the epithelial Na+ channel. J. Biol. Chem. 285, 26088–26096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kashlan O. B., Boyd C. R., Argyropoulos C., Okumura S., Hughey R. P., Grabe M., Kleyman T. R. (2010) Allosteric inhibition of the epithelial Na+ channel through peptide binding at peripheral finger and thumb domains. J. Biol. Chem. 285, 35216–35223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi S., Ghosh D. D., Okumura S., Carattino M. D., Kashlan O. B., Sheng S., Kleyman T. R. (2011) Base of the thumb domain modulates epithelial sodium channel gating. J. Biol. Chem. 286, 14753–14761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Collier D. M., Snyder P. M. (2011) Identification of epithelial Na+ channel (ENaC) intersubunit Cl-inhibitory residues suggests a trimeric αγβ channel architecture. J. Biol. Chem. 286, 6027–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Edelheit O., Hanukoglu I., Dascal N., Hanukoglu A. (2011) Identification of the roles of conserved charged residues in the extracellular domain of an epithelial sodium channel (ENaC) subunit by alanine mutagenesis. Am. J. Physiol. Renal Physiol. 300, F887–F897 [DOI] [PubMed] [Google Scholar]
- 22. Shi S., Carattino M. D., Kleyman T. R. (2012) Role of the wrist domain in the response of the epithelial sodium channel to external stimuli. J. Biol. Chem. 287, 44027–44035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen J., Kleyman T. R., Sheng S. (2013) Gain-of-function variant of the human epithelial sodium channel. Am. J. Physiol. Renal Physiol. 304, F207–F213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kashlan O. B., Blobner B. M., Zuzek Z., Tolino M., Kleyman T. R. (2015) Na+ inhibits the epithelial Na+ channel by binding to a site in an extracellular acidic cleft. J. Biol. Chem. 290, 568–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jasti J., Furukawa H., Gonzales E. B., Gouaux E. (2007) Structure of acid-sensing ion channel 1 at 1.9 Å resolution and low pH. Nature 449, 316–323 [DOI] [PubMed] [Google Scholar]
- 26. Baconguis I., Gouaux E. (2012) Structural plasticity and dynamic selectivity of acid-sensing ion channel-spider toxin complexes. Nature 489, 400–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rauh R., Diakov A., Tzschoppe A., Korbmacher J., Azad A. K., Cuppens H., Cassiman J. J., Dotsch J., Sticht H., Korbmacher C. (2010) A mutation of the epithelial sodium channel associated with atypical cystic fibrosis increases channel open probability and reduces Na+ self inhibition. J. Physiol. 588, 1211–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen J., Kleyman T. R., Sheng S. (2014) Deletion of α-subunit exon 11 of the epithelial Na+ channel reveals a regulatory module. Am. J. Physiol. Renal Physiol. 306, F561–F567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen J., Myerburg M. M., Passero C. J., Winarski K. L., Sheng S. (2011) External Cu2+ inhibits human epithelial Na+ channels by binding at a subunit interface of extracellular domains. J. Biol. Chem. 286, 27436–27446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carattino M. D., Hill W. G., Kleyman T. R. (2003) Arachidonic acid regulates surface expression of epithelial sodium channels. J. Biol. Chem. 278, 36202–36213 [DOI] [PubMed] [Google Scholar]
- 31. Firsov D., Schild L., Gautschi I., Mérillat A. M., Schneeberger E., Rossier B. C. (1996) Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc. Natl. Acad. Sci. U.S.A. 93, 15370–15375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hughey R. P., Mueller G. M., Bruns J. B., Kinlough C. L., Poland P. A., Harkleroad K. L., Carattino M. D., Kleyman T. R. (2003) Maturation of the epithelial Na+ channel involves proteolytic processing of the α- and γ-subunits. J. Biol. Chem. 278, 37073–37082 [DOI] [PubMed] [Google Scholar]
