

Abstract
Background
Necroptosis is a newly identified type of programmed cell death that differs from apoptosis. Recent studies have demonstrated that necroptosis is involved in multiple pathologies of various human diseases. Receptor-interacting protein 3 (RIP3) is known to be a critical regulator of necroptosis. This study investigated alterations in the RIP3 expression and the involvement in neural tissue damage after spinal cord injury (SCI) in mice.
Results
Immunohistochemical analysis demonstrated that the RIP3 expression was significantly increased in the lesion site after spinal cord hemisection. The increased expression of RIP3 started at 24 h, peaked at 3 days and lasted for at least 21 days after hemisection. The RIP3 expression was observed in neurons, astrocytes and oligodendrocytes. Western blot analysis also demonstrated the RIP3 protein expression significantly upregulated in the injured spinal cord. RIP3 staining using propidium iodide (PI)-labeled sections showed most of the PI-labeled cells were observed as RIP3-positive. Double staining of TUNEL and RIP3 demonstrated that TUNEL-positive cells exhibiting shrunken or fragmented nuclei, as generally observed in apoptotic cells, rarely expressed RIP3.
Conclusions
The present study first demonstrated that the expression of RIP3 is dramatically upregulated in various neural cells in the injured spinal cord and peaked at 3 days after injury. Additionally, most of the PI-labeled cells expressed RIP3 in response to neural tissue damage after SCI. The present study suggested that the upregulation of the RIP3 expression may play a role as a novel molecular mechanism in secondary neural tissue damage following SCI. However, further study is needed to clarify the specific molecular mechanism underlying the relationship between the RIP3 expression and cell death in the injured spinal cord.
Keywords: Receptor-interacting protein 3, Spinal cord injury, Necroptosis, Cell death, Necrosis, Apoptosis
Background
Necrosis was originally thought to be a purely passive and uncontrolled type of cell death. Necrosis, which is marked by cell swelling and membrane rupture, leads to inflammation via the release of intracellular signals [1–3]. In contrast, apoptosis, which is characterized by the activation of caspases and DNA fragmentation, was once considered the sole form of programmed cell death. Chromatin condensation, nuclear shrinkage and fragmentation and membrane blebbing are the result of the proteolytic activity of caspases and define the morphological characteristics of apoptosis [3–5].
“Necroptosis” is a newly identified type of programmed necrosis. Necroptosis is another form of programmed cell death that is regulated by the caspase-independent pathway and exhibits the morphological features of necrosis [6, 7]. Recent studies have shown that the receptor-interacting protein (RIP) family is specifically involved in regulating necroptosis [3]. RIP3, a member of the RIP family, is known to be a key mediator of necroptosis [8, 9]. RIP3 acts a nucleocytoplasmic shuttling protein and can locate not only in the cytoplasm but nucleus [10]. Various stimuli to cells can induce the formation of necrotic complexes that contain RIP1, TRADD, FADD and caspase-8. The interaction of RIP3 with RIP1 in the necrotic complex is an important step required for the execution of necroptosis [3, 11]. Previous studies have demonstrated that the RIP3 expression correlates with the induction of necroptosis in various types of cells [12–18]. RIP3 alone can trigger necroptosis, even in the absence of RIP1 [12, 14, 19]. Therefore, RIP3 is indispensable for necroptosis, whereas RIP1 may participate only in certain stimulus-induced types of cell necroptosis [9].
Furthermore, recent studies have shown an increased expression of RIP3 in lesions and the induction of necroptosis in various disease models [18, 20], such as that involving liver injury [14], ileitis [21], skin inflammation [22], and kidney ischemia–reperfusion injury [23]. The expression of RIP3 is also increased in retinal neurons in response to acute ischemic insults [24], and the upregulation of RIP3 contributes to the induction of necroptotic cell death in hippocampal neurons following cerebral ischemia [17].
Currently, it is considered that secondary damage after spinal cord injury (SCI) is caused by apoptosis, and most previous research related to cell death in the injured spinal cord has focused on apoptosis, not necroptosis.
In the present study, we investigated alterations in the RIP3 protein expression and the involvement of necroptosis in SCI using a spinal cord hemisection model in mice.
Methods
Animals
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Tohoku University. All efforts were made to minimize the number of animals used and to decrease their suffering. Adult female C57BL/6J mice (8–10 weeks old; Charles River, Japan Inc., Yokohama, Japan) were used in this study. The mice were housed three or four per cage and kept at a temperature of 24 °C with free access to food and water before and after surgery.
