

Background: The role of the pyranopterin component of the mononuclear molybdenum cofactor is largely unknown.
Results: Variants of pyranopterin-coordinating amino acid residues were generated, and their effects on electrochemistry/catalysis investigated.
Conclusion: The pyranopterin environment modulates molybdenum electrochemistry.
Significance: The pyranopterin coordination environment enables redox-tuning of the molybdenum atom, and facilitates molybdoenzyme reactivity toward a broad range of substrates.
Keywords: electrochemistry, electron paramagnetic resonance (EPR), electron transfer, molybdenum, reductase, charge-transfer, EPR, nitrate reductase, redox tuning
Abstract
We test the hypothesis that pyranopterin (PPT) coordination plays a critical role in defining molybdenum active site redox chemistry and reactivity in the mononuclear molybdoenzymes. The molybdenum atom of Escherichia coli nitrate reductase A (NarGHI) is coordinated by two PPT-dithiolene chelates that are defined as proximal and distal based on their proximity to a [4Fe-4S] cluster known as FS0. We examined variants of two sets of residues involved in PPT coordination: (i) those interacting directly or indirectly with the pyran oxygen of the bicyclic distal PPT (NarG-Ser719, NarG-His1163, and NarG-His1184); and (ii) those involved in bridging the two PPTs and stabilizing the oxidation state of the proximal PPT (NarG-His1092 and NarG-His1098). A S719A variant has essentially no effect on the overall Mo(VI/IV) reduction potential, whereas the H1163A and H1184A variants elicit large effects (ΔEm values of −88 and −36 mV, respectively). Ala variants of His1092 and His1098 also elicit large ΔEm values of −143 and −101 mV, respectively. An Arg variant of His1092 elicits a small ΔEm of +18 mV on the Mo(VI/IV) reduction potential. There is a linear correlation between the molybdenum Em value and both enzyme activity and the ability to support anaerobic respiratory growth on nitrate. These data support a non-innocent role for the PPT moieties in controlling active site metal redox chemistry and catalysis.
Introduction
Enzymes containing the mononuclear molybdenum cofactor (molybdenum-enzymes) support an extraordinary diversity of redox transitions spanning a reduction potential range of approximately 1 volt (1, 2). Their active sites comprise a molybdenum atom coordinated by one or two pyranopterin dithiolene chelates, with additional metal coordination being provided by oxo/sulfido groups and/or oxygen/sulfur atoms derived from amino acid side chains (3, 4). Molybdenum-enzymes fall into three major families, each of which has a distinct protein-fold: the sulfite oxidases and plant nitrate reductases (the SUOX family)2 (5); the xanthine dehydrogenases and carbon-monoxide dehydrogenases (the XDH family (6)); and enzymes related to the bacterial DMSO reductases (the DMSOR family (6, 7)). The metal atom of the SUOX and XDH families is coordinated by a single pyranopterin dithiolene chelate, whereas in the DMSOR family it is coordinated by two such moieties (6–8). The situation is further complicated by the presence of a cytosine nucleotide attached via a phosphodiester linkage to the methyl group of the pyranopterin in some members of the XDH family (9), and by the presence of a guanine nucleotide similarly attached to each pyranopterin in members of the DMSOR family (6–8). The precise function of the pyranopterin component of the molybdenum cofactor remains unclear: it clearly functions as part of the molecular scaffold holding the molybdenum atom in an optimum position for catalysis (10, 11); and it has recently been proposed that it may also modulate molybdenum reduction potentials and hence reactivity via alternate oxidation states (e.g. dihydro- versus tetrahydro- oxidation states) (8, 12–16).
Evolutionary analyses have established the presence of the DMSOR protein-fold in the “last universal common ancestor” from which all extant species evolved (17, 18). An important consequence of this is that the fold has evolved to support catalytic functions encompassing those necessary prior to and after the evolution of oxygenic photosynthetic water oxidation (19). This process likely encompassed substitutions of the protein-metal ligands, modulation of the metal coordination sphere via the bis-dithiolene chelate, and modification of the pyranopterin coordination environment. The DMSOR family is able to support a broad range of redox transitions including the following: oxidation of formate, arsenite, ethylbenzene, and dimethyl sulfide; and the reduction of polysulfide, nitrate, chlorate, trimethylamine-N-oxide, and dimethyl sulfoxide (1, 6–8). A plausible hypothesis explaining the presence of the second pyranopterin in the DMSOR family is that it adds additional facets of fine tuning to facilitate reactivity toward the observed broad range of substrates. The pyranopterin coordination environments may play a critical role in modulating dithiolene chelate electrostatics via differences in hydrogen bonding environment, conformation, and oxidation state (8, 12–15, 20, 21). Although much attention has been paid to the role of the immediate metal environment in defining catalysis (10, 22–24), little is known about the role of the pyranopterin coordination environment.
The molybdenum cofactor found in the DMSOR family is a molybdo-bis(pyranopterin guanine dinucleotide) (Mo-bisPGD)(1, 6–8). The pyranopterins are referred to as proximal and distal based on their positions relative to a conserved [4Fe-4S] cluster, known as FS0, which is present in almost every member of the DMSOR enzyme family. In enzymes for which high-resolution structures are available, the proximal pyranopterin has a more distorted conformation than that of the distal pyranopterin, consistent with the former being in a fully reduced tetrahydro-form; and with the latter having accessibility to the partially oxidized 10,10a-dihydro form (15). In two members of the family, Escherichia coli respiratory nitrate reductase (NarGHI) (25) and Aromatoleum aromaticum ethylbenzene dehydrogenase (EbdABC) (26), high-resolution protein structures reveal that the distal pyranopterin has a bicyclic structure with an open pyran ring.3 These observations raise the question of the role of the alternate open pyran ring structure in defining active site redox chemistry and catalysis. In the case of NarGHI, protein crystallography reveals that the open pyran ring oxygen participates in hydrogen bonding interactions with two conserved residues in NarG: NarG-His1163 and NarG-Ser719 (3) (Fig. 1A). The distances between the Ser719 OG proton and the pyran oxygen and between the His1163 NE2 proton and the pyran oxygen are both ∼2 Å. Closer inspection reveals the presence of a second conserved His residue (NarG-His1184) that completes a charge-transfer relay connecting the pyran oxygen with three structurally conserved water molecules at its distal end (Fig. 1). It would therefore be of interest to generate variants of NarG-Ser719, -His1163, and -His1184 and determine their effects on molybdenum electrochemistry and catalysis.
FIGURE 1.

A, charge-transfer relay in NarG. Three water molecules are conserved across a range of NarGHI structures, including those described by Protein Data Bank codes 1Q16, 1R27, 1YZ4 and 1Y5I (25, 60, 61). These waters are linked to the pyran oxygen of the bicyclic distal pyranopterin by NarG-His1184 and NarG-His1163. The OG oxygen of NarG-Ser719 is also within H-bonding distance of the pyran oxygen. The image was generated using the PyMol molecular visualization package (version 1.4.1, Schrödinger LLC). B, mechanism of charge-transfer between the pyran oxygen and the conserved water molecules. The indicated charge-transfer results in protonation of the alkoxide anion form of the pyran oxygen. The image was generated using the MarvinSketch software package (www.chemaxon.com). In both panels, some predicted hydrogens have been added for clarity.
