

Abstract
The purpose of this study is to identify protein kinase genes that modulate oxaliplatin cytotoxicity in vitro and evaluate the roles of these genes in predicting clinical outcomes in CRC patients receiving oxaliplatin-based adjuvant chemotherapy. A high-throughput RNAi screening targeting 626 human kinase genes was performed to identify kinase genes whose inhibition potentiates oxaliplatin sensitivity in CRC cells. The associations between copy numbers of the candidate genes and recurrence-free survival and overall survival were analyzed in 142 stage III CRC patients receiving first-line oxaliplatin-based adjuvant chemotherapy who were enrolled from two independent hospitals. HT-RNAi screening identified 40 kinase genes whose inhibition potentiated oxaliplatin cytotoxicity in DLD1 cells. The relative copy number (RCN) of MAP4K1 and CDKL4 were associated with increased risks of both recurrence and death. Moreover, significant genes-based risk score and the ratios of RCN of different genes can further categorize patients into subgroups with distinctly differing outcomes. The estimated AUC for the prediction models including clinical variables plus kinase biomarkers was 0.77 for the recurrence and 0.82 for the survival models. The copy numbers of MAP4K1 and CDKL4 can predict clinical outcomes in CRC patients treated with oxaliplatin-based chemotherapy.
Keywords: colorectal cancer, RNAi screening, chemotherapy, recurrence, survival
INTRODUCTION
Colorectal cancer (CRC) is the third most common malignancies and the second leading cause of cancer death worldwide [1]. The clinical outcome of CRC has significantly improved over recent decades, which can be attributed to improved surgical techniques in early disease, adjuvant therapy for locally advanced disease, and multimodality therapy for metastatic disease [2–4]. Adjuvant chemotherapy is recommended in patients with high risk stage II and stage III patients after curative resection according to the National Comprehensive Cancer Network (NCCN) Guidelines. The results from MOSAIC and NSABP C-07 clinical trial led to the approval of oxaliplatin-based regimens as the first-line chemotherapy in the adjuvant setting [3, 5]. However, three-year disease-free survival (DFS) continues to be 65% in patients with stage III CRC receiving oxaliplatin-containing adjuvant chemotherapy [6]. To identify genes involved in modulating oxaliplatin response would be instrumental in improving the efficacy of adjuvant oxaliplatin–based chemotherapy and predicting which patients are most or least likely to benefit from such treatment. To date, no predictive markers of oxaliplatin sensitivity and resistance have been clinically validated.
With the advent of high-throughput RNA interference (HT-RNAi) screen-based on large-scale gene knockdown, it has become feasible to quickly screen and validate drug modulators whose suppression can overcome resistance to chemotherapeutic agents [7]. Such studies can not only provide important insight into the mechanism of drug resistance, but also identify novel drug targets and potential combination strategies for more effective chemotherapy, and identify promising candidates to serve as biomarkers for drug response and patient stratification [8]. A number of studies utilizing HT-RNAi screen have characterized key genes contributing to the resistance to chemotherapeutic agents in patients with breast, ovarian, and other cancers [9, 10]. However, there was only one published study using HT-RNAi screen-based approach to identify modulators of oxaliplatin resistance in CRC cell lines that revealed the involvement of TP53 in oxaliplatin response [11].
Protein kinases function as key regulators of cell proliferation, differentiation and survival through the process of phosphorylation. Protein kinases are believed to play an important role in chemoresistance based on the evidence that inhibition of kinases often enhances sensitivity of cancer cells to chemotherapeutic agents including cisplatin and doxorubicin [12]. Aberrant activation of kinases was frequently observed in tumors due to somatic genome aberrations including mutations and copy number variations (CNVs). Increased copy number of aurora kinase A was associated with prognosis and response to oxaliplatin–based chemotherapy in patients with metastatic CRC [13]. However, there has not been any study published using HT-RNAi screening of the kinome to identify modulators of oxaliplatin-based chemotherapy response in locally advanced CRC. We hypothesize that CNVs of kinases are potential predictors of oxaliplatin efficacy in the adjuvant setting. To test this hypothesis, we first employed HT-RNAi screening using a lentiviral short hairpin RNA (shRNA) library against human kinome to identify potentiators of oxaliplatin response in cultured CRC cell lines. Subsequently, we used two independent CRC patient cohorts to validate the association between CNVs of “hit” genes and clinical outcomes of stage III CRC receiving oxaliplatin-based chemotherapy.
RESULTS
High-throughput RNAi screening for kinase genes modulating oxaliplatin response
We performed HT-RNAi screen using a lentiviral shRNA library that targets 626 human kinase genes. Both statistics and sensitivity parameters were used for selection of “hits” [9, 14]. Only lentiviral shRNA fulfilling the requirements of Z-score < −1.65 (equal to P < 0.05, the standard of statistical significance) and SI>0.15 (standard for sensitizing) were identified as hits. The preliminary screening, as shown in Table 1, Figure 1 and Supplementary Table 1, yielded 40 (6.4%) genes whose inhibition by shRNA potentiated DLD1 cell sensitivity to oxaliplatin, suggesting the involvement of these genes in oxaliplatin resistance.