Spinal cord hemisection model
The mice were anesthetized with 2 % sevoflurane. A 15-mm midline skin incision was made, and the laminae of T9-11 were exposed. Laminectomy was performed at the thoracic vertebra at T10, exposing the dorsal surface of the spinal cord without disrupting the dura mater. With a sharp scalpel, the spinal cord was hemi-transected on the left side only [25–28]. In mice with a compromised bladder function (a rare complication), the bladder was manual expressed twice a day until spontaneous voiding was noted. The sham-operated animals underwent the same surgical procedures, although hemisection was not applied to the spinal cord. During the surgery, the rectal temperature was monitored and maintained at 37.0 ± 0.5 °C with a heating pad.
Tissue preparation
At different time points (4 h, 24 h, 3, 7, and 21 days) after hemisection and immediately after the sham operation, the mice were overdosed via an intraperitoneal injection of 100 mg/kg sodium pentobarbital. The mice were then transcardially perfused with normal saline, followed by 4 % paraformaldehyde in 0.1 M PBS, pH 7.4. For immunohistochemical staining, the spinal cord segments containing the injured site were collected, postfixed in the same fixative overnight at 4 °C, cryoprotected in 30 % sucrose in PBS for 48 h at 4 °C and embedded in Optimal-Cutting-Temperature compound (Sakura Finetek, Japan). Serial 15-μm transverse cryostat sections obtained from around the injured site were mounted on slides. A total of 13 sequential sections were collected at 250-μm intervals that spanned 3000 μm in length along the spinal cord centered at the epicenter. The sections were used for both immunohistochemical and TUNEL staining, as described below.
Immunohistochemical staining of RIP3
For the immunohistochemical staining of RIP3, the sections were washed in PBS for 15 min, after which they were washed with PBS containing 0.3 % Tween for 10 min and blocked with 3 % milk and 5 % FBS in 0.01 M PBS for 2 h. The sections were incubated with rabbit anti-RIP3 antibodies (1:100; Sigma-Aldrich) diluted in PBS overnight at 4 °C. After rinsing with PBS, the sections were incubated with secondary antibodies. The sections were coverslipped with Vectashield containing DAPI to label the nuclei (Vector Laboratories). In each experiment, the sections were stained at the same time.
Counting and calculation of RIP3-positive cells
Following immunochemical staining of RIP3, as described above, each section was scanned using a fluorescence microscope (BX 51; Olympus, Tokyo, Japan). In order to quantify the RIP3 expression in the spinal cord, the total number of RIP3-positive cells in the injured and contralateral sides of the spinal cord was counted using the serial transverse sections at 250-μm intervals. The sections with the highest number of RIP3-positive cells on the injured side and the 250-μm rostral and caudal sections in each animal were selected. Then, the sum of the numbers in the three sections was compared between the injured side, the contralateral side and the sham group.
Western blot analysis of RIP3
The mice were killed 3 days after spinal cord hemisection and the sham operation, and their spinal cords were removed. The spinal cord was homogenized in lysis buffer. The debris was removed via centrifugation, and the protein levels in the lysates were determined with the aid of a Bio-Rad protein assay (Bio-Rad Laboratories, USA). The protein (30 μg) in the lysates was resolved using SDS–polyacrylamide gel electrophoresis (PAGE) in 15 % gels and then electrophoretically transferred to a polyvinylidene difluoride membrane. The membranes were blocked for 1 h in TBST buffer (0.01 M Tris HCl, pH 7.5, 0.15 M NaCl and 0.05 % Tween 20) containing 3 % milk and incubated with rabbit anti-RIP3 antibodies (1:100; Santa Cruz Biotechnology, USA) diluted in TBST buffer overnight at 4 °C. The membranes were incubated with secondary antibodies linked to horseradish peroxidase (1:1000; Invitrogen). The immunoreactive bands were developed using the enhanced chemiluminescence reagent (Amersham Corp). The band density was quantified using a scanned densitometric analysis and the Image Lab software program version 4.1 (Bio-Rad Laboratories). The quantity of the band density was normalized according to the level of β-tubulin and compared between the injured and uninjured spinal cord samples.
Double staining for RIP3 and various cell type markers
In order to examine the expression of RIP3 in a specific population of cells, the transverse sections obtained 3 days after hemisection were double-stained for RIP3 and various cell type markers: NeuN for neurons, GFAP for astrocytes and Olig2 for oligodendrocytes. The sections were incubated with a mixture of rabbit anti-RIP3 antibody (1:100; Sigma-Aldrich) and either goat anti-Olig2 (1:100; Santa Cruz Biotechnology), mouse anti-GFAP (1:50; Dako) or mouse anti-NeuN (1:100; Chemicon) antibodies diluted in PBS overnight at 4 °C. After rinsing with PBS, the sections were incubated with secondary antibodies, and then mounted with Vectashield containing DAPI to label the nuclei (Vector Laboratories). The specificity of the RIP3 antibody was evaluated by omission of the primary antibody in immunohistochemistry. Omitting the primary antibody (no primary immunoglobulin G control) in this protocol abrogated the staining, demonstrating the specificity of the immunoreactive staining [14, 24].