We recently examined the coordination environment of the pyranopterin piperazine nitrogens in members of the DMSOR family for which high-resolution structures are available (8, 15), revealing that all members of the family contain a His or Arg residue that bridges the two piperazine N-5 atoms. With the exception of the Thermus thermophilus polysulfide reductase (27), all DMSOR-fold enzyme structures obtained to date contain an additional residue (a His/Ser/Gln) that stabilizes the N-5 of the proximal pyranopterin in an sp3 hybridized state. Fig. 2A illustrates piperazine nitrogen coordination of the NarGHI pyranopterins. The N-10 atoms of both piperazines are hydrogen-bond donors to backbone amide oxygens (of NarG-Thr259 and NarG-Ser720). The N-5 atoms are bridged by the imidazole of NarG-His1092, with the N-5 atom of the proximal pyranopterin participating in an additional H-bond with the NE2 nitrogen of NarG-His1098. These two residues are referred to as the“bridging” and “stabilizing” histidines, respectively, and their H-bonding contacts are summarized in Fig. 2B. That almost all of the proximal N-5 atoms in the DMSOR family have two hydrogen bonding contacts favors a tetrahydro oxidation state for the proximal pyranopterin. The single H-bonding contact observed for the N-5 atoms of all the distal pyranopterins provides further support for the suggestion that these have accessibility to the 10,10a-dihydro oxidation state (8, 15). In some members of the DMSOR family (e.g. E. coli formate dehydrogenase N (FdnGHI (28)) and the periplasmic nitrate reductase from Cupriavidus necator (NapAB (29))), the bridging ligand is an Arg residue. It would therefore be of interest to generate variants of His1092 and His1098 and determine their effects on molybdenum electrochemistry and catalysis.
FIGURE 2.
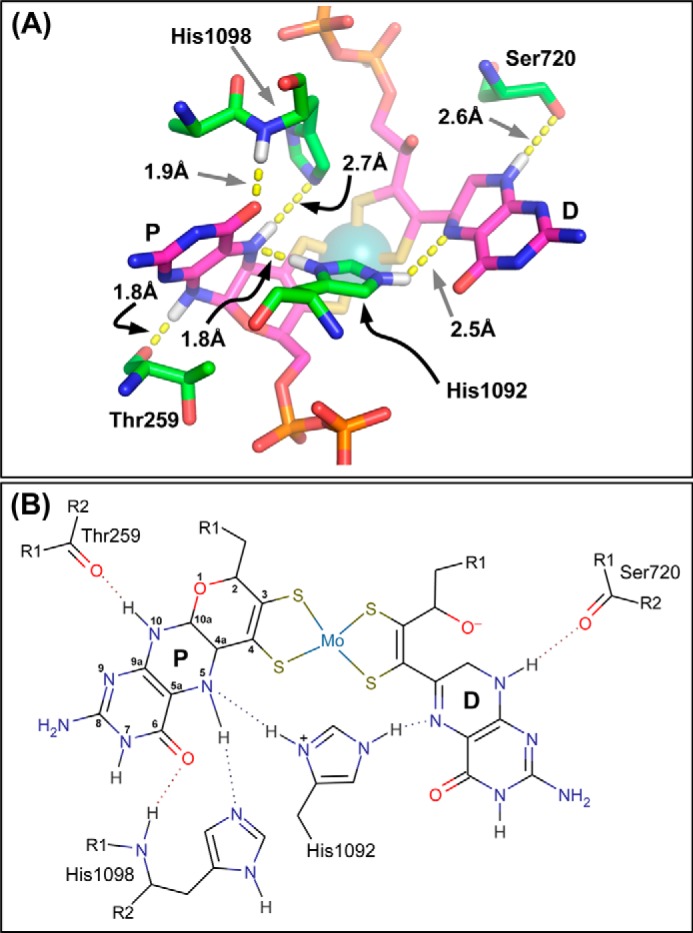
A, residues defining pyranopterin piperazine ring coordination. The image was generated using the NarGHI structure described by Protein Data Bank code 1Q16 (25). His1092 functions to bridge the two pyranopterins, with its ND1 nitrogen functioning as an H-bond donor to the proximal piperazine N-5 atom, and its NE2 nitrogen functioning as an H-bond donor to the distal piperazine N-5 atom. Note that His1092 is shown in its protonated form to facilitate H-bond donation to both piperazine N-5 atoms. For further details, see text. B, proposed H-bonding network around the piperazine rings of the two pyranopterins. The proximal pyranopterin (labeled P) is shown in its tetrahydro form, with this redox state stabilized by both the bridging His1092 and the stabilizing His1098, whereas the piperazine of the distal pyranopterin (labeled D) is shown in a form equivalent to the 10,10a-dihydro pyranopterin. In both panels, some predicted hydrogens have been added for clarity. Images were generated as described in the legend to Fig. 1.
In this paper, we tested the hypothesis that the pyranopterins of the DMSOR family of molybdoenzymes have a non-innocent role in defining molybdenum electrochemistry and catalysis. If the role of the pyranopterin dithiolene chelates is merely to contribute to a molybdenum-binding scaffold at the active site, then variants of the target residues studied herein should have little or no effect on enzyme function. If, however, the non-innocence reported for model compounds extends beyond the dithiolene chelate and into the heterocyclic ring system of the pyanopterin (13–15, 20), variants of pyranopterin-coordinating residues should have a dramatic effect on molybdenum electrochemistry and catalysis.
Experimental Procedures
Bacterial Strains and Plasmids
E. coli LCB79 (araD139 Δ(lacIPOZYA-argF) rpsL, thi Ø79(nar-lac)) (62) was used as the host for all the experiments described herein. NarGHI was expressed from the plasmid pVA700 (63).
Site-directed Mutagenesis
Mutant plasmids were generated using the QuikChange site-directed mutagenesis kit from Stratagene. DpnI was purchased from Invitrogen, and DNA purification kits were purchased from Qiagen. Mutants were verified by DNA sequencing (DNA Core Facility, Department of Biochemistry, University of Alberta). Preparation of competent cells and their transformations with plasmids were carried out as previously described (30).
Generation of a NarG-S719A Variant
A 4.5-kbp SacI-AvaI fragment of pVA700 was subcloned into pTZ18R and the resultant plasmid was used for site-directed mutagenesis. A 3.3-kbp SacI-NcoI fragment was cloned back into the pVA700 expression vector, creating pVA700/S719A.
Generation of NarG-H1163A and NarG-H1184A Variants
A 4.6-kbp EcoRI-SacII fragment of pVA700 was subcloned into pBlueScript to generate a template for site-directed mutagenesis. A 2.1-kb NcoI-SacII fragment was cloned back into the pVA700, creating pVA700/H1163A or pVA700/H1184A.
Generation of NarG-H1092A, NarG-H1092R, and NarG-H1098A Variants
The EcoRI-SacII fragment cloned into pBlueScript was used as a site-directed mutagenesis template, after which a 1.2-kbp NcoI/BstBI fragment was cloned back into the pVA700, creating pVA700/H1092A, pVA700/H1092R, or pVA700/H1098A.
Growth of Cells
Cells were grown microaerobically in 2-liter batch cultures of Terrific Broth (30) at 30 °C in the presence of 100 μg ml−1 streptomycin and ampicillin. A 10% innoculum of a stationary phase culture was used and NarGHI overexpression was induced by addition of 0.2 mm isopropyl 1-thio-β-d-galactopyranoside. Following addition of the innoculum and isopropyl 1-thio-β-d-galactopyranoside, cells were grown overnight with gentle shaking, and were harvested by centrifugation, and subsequently washed in a buffer containing 100 mm MOPS and 5 mm EDTA (pH 7.0). Cells were resuspended in buffer, and phenylmethylsulfonyl fluoride was added to a final concentration of 0.2 mm. Cell lysis was achieved by three passages through an Emulsiflex C3 microfluidizer (Avestin) at a pressure of 17,000 p.s.i. Crude membranes were prepared by differential centrifugation as previously described (31). These were resuspended in buffer and layered on top of a 55% (w/v) sucrose step (made up in buffer). Following ultracentrifugation at 40,000 rpm for 1.5 h, the floating band enriched in cytoplasmic membrane vesicles was collected. Excess sucrose was removed by two dilution and ultracentrifugation steps. The final membrane pellet was resuspended in buffer to a concentration of ∼30 mg ml−1, and flash frozen in liquid nitrogen prior to being stored at −70 °C until use. Where appropriate, buffer exchange prior to EPR analysis was achieved by dilution and re-centrifugation. Overexpression of NarGHI was evaluated by polyacrylamide gel electrophoresis (32).