- 33. Kashlan O. B., Mueller G. M., Qamar M. Z., Poland P. A., Ahner A., Rubenstein R. C., Hughey R. P., Brodsky J. L., Kleyman T. R. (2007) Small heat shock protein αA-crystallin regulates epithelial sodium channel expression. J. Biol. Chem. 282, 28149–28156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Z., Subramanya A. R., Satlin L. M., Pastor-Soler N. M., Carattino M. D., Kleyman T. R. (2013) Regulation of large-conductance Ca2+-activated K+ channels by WNK4 kinase. Am. J. Physiol. Cell Physiol. 305, C846–C853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adams A., Gottschling D. E. (1997) Methods in Yeast Genetics: A Laboratory Course Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 36. Buck T. M., Kolb A. R., Boyd C. R., Kleyman T. R., Brodsky J. L. (2010) The endoplasmic reticulum-associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones. Mol. Biol. Cell 21, 1047–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Y., Nijbroek G., Sullivan M. L., McCracken A. A., Watkins S. C., Michaelis S., Brodsky J. L. (2001) Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol. Biol. Cell 12, 1303–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hughey R. P., Bruns J. B., Kinlough C. L., Kleyman T. R. (2004) Distinct pools of epithelial sodium channels are expressed at the plasma membrane. J. Biol. Chem. 279, 48491–48494 [DOI] [PubMed] [Google Scholar]
- 39. Heidrich E., Carattino M. D., Hughey R. P., Pilewski J. M., Kleyman T. R., Myerburg M. M. (2015) Intracellular Na+ regulates epithelial Na+ channel maturation. J. Biol. Chem. 290, 11569–11577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hughey R. P., Bruns J. B., Kinlough C. L., Harkleroad K. L., Tong Q., Carattino M. D., Johnson J. P., Stockand J. D., Kleyman T. R. (2004) Epithelial sodium channels are activated by furin-dependent proteolysis. J. Biol. Chem. 279, 18111–18114 [DOI] [PubMed] [Google Scholar]
- 41. Buck T. M., Plavchak L., Roy A., Donnelly B. F., Kashlan O. B., Kleyman T. R., Subramanya A. R., Brodsky J. L. (2013) The Lhs1/GRP170 chaperones facilitate the endoplasmic reticulum-associated degradation of the epithelial sodium channel. J. Biol. Chem. 288, 18366–18380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kashlan O. B., Adelman J. L., Okumura S., Blobner B. M., Zuzek Z., Hughey R. P., Kleyman T. R., Grabe M. (2011) Constraint-based, homology model of the extracellular domain of the epithelial Na+ channel α subunit reveals a mechanism of channel activation by proteases. J. Biol. Chem. 286, 649–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carattino M. D., Sheng S., Bruns J. B., Pilewski J. M., Hughey R. P., Kleyman T. R. (2006) The epithelial Na+ channel is inhibited by a peptide derived from proteolytic processing of its α subunit. J. Biol. Chem. 281, 18901–18907 [DOI] [PubMed] [Google Scholar]
- 44. Bruns J. B., Carattino M. D., Sheng S., Maarouf A. B., Weisz O. A., Pilewski J. M., Hughey R. P., Kleyman T. R. (2007) Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the γ-subunit. J. Biol. Chem. 282, 6153–6160 [DOI] [PubMed] [Google Scholar]
- 45. Kleyman T. R., Carattino M. D., Hughey R. P. (2009) ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J. Biol. Chem. 284, 20447–20451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rossier B. C., Stutts M. J. (2009) Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu. Rev. Physiol. 71, 361–379 [DOI] [PubMed] [Google Scholar]
- 47. Shi S., Carattino M. D., Hughey R. P., Kleyman T. R. (2013) ENaC regulation by proteases and shear stress. Curr. Mol. Pharmacol. 6, 28–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gonzales E. B., Kawate T., Gouaux E. (2009) Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature 460, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3, Schrödinger, LLC, New York [Google Scholar]