RIP3 staining using propidium iodide-labeled spinal cord sections
In order to detect plasmalemma permeability, which is a hallmark of necrotic cell death, in the injured spinal cord at 3 days after hemisection, propidium iodide (PI) labeling was performed as previously described [29–31]. Briefly, PI (10 mg/mL; Sigma-Aldrich) was diluted in 0.1 mL of PBS and then intraperitoneally injected at a dose of 1 mg/kg body weight at 1 h before sacrifice. The mice were transcardially perfused, and the spinal cords were collected and sectioned as described above. For immunohistochemical detection of the RIP3 expression in the PI-labeled cells, the PI-labeled spinal cord transverse sections were stained for RIP3, as outlined above. Using a fluorescence microscope (BX 51; Olympus, Tokyo, Japan), the propidium iodide labeling was detected using emission and excitation wavelengths of 568 and 585 nm, respectively.
Double staining of RIP3 and TUNEL
In order to identify DNA fragmentation in the cells expressing RIP3, double staining of RIP3 and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) was performed using the transverse sections at 3 days after hemisection. Following immunohistochemical staining of RIP3, as described above, the TUNEL was performed using the In Situ Cell Death Detection Kit (Roche Diagnostics).
Statistical analysis
Significant differences in the number of RIP3-positive cells and the band density of the Western blots were analyzed using the unpaired t test. In all analyses, a p value of <0.05 was considered to be statistically significant.
Results
Immunohistochemical staining for RIP3
The number of cells expressing RIP3 was increased on the injured side at 3 days after spinal cord hemisection (Fig. 1). Cells expressing RIP3 were observed in both the gray and white matter of the injured side. However, all cells on the injured side did not express RIP3. On the contralateral side, the number of cells expressing RIP3 was obviously low, similar to that observed in the sham group.
Fig. 1.
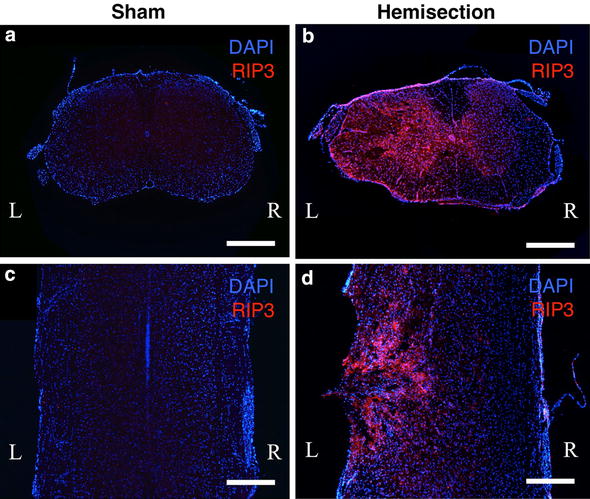
Immunohistochemical staining of RIP3 using spinal cord sections obtained 3 days after hemisection and the sham operation. The uninjured spinal cords in the sham operated mice (a, c) showed no obvious RIP3 expression. In contrast, a higher expression of RIP3 was observed on the injured side (L) than on the contralateral side (R) in the transverse (b) and coronal (d) sections after hemisection. Scale bars 500 μm
Time course of the RIP3 expression after hemisection
Representative pictures showed that the number of cells expressing RIP3 was increased on the injured side at each time point after hemisection compared to that observed in the sham group (Fig. 2). The population of cells expressing RIP3 on the injured side was relatively larger at 3 and 7 days compared to that noted at the other time points.
Fig. 2.
