

Abstract
Currently, sorafenib is the only available chemotherapeutic agent for advanced hepatocellular carcinoma (HCC), but it cannot be used in patients with liver cirrhosis (LC) or thrombocytopenia. In these cases, sorafenib is likely effective if given in combination with treatments that increase the number of platelets, such as thrombopoietin (TPO) receptor agonists. Increasing the platelet count via TPO treatment resulted in reduction of LC. Eltrombopag (EP), a TPO receptor agonist, has been reported to have antitumor effects against certain cancers, despite their lack of TPO receptor expression. We hypothesized that EP may possess antitumor activity against HCC in addition to its ability to suppress hepatic fibrosis by increasing the platelet count. In the present study, the antitumor activity of EP was examined by assessing the inhibition of cell proliferation and then ascertaining the ability of iron supplementation to reverse these effects in HepG2, Hep3B and Huh7 cells. In addition, a cell cycle assay was performed using flow cytometry, and signal transduction was evaluated by analyzing cell cycle-related protein expression. The results of EP were compared with those of the most common iron chelator, deferoxamine (DFO). The combined effect of EP and sorafenib was also assessed. The results revealed that EP exerts antitumor activity in HCC that is mediated by the modulation of intracellular iron content. EP suppressed the expression of the cell cycle-related protein cyclin D1 and elicited cell cycle arrest in the G0/G1 phase. The activity of EP was comparable to that of DFO in HCC, and EP did not compete with sorafenib at low concentrations. In conclusion, our findings suggest that EP is a good candidate chemotherapeutic agent for the treatment of HCC in patients with LC and thrombocytopenia.
Keywords: hepatocellular carcinoma, eltrombopag, deferoxamine cell cycle, iron chelate, thrombopoietin, Huh7, thrombocytopenia, cirrhosis, liver cirrhosis, liver cancer, sorafenib
Introduction
Hepatocellular carcinoma (HCC) is the most common disease that causes primary liver tumors and the fifth most common malignant disease in the world (1). HCC develops frequently in patients with liver cirrhosis (LC) (2). HCC has a poor prognosis and a high rate of recurrence, even after treatment. Currently, sorafenib is the only available chemotherapeutic agent for advanced HCC, but it cannot be used to treat patients with LC and thrombocytopenia due to the risk of bleeding (3–5). Therefore, there is an urgent need for the development of effective therapeutic agents for HCC.
Thrombopoietin (TPO) is the most important growth factor in the regulation of megakaryocyte development and platelet production (6). TPO is the endogenous ligand of the TPO receptor [(TPO-R, also known as myeloproliferative leukemia virus oncogene (MPL)], which is expressed on the surface of megakaryocytes, megakaryocyte precursors and platelets (7–10).
Eltrombopag (EP) is an oral, nonpeptide, small molecule TPO-R agonist that has proven efficacy in treating chronic immune thrombocytopenic purpura (ITP) and hepatitis C-related thrombocytopenia (11,12).
Despite concerns that some leukemia blast cells express TPO-R, EP has been reported to have no proliferative effects in patients with myelodysplastic syndromes or AML (13); instead, EP inhibits the proliferation of leukemia cell lines (14).
Additionally, recent studies have demonstrated that the antileukemic effect of EP is not related to the TPO-R pathway (15) and that EP inhibits the growth of breast, lung, and ovarian tumor cells (16). EP has been reported to increase platelet counts in a short-term clinical study that included patients with HCC (17). However, it remains unclear whether EP has an antitumor effect in HCC.
The purpose of this study was to clarify whether EP affects the growth of human HCC cells in vitro.
Materials and methods
Reagents
EP (SB-497115; Selleck Chemicals, Houston, TX, USA) was dissolved as a 1 mg/ml stock solution in distilled water and was stored in the dark at room temperature for up to 2 weeks. Deferoxamine mesylate (DFO) and ferrous ammonium citrate (FAC) were purchased from Sigma-Aldrich (St. Louis, MO, USA), and sorafenib was purchased from Invitrogen (Grand Island, NY, USA).
Cell culture
Human hepatoblastoma cell lines (HepG2 and Hep3B) and a well-differentiated human HCC cell line (Huh7) were used in these experiments. HepG2, Hep3B and Huh7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Wako Pure Chemical Industries, Ltd., Osaka, Japan) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA) and 1% penicillin/streptomycin (Invitrogen).
Cell proliferation
Following cell incubation at 37°C for 24 h, the medium was changed to 100 μl of DMEM supplemented with 10% FBS, and different concentrations (0.1, 0.4, 1, 4, 10, 40 and 100 μg/ml) of EP were added to each well. A recent double-blind, placebo-controlled, randomized dose-escalation study showed that maximum EP serum concentrations of >20 μg/ml are clinically achievable with minimal toxicity (18). The reported IC50 values of EP in other cancer cell lines range from 4.8 to 49.7 μg/ml (16). Additionally, 0.4 μg/ml EP is equivalent to 1 μM of EP. Therefore, we selected the concentrations indicated above. After a 72-h incubation at 37°C, the cells were counted based on DNA content using a bromode-oxyuridine (BrdU) assay kit (Roche Diagnostics, Penzberg, Germany) and Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Kumamoto, Japan) according to the instructions of the manufacturers.
The mechanism by which EP inhibits HCC cell growth has not been well characterized. EP reduces intracellular iron concentrations, and iron chelators exhibit anti-proliferative effects in myeloid leukemia cells (15). We investigated whether the antitumor effect of EP is secondary to its ability to deplete intracellular iron. Iron was pre-loaded into cells by treatment with 500 μg/ml FAC for 24 h prior to EP addition.