Bacterial Growth on Glycerol-Nitrate Minimal Medium
Anaerobic growth of E. coli harboring LCB79/pVA700 and mutant derivatives was assessed in a glycerol-nitrate minimal medium essentially as previously described (33, 34). Nitrate was added (as KNO3) to a final concentration of 40 mm. Growth was evaluated at 37 °C, and culture turbidity was measured using a Klett-Summerson spectrophotometer equipped with a number 66 filter. Maximal growth rates were calculated as described by Zwietering et al. (35), and were expressed as the parameter μm in units of (Klett units) h−1.
Form A Fluorescence of the Pyranopterin Cofactor
Pyranopterin was assayed by generation of its Form A fluorescent derivative following protein denaturation (36, 37), essentially as previously described (33). As starting material, 10 mg of membrane protein was used.
Redox Potentiometry and EPR Spectroscopy
Redox titrations were carried out under argon at 25 °C as previously described (38, 39) in 100 mm Tricine and 5 mm EDTA (pH 8.0). The protein concentration used was ∼30 mg ml−1. The following redox mediators were used at a concentration of 25 μm: quinhydrone, 2,6-dichloroindophenol, 1,2-naphthoquinone, toluene blue, phenazine methosulfate, thionine, methylene blue, resorufin, indigotrisulfonate, indigocarmine, anthraquinone-2-sulfonic acid, phenosafranine, and neutral red. All samples were prepared in quartz EPR tubes with a 3-mm internal diameter, and were rapidly frozen in liquid nitrogen-chilled ethanol and stored at 77 K until use. EPR spectra were recorded at 150 K using a Bruker Elexsys E500 spectrometer equipped with a Bruker SHQE microwave cavity and a Bruker ER4131 Variable Temperature Unit using liquid nitrogen as a cryogen. EPR spectra were plotted as signal intensity versus g value, with the latter being calculated from the microwave frequency and the field intensity for each sample studied. This allowed for direct comparison of the g values of the individual spectral features and corrected for minor variations in microwave frequency (Fig. 3). Potentiometric titration data were analyzed by plotting the intensity of the molybdenum peak-trough versus Eh and fitting the data to two Em values (E1 and E2) (40). Mo(V) stability constants (Kstab) were calculated as described by Ohnishi et al. (41). Fitting was performed using the Solver for Nonlinear Programming extension of Libre Office Calc. Where appropriate the statistical significance of differences in Em values was evaluated using a t test.
FIGURE 3.

Mo(V) EPR spectra of redox-poised NarGHI variants of residues involved in pyranopterin coordination. A, Ala variants of NarG-Ser719, NarG-His1163, and NarG-His1184; and B, Ala variants of His1092 and His1098, and an Arg variant of His1092. Spectra were recorded at 150 K using a microwave power of 20 milliwatts (at a frequency of ∼9.332 GHz), a modulation amplitude of 4 Gpp at a frequency of 100 kHz. Approximate g1,2,3 values and potentials at which the samples are poised are: 1.988, 1.982, 1.962 (wild-type, Eh = 150 mV); 1.988, 1.982, 1.962 (S719A, Eh = 152 mV); 1.992, 1.985, 1.964 (H1163A, Eh = 48 mV); 1.986, 1.980, 1.962 (H1184A, Eh = 103 mV); 1.978, 1.962, 1.945 (H1092A, Eh = −5 mV), 1.989, 1.984, 1.965 (His1092Arg, Eh = 146 mV); 1.992, 1.985, 1.951 (H1098A, Eh = 28 mV).
Protein Assays
Protein concentrations were assayed as described by Lowry et al. (42) modified by the inclusion of 1% (w/v) sodium dodecyl sulfate in the incubation mixture to solubilize membrane proteins (43).
Enzyme Assays
Nitrate reductase activities were measured using the nonspecific electron donor benzyl viologen (44). Assays were carried out at pH 8.0 in a buffer containing 100 mm Tricine, 0.23 mm benzyl viologen, 5 mm EDTA, and 4 mm KNO3. Benzyl viologen was reduced by adding an excess of sodium dithionite (∼0.4 mm), and the reaction was initiated by addition of a catalytic amount of enzyme.
Results and Discussion
Residues Targeted for Site-directed Mutagenesis
During nitrate reduction, the molybdenum atom of the Mo-bisPGD cycles through the IV, V, and VI oxidation states to catalyze the two-electron reduction of nitrate to nitrite (45). To investigate the effect of pyranopterin coordination on molybdenumelectrochemistry, we generated variants of two sets of pyranopterin-coordinating residues in NarG: (i) those interacting directly or indirectly with the oxygen of the open pyran ring of the distal pyranopterin (NarG-Ser719, NarG-His1163, and NarG-His1184; Fig. 1); and (ii) those involved in bridging the proximal and distal pyranopterins and stabilizing the oxidation state of the proximal pyranopterin (NarG-His1092 and NarG-His1098; Fig. 2). As judged by SDS-PAGE and Form A pyranopterin assays, each variant was expressed in a Mo-bisPGD-containing form at levels comparable with that of the wild-type. We used these variants to test the hypothesis that residues coordinating the pyranopterin moiety of the Mo-bisPGD cofactor play a critical role in defining molybdenum electrochemistry and substrate reactivity.
Impact of the Variants on the NarG Mo(V) EPR Spectrum
Of the three accessible molybdenum oxidation states, only the intermediate Mo(V) state is paramagnetic and EPR visible. It can be observed in spectra recorded of samples poised at appropriate reduction potentials. Because the NarGHI Mo(V) signal is sensitive to pH in an anion-dependent way (46, 47), we recorded spectra of samples poised at pH 8.0, wherein the so-called “high pH” form is dominant. This form exhibits a simple rhombic EPR spectrum with an absence of resolved hyperfine (1H) couplings (46), simplifying subsequent analyses of potentiometric data (see below).
Fig. 3A shows spectra of redox-poised samples containing the wild-type enzyme and variants of residues implicated in coordinating the open pyran oxygen of the distal pyranopterin (S719A, H1163A, and H1184A). These spectra are similar to those previously observed for the Mo(V) species of NarGHI (46, 47), wherein a rhombic Mo(V) species is observed with g1,2,3 values of ∼1.988, 1.982, and 1.962. As was the case in previous studies, features consistent with the presence of a small amount of the “low pH” form of the Mo(V) are observed immediately downfield from the high-pH g1 (46, 47), but these are inconsistently observed in the variants studied herein. Also in agreement with previous work (46–48), no hyperfine coupling was resolved with the isotopes of molybdenum having a nuclear spin of 5/2 (95Mo and 97Mo, with natural abundances of ∼16 and 10%, respectively). These data indicate that the S719A, H1163A, and H1184A variants do not significantly impact the molybdenum coordination sphere.
Fig. 3B shows spectra of redox-poised samples of variants of residues implicated in bridging the two pyranopterins (H1092A and H1092R), or stabilizing the proximal pyranopterin in its tetrahydro form (H1098A). Of these, the H1092A variant has the most dramatic effect on the Mo(V) EPR spectrum, shifting g2 upfield from approximately g = 1.982 (wild-type) to approximately g = 1.962 (H1092A). The H1092R variant exhibits an EPR spectrum wherein heterogeneity is observed, especially in the g3 feature. Finally, the H1098A variant exhibits a spectrum with clearly defined g1,2,3 values, with greater rhombicity than that of the wild-type. Overall, Mo(V) species are clearly visible in all of the NarGHI variants studied herein, with the H1092A variant having the greatest effect on the Mo(V) EPR spectrum (see below).
