

Abstract
Background
Aspirin and P2Y12 antagonists are antiplatelet compounds that are used clinically in patients with thrombosis. However, some patients are ‘resistant’ to antiplatelet therapy, which increases their risk of developing acute coronary syndromes. These patients often present with an underlying condition that is associated with altered levels of circulating platelet primers and platelet hyperactivity. Platelet primers cannot stimulate platelet activation, but, in combination with physiologic stimuli, significantly enhance platelet function.
Objectives
To explore the role of platelet primers in resistance to antiplatelet therapy, and to evaluate whether phosphoinositide 3-kinase (PI3K) contributes to this process.
Methods and Results
We used platelet aggregation, thromboxane A2 production and ex vivo thrombus formation as functional readouts of platelet activity. Platelets were treated with the potent P2Y12 inhibitor AR-C66096, aspirin, or a combination of both, in the presence or absence of the platelet primers insulin-like growth factor-1 (IGF-1) and thrombopoietin (TPO), or the Gz-coupled receptor ligand epinephrine. We found that platelet primers largely overcame the inhibitory effects of antiplatelet compounds on platelet functional responses. IGF-1-mediated and TPO-mediated, but not epinephrine-mediated, enhancements in the presence of antiplatelet drugs were blocked by the PI3K inhibitors wortmannin and LY294002.
Conclusions
These results demonstrate that platelet primers can contribute to antiplatelet resistance. Furthermore, our data demonstrate that there are PI3K-dependent and PI3K-independent mechanisms driving primer-mediated resistance to antiplatelet therapy.
Keywords: aspirin, drug resistance, epinephrine, insulin-like growth factor-1, P2Y12 purinoceptor antagonists
Introduction
Platelet hemostasis is a tightly regulated process mediated by various feedback control mechanisms and key signaling receptors. Disruption of these regulatory controls leads to thrombosis, which, in turn, can trigger the development of an occlusive clot and subsequent cardiovascular complications. Typically, patients who present with pathologic thrombosis undergo pharmacologic intervention with antiplatelet compounds to minimize the risk of developing acute coronary syndrome (ACS). The current ‘gold standard’ preventive measure employed by clinics to treat thrombosis involves the administration of antiplatelet drugs that target cyclooxygenase-1 (COX-1) (e.g. acetylsalicylic acid [ASA]; aspirin) and the platelet P2Y12 receptor (e.g. clopidogrel, prasugrel, or ticagrelor).
ASA irreversibly inhibits the conversion of arachidonic acid (AA) to thromboxane A2 (TxA2), an important positive feedback mediator involved in platelet activation. The inhibitory effect of ASA is achieved by acetylation of Ser529 in the active site of COX-1, blocking the enzyme's critical role in AA metabolism 1,2. Clopidogrel and prasugrel are thienopyridines whose active metabolites covalently bind to the platelet P2Y12 receptor and irreversibly inhibit ADP-mediated platelet function 3,4. ASA and platelet P2Y12 receptor antagonists may be prescribed to patients as monotherapy; however, they are also administered via a dual antiplatelet regimen as a preventive measure against thrombotic vascular events 5,6. Dual therapy remains controversial, as the PCI-CURE and CARESS trials have demonstrated major improvements in the clinical outcomes of patients receiving combined treatment 7,8, whereas other studies have shown no additional benefit of ASA treatment in the presence of strong P2Y12 receptor blockade 9.
The current antiplatelet compounds are limited in their efficacy, as various clinical studies have demonstrated a decreased response, or ‘resistance’, in certain patient subgroups 10. There are several reasons why patients may show resistance to ASA, e.g. reduced absorption/increased metabolism of ASA, the presence of TxA2-independent platelet activation pathways, and genetic polymorphisms in the genes encoding COX-1 11,12. Both clopidogrel and prasugrel are prodrugs that require conversion by hepatic cytochrome P450 (CYP) enzymes to become active metabolites in vivo 13,14. Therefore, poor bioavailability resulting from genetic polymorphisms in CYP may cause clopidogrel/prasugrel resistance 15. Furthermore, poor bioavailability may also be induced by drugs that interfere with hepatic metabolism 16. Other potential mechanisms underpinning clopidogrel resistance include conditions resulting in increased release of ADP, underdosing, and ADP-independent platelet activation pathways 17.
Patients who are resistant to antiplatelet compounds have been found to show a hyperactive platelet phenotype 18–21. It is well documented that platelet hyperactivity can be induced by elevated levels of circulating primers 22–25. Platelet primers do not stimulate platelet activation, but, in combination with physiologic stimuli, significantly enhance platelet function. The peptide hormones insulin-like growth factor-1 (IGF-1) and thrombopoietin (TPO) are known primers that enhance platelet functional responses via a phosphoinositide 3-kinase (PI3K)-dependent mechanism 26–28. In addition, catecholamines such as epinephrine can also act as platelet primers when used at suboptimal concentrations 29. Interestingly, recent pharmacogenomics studies have also linked increased expression of IGF-1 and IGF-1 receptor mRNA with ASA resistance 30. Furthermore, coronary heart disease (CHD) patients have been found to have significantly higher serum concentrations of IGF-1, a feature that is believed to contribute to coronary atherosclerosis 31,32. TPO is another common factor that has been found to be elevated in conditions associated with coronary artery disease, such as unstable angina 33,34, and epinephrine levels are increased in patients presenting with hypertension 35, and ischemic heart disease 36,37. Elevated levels of these circulating primers may therefore contribute to platelet hyperactivity and, consequently, to the resistance to antiplatelet compounds demonstrated in a population of patients with ACS. This is of particular clinical interest, as these patients are at significant risk of experiencing reoccurring major cardiovascular events 38,39.
In this study, we therefore sought to determine the role of circulating primers in resistance to antiplatelet compounds, focusing specifically on primers known to be elevated in ACS patients. We used platelet aggregation and ex vivo thrombus formation to assess the effects of the highly selective P2Y12 antagonist AR-C66096 (ARC) and ASA on platelet function in the presence or absence of the primers IGF-1 and TPO, and the Gz-coupled receptor ligand epinephrine. Our results demonstrate that: (i) platelet primers can rescue the inhibitory effects induced by P2Y12 blockade and ASA treatment; and (ii) PI3K plays a critical role in IGF-1-mediated and TPO-mediated resistance, whereas there are PI3K-independent mechanisms driving epinephrine-mediated resistance.
Materials and methods
Materials
The platelet agonists used were: protease-activated receptor 1 (PAR-1)-activating peptide (SFLLRN-NH2; Bachem, Bubendorf, Switzerland), crosslinked collagen-related peptide (CRP-XL) from R. Farndale (Department of Biochemistry, University of Cambridge, UK), and fibrillar HORM collagen (type I) derived from equine tendon (Nycomed, Konstanz, Germany). The platelet inhibitors used were: ARC tetrasodium salt (R&D Systems, Abingdon, UK), ASA (Sigma-Aldrich, Poole, UK), and wortmannin (Tocris, Bristol, UK). The platelet primers used were: long-IGF-1 recombinant protein (receptor grade – AM001; Immunological and Biochemical Test Systems, Binzwangen, Germany), epinephrine hydrochloride (Sigma-Aldrich), and recombinant human TPO (R&D Systems). d-phenylalanylprolyl-arginyl chloromethyl ketone (PPACK) was from Calbiochem (Merck Chemicals, Watford, UK), and heparin was from Sigma-Aldrich. The commercial TxA2 ELISA kit and 3,3′-dihexyloxacarbocyanine iodide (DiOC6) were from Enzo Life Sciences (Exeter, UK). All other reagents were from Sigma (Poole, UK), unless otherwise indicated.
Isolation and preparation of platelets
Venous blood was obtained from healthy volunteers with approval of the local research ethics committee at the University of Bristol. Donors provided written informed consent, and reported not having taken antiplatelet agents in the 14 days prior to donation. Blood was drawn into 4% trisodium citrate (1 : 9, v/v), and acidified with acidic citrate dextrose (1 : 7, v/v; 120 mm sodium citrate, 110 mm glucose, 80 mm citric acid). Washed platelets were isolated as previously described 40, and pelleted in the presence of 140 nm prostaglandin E1 and 0.02 U mL−1 apyrase (grade VII). Platelets were resuspended at 4 × 108 mL−1 in modified HEPES–Tyrode buffer (145 mm NaCl, 3 mm KCl, 0.5 mm Na2HPO4, 1 mm MgS04.7H2O, 10 mm HEPES, pH 7.2, 0.1% [w/v] d-glucose, and 0.02 U mL−1 apyrase), and allowed to rest at 30 °C for 30 min prior to experimentation.