Cell cycle analysis
Changes in the cell cycle after EP treatment were assessed using a Muse™ cell cycle kit (EMD Millipore, Billerica, MA, USA). The cells were cultured in 10-cm dishes with various concentrations of EP (0–100 μg/ml) for 72 h. After treatment, the cells were collected in DMEM supplemented with 1% FBS, and the Muse™ cell cycle test reagent was added. The cells were mixed by vortexing, and the reaction was allowed to proceed for 30 min at room temperature in the dark. Then, the cells were stained to determine the proportions of cells in the G0/G1, S and G2/M phases using a Muse™ cell analyzer (EMD Millipore).
Western blot analysis of transcription factors
Huh7 cells and hepatocytes were pre-cultured separately in 6-well plates for 24 h; subsequently, the medium was changed to DMEM supplemented with 10% FBS, and 0–100 μg/ml EP was added to each well. Cells were harvested 72 h after the addition of EP. For the western blot analysis, the cell lysates were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (EMD Millipore). The membranes were exposed to primary antibodies against cyclin-dependent kinase inhibitor 1A (p21/CDKN1A) and the G1/S-specific protein cyclin D1 (CCND1). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Cell Signaling Technology, Inc., Beverly, MA, USA) was used as an endogenous control. An anti-rabbit immunoglobulin G horseradish peroxidase-linked secondary antibody was used (Cell Signaling Technology, Inc.).
The most common iron chelator: DFO
To compare the effects of DFO, with those of EP, Huh7 cells were treated with DFO (0.1–100 μg/ml) for 72 h, or not treated. Cell viability was measured at 72 h using a CCK-8 assay as described above.
Evaluation of the combined effects of EP and sorafenib
Huh7 cells were exposed to combinations of EP (0–40 μg/ml) and sorafenib (4 μM) for 72 h. The effect on cell proliferation was assessed using the CCK-8 assay, and the rate of growth inhibition was calculated using the combination index (CI) according to the method reported by Chou (19). The CI was obtained using Biosoft CalcuSyn software (Biosoft, Cambridge, UK): CI=1, cumulative effect; CI<1, synergistic effect; and CI>1, antagonistic effect.
Statistical analysis
The data are presented as the mean and standard deviation. Statistical analyses were performed using the Mann-Whitney U test or one-way ANOVA, and significant results were analyzed using the Bonferroni-Dunn multiple comparisons post hoc test. In all cases, P<0.05 was considered significant.
Results
Effect of EP on human HCC cell lines
HCC cells were treated with various doses of EP for 72 h, and cell viability was assessed using the BrdU assay (Fig. 1A–C). As the dose of EP increased from 0.1 to 100 μg/ml, cell growth was inhibited in a dose-dependent manner in Huh7, HepG2 and Hep3B cells. Compared with the non-treated cells, EP significantly inhibited cell proliferation at concentrations of 40–100 μg/ml based on the BrdU assay results.
Figure 1.
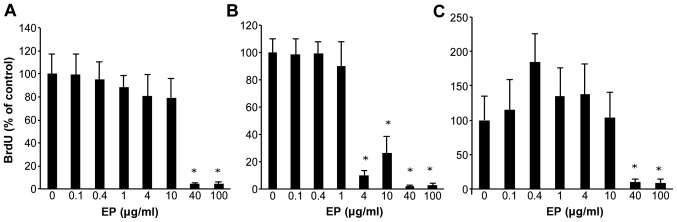
Effect of eltrombopag (EP) on human HCC cell lines. Cell lines were treated with 0–100 μg/ml EP. The inhibited cell proliferation rates are shown for (A) Huh7, (B) HepG2 and (C) Hep3B, respectively. *P<0.05 compared to 0 μg/ml EP. The values indicate ratio compared to 0 μg/ml EP as 100% control.
Preloading HCC cell lines with iron diminishes the anti-proliferative effects of EP
Huh7, HepG2 and Hep3B cells were preloaded with iron (500 μg/ml FAC) for 24 h before EP addition to clarify the antitumor mechanism of action of EP. CCK-8 cell proliferation assays were performed after 72 h. The IC50 values of EP in the HCC cell lines were 5.7 μg/ml for Huh7, 5.4 μg/ml for HepG2, and 4.7 μg/ml for Hep3B. Iron-loaded Huh7 cells were non-treated (control), or treated with EP. Preloading Huh7 cells with iron nearly abolished the anti-proliferative effects of EP (Fig. 2A–C).
Figure 2.
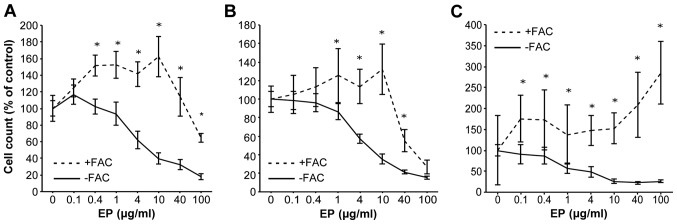
Preloading cells with iron resulted in a rescue from the antiproliferative effects of eltrombopag. Huh7 cells were untreated or treated with 0.1–100 μg/ml of EP 72 h preloaded or not with 500 μg/ml of ferric ammonium citrate (FAC) for 24 h. Cell viability was measured with a CCK-8 assay performed at 72 h. Comparison with the data without iron preloading. (A) Huh7, (B) HepG2 and (C) Hep3B. *P<0.05 compared to each dose EP not preloaded of FAC. The values indicate a ratio compared to 0 μg/ml EP as 100% control.