Influence of the NarGHI Variants on Molybdenum Electrochemistry
Molybdenum redox cycling is defined by two reduction potentials, E1 and E2, corresponding to the Mo(VI/V) and Mo(V/IV) reduction potentials, with the average of these being defined as the overall reduction potential (Em). The difference between E1 and E2 is a reflection of the stability of the intermediate Mo(V) species, and can be used to calculate its stability constant (Kstab) (41). We performed potentiometric titrations on all the variants of NarGHI studied herein, and in each case titrations were carried out on samples generated from 3 to 4 independent biological replicates. Fig. 4A shows representative titrations for the variants of residues interacting with the O-1 pyran oxygen (Fig. 1). Fig. 4B shows plots of the residuals between the experimental data and the fits. For the wild-type enzyme, we estimate values of Em and Kstab of 142 mV and 28, respectively (summarized in Fig. 4C). These values are in reasonable agreement with those previously obtained (38, 49–51). The S719A variant has a statistically insignificant effect on the Em (ΔEm = 4 mV, p = 0.57) and decreases the Kstab from 28 to 8, whereas the H1163A variant elicits a large decrease in the Em (ΔEm = −88 mV, Table 1) and a concomitant decrease in the Kstab from 28 to 1. The H1184A variant has an intermediate effect, eliciting a ΔEm of −36 mV, and decreases the Kstab, again from 28 to 1. Thus, the charge-transfer relay depicted in Fig. 1B appears to play two critical roles: (i) modulating the molybdenum reduction potentials (see below for further discussion of this); and (ii) stabilizing the Mo(V) intermediate.
FIGURE 4.
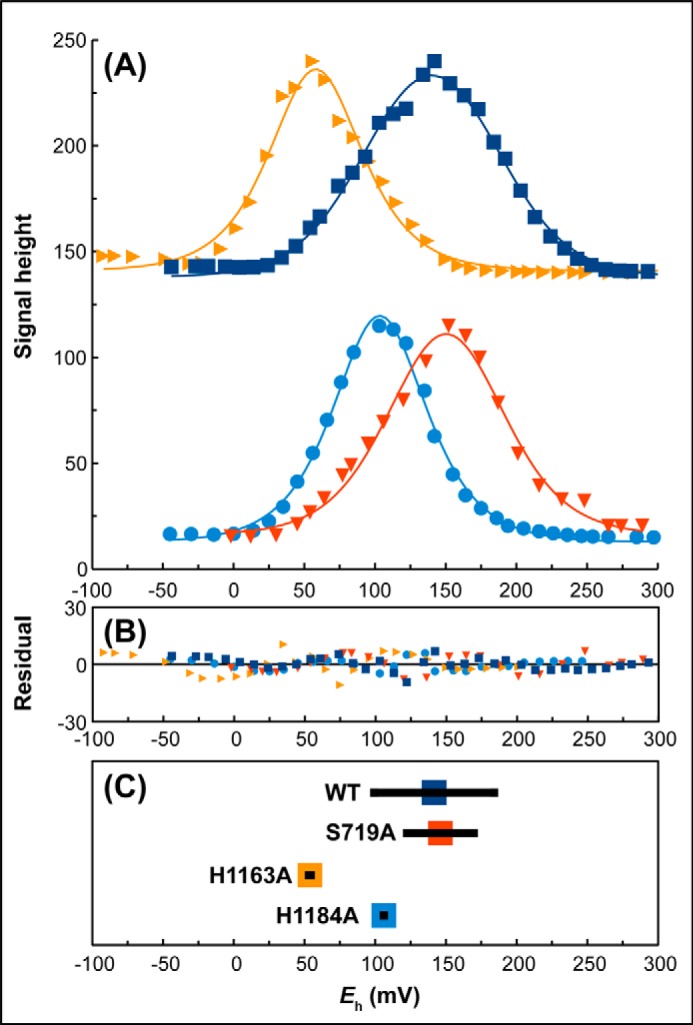
Potentiometric titrations of membranes containing variants of residues coordinating the distal pyranopterin of NarGHI. A, comparison of representative titrations of wild-type (■), S719A (▾), H1163A (▶), and H1184A (●) enzymes. Data were fit to the following parameters: wild-type, Em = 140 mV (E1 = 183 mV, E2 = 97 mV); S719A, Em = 150 mV (E1 = 176 mV, E2 = 124 mV); H1163A, Em = 58 mV (E1 = 49 mV, E2 = 68 mV); H1184A, Em = 103 mV (E1 = 96 mV, E2 = 111 mV). B, plot of residuals. C, summary of Em, E1, and E2 values for the variants based on the data presented in Table 1. Colored squares indicate the mean Em values and the horizontal lines indicate the E1 and E2 values in the cases where E1 − E2 > 0 (Kstab ≥ 1).
TABLE 1.
Effects of variants of pyranopterin-coordinating residues on molybdenum reduction potentials and enzyme activity
| Variant | Ema | ΔEmb | E1c | E2d | Kstab | Maximal growth rate | BV−:NO3− activity |
|---|---|---|---|---|---|---|---|
| mV | μm Klett units) h−1 | μmol min−1 mg−1 | |||||
| Wild-type | 142 ± 12 (n = 4) | 0 | 184 ± 10 | 99 ± 16 | 28 | 15.4 ± 0.7 (n = 3) | 48 ± 14 (n = 3) |
| S719A | 146 ± 7 (n = 3) | 4 | 170 ± 19 | 122 ± 7 | 8 | 15.5 ± 1.0 (n = 3) | 54 ± 11 (n = 3) |
| H1163A | 54 ± 3 (n = 4) | −88 | 53 ± 5 | 54 ± 9 | 1 | 6 ± 0.2 (n = 3) | 23 ± 4 (n = 3) |
| H1184A | 106 ± 5 (n = 3) | −36 | 106 ± 14 | 106 ± 5 | 1 | 11.4 ± 0.6 (n = 3) | 36 ± 1 (n = 3) |
| H1092A | −1 ± 3 (n = 3) | −143 | 35 ± 9 | −38 ± 11 | 19 | 4.4 ± 0.5 (n = 2) | 1 ± 0 (n = 3) |
| H1092R | 160 ± 7 (n = 3) | 18 | 225 ± 15 | 95 ± 2 | 171 | 12.8 ± 0.4 (n = 2) | 8.6 ± 1 (n = 3) |
| H1098A | 41 ± 4 (n = 4) | −101 | 139 ± 7 | −57 ± 2 | 1822 | 4.8 ± 0.5 (n = 2) | 16 ± 2 (n = 3) |
a Overall Mo(VI/V/IV) reduction potential at pH 8.0.
b Em (variant) minus Em (wild-type). Where appropriate, p values indicating the statistical significance of each ΔEm are given in the text.
c Mo(VI/V) reduction potential.
d Mo(V/IV) reduction potential.
Fig. 5A shows representative potentiometric titrations of the H1092A, H1092R, and H1098A variants. The H1092R variant replaces the bridging His residue with an Arg, and mimics the pyranopterin-coordination environment observed in the formate dehydrogenases and periplasmic nitrate reductases (e.g. FdnGHI and NapAB, respectively (8)). The H1092R variant elicits a modest increase of marginal statistical significance in the overall molybdenum Em (ΔEm = 18 mV, p = 0.07; Table 1), consistent with there being no change in overall charge of the bridging residue when it is changed from a His to an Arg (the pKa of the Arg guanidinium group is ∼12.5). This suggests that the imidazole pKa is significantly increased from ∼6.0 in aqueous solution to >8.0 in the bridging environment between the two pyranopterins. The H1092A variant elicits a ΔEm on the molybdenum center of approximately −143 mV along with a modest decrease in its Kstab (from 28 to 19, Table 1). A possible explanation for the large negative ΔEm is that the void between the two pyranopterins contains waters in the H1092A variant, and that these waters lack an overall positive charge, thus decreasing the overall molybdenum reduction potential. Alternatively, given that the H1092A variant has the most severe effect on the Mo(V) EPR spectrum, loss of “scaffolding” by the bridging imidazole moiety may result in a significant alteration of the geometry of dithiolene coordination that may cause the large observed ΔEm.