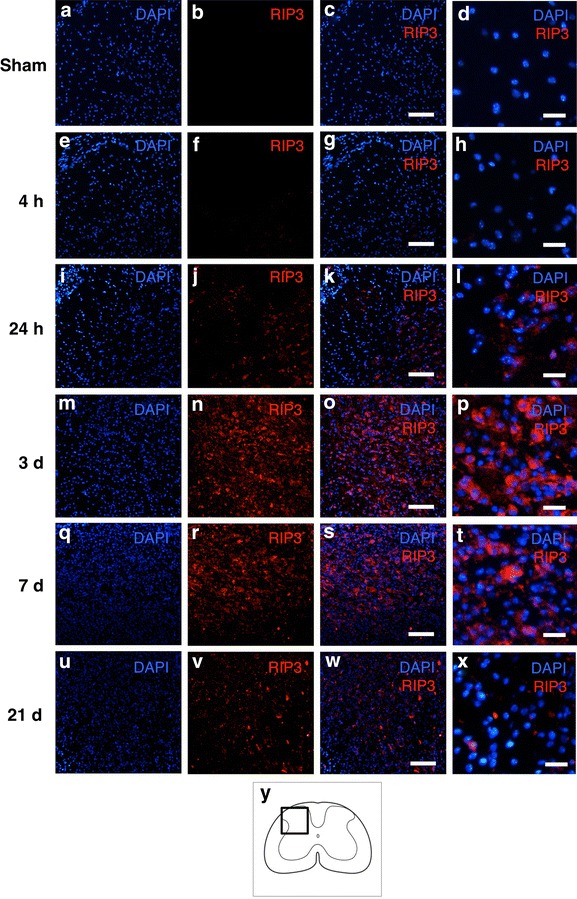
Immunohistochemical staining of RIP3 on the injured side in the transverse sections obtained at different time points. The expression of RIP3 was not observed at 4 h (e–h); however, it started to increase at 24 h (i–l), in comparison to that observed in the sham group (a–d). The population of RIP3-expressing cells was obviously larger at 3 and 7 days (m–t) than at the other time points. Scale bars 100 μm (a–c, e–g, j–k, m–o, q–s, u–w), 20 μm (d, h, l, p, t, x). The schematic drawing illustrates the location of the micrographs (y)
When counting the number of RIP3-positive cells (Fig. 3), those observed on the injured side were found to be significantly higher than those seen on the contralateral side and in the sham control group. A significant increase in the number of RIP3-positive cells was first noted at 24 h and lasted for at least 21 days (P < 0.05). The maximum number of RIP3-positive cells on the injured side was observed at 3 days after hemisection.
Fig. 3.
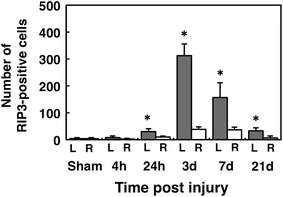
Number of RIP3-positive cells on the injured and contralateral sides at different time points. The number of RIP3-positive cells on the injured side (L) was significantly higher than that observed on the contralateral side (R) and in the sham group at 24 h and 3, 7 and 21 days. (*p < 0.05, n = 3 per each group). The values are presented as the mean ± SD
Western blot analysis of RIP3
In the Western blot analysis, the level of RIP3 proteins was significantly higher in the injured spinal cord than in the uninjured spinal cord (Fig. 4a). In the analysis of the band density, the RIP3 expression was 2.2 ± 0.5-fold higher in the injured spinal cord (Fig. 4b, P = 0.005).
Fig. 4.
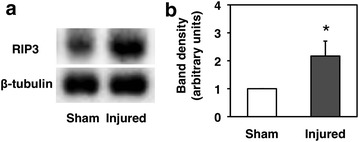
RIP3 protein expression at 3 days after hemisection. a Western blotting showed that the RIP3 protein expression in the injured spinal cord was obviously increased compared to that observed in the uninjured spinal cord after the sham operation. b A quantitative analysis of the Western blots showed that the level of RIP3 proteins in the injured spinal cord was significantly higher than that observed in the sham control samples (*p < 0.05, n = 5 per each group). The quantity of the band density was normalized according to the level of β-tubulin. The values are presented as the mean ± SD
Double staining of RIP3 and various cell type markers
In order to examine the RIP3 expression in specific populations of cells, the transverse sections obtained at 3 days after hemisection were double-stained for RIP3 and various neural cell type markers. On the double staining, the expression of RIP3 was observed in the NeuN-, GFAP- and Olig2-labeled cells (Fig. 5). The double staining demonstrated the RIP3 expression in neurons, astrocytes and oligodendrocytes. However, not all of these cells expressed RIP3.
Fig. 5.
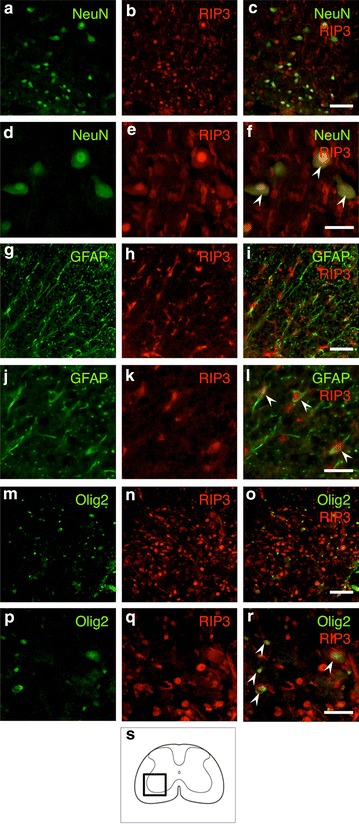
Double staining for RIP3 and various cell type markers on the injured side in the transverse sections after hemisection. The RIP3 expression was observed in the NeuN-, GFAP- and Olig2-labeled cells (arrowheads), demonstrating that the RIP3 expression was increased in neurons, astrocytes and oligodendrocytes, respectively. Scale bars: 100 μm (a–c, g–i, m–o), 50 μm (d–f, j–l, p–r). The schematic drawing illustrates the location of the micrographs (s)
RIP3 expression in the PI-labeled cells
Plasmalemma permeability, a hallmark of necrotic cell death, including necroptosis, was detected using PI-labeling in vivo. In RIP3 immunostaining using PI-labeled spinal cord sections. The number of both PI-labeled cells and RIP3-expressing cells increased on the injured side, and the PI-labeled cells were frequently observed to be RIP3-positive (Fig. 6). At a higher magnification, most of the nuclei in the PI-labeled cells expressing RIP3 were round, as generally observed in necrotic cell death.