EP elicited G1 arrest in a human HCC cell line
Huh7 cells were treated with EP (0 or 10 μg/ml) for 72 h, and the cells were monitored during the transition from the G0/G1 phase to S phase. Treatment with 10 μg/ml EP significantly induced G0/G1 phase arrest compared with control treatment (Fig. 3A).
Figure 3.

Effect of eltrombopag on cell cycle progression in Huh7 cells. (A) Cells were treated with EP (0 and 10 μg/ml) for 72 h. DNA was stained with propidium iodide, and flow cytometric analysis of cell phase distribution was performed using a Muse™ cell analyzer. (B) Cells were treated with EP (0, 1 and 10 μg/ml) for 72 h. Protein expression was determined by western blot analysis using anti-cyclin D1, anti-p21 and anti-GAPDH antibodies. SDS-PAGE was performed on acrylamide gels.
Effects of EP on cell cycle-related protein expression in an HCC cell line
Treatment with iron chelators significantly decreases CCND1 levels in various cancer cell types (20). Therefore, we measured the protein levels of CCND1 and p21/CDKN1A using western blot analysis. Treatment with EP (0, 1, or 10 μg/ml) for 72 h downregulated the protein levels of CCND1 and p21/CDKN1A in a dose-dependent manner (Fig. 3B).
Comparison of the antitumor effects of DFO and EP
Huh7 cells were treated with various doses of DFO for 72 h, and cell viability was analyzed using the CCK-8 assay. Cell proliferation was inhibited in a dose-dependent manner in Huh7 cells treated with DFO (0–100 μg/ml) (Fig. 4). EP was nearly equivalent to the most common iron chelator DFO, in terms of the inhibition of cell proliferation.
Figure 4.

Comparison of the antitumor effect of DFO and eltrombopag (EP). Huh7 cells were treated with 0.1–100 μg/ml of DFO for 72 h, or not treated. Cell viability was measured by a CCK-8 assay performed at 72 h. *P<0.05 compared to the same concentration of EP. The values indicate a ratio compared to 0 μg/ml DFO as 100% control.
Combined effect of sorafenib and EP on HCC cells
Compared with untreated cells, EP (4–100 μg/ml) significantly inhibited Huh7 cell proliferation in a dose-dependent manner, as evidenced by the CCK-8 assay results, in the presence of the antitumor agent 4 μM of sorafenib (Fig. 5A). The CI values were ~1 at 0.1–10 μg/ml EP (Fig. 5B). EP clearly did not antagonize sorafenib.
Figure 5.
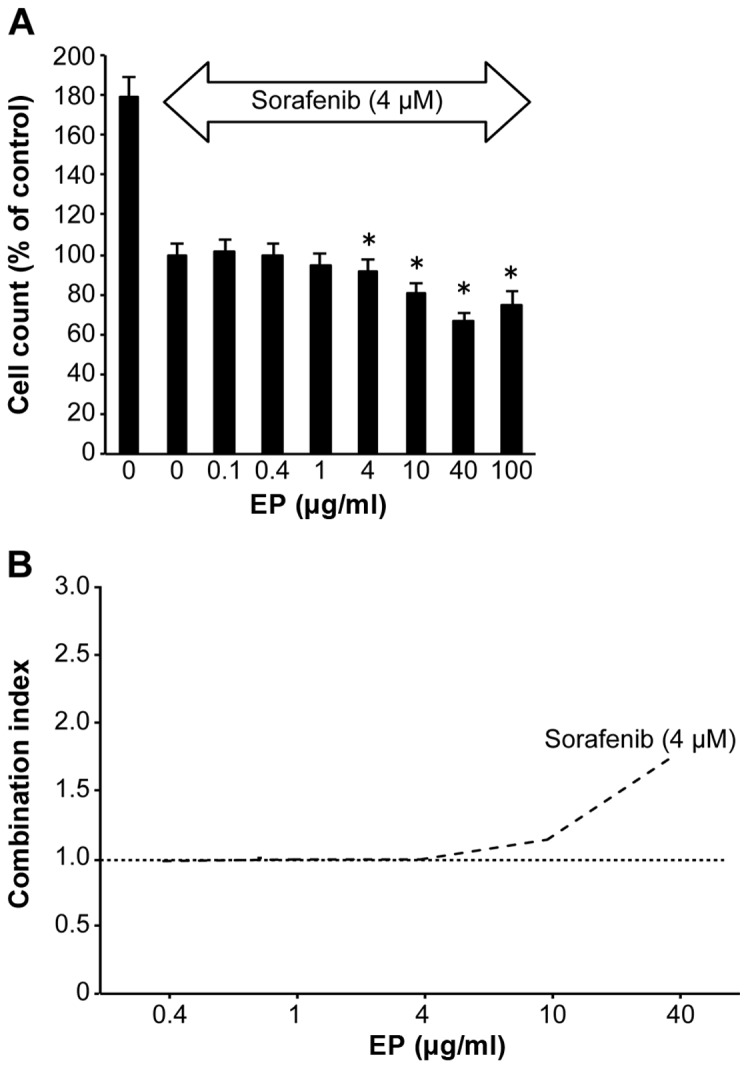
Combined effects of eltrombopag (EP) and sorafenib on HCC. (A) Huh7 cells were treated with 0.1–100 μg/ml of EP and sorafenib (0 and 4 μM), or not treated. *P<0.05 compared to 0 μg/ml EP and 4 μM sorafenib. The values indicate a ratio compared to 0 μg/ml EP and 4 μM sorafenib as 100% control. (B) Combination index values were determined using the method described by Chou (19); CI=1, cumulative effect; CI<1, synergistic effect; and CI>1, antagonistic effect.