Platelet aggregation
Platelet aggregation was performed with a Chronolog 490-4D aggregometer (Labmedics, Abingdon-on-Thames, UK) at 37 °C under continuous stirring at 1200 r.p.m. Platelets (2 × 108 mL−1) were preincubated for 10 min at 37 °C with vehicle (0.2% dimethylsulfoxide/HEPES–Tyrode buffer) or the pharmacologic inhibitors ARC (1 μm), ASA (30 μm), and ASA/ARC, with or without wortmannin (100 nm) or LY294002 (40 μm). The platelet primers IGF-1 (100 nm), TPO (50 ng mL−1) and epinephrine (5 μm) were added 5 min prior to stimulation with the PAR-1 agonist SFLLRN or the glyycoprotein (GP)VI agonist CRP-XL. Changes in light transmission were continuously monitored with aggrolink Version 4 (Chronolog Corporation, Havertown, PA, USA) for 5 min.
Measurement of TXA2 generation
TxA2 levels were measured with a commercially available colorimetric ELISA kit (Enzo Life Sciences), as previously described 41. In brief, platelet samples from the aggregation reactions were quenched at 5 min with 200 μm indomethacin and 5 mm EDTA to inhibit further production of TxA2. Samples were centrifuged for 4 min at 12 000 × g, and the supernatant was removed and stored at − 80 °C for subsequent analysis according to the manufacturer's protocol. Thromboxane B2, the stable hydrolysis product of TxA2, was used as a readout of TxA2 production.
Ex vivo thrombus formation
Thrombus formation under flow conditions was determined as previously described 26,42. In brief, anticoagulated blood drawn into 2 U mL−1 heparin and 40 μm PPACK was pretreated with vehicle (HEPES–Tyrode buffer) or ARC (1 μm) and ASA (30 μm) in the presence or absence of wortmannin (100 nm), and prelabeled with 1 μm DiOC6 for 10 min. Blood was treated with vehicle control or the platelet primers IGF-1 (5 nm or 100 nm), TPO (50 ng mL−1) and epinephrine (20 or 100 nm) for 5 min before perfusion at an arterial shear rate of 1000 s−1 for 5 min over collagen-coated coverslips (50 μg mL−1) in parallel-plate flow chambers. Phase-contrast and fluorescence images of thrombi were captured with a × 40 water immersion objective on a fluorescence microscope (BX51WI; Olmpus, Southend-on-Sea, UK) and a Rolera-XR digital camera. Chambers were flushed with HEPES–Tyrode buffer to remove non-adherent cells, and fluorescent images were taken from at least 15 random microscopic fields of view. Quantification of surface coverage was performed with imagej (National Institutes of Health, Bethesda, MD, USA).
Calcium signaling
Ca2+ measurements were performed as previously described 43. In brief, changes in intracellular Ca2+ concentration were measured by spectrofluorimetry in platelets (5 × 107 mL−1) loaded with Fura-2 at 37 °C, with stirring. Fluorescence excitation was performed at 340 nm and 380 nm with a Hitachi F-4500 (Hitachi High-Technologies, Maidenhead, UK).
Statistical analysis
Data were analyzed with graphpad prism 5 software (GraphPad Software, San Diego, CA, USA). All data are presented as mean ± standard error of the mean of at least three independent experiments. Data used in statistical analysis were tested with either a one-way or a two-way anova with a Bonferroni or Dunnett multiple comparison post hoc test.
Results
Role of P2Y12 and TxA2 in platelet aggregation mediated by PAR-1 and GPVI
To examine the role of P2Y12 and TxA2 in PAR-1-mediated and GPVI-mediated platelet aggregation, platelets were treated with ASA (TxA2 inhibitor), ARC (P2Y12/ADP inhibitor), or ASA/ARC. ASA (30 μm) had no significant effect on SFLLRN-mediated aggregation, but significantly reduced CRP-XL-mediated platelet aggregation, from 79.7% ± 1.3% to 4.0% ± 3.0% (Fig.1A,B). In agreement with previous studies, P2Y12 inhibition with ARC (1 μm) caused a significant reduction in SFLLRN-mediated aggregation 44, with the amplitude being reduced from 75.0% ± 3.6% to 38.0% ± 7.4% (Fig.1A). Similarly, CRP-XL-induced aggregation was drastically reduced from 79.7% ± 1.3% to 12.0% ± 8.5% in the presence of ARC (Fig.1B). Interestingly, combination treatment with ASA and ARC made SFLLRN-mediated aggregation reversible (Fig.1A), demonstrating the importance of TxA2 and ADP signaling in sustained platelet aggregation. ASA and ARC completely blocked CRP-XL-mediated aggregation (Fig.1B).
Figure 1.

Role of P2Y12 and thromboxane A2 in protease-activated receptor 1-mediated and glycoprotein VI-mediated platelet aggregation. (A,B) Washed platelets (2 × 108 mL−1) were pretreated with vehicle control (HEPES–Tyrode buffer) or inhibitors, i.e. 1 μm AR-C66096 (ARC), 30 μm acetylsalicylic acid (ASA), or ASA/ARC, for 10 min. Platelets were subsequently stimulated with 2 μm SFLLRN (A) or 1.5 μg mL−1 crosslinked collagen-related peptide (CRP-XL) (B), and platelet aggregation was recorded for a total of 5 min. Representative aggregation traces (Ai, Bi) and quantified percentage aggregation values (Aii, Bii) are shown. Data are mean ± standard error of the mean, n = 3–6. Statistical analysis: one-way anova was used in conjunction with Dunnett's multiple comparison test; *P < 0.05 and ***P < 0.001 as compared with vehicle control.
IGF-1, TPO and epinephrine rescue PAR-1-mediated platelet function in the presence of antiplatelet compounds
IGF-1 27, TPO 45,46 and epinephrine 29,47 are known to significantly enhance platelet functional responses to physiologic stimuli. In agreement with previous studies, we found that IGF-1, TPO and epinephrine dose-dependently increased SFLLRN-mediated platelet aggregation (Fig. S1). As IGF-1, TPO and epinephrine levels are elevated in various disease states 22,34,35,48–50 and in patients who present with CHD 31–34,36,37, we wanted to evaluate their potential contribution to antiplatelet drug resistance. Platelets were pretreated with ASA, ARC or ASA/ARC in the presence or absence of primers (Fig.2A–E). IGF-1, TPO and epinephrine were unable to activate washed platelets by themselves (Fig. S2), but rescued the inhibitory effect of ASA and/or ARC treatment on SFLLRN-mediated platelet aggregation, as demonstrated by the significant increases in the area under the aggregation curves (Fig.2E). Inhibition of platelet function by ASA was completely rescued by all primers, whereas partial rescue by IGF-1 and TPO was observed in ARC-treated and ASA/ARC-treated platelets. Epinephrine completely rescued ARC-treated platelet function, and significantly rescued the effects of ASA/ARC treatment for both high (Fig.2Eiii) and subthreshold concentrations of SFLLRN (Fig. S3). These results demonstrate the ability of IGF-1, TPO and epinephrine to rescue platelet function in the presence of the antiplatelet compounds ASA, ARC, and ASA/ARC.
Figure 2.

Insulin-like growth factor-1 (IGF-1), thrombopoietin (TPO) and epinephrine rescue protease-activated receptor 1-mediated platelet function in the presence of antiplatelet compounds. Washed platelets (2 × 108 mL−1) were pretreated with vehicle control (HEPES–Tyrode buffer) (A) or inhibitors, i.e. 1 μm AR-C66096 (ARC) (B), 30 μm acetylsalicylic acid (ASA) (C), or ASA/ARC (D), as indicated for 10 min. Platelets were subsequently incubated in the presence of vehicle control or primer, i.e. 100 nm IGF-1 (Ai–Ei), 50 ng mL−1 TPO (Aii–Eii), and 5 μm epinephrine (Aiii–Eiii), respectively, for 5 min before stimulation with 2 μm SFLLRN, and aggregation was recorded for 5 min. Representative aggregation traces (A–D) and quantified area under the curve analysis (E) are shown. Data are mean ± standard error of the mean, n = 6–8. Statistical analysis: two-way anova was used in conjunction with a Bonferroni post hoc test; *P < 0.05, **P < 0.01, ***P < 0.001.