FIGURE 5.
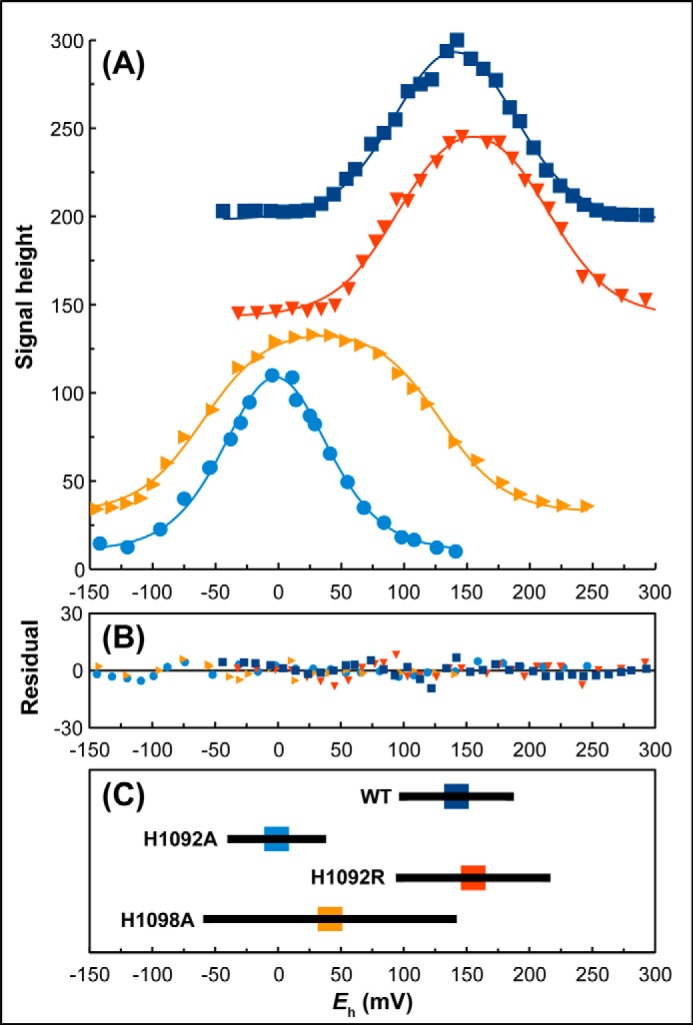
Potentiometric titrations of membranes containing variants of His1092 and His1098. A, comparison of representative titrations of wild-type (■), H1092R (▾), H1098A (▶), and H1092A (●) enzymes. Data were fit to the following parameters: wild-type, Em = 140 mV (E1 = 183 mV, E2 = 97 mV); H1092R, Em = 155 mV (E1 = 213 mV, E2 = 97 mV); H1098A, Em = 35 mV (E1 = 129 mV, E2 = −60 mV); H1092A, Em = −2 mV (E1 = 24 mV, E2 = −29 mV). B, plot of residuals. C, summary of Em, E1, and E2 values for the variants based on the data presented in Table 1. Colored squares indicate the mean Em values and the horizontal lines indicate the E1 and E2 values.
The H1098A variant elicits a ΔEm of −101 mV, along with a significant increase in Mo(V) stability, with the Kstab increasing from 28 to 1822 (Fig. 5, A and C, Table 1). Fig. 2B shows a model for how His1092 and His1098 define the H-bonding environment of the proximal pyranopterin piperazine N-5 atom. As suggested above, formal protonation of the imidazole of His1092 is supported by the relative lack of effect of the H1092R variant on the overall molybdenum Em. One of the H-bonds is eliminated in the H1098A variant, which may result in transfer of the proton from the ND1 atom to the piperazine N-5, resulting in loss of a positive charge and the observed decrease in the overall molybdenum Em.
We proposed that in the DMSOR family of enzymes, the role of the stabilizing residue (His1098 in NarG) is to maintain the N-5 atom of the proximal pyranopterin in an sp3-hybridized form, thus stabilizing the tetrahydro oxidation state (8, 15). This suggests a possible explanation for the observed increase in Mo(V) stability in the H1098A variant. Removal of the hydrogen-bonding contact provided by the His1098 imidazole may result in the 10,10a-dihydro oxidation state becoming available to the proximal pyranopterin. If the 10,10a-dihydro to tetrahydro reduction potential occurs within the range in which Mo(V) is extant, then E1 (the Mo(VI/V) reduction potential) could be shifted to the observed value of 139 mV (Table 1; see Ref. 15).
Correlation between Enzyme Activity, Cell Growth, and Molybdenum Electrochemistry
How do changes in molybdenum electrochemistry impact enzyme activity and anaerobic respiratory growth on nitrate? Specific activities in enriched membranes for the variants studied herein range from 1 to 54 μmol min−1 mg−1 for the H1092A and S719A variants, respectively (Table 1). With the exception of the H1092R variant, a strong correlation exists between overall molybdenum reduction potential and enzyme activity (r = 0.99, Fig. 6). A correlation also exists between respiratory growth rate and molybdenum reduction potential across all variants (r = 0.95, inset to Fig. 6). Thus, catalysis and growth are convincingly correlated with increased molybdenum Em. A possible explanation for this is that catalytic turnover depends on the energetics of electron transfer from FS0 (Em = −55 mV (49)) to the molybdenum.
FIGURE 6.
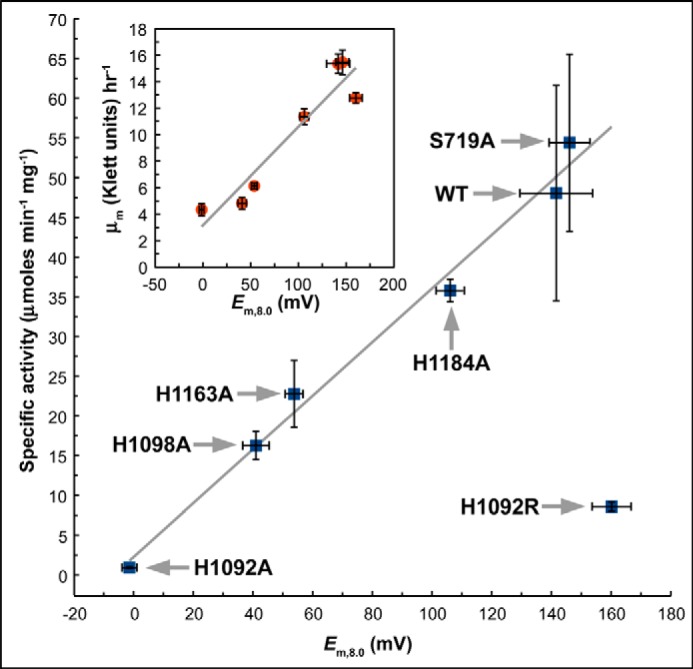
Correlation between enzyme activity and overall molybdenum reduction potential. Excluding the NarG-H1092R variant, a correlation exists between enzyme activity and overall molybdenum reduction potential (r = 0.99). Inset, correlation between growth rate and overall molybdenum reduction potential. Including the NarG-H1092R variant, a correlation exists between the maximal rate of respiratory growth on nitrate and overall molybdenum reduction potential (r = 0.95). Growth rates were calculated using the method of Zwietering et al. (35).