Discussion
Patients with advanced HCC and LC often present with thrombocytopenia. Therefore, it is difficult to use sorafenib, which currently is the only recognized effective treatment for advanced HCC (4).
EP, a second-generation TPO-R agonist that has the ability to increase platelet count, has been reported to have antitumor effects in several types of cancer (16).
The purpose of our study was to clarify the effects of EP on the growth of human HCC cells.
In the present study, EP exhibited strong antitumor activity in HCC by eliciting cell cycle arrest through iron chelation rather than through TPO-R. We concluded that EP represents a novel treatment for HCC.
TPO-R, also known as myeloproliferative leukemia virus oncogene (MPL), has been reported to be expressed in liver sinusoidal endothelial cells (LSEC) in mice and in liver progenitor cells in rats, and TPO promotes the proliferation of both of these cell types (21,22).
However, we have already reported that TPO has no proliferative effect on HCC in vitro or in vivo (23).
In contrast to TPO, the second-generation small molecule TPO-R agonist EP does not induce the production of neutralizing antibodies (24–28).
Recent studies have demonstrated that EP inhibits leukemia cell growth by depleting intracellular iron (15) and inhibits the growth of breast, lung and ovarian tumor cells (16).
These findings of tumor cell growth inhibition by EP in vitro and in vivo demonstrate that these effects of EP are TPO-R-independent.
The reported IC50 values of EP in various cell lines are 9.6–19.0 μg/ml for breast, 3.7–10.3 μg/ml for lung, and 4.8–49.7 μg/ml for ovarian cancer. These results in our study supported the previous studies that indicated IC50 of EP in tumor cells.
The observed median Cmax for EP in patients with ITP is 11.4 μg/ml at a 75-mg dose (29). The efficacy of EP in hepatitis C virus-infected patients with thrombocytopenia before the initiation of pegylated interferon and ribavirin therapy has been reported (12). A recent double-blind, placebo-controlled, randomized dose-escalation study showed that maximum serum concentrations of EP of >20 μg/ml are clinically achievable with minimal toxicity (18).
EP has three primary features: a lipophilic end, an acidic end, and a chelator backbone (30,31). These elements enable EP to reduce intracellular iron as well as possibly other polyvalent cations (24,32).
Iron (Fe) is a metal that is vital for the sustenance of life (33–35). It is an essential component of many proteins and enzymes that are involved in cell growth and replication (33,35). Cellular Fe depletion typically results in G1/S arrest (36,37), which indicates that this metal is essential for cell cycle progression as well as cell growth and division (38,39).
Compared with normal cells, neoplastic cells require a greater amount of Fe because they generally proliferate at a higher rate than their normal counterparts (39,40). This is reflected by the upregulated expression of the transferrin receptor protein 1 (TfR1) (41) and the higher rate of Fe uptake from transferrin (Tf) in cancer cells (42,43). Furthermore, neoplastic cells express high levels of ribonucleotide reductase (RR) (44,45), rendering them more susceptible to Fe chelators than normal cells (39,46). Early studies showed that the clinically utilized Fe chelator, deferoxamine (DFO), exerts some inhibitory effects on the growth of neuroblastoma and leukemia cells in culture and in clinical trials (47–51).
One mechanism by which Fe chelators exert anti-proliferative effects on tumors is through targeting molecules that are critical for regulating cell cycle progression (52,53).
Importantly, the effects of Fe chelators on CCND1 expression were determined to be due to Fe depletion, as Fe supplementation reversed these effects (20).
Furthermore, Fe depletion appears to have similar effects on both CCND1 and p21CIP1/WAF1 protein expression: it induces the ubiquitin-independent degradation of these proteins (20,54).
The following important findings were also obtained via our experiments.
In addition to DFO, the most common iron chelator (55), which is used in the clinic, recent studies have characterized new iron chelators, such as deferasirox, Dp44mT (56) and O-Trensox (57). These iron chelators induce growth inhibition in breast cancer cells and neuroblastoma (37,58). Although DFO, which exhibited a similar antitumor effect as EP in our study, is commonly used in patients, its poor membrane permeability and inability to permit redox cycling of iron are disadvantageous in terms of eliciting an anti-proliferative effect (55). Additionally, DFO is poorly absorbed in the intestine, and it must be administered via an intravenous route; furthermore, DFO has a very short plasma half-life (55). These disadvantages prompted the search for more effective chelators that are easier to administer to patients. From the results of our study, EP would be a candidate for drug as effective chelator in a clinical study.
It is well known that patients with the congenital iron overload disease hemochromatosis are more likely to develop HCC compared with the general population (2). A review by Gangaidzo and Gordeuk suggest that iron overload may contribute to the high incidence of HCC in Africa (59).
Furthermore, iron chelators have been reported to be useful for HCC treatment in vitro, in vivo and in the clinic (57,60,61).
Iron reduction therapy, such as phlebotomy or a low iron diet, is used to improve liver function and to prevent carcinogenesis in patients with chronic hepatitis C (62,63).
Conversely, it has been reported that the iron chelator DFO protects against the cytotoxic effects of sorafenib in HCC cells (64). The findings suggest that sorafenib can induce ferroptosis, which is a novel form of programmed cell death.
Thus, a potential method to attenuate the effects of sorafenib is to reduce intracellular iron levels. The cytotoxic effect of sorafenib is markedly offset when the concentration of EP increases, which is consistent with our results (Fig. 5).
