

Abstract
Purpose
This report provides the 3-year clinical outcomes from the randomized, controlled US Food and Drug Administration Investigational Device Exemption trial of the Superion® for the treatment of moderate degenerative lumbar spinal stenosis.
Patients and methods
The Superion® was evaluated in the treatment of subjects aged 45 years or older suffering from symptoms of intermittent neurogenic claudication, secondary to a confirmed diagnosis of moderate degenerative lumbar spinal stenosis at one or two contiguous levels from L1 to L5. Patients were treated between June 2008 and December 2011 at 31 investigational sites. Three hundred ninety-one subjects were included in the randomized study group consisting of 190 Superion® and 201 X-STOP® control subjects. The primary composite endpoint was individual patient success based on four components: improvement in two of three domains of the Zurich Claudication Questionnaire, no reoperations at the index level, no major implant/procedure-related complications, and no clinically significant confounding treatments.
Results
At 3 years, the proportion of subjects achieving the primary composite endpoint was greater for Superion® (63/120, 52.5%) than for X-STOP® (49/129, 38.0%) (P=0.023) and the corresponding success rates exceeded 80% for each of the individual components of the primary endpoint in the Superion® group (range: 81%–91%). Improvements in back and leg pain severity as well as back- and disease-specific functional outcomes were also maintained through 36 months.
Conclusion
The 3-year outcomes from this randomized controlled trial demonstrate durable clinical improvement consistently across all clinical outcomes for the Superion® in the treatment of patients with moderate degenerative lumbar spinal stenosis.
Keywords: InterSpinous Spacer, lumbar spinal stenosis, Superion®, neurogenic claudication
Introduction
On May 20, 2015, the US Food and Drug Administration (FDA) approved the Superion® InterSpinous Spacer (ISS) (Superion®) for commercial distribution in the United States. Not requiring concomitant surgical decompression, this is the second “stand-alone” interspinous device approved by the FDA. This pivotal regulatory decision substantiates the graduation of the Superion® device from experimental to an acceptable clinical practice modality for the treatment of intermittent symptoms of neurogenic claudication secondary to moderate degenerative lumbar spinal stenosis.
Lumbar spinal stenosis is the manifestation of arthritic degeneration of the spine resulting in bony and ligamentous encroachment of the central canal and foramina causing classic claudicant symptoms.1–3 These symptoms are often exacerbated during ambulation, standing, and trunk extension. It is estimated that 1.2 million individuals are diagnosed with lumbar spinal stenosis every year, with surgical hospitalizations increasing by 30% from 2000 to 2009.4 Over 175,000 surgeries are performed to treat spinal stenosis annually, making it the number one reason for spine surgery in the elderly population.5 In fact, stenosis is the most common indication for spine surgery in patients older than 65 years, and its prevalence is expected to rise 59% to 64 million elderly adults by the year 2025.6
The Superion® is designed for the treatment of symptoms of intermittent neurogenic claudication secondary to moderate degenerative lumbar spinal stenosis and is implanted by minimally invasive methods through a cannula.7 In contrast to direct decompression procedures, such as laminectomy or laminectomy with fusion, where the soft and bony tissues compressing the neural elements are surgically removed through an open surgical exposure, the Superion® provides minimally invasive, indirect decompression of spinal nerves, and functions by serving as a spinal extension blocker to prevent compression of neural elements in extension without the removal of tissue adjacent to the nerves.
This report provides the 3-year clinical outcomes from the randomized, controlled FDA Investigational Device Exemption trial of the Superion® for the treatment of moderate spinal stenosis.8
Materials and methods
Trial overview
The study was a prospective, multi-center, randomized controlled clinical trial comparing the Superion® to a control group consisting of the X-STOP® (X-STOP®), a legally marketed alternative with similar indications for use. The study methodology including eligibility criteria, randomization methods, sample size estimates, outcome measures, and statistical analyses have been detailed previously.9,10 Briefly, the study evaluated the use of the Superion® in the treatment of subjects aged 45 years or older suffering from moderate symptoms of intermittent neurogenic claudication, secondary to a confirmed diagnosis of moderate degenerative lumbar spinal stenosis at one or two contiguous levels from L1 to L5. Patients were treated between June 2008 and December 2011 at 31 investigational sites. Three hundred ninety-one subjects were included in the randomized study group consisting of 190 Superion® ISS and 201 X-STOP® control subjects. FDA regulatory approval was based on the 24-month outcome data in this population.
This study was approved by the Institutional Review Board at each participating site and patients provided written informed consent before any study-related procedures were performed. The trial was prospectively registered at ClinicalTrials.gov (NCT00692276).