Table 1. Functional categories of kinase hits.
| Gene Symbol | z-score | SI | Functional categories |
|---|---|---|---|
| AK7 | −3.63 | 0.26 | Metabolic regulation |
| ULK4 | −3.33 | 0.25 | MAPK signaling |
| NEK8* | −3.33 | 0.38 | Cell cycle regulation |
| TRIB1 | −3.05 | 0.17 | MAPK signaling |
| BMPR1B | −2.88 | 0.3 | TGF-β signaling |
| CERKL | −2.88 | 0.25 | Lipid signaling |
| CDK4 | −2.85 | 0.2 | Cell cycle regulation |
| PIK3CA | −2.75 | 0.24 | Lipid signaling |
| BMPR1A | −2.54 | 0.2 | TGF-β signaling |
| SPHK2* | −2.42 | 0.2 | Lipid signaling |
| NME4 | −2.39 | 0.16 | Metabolic regulation |
| PRKD3 | −2.22 | 0.26 | AGC kinase |
| DGKB* | −2.21 | 0.25 | Metabolic regulation |
| LIMK2 | −2.21 | 0.43 | Tyrosine kinase signaling |
| PGK1 | −2.21 | 0.15 | Metabolic regulation |
| PRKCD* | −2.19 | 0.18 | AGC kinase |
| DYRK4* | −2.17 | 0.18 | Tyrosine kinase signaling |
| PKN3 | −2.13 | 0.2 | PI3K-AKT signaling |
| SGK3 | −2.11 | 0.29 | PI3K-AKT signaling |
| PTK2 | −2.11 | 0.34 | Tyrosine kinase signaling |
| BMP2K | −2.09 | 0.17 | Serine/threonine kinse signaling |
| STK39* | −2.05 | 0.22 | MAPK signaling |
| MAP2K1 | −2.03 | 0.16 | MAPK signaling |
| PI4K2B | −2.02 | 0.18 | Lipid signaling |
| HK2* | −1.98 | 0.31 | Metabolic regulation |
| PRKCB | −1.94 | 0.18 | AGC kinase |
| NEK4 | −1.89 | 0.46 | Cell cycle regulation |
| STK32A | −1.86 | 0.19 | Serine/threonine-protein kinase |
| NEK9* | −1.79 | 0.17 | Cell cycle regulation |
| MAGI2 | −1.76 | 0.28 | PI3K-AKT signaling |
| LYN | −1.75 | 0.28 | MAPK signaling |
| MYLK | −1.75 | 0.36 | Calcium signaling |
| DOLK | −1.75 | 0.24 | Metabolic regulation |
| CDC7 | −1.74 | 0.17 | Cell cycle regulation |
| MAP4K1 | −1.7 | 0.27 | MAPK signaling |
| MST1R | −1.69 | 0.19 | MAPK signaling |
| AGK | −1.68 | 0.18 | Lipid signaling |
| CDKL4 | −1.67 | 0.16 | Cell cycle regulation |
| WNK1 | −1.66 | 0.23 | Serine/threonine kinse signaling |
| RIOK2 | −1.66 | 0.33 | Serine/threonine-protein kinase |
Note. Genes also found in other RNAi screens to be resistant to chemotherapy.
Figure 1. Scatter plot of z score from RNAi screen.
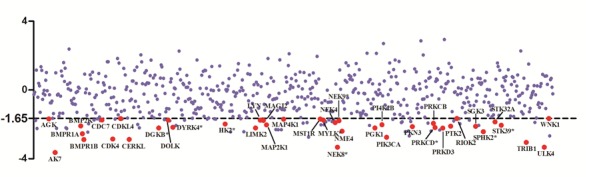
Oxaliplatin resistance screen with human kinome lenti-shRNA library was carried out. The calculated z score corresponding to each target gene was dotted and target genes with z score <-1.65 and SI>0.1 were selected as hits and showed in red solid circles.
To further explore the underlying mechanism of oxaliplatin resistance, we performed a pathway analysis of the 40 hits through the DAVID (http://david.abcc.ncifcrf.gov/) analysis tool. Several cancer-related pathways were identified. Under a stringent criterion of P < 0.01, the VEGF and ERBB signaling pathways were enriched with four key genes comprising PTK2, MAP2K1, PIK3CA and PRKCB.
Validation of kinase genes identified from HT-RNAi screening in CRC patients
We then used two independent cohorts of CRC patients receiving adjuvant oxalilplatin-based chemotherapy to evaluate whether the in vitro HT-RNAi screen-identified kinase genes are involved in oxaliplatin response in patients. Supplementary Table 2 shows the clinical characteristics of the training and validation patient cohorts. Of the 76 patients in the training set, the median age was 60 years (range, 24–79 years). The median follow-up time was 57 months. There were 19 recurrences (25%) and 28 deaths (36.8%), but the median RFS and OS had not been reached during the follow-up time. Among the 66 patients in the replication set, 19 patients (28.8%) had recurrence and 15 (22.7%) were dead. There was no significant difference between the training and validation sets with regard to age, gender, histological grade and stage.
We assessed the association of the RCN of each hit gene with recurrence and survival using a multivariate Cox model, adjusting for age, gender, tumor site and histologic grade. In the training set, a significantly increased risk of recurrence was observed for those with higher RCN of MAP4K1 (HR 1.15, 95%CI, 1.05-1.26; P=0.002) and CDKL4 (HR, 1.20, 95%CI, 1.02-1.40; P=0.02) (Supplementary Table 3 and Table 2). Higher RCN of three genes exhibited significantly increased risk of death: MAP4K1 (HR 1.11; 95%CI, 1.03-1.19; P=0.01), DOLK (HR 1.19; 95%CI, 1.00-1.41; P=0.04) and CDKL4 (HR 1.14; 95%CI, 1.00-1.29; P=0.05) (Supplementary Table 4 and Table 2).