Fig. 6.
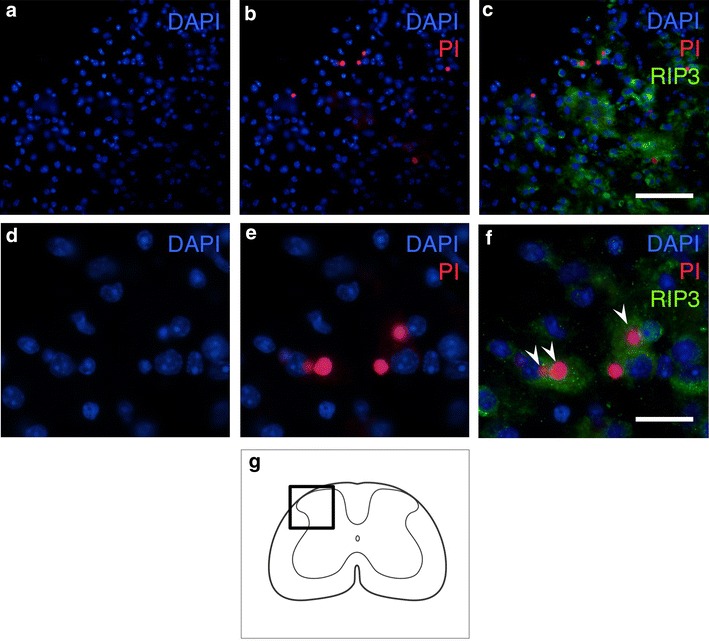
Immunostaining for RIP3 using propidium iodide (PI)-labeled sections obtained 3 days after hemisection. The representative pictures show that the PI-labeled cells were frequently observed to be RIP3-positive (a–c). At higher magnification (d–f), most of the nuclei in the PI-labeled cells expressing RIP3 (arrowheads in f) were round, which is normally observed in cells undergoing necrotic cell death, not fragmented or shrunken, as observed in apoptotic cells. Scale bars 100 μm (a–c), 20 μm (d–f). The schematic drawing illustrates the location of the micrographs (g)
Double staining of RIP3 and TUNEL
In order to detect DNA fragmentation in the cells expressing RIP3, we performed double staining of RIP3 and TUNEL. On the injured side, the numbers of TUNEL-positive and RIP3-positive cells were observed to have obviously increased (Fig. 7a–c). However, the RIP3 expression was rarely observed in the TUNEL-positive cells. Under higher magnification, most of the TUNEL-positive cells exhibiting shrunken or fragmented nuclei, as is typical of apoptotic cells, did not express RIP3 (Fig. 7d–f).
Fig. 7.
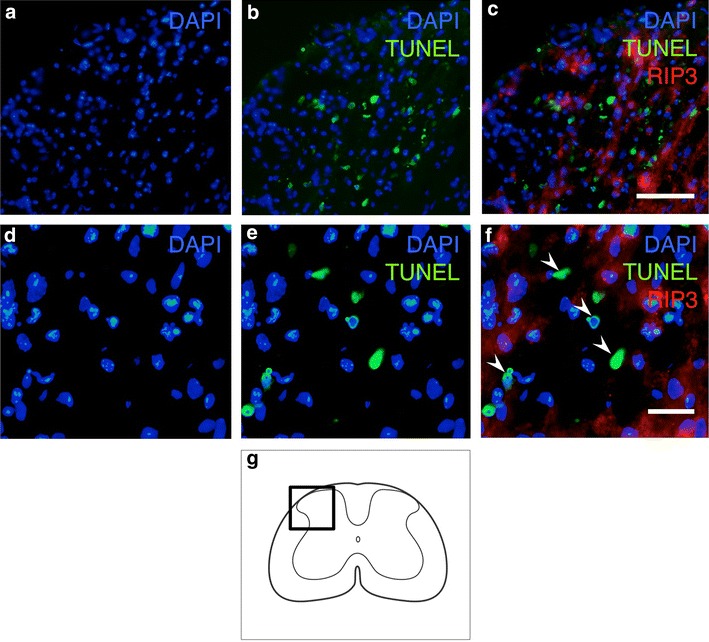
Double staining for RIP3 and TUNEL on the injured side in transverse sections obtained 3 days after hemisection. The numbers of RIP3-expressing and TUNEL-positive cells were obviously increased in the injured spinal cord (a–c). At higher magnification (d–f), the TUNEL-positive cells exhibiting shrunken or fragmented nuclei (arrowheads), a hallmark of apoptotic cell death, rarely expressed RIP3. Scale bars 100 μm (a–c), 20 μm (d–f). The schematic drawing illustrates the location of the micrographs (g)
Discussions
In the present study, the level of RIP3 proteins significantly increased on the injured site after spinal cord hemisection. An increase in the number of RIP3-positive cells at the injured site was observed starting at 24 h, peaked at 3 days and lasted for at least 21 days after injury. The RIP3 expression was observed in neurons, astrocytes and oligodendrocytes. Most of the PI-labeled cells expressed RIP3 in response to neural tissue damage after SCI. These results suggest that the expression of RIP3 was dramatically upregulated in various neural cells and may be involved in the pathological mechanism at the lesion site following SCI.