Drug repositioning, in which existing drugs are used for new purposes, is a cost-effective strategy for identifying new treatments for existing conditions, and finding use for drugs for which development has ceased; this strategy has been used to identify new usage for several drugs, including thalidomide and plerixafor (65,66). In recent years, significant progress in drug discovery technology and bioinformatics has facilitated drug repositioning, and new drug discovery tools are eliminating the innovation gap (67). This study identified EP as a candidate for drug repositioning efforts, which should lead to the novel clinical use of EP in patients with HCC and LC (Fig. 6).
Figure 6.

The schematic chart of the EP function for the regeneration and carcinoma in human liver. Platelets suppress hepatic fibrosis and promote liver regeneration. EP could be developed into a novel treatment for LC by increment of platelet counts in clinical settings. EP also exhibited antitumor activity in HCC by eliciting cell cycle arrest through iron chelation compared to without TPO-R.
EP interacts with the transmembrane domain of TPO-R. This interaction and subsequent TPO-R downstream signaling is highly species-specific and occurs only in humans and primates, not in murine cells (68).
Therefore, it is difficult to conduct an in vivo study on EP, and the lack of in vivo data represents a limitation of this study.
However, short-term EP treatment is useful and safe in combination with radio-frequency ablation in HCC patients with LC and severe thrombocytopenia, as the risk of bleeding is mitigated by the increase in platelets (69); moreover, EP does not have a proliferative effect on HCC (69). This option should be considered in clinical trials.
In conclusion, the present study demonstrated that EP is a promising and safe chemotherapeutic agent for the treatment of HCC in patients with LC and thrombocytopenia.
Acknowledgements
The present study was supported in part by grants from the Ministry of Education, Culture, Sports, and Science and Technology of Japan (MEXT).
References
- 1.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 2.Montalto G, Cervello M, Giannitrapani L, Dantona F, Terranova A, Castagnetta LA. Epidemiology, risk factors, and natural history of hepatocellular carcinoma. Ann N Y Acad Sci. 2002;963:13–20. doi: 10.1111/j.1749-6632.2002.tb04090.x. [DOI] [PubMed] [Google Scholar]
- 3.Abou-Alfa GK, Schwartz L, Ricci S, Amadori D, Santoro A, Figer A, De Greve J, Douillard JY, Lathia C, Schwartz B, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2006;24:4293–4300. doi: 10.1200/JCO.2005.01.3441. [DOI] [PubMed] [Google Scholar]
- 4.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 5.Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140:1410–1426. doi: 10.1053/j.gastro.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolber E-M, Jelkmann W. Thrombopoietin: The novel hepatic hormone. News Physiol Sci. 2002;17:6–10. doi: 10.1152/physiologyonline.2002.17.1.6. [DOI] [PubMed] [Google Scholar]
- 7.de Sauvage FJ, Carver-Moore K, Luoh SM, Ryan A, Dowd M, Eaton DL, Moore MW. Physiological regulation of early and late stages of megakaryocytopoiesis by thrombopoietin. J Exp Med. 1996;183:651–656. doi: 10.1084/jem.183.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaushansky K, Drachman JG. The molecular and cellular biology of thrombopoietin: The primary regulator of platelet production. Oncogene. 2002;21:3359–3367. doi: 10.1038/sj.onc.1205323. [DOI] [PubMed] [Google Scholar]
- 9.Deutsch VR, Tomer A. Megakaryocyte development and platelet production. Br J Haematol. 2006;134:453–466. doi: 10.1111/j.1365-2141.2006.06215.x. [DOI] [PubMed] [Google Scholar]
- 10.Kaushansky K, Broudy VC, Lin N, Jorgensen MJ, McCarty J, Fox N, Zucker-Franklin D, Lofton-Day C. Thrombopoietin, the Mp1 ligand, is essential for full megakaryocyte development. Proc Natl Acad Sci USA. 1995;92:3234–3238. doi: 10.1073/pnas.92.8.3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bussel JB, Cheng G, Saleh MN, Psaila B, Kovaleva L, Meddeb B, Kloczko J, Hassani H, Mayer B, Stone NL, et al. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N Engl J Med. 2007;357:2237–2247. doi: 10.1056/NEJMoa073275. [DOI] [PubMed] [Google Scholar]
- 12.McHutchison JG, Dusheiko G, Shiffman ML, Rodriguez-Torres M, Sigal S, Bourliere M, Berg T, Gordon SC, Campbell FM, Theodore D, et al. TPL102357 Study Group. Eltrombopag for thrombocytopenia in patients with cirrhosis associated with hepatitis C. N Engl J Med. 2007;357:2227–2236. doi: 10.1056/NEJMoa073255. [DOI] [PubMed] [Google Scholar]