Table 2. Copy number of individual genes associated with clinical outcomes of stage III CRC patients.
| Gene | Training set | Replication set | Pooled analysis | |||
|---|---|---|---|---|---|---|
| HR(95% CI)a | P | HR(95% CI)a | P | HR(95% CI)a | P | |
| Recurrence | ||||||
| MAP4K1 | 1.15(1.05-1.26) | 0.002 | 1.16(1.01-1.32) | 0.04 | 1.14(1.06-1.22) | 0.0002 |
| CDKL4 | 1.20(1.02-1.40) | 0.02 | 1.22(0.84-1.76) | 0.29 | 1.17(1.02-1.35) | 0.03 |
| Survival | ||||||
| MAP4K1 | 1.11(1.03-1.19) | 0.01 | 1.20(1.04-1.38) | 0.01 | 1.11(1.05-1.18) | 0.0005 |
| DOLK | 1.19(1.00-1.41) | 0.04 | 0.76(0.26-2.21) | 0.61 | 1.07(0.92-1.24) | 0.36 |
| CDKL4 | 1.14(1.00-1.29) | 0.05 | 1.53(1.06-2.19) | 0.02 | 1.15(1.03-1.28) | 0.01 |
Note. Adjuster for age, sex, stage and histological grade.
The significant associations between MAP4K1 and the risks of recurrence and death were confirmed in the replication set and pooled analysis (Supplementary Table 3 and 4). Patients with higher RCN of MAP4K1 showed increased risks of recurrence (HR 1.16; 95% CI, 1.01-1.32; P=0.04) and death (HR 1.20; 95% CI, 1.04-1.38; P=0.01) in the replication set. The combined HR was 1.14 (95% CI, 1.06 to1.22; P=0.0002) for recurrence and 1.11 (95%CI, 1.05-1.18; P=0.0005) for death. Although CDKL4 exhibited similar risk estimate with recurrence in the validation set, the association did not reach statistical significance. However, the increased risk of death associated with higher RCN of CDKL4 was confirmed in the replication set (HR 1.53; 95%CI, 1.06-2.19, P=0.02) and combined dataset (HR 1.15; 95% CI, 1.03-1.28, P=0.01).
The association between RCN-based risk score and overall survival
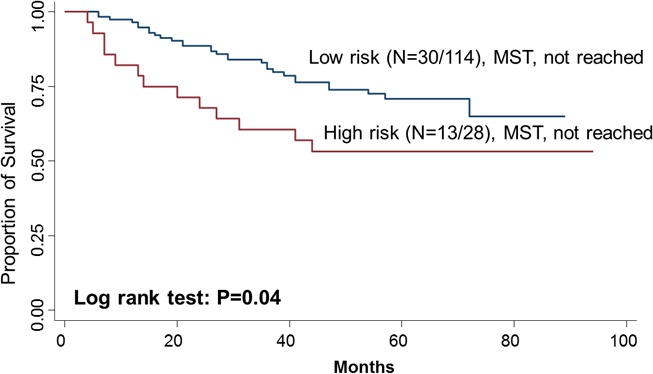
To further evaluate the predictive value of MAP4K1 and CDKL4 in survival, we performed a risk score analysis. Joint analysis of MAP4K1 and CDKL4 dichotomized the 142 patients into low-risk and high-risk group for death using the cutoff (0.45) of the risk score threshold value. The high-risk group had a 2.70-fold (95% CI, 1.37-5.33) increased risk and had a significantly shorter median survival time compared with the low-risk group (log-rank P=0.04) (Figure 2).
Figure 2. Kaplan–Meier OS curves of CRC in all patients based on the risk score of MAP4K1 and CDKL4.
The association between copy number ratios of hit genes and clinical outcomes
Since two-gene ratios have been used to evaluate cancer prognosis and provided improved risk prediction in breast cancer [15], we performed similar analyses. The RCN of each hit gene was compared to the RCN of the other 39 hit genes, which produced 780 ratios with intragroup variance. To reduce false positives, the ratios with intragroup variability below 10 (288 ratios) were used for the analyses of clinical outcomes (Supplementary Table 5). There were 29 ratios and 33 ratios significantly associated with the risk of recurrence and death, respectively (Supplementary Table 6, 7). Individual and joint analysis of the ratios of PTK2/MAGI2 and CDKL4/PTK2 showed a consistent association with the risk of recurrence in the training set, replication set and combined analysis (Supplementary Table 8). Joint analysis of these two ratios could categorize patients into two subgroups. Compared with the low-risk group, the high-risk group had a 2.91-fold (95% CI, 1.49-5.69) increased risk of recurrence. The median RFS time for the low-risk and high-risk group was >94 months and 72 months (log-rank P=0.0002), respectively (Figure 3). The ratios of PRKCD/BMP2K, MST1R/WNK1, STK39/AGK, DYRK4/SGK3 and CDKL4/CERKL were significantly associated with the risk of death in the training set, replication set and combined analysis (Supplementary Table 8). The joint analysis of these 5 ratios showed that the high-risk group was at a 5.29-fold (95% CI 2.61–10.72) increased risk of death. The median overall survival time for the low-risk and high-risk group was >94 months and 41 months (log-rank P=3.81×10−7), respectively (Figure 3).
Figure 3. Kaplan-Meier RFS and OS curves of CRC patients.