Approved indications for use
This device is indicated to treat skeletally mature patients suffering from pain, numbness, and/or cramping in the legs (intermittent neurogenic claudication) secondary to a diagnosis of moderate degenerative lumbar spinal stenosis, with or without Grade 1 spondylolisthesis, confirmed by X-ray, magnetic resonance imaging, and/or computed tomography evidence of thickened ligamentum flavum, narrowed lateral recess, and/or central canal or foraminal narrowing. The Superion® is indicated for those patients with impaired physical function who experience relief in flexion from symptoms of leg/buttock/groin pain, numbness, and/or cramping, with or without back pain, and who have undergone at least 6 months of nonoperative treatment. The Superion® may be implanted at one or two adjacent lumbar levels in patients in whom treatment is indicated at no more than two levels, from L1 to L5.
For this intended use, moderate degenerative lumbar spinal stenosis is defined as follows:
- A reduction of 25%–50% in the central canal and/or nerve root canal (subarticular and neuroforaminal) compared to the adjacent levels on radiographic studies, with radio-graphic confirmation of any one of the following:
- ○ Evidence of thecal sac and/or cauda equina compression
- ○ Evidence of nerve root impingement (displacement or compression) by either osseous or nonosseous elements
- ○ Evidence of hypertrophic facets with canal encroachment.
- And associated with the following clinical signs:
- ○ Presents with moderately impaired physical function defined as a score of ≥2.0 of the Zurich Claudication Questionnaire (ZCQ)
- ○ Ability to sit for 50 minutes without pain and to walk 50 feet or more.
Primary and secondary outcomes
The primary composite endpoint of the investigation as mandated by FDA was individual patient success, which required the patient to meet all of the following criteria at 24 months:
- Clinically significant improvement in outcomes compared to baseline, as determined by meeting the criterion for at least two of three domains of ZCQ.
- ≥0.5 point improvement in physical function
- ≥0.5 point improvement in symptom severity
- Score of ≤2.5 points on patient satisfaction domain
No reoperations, removals, revisions, or supplemental fixation at the index level(s).
- No major implant or procedure-related complications.
- No dislodgement, migration, or deformation
- No new or persistent worsened neurological deficit at the index level
- No spinous process fractures
- No deep infection, death, or other permanent device attributed disability
- No clinically significant confounding treatments:
- No epidural injections, nerve block procedures at index level, spinal cord stimulators, or rhizotomies.
Secondary outcomes included leg and back pain severity assessed on a 100 mm visual analog scale, the Oswestry Disability Index (ODI), and the number of patients that required reoperation, revision or implant removal.
Three-year evaluation
The primary composite endpoint and all secondary outcomes were re-evaluated at the 36-month follow-up interval. In all, 90.2% and 91.4% of the Superion® and X-STOP® study subjects, respectively, were available at this interval. Statistical analysis was performed by an independent biostatistical firm who received all data for analysis directly from a study-specific electronic database. All outcomes were reported using a modified intention-to-treat population, which included all randomized patients who began anesthesia on the implant date. Minimal clinically important changes were defined as 20 mm or more improvement in pain scores and a 15% point or more improvement in ODI. Frequency distributions were compared between groups using Fisher’s exact test, two-tailed.
Results
All subject background characteristics and operative details for the originally randomized inception cohort have been published previously.9 Based on achieving the a priori specified 24-month primary endpoint, the two devices were demonstrated to be statistically noninferior as per the initial trial hypothesis, satisfying the FDA regulatory requirements for approval.
At 36 months, the proportion of subjects achieving the primary composite endpoint was greater for Superion® (63/120, 52.5%) than for X-STOP® (49/129, 38.0%) (P=0.023) (Table 1). The subjects implanted with the Superion® showed no degradation in clinical success compared to the 24-month endpoint analysis (53%), whereas X-STOP® subjects showed a modest degradation over the same timeframe (50%). As shown in Table 1, the corresponding 36-month success rates exceeded 80% for each of the individual components of the primary endpoint in the Superion® group. Specifically, the success rates were 88%, 81%, 91%, and 87% for improvement in two of three domains of the ZCQ, no reoperations at the index level, no major implant/procedure-related complications, and no clinically significant confounding treatments, respectively.
Table 1.