- 13.Will B, Kawahara M, Luciano JP, Bruns I, Parekh S, Erickson-Miller CL, Aivado MA, Verma A, Steidl U. Effect of the nonpeptide thrombopoietin receptor agonist Eltrombopag on bone marrow cells from patients with acute myeloid leukemia and myelodysplastic syndrome. Blood. 2009;114:3899–3908. doi: 10.1182/blood-2009-04-219493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erickson-Miller CL, Kirchner J, Aivado M, May R, Payne P, Chadderton A. Reduced proliferation of non-megakaryocytic acute myelogenous leukemia and other leukemia and lymphoma cell lines in response to eltrombopag. Leuk Res. 2010;34:1224–1231. doi: 10.1016/j.leukres.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Roth M, Will B, Simkin G, Narayanagari S, Barreyro L, Bartholdy B, Tamari R, Mitsiades CS, Verma A, Steidl U. Eltrombopag inhibits the proliferation of leukemia cells via reduction of intracellular iron and induction of differentiation. Blood. 2012;120:386–394. doi: 10.1182/blood-2011-12-399667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erickson-Miller CL, Pillarisetti K, Kirchner J, Figueroa DJ, Ottesen L, Martin AM, Liu Y, Kamel YM, Messam C. Low or undetectable TPO receptor expression in malignant tissue and cell lines derived from breast, lung, and ovarian tumors. BMC Cancer. 2012;12:405. doi: 10.1186/1471-2407-12-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawaguchi T, Komori A, Seike M, Fujiyama S, Watanabe H, Tanaka M, Sakisaka S, Nakamuta M, Sasaki Y, Oketani M, et al. Efficacy and safety of eltrombopag in Japanese patients with chronic liver disease and thrombocytopenia: A randomized, open-label, phase II study. J Gastroenterol. 2012;47:1342–1351. doi: 10.1007/s00535-012-0600-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matthys G, Park JW, McGuire S, Wire MB, Bowen C, Williams D, Jenkins J, Peng B. Clinical pharmacokinetics, platelet response, and safety of eltrombopag at supratherapeutic doses of up to 200 mg once daily in healthy volunteers. J Clin Pharmacol. 2011;51:301–308. doi: 10.1177/0091270010368677. [DOI] [PubMed] [Google Scholar]
- 19.Chou T-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 20.Nurtjahja-Tjendraputra E, Fu D, Phang JM, Richardson DR. Iron chelation regulates cyclin D1 expression via the proteasome: A link to iron deficiency-mediated growth suppression. Blood. 2007;109:4045–4054. doi: 10.1182/blood-2006-10-047753. [DOI] [PubMed] [Google Scholar]
- 21.Cardier JE, Dempsey J. Thrombopoietin and its receptor, c-mpl, are constitutively expressed by mouse liver endothelial cells: Evidence of thrombopoietin as a growth factor for liver endothelial cells. Blood. 1998;91:923–929. [PubMed] [Google Scholar]
- 22.Schmelzer E, Deiwick A, Bruns H, Fiegel HC, Bader A. Thrombopoietin is a growth factor for rat hepatic progenitors. Eur J Gastroenterol Hepatol. 2008;20:209–216. doi: 10.1097/MEG.0b013e3282f246e6. [DOI] [PubMed] [Google Scholar]
- 23.Nozaki R, Murata S, Nowatari T, Maruyama T, Ikeda N, Kawasaki T, Fukunaga K, Ohkohchi N. Effects of thrombopoietin on growth of hepatocellular carcinoma: Is thrombopoietin therapy for liver disease safe or not? Hepatol Res. 2013;43:610–620. doi: 10.1111/hepr.12006. [DOI] [PubMed] [Google Scholar]
- 24.Erickson-Miller CL, DeLorme E, Tian SS, Hopson CB, Stark K, Giampa L, Valoret EI, Duffy KJ, Luengo JL, Rosen J, et al. Discovery and characterization of a selective, nonpeptidyl thrombopoietin receptor agonist. Exp Hematol. 2005;33:85–93. doi: 10.1016/j.exphem.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Erickson-Miller C, Delorme E, Giampa L, Hopson C, Valoret E, Tian SS, Miller SG, Keenan R, Rosen J, Dillon S, et al. Biological activity and selectivity for Tpo receptor of the orally bioavailable, small molecule Tpo receptor agonist, SB-497115. Blood (ASH Annual Meeting Abstracts) 2004;104:2912. [Google Scholar]
- 26.Kalota A, Brennan K, Erickson-Miller CL, Danet G, Carroll M, Gewirtz AM. Effects of SB559457, a novel small molecule thrombopoietin receptor (TpoR) agonist, on human hematopoietic cell growth and differentiation. Blood (ASH Annual Meeting Abstracts) 2004;104:2913. [Google Scholar]
- 27.Safonov IG, Heerding DA, Keenan RM, Price AT, Erickson-Miller CL, Hopson CB, Levin JL, Lord KA, Tapley PM. New benzimidazoles as thrombopoietin receptor agonists. Bioorg Med Chem Lett. 2006;16:1212–1216. doi: 10.1016/j.bmcl.2005.11.096. [DOI] [PubMed] [Google Scholar]
- 28.Luengo JI, Duffy KJ, Shaw AN, Delorme E, Wiggall KJ, Giampa L, Liu N, Smith H, Tian SS, Miller SG, et al. Discovery of SB-497115, a small-molecule thrombopoietin (TPO) receptor agonist for the treatment of thrombocytopenia. Blood (ASH Annual Meeting Abstracts) 2004;104:2910. [Google Scholar]
- 29.Peeters K, Stassen J-M, Collen D, Van Geet C, Freson K. Emerging treatments for thrombocytopenia: Increasing platelet production. Drug Discov Today. 2008;13:798–806. doi: 10.1016/j.drudis.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Duffy KJ, Shaw AN, Delorme E, Dillon SB, Erickson-Miller C, Giampa L, Huang Y, Keenan RM, Lamb P, Liu N, et al. Identification of a pharmacophore for thrombopoietic activity of small, non-peptidyl molecules. 1. Discovery and optimization of salicylaldehyde thiosemicarbazone thrombopoietin mimics. J Med Chem. 2002;45:3573–3575. doi: 10.1021/jm025535c. [DOI] [PubMed] [Google Scholar]