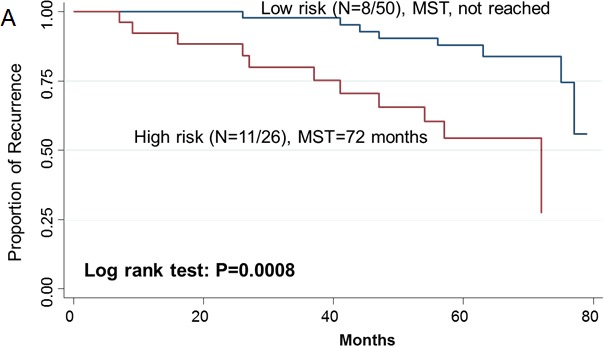
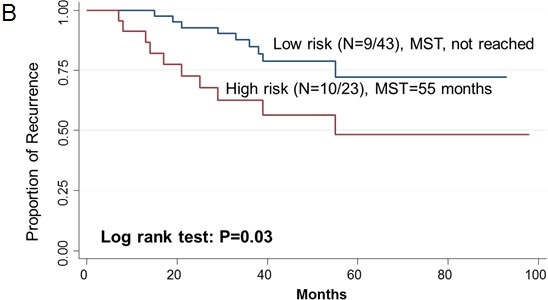
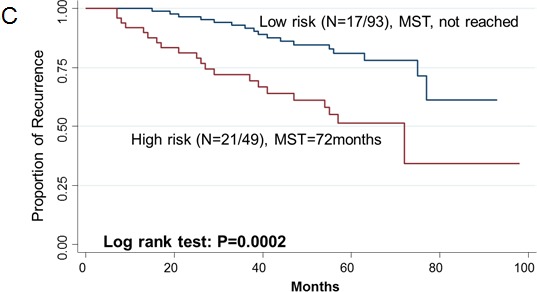
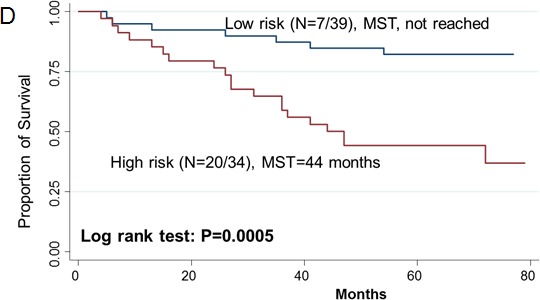
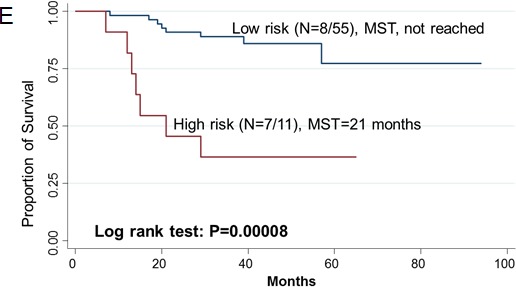
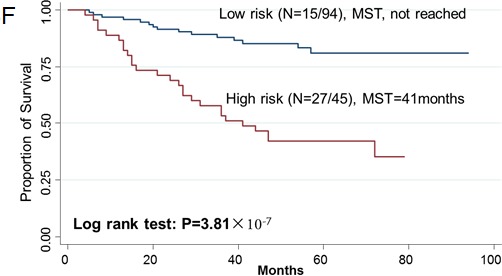
Kaplan–Meier RFS curves of patients based on joint ratios of PTK2/MAGI2 and CDKL4/PTK2 in training set (A), replication set (B), and combined set (C). Kaplan–Meier OS curves of patients based on the joint ratios of PRKCD/BMP2K, MST1R/WNK1, STK39/AGK, DYRK4/SGK3 and CDKL4/CERKL in training set (D), replication set (E), and combined set (F). MST, median event-free survival times.
ROC model for evaluating the prediction ability of clinical outcome
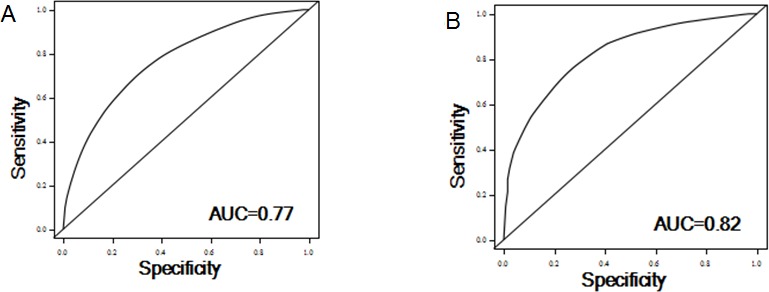
To evaluate the discriminatory ability, we constructed ROC and computed the AUC for risk prediction models of recurrence and death. For the prediction ability of recurrence, when we incorporated clinical variables (age, sex, stage and histological grade), effect of MAP4K1 and joint ratios of PTK2/MAGI2 and CDKL4/PTK2, the AUC for all patients (n =142) was 0.77 (0.62-0.74). The sensitivity and specificity was 78% and 65%, respectively. For survival prediction, the AUC reached 0.82 (0.71-0.85), when we included clinical variables, combined risk score of MAP4K1 and CDKL4, and combined ratios of PRKCD/BMP2K, MST1R/WNK1, STK39/AGK, DYRK4/SGK3 and CDKL4/CERKL in the model. The sensitivity and specificity was 85% and 69%, respectively (Figure 4).
Figure 4. ROC curve analysis for the risk of recurrence (A) and survival (B) based on the prediction model.