Comparative 36-month success rates between Superion® and X-STOP® overall and for each primary endpoint component
| Number and percentage meeting criteria
|
P-value* | ||||||
|---|---|---|---|---|---|---|---|
| Superion® ISS
|
X-STOP®
|
||||||
| N | n | % | N | n | % | ||
| 1) ZCQ Responder (at least two of three ZCQ domains) | 81 | 71 | 87.7 | 75 | 63 | 84.0 | 0.65 |
| 2) No reoperations, revisions, removals or supplemental fixation at the index level(s) | 138 | 112 | 81.2 | 148 | 118 | 79.7 | 0.77 |
| 3) No major device- or procedure-related complications | 138 | 125 | 90.6 | 148 | 126 | 85.1 | 0.21 |
| 4) No clinically significant confounding treatments | 138 | 120 | 87.0 | 148 | 118 | 79.7 | 0.11 |
| Composite clinical success | 120 | 63 | 52.5 | 129 | 49 | 38.0 | 0.023 |
Note:
Fisher’s exact test, two-tailed.
Abbreviations: ISS, InterSpinous Spacer; ZCQ, Zurich Claudication Questionnaire.
Table 2 provides the 36-month success rates for pain severity as well as back- and disease-specific outcomes based on the minimal clinically important difference criteria for each variable. Five of six comparisons qualitatively favored treatment with the Superion® device; however, only the leg pain results achieved statistical significance. Inspection of the line graphs for each outcome captures both the durability of the Superion® results and the modest degradation in X-STOP® results between 24 and 36 months for the ZCQ (Figure 1) as well as for back and leg pain severity (Figure 2) and back function (Figure 3).
Table 2.
Comparative 36 Month Success Rates between Superion® and X-STOP® for Primary and Secondary Clinical Outcomes
| 36-month clinical outcomes | Superion® ISS | X-STOP® | P-value* |
|---|---|---|---|
| Pain | |||
| VAS back: ≥20 mm decrease | 76.8% (63/82) | 69.7% (53/76) | 0.37 |
| VAS leg (worse): ≥20 mm decrease | 84.1% (69/82) | 69.7% (53/76) | 0.037 |
| Back and stenosis-related outcomes | |||
| ZCQ physical function: ≥0.5 point decrease | 80.5% (66/82) | 77.9% (60/77) | 0.70 |
| ZCQ symptom severity: ≥0.5 point decrease | 82.9% (68/82) | 75.3% (58/77) | 0.25 |
| ZCQ patient satisfaction: ≤2.5 points | 91.5% (75/82) | 88.3% (68/77) | 0.60 |
| ODI: ≥15 point decrease | 69.5% (57/82) | 71.4% (55/77) | 0.86 |
Note:
Fisher’s exact test, two-tailed.
Abbreviations: ISS, InterSpinous Spacer; ODI, Oswestry Disability Index; VAS, visual analog scale; ZCQ, Zurich Claudication Questionnaire.
Figure 1.
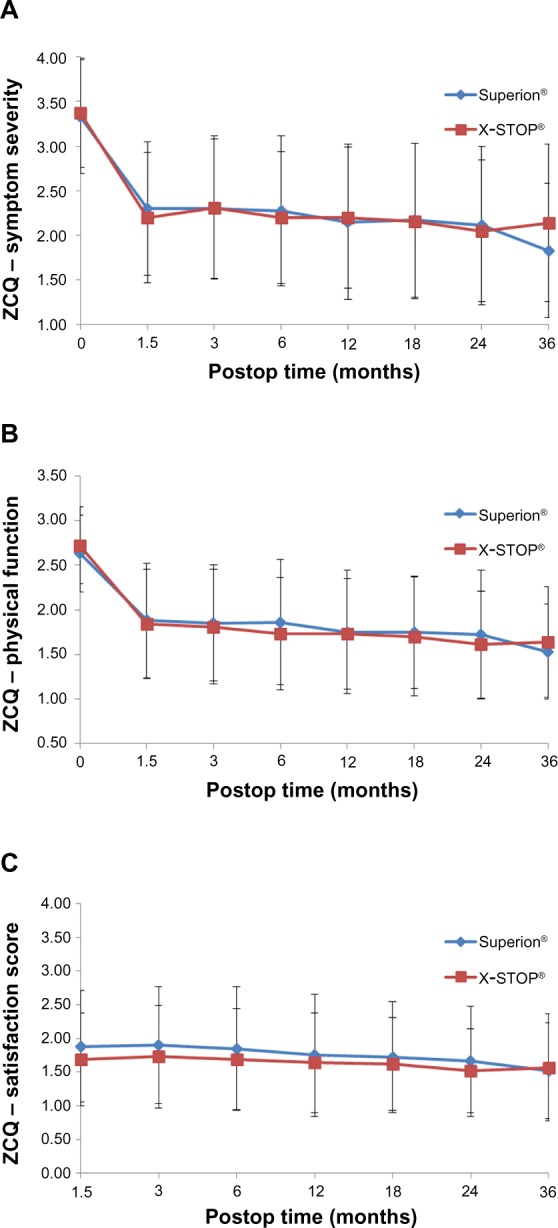
Time course of results for each sub-domain of the ZCQ.