- 31.Duffy KJ, Price AT, Delorme E, Dillon SB, Duquenne C, Erickson-Miller C, Giampa L, Huang Y, Keenan RM, Lamb P, et al. Identification of a pharmacophore for thrombopoietic activity of small, non-peptidyl molecules. 2. Rational design of naphtho[1,2-d]imidazole thrombopoietin mimics. J Med Chem. 2002;45:3576–3578. doi: 10.1021/jm0255365. [DOI] [PubMed] [Google Scholar]
- 32.Williams DD, Peng B, Bailey CK, Wire MB, Deng Y, Park JW, Collins DA, Kapsi SG, Jenkins JM. Effects of food and antacids on the pharmacokinetics of eltrombopag in healthy adult subjects: Two single-dose, open-label, randomized-sequence, crossover studies. Clin Ther. 2009;31:764–776. doi: 10.1016/j.clinthera.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 33.Hershko C. Control of disease by selective iron depletion: A novel therapeutic strategy utilizing iron chelators. Baillieres Clin Haematol. 1994;7:965–1000. doi: 10.1016/S0950-3536(05)80133-7. [DOI] [PubMed] [Google Scholar]
- 34.Buss JL, Greene BT, Turner J, Torti FM, Torti SV. Iron chelators in cancer chemotherapy. Curr Top Med Chem. 2004;4:1623–1635. doi: 10.2174/1568026043387269. [DOI] [PubMed] [Google Scholar]
- 35.Andrews NC. Disorders of iron metabolism. N Engl J Med. 1999;341:1986–1995. doi: 10.1056/NEJM199912233412607. [DOI] [PubMed] [Google Scholar]
- 36.Lucas JJ, Szepesi A, Domenico J, Takase K, Tordai A, Terada N, Gelfand EW. Effects of iron-depletion on cell cycle progression in normal human T lymphocytes: Selective inhibition of the appearance of the cyclin A-associated component of the p33cdk2 kinase. Blood. 1995;86:2268–2280. [PubMed] [Google Scholar]
- 37.Brodie C, Siriwardana G, Lucas J, Schleicher R, Terada N, Szepesi A, Gelfand E, Seligman P. Neuroblastoma sensitivity to growth inhibition by deferrioxamine: Evidence for a block in G1 phase of the cell cycle. Cancer Res. 1993;53:3968–3975. [PubMed] [Google Scholar]
- 38.Kwok JC, Richardson DR. The iron metabolism of neoplastic cells: Alterations that facilitate proliferation? Crit Rev Oncol Hematol. 2002;42:65–78. doi: 10.1016/S1040-8428(01)00213-X. [DOI] [PubMed] [Google Scholar]
- 39.Le NTV, Richardson DR. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim Biophys Acta. 2002;1603:31–46. doi: 10.1016/s0304-419x(02)00068-9. [DOI] [PubMed] [Google Scholar]
- 40.Kalinowski DS, Richardson DR. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol Rev. 2005;57:547–583. doi: 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- 41.Larrick JW, Cresswell P. Modulation of cell surface iron transferrin receptors by cellular density and state of activation. J Supramol Struct. 1979;11:579–586. doi: 10.1002/jss.400110415. [DOI] [PubMed] [Google Scholar]
- 42.Richardson DR, Baker E. The uptake of iron and transferrin by the human malignant melanoma cell. Biochim Biophys Acta. 1990;1053:1–12. doi: 10.1016/0167-4889(90)90018-9. [DOI] [PubMed] [Google Scholar]
- 43.Richardson DR, Baker E. The effect of desferrioxamine and ferric ammonium citrate on the uptake of iron by the membrane iron-binding component of human melanoma cells. Biochim Biophys Acta. 1992;1103:275–280. doi: 10.1016/0005-2736(92)90097-6. [DOI] [PubMed] [Google Scholar]
- 44.Elford HL, Freese M, Passamani E, Morris HP. Ribonucleotide reductase and cell proliferation. I. Variations of ribonucleotide reductase activity with tumor growth rate in a series of rat hepatomas. J Biol Chem. 1970;245:5228–5233. [PubMed] [Google Scholar]
- 45.Takeda E, Weber G. Role of ribonucleotide reductase in expression in the neoplastic program. Life Sci. 1981;28:1007–1014. doi: 10.1016/0024-3205(81)90746-3. [DOI] [PubMed] [Google Scholar]
- 46.Witt L, Yap T, Blakley RL. Regulation of ribonucleotide reductase activity and its possible exploitation in chemotherapy. Adv Enzyme Regul. 1978;17:157–171. doi: 10.1016/0065-2571(79)90012-8. [DOI] [PubMed] [Google Scholar]
- 47.Donfrancesco A, Deb G, Dominici C, Pileggi D, Castello MA, Helson L. Effects of a single course of deferoxamine in neuroblastoma patients. Cancer Res. 1990;50:4929–4930. [PubMed] [Google Scholar]
- 48.Estrov Z, Tawa A, Wang XH, Dubé ID, Sulh H, Cohen A, Gelfand EW, Freedman MH. In vitro and in vivo effects of deferoxamine in neonatal acute leukemia. Blood. 1987;69:757–761. [PubMed] [Google Scholar]
- 49.Kaplinsky C, Estrov Z, Freedman MH, Gelfand EW, Cohen A. Effect of deferoxamine on DNA synthesis, DNA repair, cell proliferation, and differentiation of HL-60 cells. Leukemia. 1987;1:437–441. [PubMed] [Google Scholar]
- 50.Blatt J, Taylor SR, Stitely S. Mechanism of antineuroblastoma activity of deferoxamine in vitro. J Lab Clin Med. 1988;112:433–436. [PubMed] [Google Scholar]
- 51.Blatt J, Stitely S. Antineuroblastoma activity of desferoxamine in human cell lines. Cancer Res. 1987;47:1749–1750. [PubMed] [Google Scholar]