DISCUSSION
To our knowledge, this is the first study to identify modulators of oxaliplatin response by a combination of HT-RNAi functional screen of human kinases and copy number analysis of human patient samples. In this study, we found 40 kinase genes responsible for oxaliplatin response through the kinome HT-RNAi screen, 9 of which (22.5%) have been reported to be involved in chemoresistance in other RNAi screens [7, 14]. Further, we found that high copy number of MAP4K1 showed consistent association with both recurrence and survival for patients with stage III CRC patients receiving oxaliplatin-based chemotherapy, highlighting the important role of MAP4K1 in modulating oxaliplatin response and its potential as a biomarker for the efficacy of oxaliplatin-based chemotherapy.
MAP4K1 exerts its biological function upon extracellular stimulation by activating MAP3 kinases which in turn phosphorylate JNK, leading to the activation of MAPK pathway [16]. JNK has been reported in colon and other cancer cells to sustain multidrug resistance (MDR), and inhibition of JNK enhanced the apoptosis of colon cancer cells treated with chemotherapy agents such as nocodazole, camptothecin and 5-FU [17–19]. Enforced MAP4K1 expression was also shown to stimulate NFκB pathway which impeded apoptosis, resulting in resistance to cytotoxic drugs in cancer cell lines [20, 21]. Therefore, the function of MAP4K1 contributing to MDR and anti-apoptosis would account for the observation that CRC patients with high copy number of MAP4K1 had a worse clinical outcome. This finding was also supported by recent studies on another MAP4 kinase-MAP4K4, whose high expression was found to be associated with poor prognosis in hepatocellular carcinoma and pancreatic ductal adenocarcinoma [22, 23].
Our study also showed that a significant association of CDKL4 with both recurrence and death. CDKL4 belongs to cyclin-dependent protein kinase (CDK) family responsible for cell cycle progression. In colon cancer cells, CDK inhibitors were demonstrated to inhibit cell proliferation and induce cell cycle arrest and apoptosis [24]. Furthermore, inhibition of CDKs enhanced cell death in response to chemotherapy through downregulation of anti-apoptotic proteins including XIAP, BCL2, MCL-1 and suppression of survivin phposphorylation [25]. In a phase I study of CDK inhibitor (Flavopiridol) administered with oxaliplatin-based chemotherapy in platinum-refractory germ cell tumors, 33% of patients showed a partial response [26]. Consistent with this, our HT-RNAi screen results showed increased colon cancer cell death after targeting CDKL4 in the presence of oxaliplatin.
The present study also showed that the risk score of MAP4K1 and CDKL4 could affect CRC overall survival. Activated JNK driven by MAP4 kinases is able to phosphorylate c-Jun, which subsequently promote gene transcription responsible for G1/S and G2/M transition [27, 28]. Therefore, MAP4K1 possibly coordinated with CDKL4 to achieve cumulative effect on cell cycle progression. We also evaluated the effect of copy number ratios of hit genes on clinical outcomes. Among them, MST1R/WNK1, STK39/AGK, DYRK4/SGK3 and CDKL4/PTK2 have cumulative effect on overall survival. Interestingly, all these hit genes have been implicated in MAPK and PI3K signaling pathways. In vitro experiments have demonstrated the complex interactions between components of the PI3K and MAPK pathway, providing a rationale for the combined inhibition of both pathways to confer enhanced sensitivity to chemotherapeutics agents [29].
When we incorporated all the potential predictive markers revealed in this study into a multivariate model, we obtained excellent prediction efficacy with an AUC of 0.77 for recurrence and 0.82 for survival, highlighting the importance of considering biomarkers in prediction of clinical outcomes. The risk models to predict clinical efficacy may allow clinicians to identify patients at high risk of recurrence and survival before the start of therapy and make better-informed clinical decisions and follow up surveillance for patients.
Furthermore, a pathway analysis revealed that the VEGF and ERBB signaling pathway genes were enriched in modulating oxaliplatin response, suggesting drugs targeting these two pathways may enhance the sensitivity of CRC cells to oxaliplatin. Indeed, targeted therapies by Avastin and Cetuximab have now been used in combination with oxaliplatin-based chemotherapy to treat metastatic CRC [30, 31]. Finally, our study uncovered several potential drug targets that may overcome oxaliplatin resistance and improve therapy. This is particularly significant as acquired resistance to Avastin and Cetuximab ultimately takes place after a period of usage.
In conclusion, using HT-RNAi screen followed by patient cohort validation, we have identified several plausible kinase genes that are associated with oxaliplatin resistance in CRC cell lines. These genes are potential drug targets to overcome oxiliplatin resistance and their copy numbers may serve as biomarkers for clinical outcomes in CRC patients receiving oxaliplatin-based chemotherapy.
MATERIALS AND METHODS
Cell lines
DLD1 was purchased from American Type Culture Collection (ATCC, United States). 293FT was obtained from life technologies (United States). DLD1 was cultured in RPMI-1640 medium supplemented with 10% FBS. 293FT cells were maintained in DMEM supplemented with 10% FBS, 0.1mmol/L MEM non-essential amino acids, 6 mmol L-glutamine, 1mmol/L sodium pyruvate and 1% penicillin/streptomycin, 500μg/mL geneticin.
Lentivial shRNA library construction
The shRNA sequences targeting 626 human kinase genes (Supplementary Table 1) were generated using the design tool from the Broad Institute RNAi Consortium (http://www.broadinstitute.org/rnai/public/). Four pairs of shRNA oligonucleotides for each gene were selected, chemically-synthesized and then constructed into pLvUCTP lentiviral vector which was modified from FuGW as we previously described [32]. The library construction method was modified from the protocol described elsewhere [33]. Briefly, four lentiviral shRNA vectors for each gene were prepared and mixed equally. Transfections were carried out in 24-well plates by adding the mixture of shRNA vectors, packaging plasmids and FUGENE (Roche, Switzerland). Lentiviral supernatants were harvested at 24, 48 and 60 h post-transfection and purified by passaging into 0.45 μm filter (Millipore, Germany). Collected lentiviruses were stored at −80°C for long storage.