IGF-1, TPO and epinephrine rescue GPVI-mediated platelet function in the presence of antiplatelet compounds
Next, we addressed whether primer-mediated resistance to antiplatelet compounds was a PAR-1-specific event. Therefore, we replicated the aggregation studies with an agonist that targets GPVI, i.e. CRP-XL, and pretreated platelets with ASA, ARC or ASA/ARC before spiking with primer (Fig.3A–E). We found that ASA, ARC and ASA/ARC significantly inhibited CRP-XL-mediated platelet aggregation. Similarly to SFLLRN-mediated aggregation, pretreatment with IGF-1, TPO or epinephrine significantly reduced the inhibitory effects of individual treatments with the antiplatelet compounds (Fig.3B–C). Interestingly, epinephrine was the only primer that was able to significantly rescue the inhibitory effect of ASA/ARC treatment (increase from 0.6% ± 0.47% to 73.4% ± 19.3%; area under the curve analysis) (Fig.3Aiii–Eiii). These results demonstrate the ability of primers to rescue the inhibitory effects of individual antiplatelet compounds on CRP-XL-induced GPVI signaling.
Figure 3.
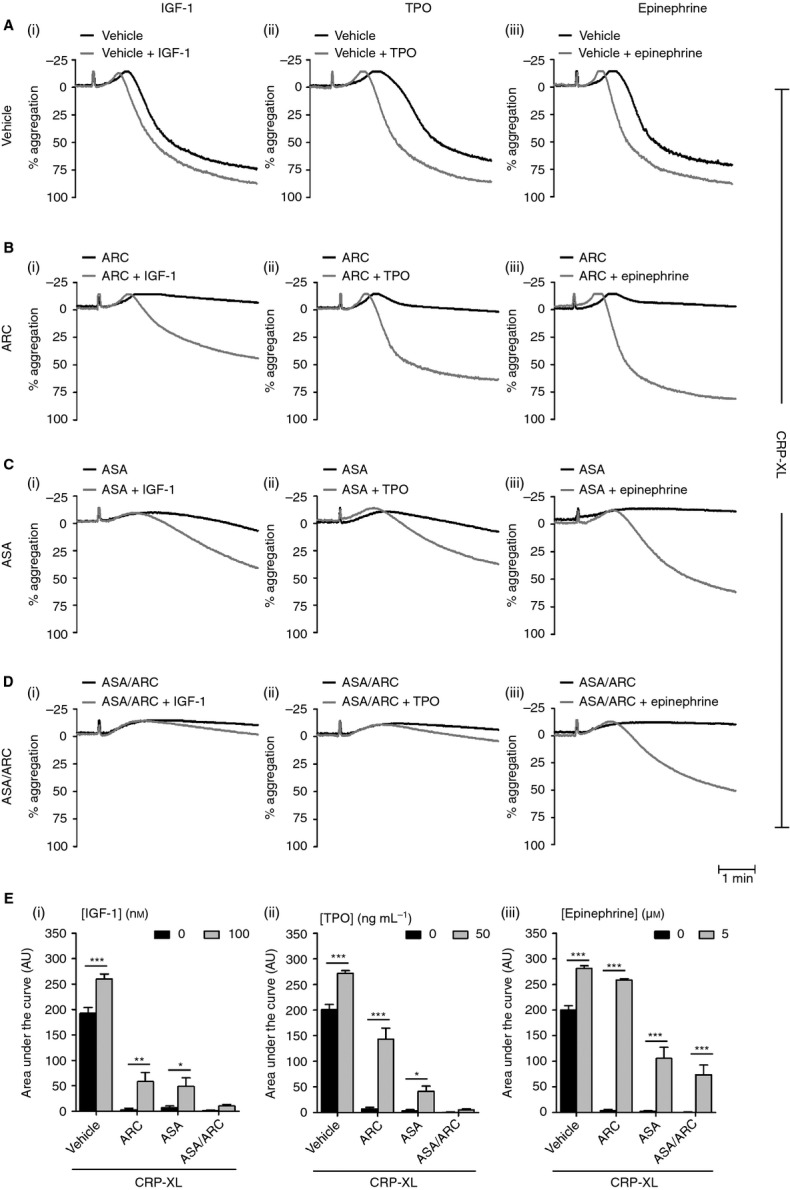
Insulin-like growth factor-1 (IGF-1), thrombopoietin (TPO) and epinephrine rescue glycoprotein VI-mediated platelet function in the presence of antiplatelet compounds. Washed platelets (2 × 108 mL−1) were pretreated with vehicle control (HEPES–Tyrode buffer (A) or inhibitors, i.e. 1 μm AR-C66096 (ARC) (B), 30 μm acetylsalicylic acid (ASA) (C) or ASA/ARC (D), as indicated for 10 min. Platelets were subsequently incubated in the presence of vehicle control or primer, i.e. 100 nm IGF-1 (Ai–Ei), 50 ng mL−1 TPO (Aii–Eii), and 5 μm epinephrine (Aiii–Eiii), respectively, for 5 min. Platelets were then stimulated with crosslinked collagen-related peptide (0.4–2 μg mL−1) to induce an approximately 80% aggregation response, and aggregation was recorded for 5 min. Representative aggregation traces (A–D) and quantified area under the curve analysis (E) are shown. Data are mean ± standard error of the mean, n = 6–8. Statistical analysis: two-way anova was used in conjunction with a Bonferroni post hoc test; *P < 0.05, **P < 0.01, ***P < 0.001.
The role of TxA2 formation in primer-mediated rescue of the effect of antiplatelet drugs
Antagonism of platelet P2Y12 receptors can inhibit platelet activation by inhibiting TxA2 production and dampening platelet responses following TxA2 receptor activation 51–53. Similarly, ASA blocks COX-1 activity and subsequent generation of TxA2 9. Given the important role of TxA2 in platelet activation, we were interested in investigating whether: (i) primers can increase PAR-1-mediated TxA2 formation; and (ii) whether this contributes to primer-mediated rescue of PAR-1-mediated platelet function, in particular in the presence of ARC. As expected, ASA and ASA/ARC blocked TxA2 production under all conditions. Interestingly, we found that IGF-1, TPO and epinephrine significantly elevated TxA2 production in vehicle-treated platelets (Fig.4A–C). However, ARC treatment blocked IGF-1-mediated and TPO-mediated enhancement of TxA2 production. In contrast, epinephrine was able to enhance TxA2 production in the presence of ARC.
Figure 4.
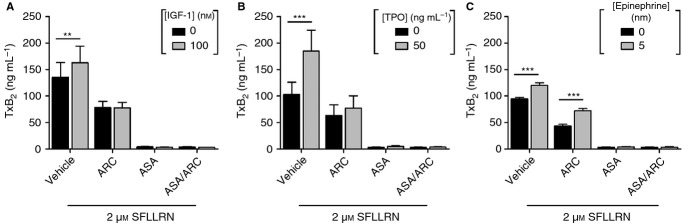
The role of thromboxane A2 (TxA2) formation in primer-mediated rescue of the effect of antiplatelet drugs. (A) Insulin-like growth factor-1 (IGF-1). (B) Thrombopoietin (TPO). (C) Epinephrine. TxA2 production was assessed with a thromboxane B2 (TxB2) ELISA with releasates generated from the samples shown in Fig.2A–D). Data are presented as mean ± standard error of the mean, n = 3–4. Statistical analysis: two-way anova was used in conjunction with a Bonferroni post hoc test; **P < 0.01, ***P < 0.001. ARC, AR-C66096; ASA, acetylsalicylic acid.