Traditionally, secondary damage of neural tissue following SCI has been considered to be caused by apoptosis, not necroptosis. Most previous research related to cell death in the injured spinal cord has focused on apoptosis. In the present study, both immunohistochemistry and Western blot analysis demonstrated that the RIP3 expression was significantly upregulated at the lesion site after SCI. Importantly, the upregulation of the RIP3 protein was observed starting at 24 h, after which it peaked at 3 days and lasted for 21 days after injury. The time course of the RIP3 expression is quite similar to that of secondary damage following SCI [32–34]. Additionally, we have previously confirmed that the number of TUNEL-positive cells peaked at 3 days in the thoracic spinal cord hemisection model in mice (data not shown). The TUNEL-positivity potentially indicates apoptotic cell death as well as necrotic cell death if the morphological characteristics of the cells are ignored [35]. Whalen et al. [31] suggested that different phenotypes of cell death can occur at the lesion site after the CNS injury. These findings suggest that the increased expression of RIP3 in the injured spinal cord may contribute to secondary neural tissue damage and is possibly associated with not only apoptosis, but also necrotic cell death after SCI.
Recent studies have demonstrated that necroptosis plays a role in various pathological conditions in the CNS. Necroptosis can contribute to neural tissue damage in the model of adult brain ischemia [17, 36, 37], neonatal hypoxia–ischemia [38], traumatic brain injury [29] and neurodegenerative diseases [39, 40]. Retinal ischemia [24, 41] and photoreceptor loss-associated retinal disorders [42] also involve neuronal necroptosis. Previous studies have shown that apoptosis is associated with peculiar morphological traits, including chromatin condensation and nuclear shrinkage and fragmentation, as well as blebbing of the intact plasma membrane and shedding of vacuoles containing cytoplasmic portions known as apoptotic bodies [1, 4, 5]. On the other hand, necroptosis involves a similar morphology to that of necrosis, such as minor ultrastructural modifications of the nucleus (dilatation of the nuclear membrane), osmotic swelling of organelles, an increased cell volume and breakdown of the plasma membrane [3, 11]. In the present study, most of the PI-positive cells expressed RIP3 in the injured spinal cord and contained round nuclei, which is generally observed in cells undergoing necrotic cell death. Additionally, the TUNEL-positive cells exhibiting shrunken or fragmented nuclei rarely expressed RIP3 at the lesion site. Based on the increased expression of RIP3 and the morphology of the nuclei in these cells, necroptosis may be one of the several types of cell deaths occurring in the injured spinal cord.
However, it has been reported that the PI- or TUNEL-positivity cannot perfectly differentiate between necrotic and apoptotic cells. A previous study suggested that PI-positive cells can occasionally be TUNEL-positive after CNS injury [31]. The PI-positivity is a hallmark of necrosis, but cannot be used by itself to identify necrotic cells because PI can enter cells with activated pannexin channels [43]. The TUNEL-positivity potentially indicates apoptotic cell death as well as necrotic cell death [35]. Additionally, the PI- or TUNEL-positivity did not perfectly reflect the RIP3 expression in the lesion site after hemisection in this study. Thus, from the data presented in this study, no firm conclusion about cell death in the RIP3-positive cells in the injured spinal cord can be made. The precise molecular mechanism underlying the relationship between RIP3 expression and PI- and/or TUNEL-positivity in neural tissue damage requires further clarification.