HT-RNAi screen and hits selection and validation
For HT-RNAi screen, we used DLD1 cell line because our previous study has shown that DLD1 was the most resistant to oxaliplatin among 17 CRC cell lines [32]. DLD1 cells were seeded in 96-well plates at the density of 2×103 cells per well. The next day, medium was removed and replaced with fresh medium supplemented with 8 μg/ml polybrene (Sigma, United States), and then lentiviral shRNAs were added to cells at a multiplicity of infection (MOI) of 5 in triplicates. Equal amount of lentiviral scramble shRNA was placed in each plate as a control [32]. After 24 h, cells were treated with equal volume of vehicle control (water) and 10 μM oxaliplatin (Sigma, United States) solution; this concentration resulted in an average of 40% cell growth inhibition and was close to the plasma concentration in CRC in our previous study [32]. At 48 h post-infection, cells were subjected to MTT assay. Z-score and sensitivity index (SI) value were utilized as the criteria for hits selection. The calculation of Z score and SI were performed according to the protocols described previously [9, 14]. The Z score and SI of lentiviral shRNA-treated cells were normalized to those of the scramble shRNA-treated cells.
CRC patient cohort and data collection
142 histologically confirmed stage III colorectal adenocarcinoma patients were recruited between June 2006 and December 2011, with follow-up to January 2014. Among them, 76 patients were recruited from RuiJin Hospital affiliated to Shanghai Jiaotong University and used as the training set, and 66 patients were enrolled from The Tenth People's Hospital affiliated to Shanghai Tongji University and used as the validation set. All the patients were newly diagnosed cases (diagnosed within 1 year of enrollment) and none had undergone chemotherapy or radiotherapy prior to study enrollment. All the patients underwent radical resection and first-line oxaliplatin-based adjuvant chemotherapy for at least of six cycles. Patients were staged according to the American Joint Committee on Cancer-tumor-node metastasis staging system (TNM). We reviewed patients' medical records to collect clinical information including date of diagnosis, clinical stage, tumor location, histological grade, pathological stage and treatment. The information of recurrence/death was obtained from the medical records or telephone follow-up. Paraffin-embedded (FFPE) specimens were provided by pathological departments. The study was approved by the Human Research Ethics Committees of Shanghai Jiaotong University and TongJi University.
DNA extraction from FFPE samples
Manual macrodissection of tumor samples were performed according to the protocol described elsewhere [34, 35]. 50-100 tissue sections (2 μm thick per each) were sliced from each FFPE sample. Hematoxylin and eosin (H&E)-stained tissue sections were reviewed by three pathologists. Manual macrodissection was performed only when three pathologists reached consensus. The tumor samples were collected by macrodissection of area with over 70% neoplastic cells and subjected to DNA extraction using DNA FFPE kit (Qiagen, German).
Quantitative PCR and copy number analysis
20 ng of genomic DNA from each sample was subjected to Quantitative PCR using the SYBR Green kit (Qiagen, Germany). The PCR program was 15 s of denaturation at 95°C, 20 s of primer annealing at 58°C and 20 s of extension at 72°C. GAPDH was used as the reference gene. Each assay was run in duplicates. Data points that generated duplicated Ct values with over one cycle variance were excluded from analysis. The Ct value obtained from each sample was normalized to the mean Ct of all samples and then subjected to analysis with the 2−ΔΔCt method [36]. Briefly, the mean Ct for each gene in all samples was firstly calculated and ΔCt was computed by subtracting individual Ct from mean Ct. ΔΔCt was then computed by subtracting ΔCt of a target gene from ΔCt of the reference gene (GAPDH). Finally, 2−ΔΔCt was calculated and defined as relative copy number (RCN) of each target gene [36].
Statistical analysis
Statistical analyses were conducted using Intercooled STATA software, version 10 (College Station). Chi-square (x2) analysis was used to evaluate differences in patient characteristics of categorical variables and Student's t test was used to assess the difference of continuous variables. The study endpoints were recurrence-free survival (RFS) and overall survival (OS). RFS was defined as the time from pathologic diagnosis until first recurrence. OS was calculated from pathologic diagnosis to death. Due to the challenge in the normalization of gene copy number, we followed the procedure described in Boeri et al [37] to compute the ratio of RCN between high copy number genes and restricted the analysis to 278 ratios with minimal individual variation with intra-group variance ≤10 to reduce the potential false positive findings. To assess the association of RCN with the recurrence and survival of CRC, multivariate Cox proportional hazards model was used to estimate HRs and their 95% confidence intervals (CI) in the training set, the replication set and pool set, adjusting for age, gender, stage and histological grade. Kaplan–Meier method was used to plot RFS and OS curves with the log-rank test to compare curves. We also evaluated the combined effects of the significant genes based on their RCN. For the ratios exhibiting significant association with recurrence or survival in both the training and validation data, we constructed the risk scores from all the possible combinations of the ratios. The risk score of combined genes for each patient was derived by linear combination of the product of reference-normalized copy number of each gene by its Cox regression corresponding coefficient. We also dichotomized patients into two groups using a cut off value of the ratio generated by the spline regression method. We constructed receiver operating characteristic (ROC) curves and calculated the area under the curve (AUC) to evaluate the specificity and sensitivity of predicting recurrence and survival. All P values were 2-sided and a P ≤ 0.05 was considered statistically significant.