PI3K plays a critical role in primer-mediated resistance to dual antiplatelet therapy
Various platelet primers have been shown to enhance platelet function via a PI3K-dependent signaling mechanism 26–29,54,55. To investigate whether primer-mediated resistance in SFLLRN-stimulated platelets was mediated by PI3K, we treated platelets with ASA, ARC and/or ASA/ARC in the presence of the pan-PI3K inhibitors wortmannin (Fig.5) and LY294002 (Fig. S4). The results indicated that IGF-1-mediated and TPO-mediated resistance are driven primarily by PI3K, as the addition of wortmannin or LY294002 ablated primer-mediated enhancements in the presence of the antiplatelet compounds (Fig.5Fi,ii; Fig. S4Di,ii)). Interestingly, epinephrine was still able to significantly enhance platelet aggregation in the presence of wortmannin and LY294002 when platelets were treated with various antiplatelet combinations (Fig.5Aiii–Diii; Fig. S4Diii). However, the functional enhancements achieved were more reversible in the presence of the PI3K inhibitors, particularly in the presence of wortmannin, as reflected by a reduction in the rescue effect of epinephrine on ASA/ARC-treated platelets (compare Fig.2Eiii and Fig.5Fiii for area under the curve analysis). Wortmannin reduced aggregation amplitude (t = 5 min) in the presence of epinephrine, from 52% ± 3.7% to 8% ± 0.75% (n = 3–8). These results demonstrate that PI3K contributes to the later stages of epinephrine-mediated rescue of platelet aggregation; however, epinephrine-mediated resistance appears to be largely PI3K-independent.
Figure 5.

Phosphoinositide 3-kinase plays a critical role in primer-mediated resistance to dual antiplatelet therapy. Washed platelets (2 × 108 mL−1) were pretreated with vehicle control (dimethylsulfoxide [DMSO]) (A) or inhibitors, i.e. 100 nm wortmannin (WTM) (B), 1 μm AR-C66096 (ARC) (C), 30 μm acetylsalicylic acid (ASA) (D), or ASA/ARC (E), as indicated for 10 min. Platelets were subsequently incubated in the presence of vehicle control or primer, i.e. 100 nm insulin-like growth factor-1 (IGF-1) (Ai–Ei), 50 ng mL−1 thrombopoietin (TPO) (Aii–Eii), and 5 μm epinephrine (Aiii–Eiii), respectively, for 5 min before stimulation with 2 μm SFLLRN, and aggregation was recorded for 5 min. Representative aggregation traces (A–E) and quantified area under the curve analysis (F) are shown. Data are mean ± standard error of the mean, n = 3. Statistical analysis: two-way anova was used in conjunction with a Bonferroni post hoc test; **P < 0.01, ***P < 0.001.
Ex vivo thrombus formation on collagen is reduced by dual antiplatelet therapy; a process that is rescued by IGF-1 and epinephrine
A recent study has demonstrated that blood from patients receiving dual antiplatelet therapy with P2Y12 inhibitors and ASA have a decreased rate of thrombus formation over collagen 56. In agreement with this, we found that exogenous addition of ASA and ARC resulted in a significant reduction in collagen-mediated thrombus formation, as demonstrated by a reduction in the area covered by platelets and the average thrombus size as compared with vehicle control (Fig.6A–C). Platelets pretreated with the antiplatelet compounds also appeared to form thrombi with a loosely packed platelet morphology, similarly to previous findings 57. Interestingly, we found that pretreatment of blood with epinephrine or IGF-1, but not with TPO, reversed the inhibitory effects of the dual antiplatelet compounds, as demonstrated by the complete rescue of the area covered with thrombi (Fig.6A–C). Epinephrine and IGF-1 treatment also increased the average thrombus size, although not to the level of the vehicle control (Fig.6A–C). These results demonstrate that epinephrine and IGF-1 are able to reverse the inhibitory effects of dual antiplatelet treatment and affect the morphology of thrombi.
Figure 6.

Ex vivo thrombus formation on collagen is reduced by dual antiplatelet therapy, a process that is rescued by insulin-like growth factor-1 (IGF-1) and epinephrine. Fluorescently labeled (dihexyloxacarbocyanine iodide [DiOC6]) whole blood anticoagulated with 2 U mL−1 heparin and 40 μm d-phenylalanylprolyl-arginyl chloromethyl ketone was pretreated with vehicle control or 1 μm AR-C66096 (ARC) and 30 μm acetylsalicylic acid (ASA) in combination for 10 min. Blood samples were subsequently preincubated with vehicle control or primer, i.e. 100 nm epinephrine (A), 100 nm IGF-1 (B), and 50 ng mL−1 thrombopoietin (TPO) (C), as indicated for 5 min. Samples were then perfused at an arterial shear rate of 1000 s−1 over fibrillar collagen (50 μg mL−1) for 5 min. Samples were washed with HEPES–Tyrode buffer for 2 min to remove non-adherent cells. Representative fluorescent images are shown, along with quantitative analysis of surface area covered (%) with thrombi and the average thrombus size (AU) Data represent the average results taken from ≥ 15 random microscopic fields per experiment (n = 4–5). Statistical analysis: one-way anova was used in conjunction with a Bonferroni post hoc test; *P < 0.05, **P < 0.01, ***P < 0.001.
PI3K plays a critical role in IGF-1-mediated rescue of ex vivo thrombus formation following inhibition with ASA/ARC treatment
To gain some mechanistic insights into the resistance induced by IGF-1 and epinephrine on thrombus formation in whole blood treated with ASA/ARC, we treated samples with the PI3K inhibitor wortmannin (Fig.7). The results demonstrated that epinephrine-mediated resistance to ASA/ARC was largely independent of PI3K activity, as epinephrine was still able to enhance ex vivo thrombus formation in the presence of wortmannin (Fig.7A). In contrast, IGF-1-mediated resistance to ASA/ARC was PI3K-dependent (Fig.7B).
Figure 7.

Phosphoinositide 3-kinase plays a critical role in insulin-like growth factor-1 (IGF-1)-mediated rescue of ex vivo thrombus formation following inhibition with acetylsalicylic acid (ASA)/AR-C66096 (ARC) treatment. Fluorescently labeled (dihexyloxacarbocyanine iodide) whole blood anticoagulated with 2 U mL−1 heparin and 40 μm d-phenylalanylprolyl-arginyl chloromethyl ketone was pretreated with vehicle control or 1 μm ARC and 30 μm ASA in combination in the presence or absence of 100 nm wortmannin (WTM) for a total of 10 min. Blood samples were subsequently preincubated with vehicle control or primer, i.e. 5 μm epinephrine (A) or 100 nm IGF-1 (B), as indicated for a total of 5 min. Samples were then perfused at an arterial shear rate of 1000 s−1 over fibrillar collagen (50 μg mL−1) for 5 min. Samples were washed with HEPES–Tyrode buffer for 2 min to remove non-adherent cells. Representative fluorescent images are shown, along with quantitative analysis of surface area covered (%) with thrombi. Data represent the average results taken from ≥ 15 random microscopic fields per experiment (n = 3–5). Statistical analysis: one-way anova was used in conjunction with a Bonferroni post hoc test; **P < 0.01, ***P < 0.001.
Synergistic effects of IGF-1 and epinephrine on primer-mediated resistance to dual antiplatelet therapy during ex vivo thrombus formation
It is likely that patients who are susceptible to cardiovascular complications will present with elevations in multiple combinations of circulating primers in vivo. We treated whole blood with more physiologic concentrations of IGF-1 (5 nm) and epinephrine (20 nm), alone or in combination, to assess the effects of combined primer treatments on antiplatelet resistance. We found that 5 nm IGF-1 alone was unable to enhance thrombus formation in the presence of ASA/ARC, whereas 20 nm epinephrine was still able to significantly increase the area covered with thrombi (Fig.8). Interestingly, combined treatment with 5 nm IGF-1 and 20 nm epinephrine had synergistic rescuing effects, with full recovery of the area covered with thrombi in the presence of ASA/ARC. Furthermore, the morphology of the thrombi formed was similar to that of the thrombi achieved with the vehicle control. The synergistic effect of IGF-1 treatment in combination with epinephrine was blocked by wortmannin, with the area covered by thrombi being comparable to the rescue achieved with epinephrine alone. This further confirms the important role of PI3K in IGF-1-mediated resistance to antiplatelet therapy.