- 52.Gao J, Richardson DR. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents, IV: The mechanisms involved in inhibiting cell-cycle progression. Blood. 2001;98:842–850. doi: 10.1182/blood.V98.3.842. [DOI] [PubMed] [Google Scholar]
- 53.Le NTV, Richardson DR. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: A link between iron metabolism and proliferation. Blood. 2004;104:2967–2975. doi: 10.1182/blood-2004-05-1866. [DOI] [PubMed] [Google Scholar]
- 54.Fu D, Richardson DR. Iron chelation and regulation of the cell cycle: 2 mechanisms of posttranscriptional regulation of the universal cyclin-dependent kinase inhibitor p21CIP1/WAF1 by iron depletion. Blood. 2007;110:752–761. doi: 10.1182/blood-2007-03-076737. [DOI] [PubMed] [Google Scholar]
- 55.Keberle H. The Biochemistry of desferrioxamine and its relation to iron metabolism. Ann NY Acad Sci. 1964;119:758–768. doi: 10.1111/j.1749-6632.1965.tb54077.x. [DOI] [PubMed] [Google Scholar]
- 56.Whitnall M, Howard J, Ponka P, Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci USA. 2006;103:14901–14906. doi: 10.1073/pnas.0604979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rakba N, Loyer P, Gilot D, Delcros JG, Glaise D, Baret P, Pierre JL, Brissot P, Lescoat G. Antiproliferative and apoptotic effects of O-Trensox, a new synthetic iron chelator, on differentiated human hepatoma cell lines. Carcinogenesis. 2000;21:943–951. doi: 10.1093/carcin/21.5.943. [DOI] [PubMed] [Google Scholar]
- 58.Rao VA, Klein SR, Agama KK, Toyoda E, Adachi N, Pommier Y, Shacter EB. The iron chelator Dp44mT causes DNA damage and selective inhibition of topoisomerase IIalpha in breast cancer cells. Cancer Res. 2009;69:948–957. doi: 10.1158/0008-5472.CAN-08-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gangaidzo IT, Gordeuk VR. Hepatocellular carcinoma and African iron overload. Gut. 1995;37:727–730. doi: 10.1136/gut.37.5.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ba Q, Hao M, Huang H, Hou J, Ge S, Zhang Z, Yin J, Chu R, Jiang H, Wang F, et al. Iron deprivation suppresses hepatocellular carcinoma growth in experimental studies. Clin Cancer Res. 2011;17:7625–7633. doi: 10.1158/1078-0432.CCR-10-3099. [DOI] [PubMed] [Google Scholar]
- 61.Yamasaki T, Terai S, Sakaida I. Deferoxamine for advanced hepatocellular carcinoma. N Engl J Med. 2011;365:576–578. doi: 10.1056/NEJMc1105726. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka H, Fujita N, Sugimoto R, Urawa N, Horiike S, Kobayashi Y, Iwasa M, Ma N, Kawanishi S, Watanabe S, et al. Hepatic oxidative DNA damage is associated with increased risk for hepatocellular carcinoma in chronic hepatitis C. Br J Cancer. 2008;98:580–586. doi: 10.1038/sj.bjc.6604204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kato J, Kobune M, Nakamura T, Kuroiwa G, Takada K, Takimoto R, Sato Y, Fujikawa K, Takahashi M, Takayama T, et al. Normalization of elevated hepatic 8-hydroxy-2′-deoxyguanosine levels in chronic hepatitis C patients by phlebotomy and low iron diet. Cancer Res. 2001;61:8697–8702. [PubMed] [Google Scholar]
- 64.Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Mazière JC, Chauffert B, Galmiche A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013;133:1732–1742. doi: 10.1002/ijc.28159. [DOI] [PubMed] [Google Scholar]
- 65.Musto P, D'Auria F, Pietrantuono G, Bringhen S, Morabito F, Di Raimondo F, Pozzi S, Sacchi S, Boccadoro M, Palumbo A Gruppo Italiano Malatte Ematologiche dell'Adulto Multiple Myeloma Working Party; Italian Myeloma Network and Gruppo Italiano Studio Linfomi. Role of thalidomide in previously untreated patients with multiple myeloma. Expert Rev Anticancer Ther. 2008;8:1569–1580. doi: 10.1586/14737140.8.10.1569. [DOI] [PubMed] [Google Scholar]
- 66.Mark TM, Reid W, Niesvizky R, Gergis U, Pearse R, Mayer S, Greenberg J, Coleman M, Van Besien K, Shore T. A phase 1 study of bendamustine and melphalan conditioning for autologous stem cell transplantation in multiple myeloma. Biol Blood Marrow Transplant. 2013;19:831–837. doi: 10.1016/j.bbmt.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sleigh SH, Barton DCL. Repurposing strategies for therapeutics. Pharmaceut Med. 2010;24:151–159. [Google Scholar]
- 68.Erickson-Miller CL, Delorme E, Tian S-S, Hopson CB, Landis AJ, Valoret EI, Sellers TS, Rosen J, Miller SG, Luengo JI, et al. Preclinical activity of eltrombopag (SB-497115), an oral, nonpeptide thrombopoietin receptor agonist. Stem Cells. 2009;27:424–430. doi: 10.1634/stemcells.2008-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kawaguchi T, Nakano M, Satani M, Sumie S, Yamada S, Amano K, Kuromatsu R, Sata M. Usefulness of short-term eltrombopag treatment as a supportive treatment in hepatocellular carcinoma patients with cirrhosis and severe thrombocytopenia: A report of two cases. Oncol Lett. 2014;7:2130–2134. doi: 10.3892/ol.2014.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]