SUPPLEMENTARY MATERIAL TABLES
Acknowledgments
This work was supported by the grant from National Natural Science Foundation of China (81272480) and Natural Science Foundation of Shanghai (11DZ1910200, 12ZR1418800 and 11nm0503800).
Footnotes
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Hurwitz HI, Fehrenbacher L, Hainsworth JD, Heim W, Berlin J, Holmgren E, Hambleton J, Novotny WF, Kabbinavar F. Bevacizumab in combination with fluorouracil and leucovorin: an active regimen for first-line metastatic colorectal cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:3502–3508. doi: 10.1200/JCO.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 3.Andre T, Boni C, Navarro M, Tabernero J, Hickish T, Topham C, Bonetti A, Clingan P, Bridgewater J, Rivera F, de Gramont A. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:3109–3116. doi: 10.1200/JCO.2008.20.6771. [DOI] [PubMed] [Google Scholar]
- 4.Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, Rougier P, Ciardiello F. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:2011–2019. doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]
- 5.Kuebler JP, Wieand HS, O'Connell MJ, Smith RE, Colangelo LH, Yothers G, Petrelli NJ, Findlay MP, Seay TE, Atkins JN, Zapas JL, Goodwin JW, Fehrenbacher L, Ramanathan RK, Conley BA, Flynn PJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25:2198–2204. doi: 10.1200/JCO.2006.08.2974. [DOI] [PubMed] [Google Scholar]
- 6.Uncu D, Aksoy S, Cetin B, Yetisyigit T, Ozdemir N, Berk V, Dane F, Inal A, Harputluoglu H, Budakoglu B, Koca D, Sevinc A, Cihan S, Durnali AG, Ozkan M, Ozturk MA, et al. Results of adjuvant FOLFOX regimens in stage III colorectal cancer patients: retrospective analysis of 667 patients. Oncology. 2013;84:240–245. doi: 10.1159/000336902. [DOI] [PubMed] [Google Scholar]
- 7.MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nature cell biology. 2005;7:591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- 8.Khan O, Fotheringham S, Wood V, Stimson L, Zhang C, Pezzella F, Duvic M, Kerr DJ, La Thangue NB. HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:6532–6537. doi: 10.1073/pnas.0913912107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iorns E, Turner NC, Elliott R, Syed N, Garrone O, Gasco M, Tutt AN, Crook T, Lord CJ, Ashworth A. Identification of CDK10 as an important determinant of resistance to endocrine therapy for breast cancer. Cancer cell. 2008;13:91–104. doi: 10.1016/j.ccr.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Stevenson L, Allen WL, Turkington R, Jithesh PV, Proutski I, Stewart G, Lenz HJ, Van Schaeybroeck S, Longley DB, Johnston PG. Identification of galanin and its receptor GalR1 as novel determinants of resistance to chemotherapy and potential biomarkers in colorectal cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:5412–5426. doi: 10.1158/1078-0432.CCR-12-1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harradine KA, Kassner M, Chow D, Aziz M, Von Hoff DD, Baker JB, Yin H, Pelham RJ. Functional genomics reveals diverse cellular processes that modulate tumor cell response to oxaliplatin. Molecular cancer research : MCR. 2011;9:173–182. doi: 10.1158/1541-7786.MCR-10-0412. [DOI] [PubMed] [Google Scholar]
- 12.Zwang Y, Yarden Y. p38 MAP kinase mediates stress-induced internalization of EGFR: implications for cancer chemotherapy. The EMBO journal. 2006;25:4195–4206. doi: 10.1038/sj.emboj.7601297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dotan E, Meropol NJ, Zhu F, Zambito F, Bove B, Cai KQ, Godwin AK, Golemis EA, Astsaturov I, Cohen SJ. Relationship of increased aurora kinase A gene copy number, prognosis and response to chemotherapy in patients with metastatic colorectal cancer. British journal of cancer. 2012;106:748–755. doi: 10.1038/bjc.2011.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, Elustondo F, Chang J, Temple J, Ahmed AA, Brenton JD, Downward J, Nicke B. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer cell. 2007;11:498–512. doi: 10.1016/j.ccr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 15.Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, Muir B, Mohapatra G, Salunga R, Tuggle JT, Tran Y, Tran D, Tassin A, Amon P, Wang W, Enright E, et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer cell. 2004;5:607–616. doi: 10.1016/j.ccr.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 16.Hu MC, Qiu WR, Wang X, Meyer CF, Tan TH. Human HPK1, a novel human hematopoietic progenitor kinase that activates the JNK/SAPK kinase cascade. Genes & development. 1996;10:2251–2264. doi: 10.1101/gad.10.18.2251. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Shi X, Zhang QJ, Hampong M, Paddon H, Wahyuningsih D, Pelech S. Nocodazole-induced p53-dependent c-Jun N-terminal kinase activation reduces apoptosis in human colon carcinoma HCT116 cells. The Journal of biological chemistry. 2002;277:43648–43658. doi: 10.1074/jbc.M203214200. [DOI] [PubMed] [Google Scholar]
- 18.Sui H, Zhou S, Wang Y, Liu X, Zhou L, Yin P, Fan Z, Li Q. COX-2 contributes to P-glycoprotein-mediated multidrug resistance via phosphorylation of c-Jun at Ser63/73 in colorectal cancer. Carcinogenesis. 2011;32:667–675. doi: 10.1093/carcin/bgr016. [DOI] [PubMed] [Google Scholar]