Figure 8.

Synergistic effects of insulin-like growth factor-1 (IGF-1) and epinephrine on primer-mediated resistance to dual antiplatelet therapy during ex vivo thrombus formation. Fluorescently labeled (dihexyloxacarbocyanine iodide) whole blood anticoagulated with 2 U mL−1 heparin and 40 μm d-phenylalanylprolyl-arginyl chloromethyl ketone was pretreated with vehicle control or 1 μm AR-C66096 (ARC) and 30 μm acetylsalicylic acid (ASA) in combination in the presence or absence of 100 nm wortmannin (WTM) for a total of 10 min. Blood samples were subsequently preincubated with vehicle control or primer, i.e. 5 nm IGF-1, 20 nm epinephrine, or a combination of both, for a total of 5 min. Samples were then perfused at an arterial shear rate of 1000 s−1 over fibrillar collagen (50 μg mL−1) for 5 min. Samples were washed with HEPES–Tyrode buffer for 2 min to remove non-adherent cells. Representative fluorescent images are shown, along with quantitative analysis of surface area covered (%) with thrombi. Data represent the average results taken from ≥ 15 random microscopic fields per experiment (n = 3–5). Statistical analysis: one-way anova was used in conjunction with a Bonferroni post hoc test; *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Aspirin and P2Y12 antagonists are commonly administered to patients at risk for thrombosis. Certain patient populations show resistance to these antiplatelet compounds, and are at risk of developing subsequent cardiovascular complications 58. In this study, we determined the role of platelet primers in resistance to antiplatelet therapy. We found that the primers IGF-1, TPO, and epinephrine, which are known to be elevated in patients with or at risk of ACS, were able to overcome the inhibitory effects of antiplatelet drugs on platelet functional responses 31–34,36,37. Furthermore, we have demonstrated that IGF-1-mediated and TPO-mediated resistance to antiplatelet drugs is PI3K-dependent, as pan-PI3K inhibitors blocked this resistance. Interestingly, PI3K inhibition did not block epinephrine-mediated resistance, revealing a PI3K-independent mechanism of resistance.
One of the proposed mechanisms by which antiplatelet resistance develops is via activation of alternative signaling pathways that are ADP-independent and/or TxA2-independent 59. Interestingly, we found that platelet primers were able to enhance functional responses in the presence of ADP and TxA2 inhibitors. This demonstrates that there are ADP-independent and TxA2-independent mechanisms driving primer-mediated resistance to antiplatelet compounds. Indeed, several studies found priming effects of IGF-1, TPO and epinephrine on platelet function under conditions where the ADP receptor P2Y12 was blocked, confirming that ADP release is not essential in this process 27,28,60,61. In contrast, other studies have shown that the effect of TPO on platelet function is, at least partially, mediated by an increase in TxA2 production 62. Here, we have demonstrated that IGF-1, TPO and epinephrine all increase SFLLRN-mediated TxA2 production. However, it is unlikely that this increase in TxA2 formation contributes to primer-mediated resistance to antiplatelet compounds, because: (i) IGF-1-mediated and TPO-mediated increases in TxA2 formation were blocked by ARC; and (ii) although epinephrine-mediated increases in TxA2 formation were still present in ARC-treated platelets, the finding that ASA had little effect on epinephrine-mediated rescue of the inhibitory effect of ARC (Fig.2Eiii) suggests that increased TxA2 formation plays a minor role in this context. These findings confirm that primer-mediated resistance to ARC is largely independent of TxA2 formation.
We translated our studies into a more physiologic setting to assess the effects of treatment of whole blood with ASA/ARC on ex vivo thrombus formation. ASA/ARC treatment significantly reduced the area covered with thrombi and the average thrombus size. Notably, ASA/ARC treatment had a distinct effect on thrombus morphology, whereby individual platelets could be clearly identified. This observation is in agreement with other studies, which have also shown the ability of the P2Y12 inhibitor AR-C69931MX (cangrelor) to reduce platelet thrombus height 63. Furthermore, platelets from P2Y12-deficient mice that have been treated with aspirin show a similar arrangement of loosely packed thrombi during flow studies 57. As TxA2 and ADP play an important paracrine/autocrine role that is involved in stabilizing thrombi 64,65, it may not be surprising that aspirin and P2Y12 blockade gives rise to a loosely packed platelet morphology.
Epinephrine and IGF-1 rescued the inhibitory effects of ASA/ARC, significantly increasing not only the area covered with thrombi, but also the thrombus size. Although epinephrine-treated platelets formed larger thrombi, they retained their loosely packed morphology. Conversely, IGF-1 treatment appeared to give rise to thrombi that were more comparable to those obtained with the vehicle control, with the identification of individual platelets being more difficult. TPO was unable to rescue the inhibitory effects of ASA and P2Y12 blockade, demonstrating that TPO-mediated potentiation of GPVI platelet function is reliant on TxA2 and ADP signaling.
Numerous platelet primers, including matrix metalloproteinase-2, Gas6, ephrin B, IGF-1, TPO, and epinephrine, have been reported to enhance platelet function via a PI3K-dependent signaling mechanism 26–29,54,55. To establish the role of PI3K in primer-mediated resistance to antiplatelet compounds, we treated platelets with pan-PI3K inhibitors, and we observed complete blockade of IGF-1-mediated and TPO-mediated potentiation of SFLLRN-induced aggregation in the presence of ASA/ARC. This observation is consistent with previous studies that demonstrated an important role of PI3K in IGF-1-mediated and TPO-mediated potentiation of platelet function 26–28,61,66, and reveals a critical role of PI3K in IGF-1-mediated and TPO-mediated resistance to ASA and P2Y12 antagonism. It is likely that the p110α isoform of PI3K drives IGF-1-mediated resistance to antiplatelet compounds, as previous studies have revealed an important role of this isoform in IGF-1-mediated potentiation of platelet function 26,27,61. Interestingly, PI3K inhibitors were unable to block epinephrine-mediated enhancement of platelet function in the presence of ASA/ARC, revealing a PI3K-independent mechanism for epinephrine-mediated resistance. Epinephrine binds to the α2A-adrenergic receptor, and stimulates activation of Gz signaling pathways, which may substitute for Gi-coupled P2Y12 signaling during platelet activation 67. Therefore, epinephrine may compensate for blockade of ADP, resulting in resistance to P2Y12 antagonists. It is of note that several studies found a correlation between exaggerated platelet responses to low doses of epinephrine and platelet hyperactivity 68,69, indicating that epinephrine may contribute to platelet hyperactivity. Epinephrine has also been found to potentiate Ca2+ release in platelets; therefore, we explored the role that Ca2+ may play in epinephrine-mediated resistance to antiplatelet compounds 70. Our results demonstrated that elevations in intracellular Ca2+ did not contribute to epinephrine-mediated resistance or to the resistance induced by IGF-1 and TPO (Fig. S5).
IGF-1-mediated enhancement of thrombus formation in the presence of ASA/ARC was blocked by wortmannin. However, epinephrine was still able to rescue the inhibitory effects of the antiplatelet compounds in the presence of wortmannin. This reinforces the role of PI3K in IGF-1-mediated resistance, and further demonstrates the PI3K-independent mechanisms driving epinephrine-mediated resistance. Interestingly, combined treatment with more physiologic levels of IGF-1 (5 nm) and epinephrine (20 nm) gave rise to synergistic rescuing effects in the presence of ASA/ARC. Although IGF-1 (5 nm) was unable to rescue the inhibitory effects of ASA/ARC alone, it was able to enhance the rescuing effects of epinephrine (20 nm) and return the morphology of the thrombi to that seen with the vehicle. This was a particularly interesting observation, as it is likely that patients who present with ACS or who are susceptible to ACS will have elevations in multiple circulating primers. Primers may act independently to make patients resistant to antiplatelet therapy; however, it is more likely that combinations of primers have additive or synergistic effects that put patients at higher risk for thrombotic vascular events.