It is notable that the NarGHI catalytic activity appears to be dependent on the overall molybdenum Em, but not on the Mo(V) Kstab (Table 1). Protein film voltammetry studies of NarGHI (45) and two other members of the DMSOR family (the periplasmic nitrate reductase from Rhodobactersphaeroides, NapAB (52), and the Me2SO reductase from E. coli (53)) demonstrated that enzyme turnover decreases below a reduction potential referred to as Eswitch. It was therefore proposed that observation of an Eswitch arises because substrate binds preferentially to the Mo(V) form of the cofactor (45, 52, 53). In the case of NarGHI, this is corroborated by the observation of a stable nitrate adduct to the Mo(V) (47). If the mechanism of nitrate reduction proceeds via substrate binding to a Mo(V) species, we would expect a variant that increases the Mo(V) Kstab to retain significant catalytic activity. The H1098A variant exhibits a very large Mo(V) Kstab (∼1822; Table 1), but has low specific activity and growth rate compared with the wild-type (Fig. 6).
As previously noted (15), an alternative explanation for the observed Eswitch is that it controls a reduction of the distal pyranopterin from an oxidation state equivalent to the 10,10a-dihydro form to one equivalent to the tetrahydro form. This provides a possible explanation for why the H1092R variant supports respiratory growth on nitrate, but has decreased enzyme activity in our in vitro assay (Fig. 6). It is possible that a significant positive ΔEm is elicited on the Eswitch, resulting in lower activity measured using benzyl viologen as electron donor (Em = −374 mV (54)). NarGHI is able to function in vivo with either ubiquinol or menaquinol as electron donor (55), and these have reduction potentials of +110 and −80 mV, respectively (2), rendering them both less likely to reduce the distal pyranopterin than the artificial electron donor benzyl viologen.
Role of the His1163/His1184 Charge-transfer Relay in NarGHI Maturation and in Modulation of Molybdenum Electrochemistry
NarGHI maturation is a carefully orchestrated process involving the NarJ system-specific chaperone (56) and the twin arginine translocase (57). The process ensures assembly of correctly folded enzyme with a complete complement of iron-sulfur clusters and hemes (56, 58). Insertion of the Mo-bisPGD cofactor depends on the presence of the FS0 [4Fe-4S] cluster (56, 58). With the exception of EbdABC (26), NarGHI is the only enzyme examined to date that has a bicyclic distal pyranopterin (25). Closer examination of the charge-transfer relay comprising His1163 and His1184 (Fig. 1) suggests a possible mechanism for pyran ring opening (Fig. 7), which proceeds as follows. The His1163/His1184 charge-transfer relay catalyzes elimination of the C-4a proton (Fig. 7A), generating an open ring-form with a piperazine ring in a form equivalent to the 5,10-dihydro form (Fig. 7B) in a mechanism essentially identical to that previously described (59). This form rearranges to the lowest energy tautomer (equivalent to the 10,10a-dihydro form (15, 59)) shown in Fig. 7C. A mechanism essentially identical to that presented in Fig. 7, A–C, can be proposed for ring-opening of the distal pyranopterin in EbdABC, with the distinction that ring opening would be catalyzed by a guanidinium side chain rather than by an imidazole. The equilibrium between the structures shown in Fig. 7, C and D, illustrates how the formal change on the pyran oxygen (and the overall molybdenum reduction potential) can be modulated by the His1163/His1184 charge-transfer relay. Stabilization of a the pyran oxygen in its alkoxide form would decrease the overall molybdenum reduction potential, whereas formal protonation to the hydroxyl form would increase the overall molybdenum reduction potential.
FIGURE 7.

Proposed mechanism of pyran ring opening of the distal pyranopterin of NarGHI. A, the electron transfer relay comprising NarG-His1163 and NarG-His1184 catalyzes proton abstraction from the C-4a atom and pyran ring-opening. The product of this reaction has a piperazine oxidation state and structure equivalent to the 5,10-dihydro pyranopterin (B). A tautomerization reaction results in a form equivalent to the 10,10a-dihydro pyranopterin with a protonated oxygen equivalent to O-1. The pterin core of this form is equivalent to the lowest energy dihydropterin tautomer (15, 59). C, the NarG-His1163/His1184 charge-transfer relay functions to modulate an equilibrium between the protonated (hydroxyl) and deprotonated (alkoxide) forms of the bicyclic distal pyranopterin, with the protonated form having a higher overall predicted Mo(VI/VIV) reduction potential than the deprotonated form (D).
In the context of the model presented in Fig. 7, C and D, for charge-transfer-induced molybdenum reduction potential modulation, it is notable that the H1163A variant elicits a large ΔEm that is approximately double that elicited by the H1184A variant. This is consistent with the entire charge-transfer relay being involved in molybdenum reduction potential modulation. It is possible to speculate that the H1163A variant may impact an equilibrium between the open and closed pyran ring forms of the distal pyranopterin, and we are currently addressing this issue using protein crystallography.
Conclusions
We have demonstrated the importance of the pyranopterin coordination environment in defining molybdenum electrochemistry and substrate reactivity, with enzyme activity correlating with increasing overall molybdenum reduction potential. Our studies demonstrate the importance of factors beyond the immediate molybdenum coordination sphere in defining enzyme activity.
Author Contributions
S. W. designed, performed, and analyzed the experiments presented in the manuscript. R. A. R. wrote the paper and contributed to the data analyses. J. H. W. conceived and coordinated the study.
Acknowledgments
We thank Justin Fedor for critical reading of the manuscript and Shannon Murphy for providing technical support.
This work was supported by Canadian Institutes of Health Research Grant MOP15292, the Canada Foundation for Innovation, the Alberta Heritage Foundation for Medical Research, and the Natural Science and Engineering Research Council-Collaborative Research and Training Experience Program. The authors declare that they have no conflicts of interest with the contents of this article.
The bicyclic form is therefore formally a pterin rather than a pyranopterin. However, for the sake of brevity, it is referred to as a pyranopterin herein.
- SUOX
- sulfite oxidase
- DmsABC
- E. coli Me2SO reductase
- DMSOR
- Me2SO reductase
- EbdABC
- A. aromaticum ethylbenzene dehydrogenase
- Mo-bisPGD
- molybdo-bis(pyranopterin guanine dinucleotide)
- NarGHI
- E. coli nitrate reductase A
- XDH
- xanthine dehydrogenase
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- Em
- midpoint potential.