- 19.Sui X, Kong N, Wang X, Fang Y, Hu X, Xu Y, Chen W, Wang K, Li D, Jin W, Lou F, Zheng Y, Hu H, Gong L, Zhou X, Pan H, et al. JNK confers 5-fluorouracil resistance in p53-deficient and mutant p53-expressing colon cancer cells by inducing survival autophagy. Scientific reports. 2014;4:4694. doi: 10.1038/srep04694. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Wang CY, Cusack JC, Jr, Liu R, Baldwin AS., Jr Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nature medicine. 1999;5:412–417. doi: 10.1038/7410. [DOI] [PubMed] [Google Scholar]
- 21.Brenner D, Golks A, Kiefer F, Krammer PH, Arnold R. Activation or suppression of NFkappaB by HPK1 determines sensitivity to activation-induced cell death. The EMBO journal. 2005;24:4279–4290. doi: 10.1038/sj.emboj.7600894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang JJ, Wang H, Rashid A, Tan TH, Hwang RF, Hamilton SR, Abbruzzese JL, Evans DB. Expression of MAP4K4 is associated with worse prognosis in patients with stage II pancreatic ductal adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:7043–7049. doi: 10.1158/1078-0432.CCR-08-0381. [DOI] [PubMed] [Google Scholar]
- 23.Liu AW, Cai J, Zhao XL, Jiang TH, He TF, Fu HQ, Zhu MH, Zhang SH. ShRNA-targeted MAP4K4 inhibits hepatocellular carcinoma growth. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:710–720. doi: 10.1158/1078-0432.CCR-10-0331. [DOI] [PubMed] [Google Scholar]
- 24.Lee DE, Lee KW, Jung SK, Lee EJ, Hwang JA, Lim TG, Kim BY, Bode AM, Lee HJ, Dong Z. 6,7,4′-trihydroxyisoflavone inhibits HCT-116 human colon cancer cell proliferation by targeting CDK1 and CDK2. Carcinogenesis. 2011;32:629–635. doi: 10.1093/carcin/bgr008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24:1770–1783. doi: 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- 26.Rathkopf D, Dickson MA, Feldman DR, Carvajal RD, Shah MA, Wu N, Lefkowitz R, Gonen M, Cane LM, Dials HJ, Winkelmann JL, Bosl GJ, Schwartz GK. Phase I study of flavopiridol with oxaliplatin and fluorouracil/leucovorin in advanced solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:7405–7411. doi: 10.1158/1078-0432.CCR-09-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du L, Lyle CS, Obey TB, Gaarde WA, Muir JA, Bennett BL, Chambers TC. Inhibition of cell proliferation and cell cycle progression by specific inhibition of basal JNK activity: evidence that mitotic Bcl-2 phosphorylation is JNK-independent. The Journal of biological chemistry. 2004;279:11957–11966. doi: 10.1074/jbc.M304935200. [DOI] [PubMed] [Google Scholar]
- 28.Ouafik L, Berenguer-Daize C, Berthois Y. Adrenomedullin promotes cell cycle transit and up-regulates cyclin D1 protein level in human glioblastoma cells through the activation of c-Jun/JNK/AP-1 signal transduction pathway. Cellular signalling. 2009;21:597–608. doi: 10.1016/j.cellsig.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M, Heynck S, Stuckrath I, Weiss J, Fischer F, Michel K, Goel A, Regales L, Politi KA, Perera S, Getlik M, et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:18351–18356. doi: 10.1073/pnas.0907325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lenz HJ, Van Cutsem E, Khambata-Ford S, Mayer RJ, Gold P, Stella P, Mirtsching B, Cohn AL, Pippas AW, Azarnia N, Tsuchihashi Z, Mauro DJ, Rowinsky EK. Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidines. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24:4914–4921. doi: 10.1200/JCO.2006.06.7595. [DOI] [PubMed] [Google Scholar]
- 31.Loupakis F, Cremolini C, Masi G, Lonardi S, Zagonel V, Salvatore L, Cortesi E, Tomasello G, Ronzoni M, Spadi R, Zaniboni A, Tonini G, Buonadonna A, Amoroso D, Chiara S, Carlomagno C, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. The New England journal of medicine. 2014;371:1609–1618. doi: 10.1056/NEJMoa1403108. [DOI] [PubMed] [Google Scholar]
- 32.Zhang YJ, Li AJ, Han Y, Yin L, Lin MB. Inhibition of Girdin enhances chemosensitivity of colorectal cancer cells to oxaliplatin. World journal of gastroenterology : WJG. 2014;20:8229–8236. doi: 10.3748/wjg.v20.i25.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, Carpenter AE, Foo SY, Stewart SA, Stockwell BR, Hacohen N, Hahn WC, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 34.Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, Navratilova M, Szeszenia-Dabrowska N, Mates D, Mukeria A, Holcatova I, Schmidt LS, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:4726–4734. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kotoula V, Charalambous E, Biesmans B, Malousi A, Vrettou E, Fountzilas G, Karkavelas G. Targeted KRAS mutation assessment on patient tumor histologic material in real time diagnostics. PloS one. 2009;4:e7746. doi: 10.1371/journal.pone.0007746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Boeri M, Verri C, Conte D, Roz L, Modena P, Facchinetti F, Calabro E, Croce CM, Pastorino U, Sozzi G. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3713–3718. doi: 10.1073/pnas.1100048108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.