In conclusion, we have demonstrated the ability of circulating primers to overcome the inhibitory effects of ASA and/or ARC. Changes in plasma levels of primers therefore predispose patients to antiplatelet resistance and thrombotic events. We have shown that there are PI3K-dependent mechanisms driving IGF-1-mediated and TPO-mediated resistance, whereas PI3K-independent mechanisms drive epinephrine-mediated resistance. The PI3K pathway and the α2A-adrenergic receptor may be promising drug targets to combat the insufficient inhibition induced by current antiplatelet therapies.
Addendum
T. A. Blair designed and performed experiments, analyzed the results, and wrote the manuscript. S. F. Moore designed and performed initial experiments, contributed to discussion, and edited the manuscript. I. Hers conceived the experiments, supervised the project, and wrote the manuscript.
Acknowledgments
This work was supported by the British Heart Foundation (grants FS/12/3/29232 and PG/10/100/28658). We thank E. Aitken for technical support during this work. We also acknowledge S. England and T. Allan for performing preliminary work for the study.
Disclosure of Conflict of Interests
The authors state that they have no conflict of interest.
Supporting Information
Fig. S1. IGF-1, TPO and epinephrine potentiate platelet aggregation in a dose-dependent manner.
Fig. S2. IGF-1, TPO and epinephrineA do not act as agonists in the washed platelet system.
Fig. S3. Epinephrine and combined primer treatments rescue PAR-1-mediated platelet aggregation induced by subthreshold SFLLRN in the presence of antiplatelet compounds.
Fig. S4. The PI3K inhibitor LY294002 reveals a critical role of PI3K in IGF-1-mediated and TPO-mediated resistance to dual antiplatelet therapy.
Fig. S5. Primer-mediated resistance to antiplatelet drugs is not driven by Ca2+ signaling.
References
- Roth GJ, Majerus PW. The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J Clin Investig. 1975;56:624–32. doi: 10.1172/JCI108132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrono C, Baigent C, Hirsh J, Roth G American College of Chest Physicians. Antiplatelet drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition) Chest. 2008;133:199S–233S. doi: 10.1378/chest.08-0672. [DOI] [PubMed] [Google Scholar]
- Savi P, Labouret C, Delesque N, Guette F, Lupker J, Herbert JM. P2y(12), a new platelet ADP receptor, target of clopidogrel. Biochem Biophys Res Commun. 2001;283:379–83. doi: 10.1006/bbrc.2001.4816. [DOI] [PubMed] [Google Scholar]
- Weber AA, Reimann S, Schror K. Specific inhibition of ADP-induced platelet aggregation by clopidogrel in vitro. Br J Pharmacol. 1999;126:415–20. doi: 10.1038/sj.bjp.0702276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saw J, Madsen EH, Chan S, Maurer-Spurej E. The ELAPSE (Evaluation of Long-Term Clopidogrel Antiplatelet and Systemic Anti-Inflammatory Effects) study. J Am Coll Cardiol. 2008;52:1826–33. doi: 10.1016/j.jacc.2008.08.047. [DOI] [PubMed] [Google Scholar]
- Antithrombotic Trialists Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SR, Yusuf S, Peters RJ, Bertrand ME, Lewis BS, Natarajan MK, Malmberg K, Rupprecht H, Zhao F, Chrolavicius S, Copland I, Fox KA. Clopidogrel in Unstable angina to prevent Recurrent Events trial I. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet. 2001;358:527–33. doi: 10.1016/s0140-6736(01)05701-4. [DOI] [PubMed] [Google Scholar]
- Markus HS, Droste DW, Kaps M, Larrue V, Lees KR, Siebler M, Ringelstein EB. Dual antiplatelet therapy with clopidogrel and aspirin in symptomatic carotid stenosis evaluated using Doppler embolic signal detection – The Clopidogrel and Aspirin for Reduction of Emboli in Symptomatic Carotid Stenosis (CARESS) trial. Circulation. 2005;111:2233–40. doi: 10.1161/01.CIR.0000163561.90680.1C. [DOI] [PubMed] [Google Scholar]
- Armstrong PC, Leadbeater PD, Chan MV, Kirkby NS, Jakubowski JA, Mitchell JA, Warner TD. In the presence of strong P2Y12 receptor blockade, aspirin provides little additional inhibition of platelet aggregation. J Thromb Haemost. 2011;9:552–61. doi: 10.1111/j.1538-7836.2010.04160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gum PA, Kottke-Marchant K, Poggio ED, Gurm H, Welsh PA, Brooks L, Sapp SK, Topol EJ. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol. 2001;88:230–5. doi: 10.1016/s0002-9149(01)01631-9. [DOI] [PubMed] [Google Scholar]
- Kasotakis G, Pipinos II, Lynch TG. Current evidence and clinical implications of aspirin resistance. J Vasc Surg. 2009;50:1500–10. doi: 10.1016/j.jvs.2009.06.023. [DOI] [PubMed] [Google Scholar]
- Goodman T, Sharma P, Ferro A. The genetics of aspirin resistance. Int J Clin Pract. 2007;61:826–34. doi: 10.1111/j.1742-1241.2007.01344.x. [DOI] [PubMed] [Google Scholar]
- Clarke TA, Waskell LA. The metabolism of clopidogrel is catalyzed by human cytochrome P450 3A and is inhibited by atorvastatin. Drug Metab Dispos. 2003;31:53–9. doi: 10.1124/dmd.31.1.53. [DOI] [PubMed] [Google Scholar]
- Pereillo JM, Maftouh M, Andrieu A, Uzabiaga MF, Fedeli O, Savi P, Pascal M, Herbert JM, Maffrand JP, Picard C. Structure and stereochemistry of the active metabolite of clopidogrel. Drug Metab Dispos. 2002;30:1288–95. doi: 10.1124/dmd.30.11.1288. [DOI] [PubMed] [Google Scholar]
- Hou X, Shi J, Sun H. Gene polymorphism of cytochrome P450 2C19*2 and clopidogrel resistance reflected by platelet function assays: a meta-analysis. Eur J Clin Pharmacol. 2014;70:1041–7. doi: 10.1007/s00228-014-1714-x. [DOI] [PubMed] [Google Scholar]
- Feher G, Koltai K, Alkonyi B, Papp E, Keszthelyi Z, Kesmarky G, Toth K. Clopidogrel resistance: role of body mass and concomitant medications. Int J Cardiol. 2007;120:188–92. doi: 10.1016/j.ijcard.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Nguyen TA, Diodati JG, Pharand C. Resistance to clopidogrel: a review of the evidence. J Am Coll Cardiol. 2005;45:1157–64. doi: 10.1016/j.jacc.2005.01.034. [DOI] [PubMed] [Google Scholar]
- Macchi L, Christiaens L, Brabant S, Sorel N, Allal J, Mauco G, Brizard A. Resistance to aspirin in vitro is associated with increased platelet sensitivity to adenosine diphosphate. Thromb Res. 2002;107:45–9. doi: 10.1016/s0049-3848(02)00210-4. [DOI] [PubMed] [Google Scholar]
- Dichiara J, Bliden KP, Tantry US, Chaganti SK, Kreutz RP, Gesheff TB, Kreutz Y, Gurbel PA. Platelet function measured by VerifyNow identifies generalized high platelet reactivity in aspirin treated patients. Platelets. 2007;18:414–23. doi: 10.1080/09537100701206824. [DOI] [PubMed] [Google Scholar]
- Furman MI, Frelinger AL, 3rd, Michelson AD. GPIIb/IIIa inhibitor-induced dethrombosis. J Thromb Thrombolysis. 2004;18:11–17. doi: 10.1007/s11239-004-0168-x. [DOI] [PubMed] [Google Scholar]
- Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, Ramirez C, Sabate M, Jimenez-Quevedo P, Hernandez R, Moreno R, Escaned J, Alfonso F, Banuelos C, Costa MA, Bass TA, Macaya C. Platelet function profiles in patients with type 2 diabetes and coronary artery disease on combined aspirin and clopidogrel treatment. Diabetes. 2005;54:2430–5. doi: 10.2337/diabetes.54.8.2430. [DOI] [PubMed] [Google Scholar]