Notes: (A) Symptom severity. (B) Physical function, and (C) Patient satisfaction.
Abbreviations: Postop, postoperative; ZCQ, Zurich Claudication Questionnaire.
Figure 2.
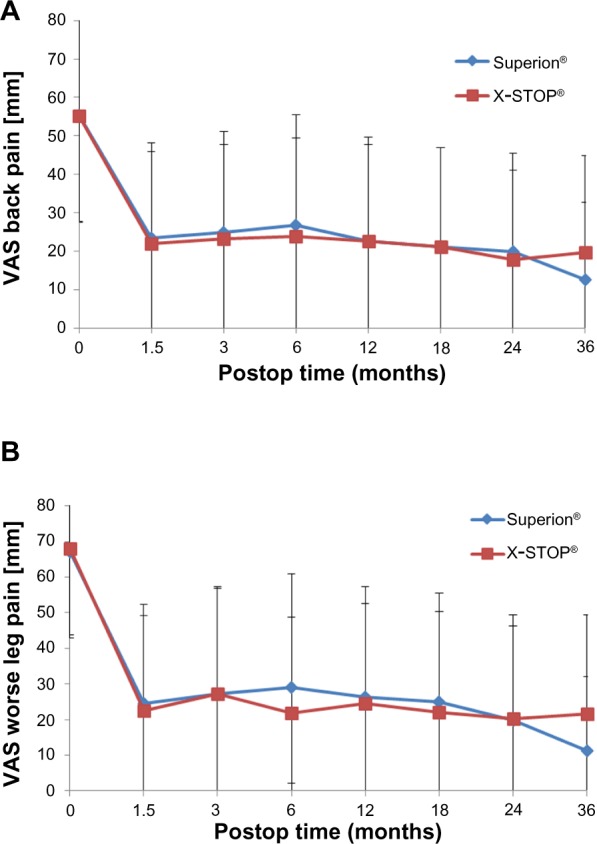
Time course of results for pain severity.
Notes: (A) Back pain. (B) Leg pain.
Abbreviations: VAS, visual analog scale; Postop, postoperative.
Figure 3.
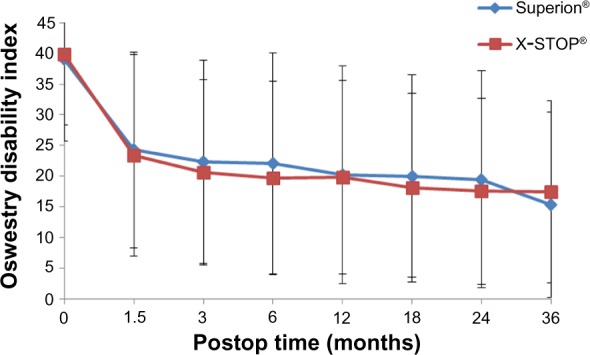
Time course of results for ODI.
Abbreviations: ODI, Oswestry Disability Index; Postop, postoperative.
Comparing the 24-month data with the 36-month data, there was a higher increase in X-STOP® reoperations, revisions, and removals (n=15 out of 44 total) compared to the Superion® device (n=11 out of 49 total).
Discussion
With its recent regulatory approval, the Superion® becomes the only “stand-alone” interspinous device on the US market available to patients for the treatment of moderate spinal stenosis. While the X-STOP® received FDA regulatory approval in 2005, the manufacturer (Medtronic, Inc., Minneapolis, MN, USA) recently (2015) elected voluntarily to cease sale and distribution of the implant. This leaves the Superion® as the de facto clinical option for physicians and their patients seeking a minimally invasive alternative to laminectomy for claudicant symptoms refractory to conservative care.
Importantly, the Superion® implantation procedure does not cause substantial alterations or disruptions to the spinal anatomy which likely reduces the complexity of future surgical options in the event that revision becomes necessary to address progressive degenerative changes and/or reemergence of symptoms. If device removal is required, the implant can be removed via the same minimally invasive access as the original implantation procedure. This suggests that the Superion® device may be considered a reasonable “first line” option in the continuum of care for the treatment of moderate lumbar spinal stenosis.