References
- 1. Grimaldi S., Schoepp-Cothenet B., Ceccaldi P., Guigliarelli B., Magalon A. (2013) The prokaryotic Mo/W-bisPGD enzymes family: a catalytic workhorse in bioenergetic. Biochim. Biophys. Acta 1827, 1048–1085 [DOI] [PubMed] [Google Scholar]
- 2. Unden G., Bongaerts J. (1997) Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim. Biophys. Acta 1320, 217–234 [DOI] [PubMed] [Google Scholar]
- 3. Moura J. J., Brondino C. D., Trincão J., Romão M. J. (2004) Mo and W bis-MGD enzymes: nitrate reductases and formate dehydrogenases. J. Biol. Inorg. Chem. 9, 791–799 [DOI] [PubMed] [Google Scholar]
- 4. Gonzalez P. J., Rivas M. G., Mota C. S., Brondino C. D., Moura I., Moura J. J. (2013) Periplasmic nitrate reductases and formate dehydrogenases: biological control of the chemical properties of Mo and W for fine tuning of reactivity, substrate specificity and metabolic role. Coord. Chem. Rev. 257, 315–331 [Google Scholar]
- 5. Workun G. J., Moquin K., Rothery R. A., Weiner J. H. (2008) Evolutionary persistence of the molybdopyranopterin-containing sulfite oxidase protein fold. Microbiol. Mol. Biol. Rev. 72, 228–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Magalon A., Fedor J. G., Walburger A., Weiner J. H. (2011) Molybdenum enzymes in bacteria and their maturation. Coord. Chem. Rev. 255, 1159–1178 [Google Scholar]
- 7. Rothery R. A., Workun G. J., Weiner J. H. (2008) The prokaryotic complex iron-sulfur molybdoenzyme family. Biochim. Biophys. Acta 1778, 1897–1929 [DOI] [PubMed] [Google Scholar]
- 8. Rothery R. A., Weiner J. H. (2015) Shifting the metallocentric molybdoenzyme paradigm: the importance of pyranopterin coordination. J. Biol. Inorg. Chem. 20, 349–372 [DOI] [PubMed] [Google Scholar]
- 9. Rebelo J. M., Dias J. M., Huber R., Moura J. J., Romão M. J. (2001) Structure refinement of the aldehyde oxidoreductase from Desulfovibrio gigas (MOP) at 1.28 Å. J. Biol. Inorg. Chem. 6, 791–800 [DOI] [PubMed] [Google Scholar]
- 10. Pushie M. J., George G. N. (2011) Spectroscopic studies of molybdenum and tungsten enzymes. Coord. Chem. Rev. 255, 1055–1084 [Google Scholar]
- 11. Mtei R. P., Perera E., Mogesa B., Stein B., Basu P., Kirk M. L. (2011) A valence bond description of dizwitterionic dithiolene character in an oxomolybdenum-bis(dithione). Eur. J. Inorg. Chem. 2011, 5467–5470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mtei R. P., Lyashenko G., Stein B., Rubie N., Hille R., Kirk M. L. (2011) Spectroscopic and electronic structure studies of a dimethyl sulfoxide reductase catalytic intermediate: implications for electron- and atom-transfer reactivity. J. Am. Chem. Soc. 133, 9762–9774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matz K. G., Mtei R. P., Leung B., Burgmayer S. J., Kirk M. L. (2010) Noninnocent dithiolene ligands: a new oxomolybdenum complex possessing a donor-acceptor dithiolene ligand. J. Am. Chem. Soc. 132, 7830–7831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matz K. G., Mtei R. P., Rothstein R., Kirk M. L., Burgmayer S. J. (2011) Study of molybdenum(4+) quinoxalyldithiolenes as models for the noninnocent pyranopterin in the molybdenum cofactor. Inorg. Chem. 50, 9804–9815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rothery R. A., Stein B., Solomonson M., Kirk M. L., Weiner J. H. (2012) Pyranopterin conformation defines the function of molybdenum and tungsten enzymes. Proc. Natl. Acad. Sci. U.S.A. 109, 14773–14778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jacques J. G., Fourmond V., Arnoux P., Sabaty M., Etienne E., Grosse S., Biaso F., Bertrand P., Pignol D., Léger C., Guigliarelli B., Burlat B. (2014) Reductive activation in periplasmic nitrate reductase involves chemical modifications of the Mo-cofactor beyond the first coordination sphere of the metal ion. Biochim. Biophys. Acta 1837, 277–286 [DOI] [PubMed] [Google Scholar]
- 17. Baymann F., Lebrun E., Brugna M., Schoepp-Cothenet B., Giudici-Orticoni M.-T., Nitschke W. (2003) The redox protein construction kit: pre-last universal common ancestor evolution of energy-conserving enzymes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 267–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schoepp-Cothenet B., van Lis R., Philippot P., Magalon A., Russell M. J., Nitschke W. (2012) The ineluctable requirement for the trans-iron elements molybdenum and/or tungsten in the origin of life. Sci. Rep. 2, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dietrich L. E., Tice M. M., Newman D. K. (2006) The co-evolution of life and Earth. Curr. Biol. 16, R395-R400 [DOI] [PubMed] [Google Scholar]
- 20. Basu P., Burgmayer S. J. (2011) Pterin chemistry and its relationship to the molybdenum cofactor. Coord. Chem. Rev. 255, 1016–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Helton M. E., Gebhart N. L., Davies E. S., McMaster J., Garner C. D., Kirk M. L. (2001) Thermally driven intramolecular charge transfer in an oxo-molybdenum dithiolate complex. J. Am. Chem. Soc. 123, 10389–10390 [DOI] [PubMed] [Google Scholar]
- 22. Feng C., Tollin G., Enemark J. H. (2007) Sulfite oxidizing enzymes. Biochim. Biophys. Acta 1774, 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cerqueira N. M., Gonzalez P. J., Brondino C. D., Romão M. J., Romão C. C., Moura I., Moura J. J. (2009) The effect of the sixth sulfur ligand in the catalytic mechanism of periplasmic nitrate reductase. J. Comput. Chem. 30, 2466–2484 [DOI] [PubMed] [Google Scholar]
- 24. Hille R., Hall J., Basu P. (2014) The mononuclear molybdenum enzymes. Chem. Rev. 114, 3963–4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bertero M. G., Rothery R. A., Palak M., Hou C., Lim D., Blasco F., Weiner J. H., Strynadka N. C. (2003) Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nat. Struct. Biol. 10, 681–687 [DOI] [PubMed] [Google Scholar]
- 26. Kloer D. P., Hagel C., Heider J., Schulz G. E. (2006) Crystal structure of ethylbenzene dehydrogenase from Aromatoleum aromaticum. Structure 14, 1377–1388 [DOI] [PubMed] [Google Scholar]
- 27. Jormakka M., Yokoyama K., Yano T., Tamakoshi M., Akimoto S., Shimamura T., Curmi P., Iwata S. (2008) Molecular mechanism of energy conservation in polysulfide respiration. Nat. Struct. Mol. Biol. 15, 730–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jormakka M., Törnroth S., Byrne B., Iwata S. (2002) Molecular basis of proton motive force generation: structure of formate dehydrogenase-N. Science 295, 1863–1868 [DOI] [PubMed] [Google Scholar]
- 29. Coelho C., González P. J., Moura J. G., Moura I., Trincão J., João Romão M. (2011) The crystal structure of Cupriavidus necator nitrate reductase in oxidized and partially reduced states. J. Mol. Biol. 408, 932–948 [DOI] [PubMed] [Google Scholar]
- 30. Sambrook J., Russell D. W. (2001) Molecular Cloning: a Laboratory Manual, 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 31. Cammack R., Weiner J. H. (1990) Electron paramagnetic resonance spectroscopic characterization of dimethyl sulfoxide reductase of Escherichia coli. Biochemistry 29, 8410–8416 [DOI] [PubMed] [Google Scholar]
- 32. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 33. Rothery R. A., Grant J. L., Johnson J. L., Rajagopalan K. V., Weiner J. H. (1995) Association of molybdopterin guanine dinucleotide with Escherichia coli dimethyl sulfoxide reductase: effect of tungstate and a mob mutation. J. Bacteriol. 177, 2057–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rothery R. A., Weiner J. H. (1991) Alteration of the iron-sulfur cluster composition of Escherichia coli dimethyl sulfoxide reductase by site-directed mutagenesis. Biochemistry 30, 8296–8305 [DOI] [PubMed] [Google Scholar]