- Lupia E, Bosco O, Mariano F, Dondi AE, Goffi A, Spatola T, Cuccurullo A, Tizzani P, Brondino G, Stella M, Montrucchio G. Elevated thrombopoietin in plasma of burned patients without and with sepsis enhances platelet activation. J Thromb Haemost. 2009;7:1000–8. doi: 10.1111/j.1538-7836.2009.03348.x. [DOI] [PubMed] [Google Scholar]
- Stolla MC, Li D, Lu L, Woulfe DS. Enhanced platelet activity and thrombosis in a murine model of type I diabetes are partially insulin-like growth factor 1-dependent and phosphoinositide 3-kinase-dependent. J Thromb Haemost. 2013;11:919–29. doi: 10.1111/jth.12170. [DOI] [PubMed] [Google Scholar]
- Wang JS, Cheng LJ. Effect of strenuous, acute exercise on alpha2-adrenergic agonist-potentiated platelet activation. Arterioscler Thromb Vasc Biol. 1999;19:1559–65. doi: 10.1161/01.atv.19.6.1559. [DOI] [PubMed] [Google Scholar]
- Gresele P, Falcinelli E, Momi S. Potentiation and priming of platelet activation: a potential target for antiplatelet therapy. Trends Pharmacol Sci. 2008;29:352–60. doi: 10.1016/j.tips.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Blair TA, Moore SF, Williams CM, Poole AW, Vanhaesebroeck B, Hers I. Phosphoinositide 3-kinases p110alpha and p110beta have differential roles in insulin-like growth factor-1-mediated Akt phosphorylation and platelet priming. Arterioscler Thromb Vasc Biol. 2014;34:1681–8. doi: 10.1161/ATVBAHA.114.303954. [DOI] [PubMed] [Google Scholar]
- Hers I. Insulin-like growth factor-1 potentiates platelet activation via the IRS/PI3Kalpha pathway. Blood. 2007;110:4243–52. doi: 10.1182/blood-2006-10-050633. [DOI] [PubMed] [Google Scholar]
- Pasquet JM, Gross BS, Gratacap MP, Quek L, Pasquet S, Payrastre B, van Willigen G, Mountford JC, Watson SP. Thrombopoietin potentiates collagen receptor signaling in platelets through a phosphatidylinositol 3-kinase-dependent pathway. Blood. 2000;95:3429–34. [PubMed] [Google Scholar]
- Selheim F, Froyset AK, Strand I, Vassbotn FS, Holmsen H. Adrenaline potentiates PI 3-kinase in platelets stimulated with thrombin and SFRLLN: role of secreted ADP. FEBS Lett. 2000;485:62–6. doi: 10.1016/s0014-5793(00)02188-8. [DOI] [PubMed] [Google Scholar]
- Luchessi AD, Silbiger VN, Hirata RD, Lima-Neto LG, Cavichioli D, Iniguez A, Bravo M, Bastos G, Sousa AG, Brion M, Carracedo A, Hirata MH. Pharmacogenomics of anti-platelet therapy focused on peripheral blood cells of coronary arterial disease patients. Clin Chim Acta. 2013;425:9–17. doi: 10.1016/j.cca.2013.06.021. [DOI] [PubMed] [Google Scholar]
- Fischer F, Schulte H, Mohan S, Tataru MC, Kohler E, Assmann G, von Eckardstein A. Associations of insulin-like growth factors, insulin-like growth factor binding proteins and acid-labile subunit with coronary heart disease. Clin Endocrinol. 2004;61:595–602. doi: 10.1111/j.1365-2265.2004.02136.x. [DOI] [PubMed] [Google Scholar]
- Bayes-Genis A, Conover CA, Schwartz RS. The insulin-like growth factor axis: a review of atherosclerosis and restenosis. Circ Res. 2000;86:125–30. doi: 10.1161/01.res.86.2.125. [DOI] [PubMed] [Google Scholar]
- Senaran H, Ileri M, Altinbas A, Kosar A, Yetkin E, Ozturk M, Karaaslan Y, Kirazli S. Thrombopoietin and mean platelet volume in coronary artery disease. Clin Cardiol. 2001;24:405–8. doi: 10.1002/clc.4960240511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupia E, Bosco O, Bergerone S, Dondi AE, Goffi A, Oliaro E, Cordero M, Del Sorbo L, Trevi G, Montrucchio G. Thrombopoietin contributes to enhanced platelet activation in patients with unstable angina. J Am Coll Cardiol. 2006;48:2195–203. doi: 10.1016/j.jacc.2006.04.106. [DOI] [PubMed] [Google Scholar]
- Jacobs MC, Lenders JW, Willemsen JJ, Thien T. Adrenomedullary secretion of epinephrine is increased in mild essential hypertension. Hypertension. 1997;29:1303–8. doi: 10.1161/01.hyp.29.6.1303. [DOI] [PubMed] [Google Scholar]
- Slavikova J, Kuncova J, Topolcan O. Plasma catecholamines and ischemic heart disease. Clin Cardiol. 2007;30:326–30. doi: 10.1002/clc.20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsberg RP, Cryer PE, Roberts R. Serial plasma catecholamine response early in the course of clinical acute myocardial infarction: relationship to infarct extent and mortality. Am Heart J. 1981;102:24–9. doi: 10.1016/0002-8703(81)90408-7. [DOI] [PubMed] [Google Scholar]
- Pamukcu B, Oflaz H, Oncul A, Umman B, Mercanoglu F, Ozcan M, Meric M, Nisanci Y. The role of aspirin resistance on outcome in patients with acute coronary syndrome and the effect of clopidogrel therapy in the prevention of major cardiovascular events. J Thromb Thrombolysis. 2006;22:103–10. doi: 10.1007/s11239-006-8952-4. [DOI] [PubMed] [Google Scholar]
- Matetzky S, Shenkman B, Guetta V, Shechter M, Beinart R, Goldenberg I, Novikov I, Pres H, Savion N, Varon D, Hod H. Clopidogrel resistance is associated with increased risk of recurrent atherothrombotic events in patients with acute myocardial infarction. Circulation. 2004;109:3171–5. doi: 10.1161/01.CIR.0000130846.46168.03. [DOI] [PubMed] [Google Scholar]
- Hunter RW, Hers I. Insulin/IGF-1 hybrid receptor expression on human platelets: consequences for the effect of insulin on platelet function. J Thromb Haemost. 2009;7:2123–30. doi: 10.1111/j.1538-7836.2009.03637.x. [DOI] [PubMed] [Google Scholar]
- Walsh TG, Harper MT, Poole AW. SDF-1alpha is a novel autocrine activator of platelets operating through its receptor CXCR4. Cell Signal. 2014;27:37–46. doi: 10.1016/j.cellsig.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilio K, Harper MT, Cosemans JM, Konopatskaya O, Munnix IC, Prinzen L, Leitges M, Liu Q, Molkentin JD, Heemskerk JW, Poole AW. Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. J Biol Chem. 2010;285:23410–19. doi: 10.1074/jbc.M110.136176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper MT, Poole AW. Chloride channels are necessary for full platelet phosphatidylserine exposure and procoagulant activity. Cell Death Dis. 2013;4:e969. doi: 10.1038/cddis.2013.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Foster C, Lecchi A, Quinton TM, Prosser DM, Jin J, Cattaneo M, Kunapuli SP. Protease-activated receptors 1 and 4 do not stimulate G(i) signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of G(i) signaling. Blood. 2002;99:3629–36. doi: 10.1182/blood.v99.10.3629. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Arai T, Tanaka T, Yamaoka G, Kiuchi H, Kajikawa T, Kawanishi K, Ohnishi H, Yamaguchi M, Takahara J, Irino S. Thrombopoietin modulates platelet activation in vitro through protein-tyrosine phosphorylation. Stem Cells. 1996;14:439–44. doi: 10.1002/stem.140439. [DOI] [PubMed] [Google Scholar]
- Oda A, Miyakawa Y, Druker BJ, Ozaki K, Yabusaki K, Shirasawa Y, Handa M, Kato T, Miyazaki H, Shimosaka A, Ikeda Y. Thrombopoietin primes human platelet aggregation induced by shear stress and by multiple agonists. Blood. 1996;87:4664–70. [PubMed] [Google Scholar]