The durable clinical results achieved with the Superion® in the current study are further reflected in a low conversion rate to surgical decompression of only 14% (26/190) at 3 years. This finding may have a profound effect on the health economics and societal costs of treating the increasing number of patients suffering from spinal stenosis. Indeed, approximately 40% of patients treated conservatively to alleviate early signs of spinal stenosis ultimately require decompression surgery within 10 years due to persistently worsening symptoms.11 Use of an InterSpinous Spacer at the appropriate juncture in the continuum of care may obviate the need for decompression surgery in the majority of patients carefully selected in accordance with the approved indications for use.
Conclusion
The 3-year outcomes from this randomized controlled trial demonstrate durable clinical improvement consistently across all clinical outcomes for the Superion® in the treatment of patients with moderate spinal stenosis. At this follow-up interval, a success rate in excess of 80% was maintained in all the four components of the primary endpoint.
Acknowledgments
The authors wish to thank Greg Maislin for data management support and for conducting all statistical analyses. Financial support for this work was provided by VertiFlex, Inc. (San Clemente, CA, USA).
Footnotes
Author contributions
All authors contributed to the study design, conception, and execution as well as data interpretation, drafting of the manuscript, and provided critical revision of the manuscript for intellectual content. All authors read and provided final approval of the version to be published. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy and integrity of any part of the work are appropriately investigated and resolved.
Disclosure
JB is an independent advisor to VertiFlex. The authors report no other conflicts of interest in this work.
References
- 1.Atlas SJ, Delitto A. Spinal stenosis: surgical versus nonsurgical treatment. Clin Orthop Relat Res. 2006;443:198–207. doi: 10.1097/01.blo.0000198722.70138.96. [DOI] [PubMed] [Google Scholar]
- 2.Ciol MA, Deyo RA, Howell E, Kreif S. An assessment of surgery for spinal stenosis: time trends, geographic variations, complications, and reoperations. J Am Geriatr Soc. 1996;44(3):285–290. doi: 10.1111/j.1532-5415.1996.tb00915.x. [DOI] [PubMed] [Google Scholar]
- 3.Katz JN, Harris MB. Clinical practice. Lumbar spinal stenosis. N Engl J Med. 2008;358(8):818–825. doi: 10.1056/NEJMcp0708097. [DOI] [PubMed] [Google Scholar]
- 4.Skolasky RL, Maggard AM, Thorpe RJ, Jr, Wegener ST, Riley LH., 3rd United States hospital admissions for lumbar spinal stenosis: racial and ethnic differences, 2000 through 2009. Spine (Phila Pa 1976) 2013;38(26):2272–2278. doi: 10.1097/BRS.0b013e3182a3d392. [DOI] [PubMed] [Google Scholar]
- 5.Deyo RA, Mirza SK, Martin BI, Kreuter W, Goodman DC, Jarvik JG. Trends, major medical complications, and charges associated with surgery for lumbar spinal stenosis in older adults. JAMA. 2010;303(13):1259–1265. doi: 10.1001/jama.2010.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deyo RA. Treatment of lumbar spinal stenosis: a balancing act. Spine J. 2010;10(7):625–627. doi: 10.1016/j.spinee.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 7.Loguidice V, Bini W, Shabat S, Miller LE, Block JE. Rationale, design and clinical performance of the Superion(R) Interspinous Spacer: a minimally invasive implant for treatment of lumbar spinal stenosis. Expert Rev Med Devices. 2011;8(4):419–426. doi: 10.1586/erd.11.24. [DOI] [PubMed] [Google Scholar]
- 8.Miller LE, Block JE. Interspinous spacer implant in patients with lumbar spinal stenosis: preliminary results of a multicenter, randomized, controlled trial. Pain Res Treat. 2012;2012:823509. doi: 10.1155/2012/823509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel VV, Whang PG, Haley TR, et al. Superion interspinous process spacer for intermittent neurogenic claudication secondary to moderate lumbar spinal stenosis: two-year results from a randomized controlled FDA-IDE pivotal trial. Spine (Phila Pa 1976) 2015;40(5):275–282. doi: 10.1097/BRS.0000000000000735. [DOI] [PubMed] [Google Scholar]
- 10.Patel VV, Whang PG, Haley TR, et al. Two-year clinical outcomes of a multicenter randomized controlled trial comparing two interspinous spacers for treatment of moderate lumbar spinal stenosis. BMC Musculoskelet Disord. 2014;15:221. doi: 10.1186/1471-2474-15-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atlas SJ, Keller RB, Wu YA, Deyo RA, Singer DE. Long-term outcomes of surgical and nonsurgical management of lumbar spinal stenosis: 8 to 10 year results from the maine lumbar spine study. Spine (Phila Pa 1976) 2005;30(8):936–943. doi: 10.1097/01.brs.0000158953.57966.c0. [DOI] [PubMed] [Google Scholar]