- 35. Zwietering M. H., Jongenburger I., Rombouts F. M., van't Riet K. (1990) Modeling of the bacterial growth curve. Appl. Environ. Microbiol. 56, 1875–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johnson J. L., Indermaur L. W., Rajagopalan K. V. (1991) Molybdenum cofactor biosynthesis in Escherichia coli: requirement of the chlB gene product for the formation of molybdopterin guanine dinucleotide. J. Biol. Chem. 266, 12140–12145 [PubMed] [Google Scholar]
- 37. Johnson J. L., Rajagopalan K. V. (1982) Structural and metabolic relationship between the molybdenum cofactor and urothione. Proc. Natl. Acad. Sci. U.S.A. 79, 6856–6860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rothery R. A., Magalon A., Giordano G., Guigliarelli B., Blasco F., Weiner J. H. (1998) The molybdenum cofactor of Escherichia coli nitrate reductase A (NarGHI): effect of a mobAB mutation and interactions with [Fe-S] clusters. J. Biol. Chem. 273, 7462–7469 [DOI] [PubMed] [Google Scholar]
- 39. Rothery R. A., Trieber C. A., Weiner J. H. (1999) Interactions between the molybdenum cofactor and iron-sulfur clusters of Escherichia coli dimethyl sulfoxide reductase. J. Biol. Chem. 274, 13002–13009 [DOI] [PubMed] [Google Scholar]
- 40. Hastings S. F., Kaysser T. M., Jiang F., Salerno J. C., Gennis R. B., Ingledew W. J. (1998) Identification of a stable semiquinone intermediate in the purified and membrane bound ubiquinol oxidase-cytochrome bd from Escherichia coli. Eur. J. Biochem. 255, 317–323 [DOI] [PubMed] [Google Scholar]
- 41. Ohnishi T., King T. E., Salerno J. C., Blum H., Bowyer J. R., Maida T. (1981) Thermodynamic and electron paramagnetic resonance characterization of flavin in succinate dehydrogenase. J. Biol. Chem. 256, 5577–5582 [PubMed] [Google Scholar]
- 42. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 43. Markwell M. A., Haas S. M., Bieber L. L., Tolbert N. E. (1978) A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal. Biochem. 87, 206–210 [DOI] [PubMed] [Google Scholar]
- 44. Morpeth F. F., Boxer D. H. (1985) Kinetic analysis of respiratory nitrate reductase from Escherichia coli K12. Biochemistry 24, 40–46 [DOI] [PubMed] [Google Scholar]
- 45. Elliott S. J., Hoke K. R., Heffron K., Palak M., Rothery R. A., Weiner J. H., Armstrong F. A. (2004) Voltammetric studies of the catalytic mechanism of the respiratory nitrate reductase from Escherichia coli: how nitrate reduction and inhibition depend on the oxidation state of the active site. Biochemistry 43, 799–807 [DOI] [PubMed] [Google Scholar]
- 46. Vincent S. P., Bray R. C. (1978) Electron-paramagnetic-resonance studies on nitrate reductase from Escherichia coli K12. Biochem. J. 171, 639–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. George G. N., Bray R. C., Morpeth F. F., Boxer D. H. (1985) Complexes with halide and other anions of the molybdenum centre of nitrate reductase from Escherichia coli. Biochem. J. 227, 925–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. George G. N., Turner N. A., Bray R. C., Morpeth F. F., Boxer D. H., Cramer S. P. (1989) X-ray-absorption and electron-paramagnetic-resonance spectroscopic studies of the environment of molybdenum in high-pH and low-pH forms of Escherichia coli nitrate reductase. Biochem. J. 259, 693–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rothery R. A., Bertero M. G., Cammack R., Palak M., Blasco F., Strynadka N. C., Weiner J. H. (2004) The catalytic subunit of Escherichia coli nitrate reductase A contains a novel [4Fe-4S] cluster with a high-spin ground state. Biochemistry 43, 5324–5333 [DOI] [PubMed] [Google Scholar]
- 50. Vincent S. P. (1979) Oxidation-reduction potentials of molybdenum and iron-sulphur centres in nitrate reductase from Escherichia coli. Biochem. J. 177, 757–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guigliarelli B., Asso M., More C., Augier V., Blasco F., Pommier J., Giordano G., Bertrand P. (1992) EPR and redox characterization of iron-sulfur centers in nitrate reductases A and Z from Escherichia coli: evidence for a high-potential and a low-potential class and their relevance in the electron-transfer mechanism. Eur. J. Biochem. 207, 61–68 [DOI] [PubMed] [Google Scholar]
- 52. Frangioni B., Arnoux P., Sabaty M., Pignol D., Bertrand P., Guigliarelli B., Léger C. (2004) In Rhodobacter sphaeroides respiratory nitrate reductase, the kinetics of substrate binding favors intramolecular electron transfer. J. Am. Chem. Soc. 126, 1328–1329 [DOI] [PubMed] [Google Scholar]
- 53. Heffron K., Léger C., Rothery R. A., Weiner J. H., Armstrong F. A. (2001) Determination of an optimal potential window for catalysis by E. coli dimethyl sulfoxide reductase and hypothesis on the role of Mo(V) in the reaction pathway. Biochemistry 40, 3117–3126 [DOI] [PubMed] [Google Scholar]
- 54. Wardman P. (1991) The reduction potential of benzyl viologen: an important reference compound for oxidant/radical redox couples. Free Radic. Res. Commun. 14, 57–67 [DOI] [PubMed] [Google Scholar]
- 55. Wallace B. J., Young I. G. (1977) Role of quinones in electron transport to oxygen and nitrate in Escherichia coli. Studies with a ubiA− menA− double quinone mutant. Biochim. Biophys. Acta 461, 84–100 [DOI] [PubMed] [Google Scholar]
- 56. Lanciano P., Vergnes A., Grimaldi S., Guigliarelli B., Magalon A. (2007) Biogenesis of a respiratory complex is orchestrated by a single accessory protein. J. Biol. Chem. 282, 17468–17474 [DOI] [PubMed] [Google Scholar]
- 57. Chan C. S., Howell J. M., Workentine M. L., Turner R. J. (2006) Twin-arginine translocase may have a role in the chaperone function of NarJ from Escherichia coli. Biochem. Biophys. Res. Commun. 343, 244–251 [DOI] [PubMed] [Google Scholar]
- 58. Rothery R. A., Bertero M. G., Spreter T., Bouromand N., Strynadka N. C., Weiner J. H. (2010) Protein crystallography reveals a role for the FS0 cluster of Escherichia coli nitrate reductase A (NarGHI) in enzyme maturation. J. Biol. Chem. 285, 8801–8807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Greatbanks S. P., Hillier I. H., Garner C. D., Joule J. A. (1997) The relative stabilities of dihydropterins: a comment on the structure of Moco, the cofactor of the oxomolybdoenzymes. J. Chem. Soc. Perkin Trans. 2, 1529–1534 [Google Scholar]
- 60. Jormakka M., Richardson D., Byrne B., Iwata S. (2004) Architecture of NarGH reveals a structural classification of Mo-bisMGD enzymes. Structure 12, 95–104 [DOI] [PubMed] [Google Scholar]
- 61. Bertero M. G., Rothery R. A., Boroumand N., Palak M., Blasco F., Ginet N., Weiner J. H., Strynadka N. C. (2005) Structural and biochemical characterization of a quinol binding site of Escherichia coli nitrate reductase A. J. Biol. Chem. 280, 14836–14843 [DOI] [PubMed] [Google Scholar]
- 62. Pascal M. C., Burini J. F., Ratouchniak J., Chippaux M. (1982) Regulation of the nitrate reductase operon: effect of mutations in chlA, B, D and E genes. Mol. Gen. Genet. 188, 103–106 [DOI] [PubMed] [Google Scholar]
- 63. Guigliarelli B., Magalon A., Asso M., Bertrand P., Frixon C., Giordano G., Blasco F. (1996) Complete coordination of the four Fe-S centers of the β subunit from Escherichia coli nitrate reductase: physiological, biochemical, and EPR characterization of site-directed mutants lacking the highest or lowest potential [4Fe-4S] clusters. Biochemistry 35, 4828–4836 [DOI] [PubMed] [Google Scholar]