- Mills DC, Roberts GC. Effects of adrenaline on human blood platelets. J Physiol. 1967;193:443–53. doi: 10.1113/jphysiol.1967.sp008369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam SY, Lee EJ, Kim KR, Cha BS, Song YD, Lim SK, Lee HC, Huh KB. Effect of obesity on total and free insulin-like growth factor (IGF)-1, and their relationship to IGF-binding protein (BP)-1, IGFBP-2, IGFBP-3, insulin, and growth hormone. Int J Obes Relat Metab Disord. 1997;21:355–9. doi: 10.1038/sj.ijo.0800412. [DOI] [PubMed] [Google Scholar]
- Godau J, Herfurth M, Kattner B, Gasser T, Berg D. Increased serum insulin-like growth factor 1 in early idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry. 2010;81:536–8. doi: 10.1136/jnnp.2009.175752. [DOI] [PubMed] [Google Scholar]
- Walsh LA, Damjanovski S. IGF-1 increases invasive potential of MCF 7 breast cancer cells and induces activation of latent TGF-beta1 resulting in epithelial to mesenchymal transition. Cell Commun Signal. 2011;9:10. doi: 10.1186/1478-811X-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhang G, Le Breton GC, Gao X, Malik AB, Du X. Two waves of platelet secretion induced by thromboxane A2 receptor and a critical role for phosphoinositide 3-kinases. J Biol Chem. 2003;278:30725–31. doi: 10.1074/jbc.M301838200. [DOI] [PubMed] [Google Scholar]
- Paul BZ, Jin J, Kunapuli SP. Molecular mechanism of thromboxane A(2)-induced platelet aggregation. Essential role for p2t(ac) and alpha(2a) receptors. J Biol Chem. 1999;274:29108–14. doi: 10.1074/jbc.274.41.29108. [DOI] [PubMed] [Google Scholar]
- Armstrong PC, Dhanji AR, Tucker AT, Mitchell JA, Warner TD. Reduction of platelet thromboxane A2 production ex vivo and in vivo by clopidogrel therapy. J Thromb Haemost. 2010;8:613–15. doi: 10.1111/j.1538-7836.2009.03714.x. [DOI] [PubMed] [Google Scholar]
- Falcinelli E, Guglielmini G, Torti M, Gresele P. Intraplatelet signaling mechanisms of the priming effect of matrix metalloproteinase-2 on platelet aggregation. J Thromb Haemost. 2005;3:2526–35. doi: 10.1111/j.1538-7836.2005.01614.x. [DOI] [PubMed] [Google Scholar]
- Jackson SF, Schoenwaelder SM. Type I phosphoinositide 3-kinases: potential antithrombotic targets? Cell Mol Life Sci. 2006;63:1085–90. doi: 10.1007/s00018-006-6001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucitt MB, O'Brien S, Cowman J, Meade G, Basabe-Desmonts L, Somers M, Kent N, Ricco AJ, Kenny D. Assaying the efficacy of dual-antiplatelet therapy: use of a controlled-shear-rate microfluidic device with a well-defined collagen surface to track dynamic platelet adhesion. Anal Bioanal Chem. 2013;405:4823–34. doi: 10.1007/s00216-013-6897-y. [DOI] [PubMed] [Google Scholar]
- Andre P, LaRocca T, Delaney SM, Lin PH, Vincent D, Sinha U, Conley PB, Phillips DR. Anticoagulants (thrombin inhibitors) and aspirin synergize with P2Y12 receptor antagonism in thrombosis. Circulation. 2003;108:2697–703. doi: 10.1161/01.CIR.0000093279.36628.12. [DOI] [PubMed] [Google Scholar]
- Sofi F, Marcucci R, Gori AM, Giusti B, Abbate R, Gensini GF. Clopidogrel non-responsiveness and risk of cardiovascular morbidity. An updated meta-analysis. Thromb Haemost. 2010;103:841–8. doi: 10.1160/TH09-06-0418. [DOI] [PubMed] [Google Scholar]
- Topcuoglu MA, Arsava EM, Ay H. Antiplatelet resistance in stroke. Expert Rev Neurother. 2011;11:251–63. doi: 10.1586/ern.10.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenegard M, Vretenbrant-Oberg K, Nylander M, Desilets S, Lindstrom EG, Larsson A, Ramstrom I, Ramstrom S, Lindahl TL. The ATP-gated P2X1 receptor plays a pivotal role in activation of aspirin-treated platelets by thrombin and epinephrine. J Biol Chem. 2008;283:18493–504. doi: 10.1074/jbc.M800358200. [DOI] [PubMed] [Google Scholar]
- Kim S, Garcia A, Jackson SP, Kunapuli SP. Insulin-like growth factor-1 regulates platelet activation through PI3-K alpha isoform. Blood. 2007;110:4206–13. doi: 10.1182/blood-2007-03-080804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Os E, Wu YP, Pouwels JG, Ijsseldijk MJ, Sixma JJ, Akkerman JW, De Groot PG, van Willigen G. Thrombopoietin increases platelet adhesion under flow and decreases rolling. Br J Haematol. 2003;121:482–90. doi: 10.1046/j.1365-2141.2003.04292.x. [DOI] [PubMed] [Google Scholar]
- Goto S, Tamura N, Ishida H, Ruggeri ZM. Dependence of platelet thrombus stability on sustained glycoprotein IIb/IIIa activation through adenosine 5′-diphosphate receptor stimulation and cyclic calcium signaling. J Am Coll Cardiol. 2006;47:155–62. doi: 10.1016/j.jacc.2005.08.055. [DOI] [PubMed] [Google Scholar]
- van Gestel MA, Reitsma S, Slaaf DW, Heijnen VV, Feijge MA, Lindhout T, van Zandvoort MA, Elg M, Reneman RS, Heemskerk JW, Egbrink MG. Both ADP and thrombin regulate arteriolar thrombus stabilization and embolization, but are not involved in initial hemostasis as induced by micropuncture. Microcirculation. 2007;14:193–205. doi: 10.1080/10739680601139294. [DOI] [PubMed] [Google Scholar]
- Stegner D, Nieswandt B. Platelet receptor signaling in thrombus formation. J Mol Med. 2011;89:109–21. doi: 10.1007/s00109-010-0691-5. [DOI] [PubMed] [Google Scholar]
- Kojima H, Shinagawa A, Shimizu S, Kanada H, Hibi M, Hirano T, Nagasawa T. Role of phosphatidylinositol-3 kinase and its association with Gab1 in thrombopoietin-mediated up-regulation of platelet function. Exp Hematol. 2001;29:616–22. doi: 10.1016/s0301-472x(01)00623-3. [DOI] [PubMed] [Google Scholar]
- Kim S, Jin J, Kunapuli SP. Akt activation in platelets depends on Gi signaling pathways. J Biol Chem. 2004;279:4186–95. doi: 10.1074/jbc.M306162200. [DOI] [PubMed] [Google Scholar]
- Hayes C, Kitahara S, Tcherniantchouk O. Decreased threshold of aggregation to low-dose epinephrine is evidence of platelet hyperaggregability in patients with thrombosis. Hematol Rep. 2014;6:5326. doi: 10.4081/hr.2014.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee DL, Bergeron AL, Sun CW, Dong JF, Bray PF. Platelet hyperreactivity generalizes to multiple forms of stimulation. J Thromb Haemost. 2006;4:2043–50. doi: 10.1111/j.1538-7836.2006.02089.x. [DOI] [PubMed] [Google Scholar]
- Keularts IM, van Gorp RM, Feijge MA, Vuist WM, Heemskerk JW. alpha(2A)-adrenergic receptor stimulation potentiates calcium release in platelets by modulating cAMP levels. J Biol Chem. 2000;275:1763–72. doi: 10.1074/jbc.275.3.1763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. IGF-1, TPO and epinephrine potentiate platelet aggregation in a dose-dependent manner.
Fig. S2. IGF-1, TPO and epinephrineA do not act as agonists in the washed platelet system.
Fig. S3. Epinephrine and combined primer treatments rescue PAR-1-mediated platelet aggregation induced by subthreshold SFLLRN in the presence of antiplatelet compounds.
Fig. S4. The PI3K inhibitor LY294002 reveals a critical role of PI3K in IGF-1-mediated and TPO-mediated resistance to dual antiplatelet therapy.
Fig. S5. Primer-mediated resistance to antiplatelet drugs is not driven by Ca2+ signaling.