Previous studies have suggested that necroptosis is induced in various neural cells. The RIP3 expression in retinal neurons is upregulated in response to acute ischemic insults [24], and cerebral ischemia induces necroptotic cell death in hippocampal neurons [17]. In addition, necroptosis drives motor neuron death in amyotrophic lateral sclerosis ALS [40], Furthermore, hemin induces necroptotic cell death in cortical astrocyte cultures [44]. Necroptosis is induced in cultured rat astrocytes by the administration of staurosporine [45] Necroptosis contributes to arachidonic acid-induced oxidative cell death in primary oligodendrocyte precursor cultures [46]. According to our results, the expression of RIP3 is observed in neurons, astrocytes and oligodendrocytes in the injured spinal cord. These findings suggested that necroptosis may occur in various neural cells and may contribute to multiple pathological mechanisms after SCI. However, the molecular mechanisms defining the features of necroptosis are not fully understood, and the precise pathological mechanisms induced by necroptosis after SCI remain unclear. Further studies to clarify the molecular and biological mechanisms of necroptosis will hopefully lead to the development of novel therapeutic strategies for the treatment of SCI.
Conclusion
In the present study, the expression of RIP3 is dramatically increased at the lesion site after SCI. The increased RIP3 expression was observed in neurons, astrocytes and oligodendrocytes. Most of the PI-labeled cells expressed RIP3 in response to neural tissue damage. This study is the first to provide evidence supporting the increased expression of RIP3 involved in neural tissue damage after SCI.
Authors’ contributions
HK and HO designed the experiment, HK, ST and KY performed the experiment and analyzed the data, HK drafted the manuscript, HK and HO revised the manuscript and participated in paper modification. All authors read and approved the final manuscript.
Acknowledgements
We thank Ms. Michiko Fukuyama and Mr. Hideki Yamamoto for their technical assistance and the animal care team at the Institute for Animal Experimentation at Tohoku University for providing the animal care in this study.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Haruo Kanno, Email: kanno-h@isis.ocn.ne.jp.
Hiroshi Ozawa, Email: hozawa@med.tohoku.ac.jp.
Satoshi Tateda, Email: tateda3104@gmail.com.
Kenichiro Yahata, Email: impreza_51@yahoo.co.jp.
Eiji Itoi, Email: itoi-eiji@med.tohoku.ac.jp.
References
- 1.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol. 1990;181(3):195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 2.Puyal J, Ginet V, Clarke P. Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: A challenge for neuroprotection. Prog Neurobiol. 2013;105:24–48. doi: 10.1016/j.pneurobio.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Vandenabeele P, Galluzzi L, Vanden B, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 4.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361(16):1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hitomi J, Christofferson D, Ng A, Yao J, Degterev A, Xavier R, Yuan J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Degterev A, Hitomi J, Germscheid M, Ch’en I, Korkina O, Teng X, Abbott D, Cuny G, Yuan C, Wagner G, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galluzzi L, Vitale I, Abrams J, Alnemri E, Baehrecke E, Blagosklonny M, Dawson T, Dawson V, El-Deiry W, Fulda S, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han J, Zhong C, Zhang D. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol. 2011;12:1143–1149. doi: 10.1038/ni.2159. [DOI] [PubMed] [Google Scholar]
- 10.Yang Y, Ma J, Chen Y, Wu M. Nucleocytoplasmic shuttling of receptor-interacting protein 3 (RIP3): identification of novel nuclear export and import signals in RIP3. J Biol Chem. 2004;279(37):38820–38829. doi: 10.1074/jbc.M401663200. [DOI] [PubMed] [Google Scholar]
- 11.Galluzzi L, Vanden B, Vanlangenakker N, Buettner S, Eisenberg T, Vandenabeele P, Madeo F, Kroemer G. Programmed necrosis from molecules to health and disease. Int Rev Cell Mol Biol. 2011;289:1–35. doi: 10.1016/B978-0-12-386039-2.00001-8. [DOI] [PubMed] [Google Scholar]
- 12.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 13.Moriwaki K, Chan FKM. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27:1640–1649. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roychowdhury S, McMullen M, Pisano S, Liu X, Nagy L. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57:1773–1783. doi: 10.1002/hep.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim L. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013;4:e716. doi: 10.1038/cddis.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin J, Li H, Yang M, Ren J, Huang Z, Han F, Huang J, Ma J, Zhang D, Zhang Z, et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013;3(1):200–210. doi: 10.1016/j.celrep.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Vieira M, Fernandes J, Carreto L, Anuncibay-Soto B, Santos M, Han J, Fernandez-Lopez A, Duarte CB, Carvalho AL, Santos AE. Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3. Neurobiol Dis. 2014;68:26–36. doi: 10.1016/j.nbd.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 18.Khan N, Lawlor KE, Murphy JM, Vince JE. More to life than death: molecular determinants of necroptotic and non-necroptotic RIP3 kinase signaling. Curr Opin Immunol. 2014;26:76–89. doi: 10.1016/j.coi.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 19.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7(4):302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith CCT, Yellon DM. Necroptosis, necrostatins and tissue injury. J Cell Mol Med. 2011;15:1797–1806. doi: 10.1111/j.1582-4934.2011.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Günther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner M, Hedrick S, Tenzer S, Neurath M, et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonnet M, Preukschat D, Welz P, van Loo G, Ermolaeva M, Bloch W, Haase I, Pasparakis M. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. 2011;35:572–582. doi: 10.1016/j.immuni.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 23.Linkermann A, Bräsen J, Darding M, Jin M, Sanz A, Heller J, De Zen F, Weinlich R, Ortiz A, Walczak H, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2013;110:12024–12029. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang J, Shang L, Zhang M, Wang H, Chen D, Tong J, Huang H, Yan X, Zeng L, Xiong K. Differential neuronal expression of receptor interacting protein 3 in rat retina: involvement in ischemic stress response. BMC Neurosci. 2013;14:16. doi: 10.1186/1471-2202-14-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanno H, Ozawa H, Sekiguchi A, Yamaya S, Itoi E. Induction of autophagy and autophagic cell death in damaged neural tissue after acute spinal cord injury in mice. Spine. 2011;36(22):E1427–E1434. doi: 10.1097/BRS.0b013e3182028c3a. [DOI] [PubMed] [Google Scholar]
- 26.Kanno H, Ozawa H, Sekiguchi A, Itoi E. Spinal cord injury induces upregulation of Beclin 1 and promotes autophagic cell death. Neurobiol Dis. 2009;33(2):143–148. doi: 10.1016/j.nbd.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 27.Dong H, Fazzaro A, Xiang C, Korsmeyer SJ, Jacquin MF, McDonald JW. Enhanced oligodendrocyte survival after spinal cord injury in Bax-deficient mice and mice with delayed Wallerian degeneration. J Neurosci Off J Soc Neurosci. 2003;23(25):8682–8691. doi: 10.1523/JNEUROSCI.23-25-08682.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalderon N, Fuks Z. Structural recovery in lesioned adult mammalian spinal cord by x-irradiation of the lesion site. Proc Natl Acad Sci USA. 1996;93(20):11179–11184. doi: 10.1073/pnas.93.20.11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.You Z, Savitz S, Yang J, Degterev A, Yuan J, Cuny G, Moskowitz M, Whalen M. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28:1564–1573. doi: 10.1038/jcbfm.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu X, Tao L, Tejima-Mandeville E, Qiu J, Park J, Garber K, Ericsson M, Lo E, Whalen M. Plasmalemma permeability and necrotic cell death phenotypes after intracerebral hemorrhage in mice. Stroke. 2012;43:524–531. doi: 10.1161/STROKEAHA.111.635672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whalen M, Dalkara T, You Z, Qiu J, Bermpohl D, Mehta N, Suter B, Bhide P, Lo E, Ericsson M, et al. Acute plasmalemma permeability and protracted clearance of injured cells after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28:490–505. doi: 10.1038/sj.jcbfm.9600544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Citron BA, Arnold PM, Sebastian C, Qin F, Malladi S, Ameenuddin S, Landis ME, Festoff BW. Rapid upregulation of caspase-3 in rat spinal cord after injury: mRNA, protein, and cellular localization correlates with apoptotic cell death. Exp Neurol. 2000;166(2):213–226. doi: 10.1006/exnr.2000.7523. [DOI] [PubMed] [Google Scholar]
- 33.Lu J, Ashwell KW, Waite P. Advances in secondary spinal cord injury: role of apoptosis. Spine. 2000;25(14):1859–1866. doi: 10.1097/00007632-200007150-00022. [DOI] [PubMed] [Google Scholar]
- 34.Yong C, Arnold PM, Zoubine MN, Citron BA, Watanabe I, Berman NE, Festoff BW. Apoptosis in cellular compartments of rat spinal cord after severe contusion injury. J Neurotrauma. 1998;15(7):459–472. doi: 10.1089/neu.1998.15.459. [DOI] [PubMed] [Google Scholar]
- 35.Beattie MS, Farooqui AA, Bresnahan JC. Review of current evidence for apoptosis after spinal cord injury. J Neurotrauma. 2000;17(10):915–925. doi: 10.1089/neu.2000.17.915. [DOI] [PubMed] [Google Scholar]
- 36.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny G, Mitchison T, Moskowitz M, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 37.Xu X, Chua K, Chua C, Liu C, Hamdy R, Chua B. Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Res. 2010;1355:189–194. doi: 10.1016/j.brainres.2010.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Northington F, Chavez-Valdez R, Graham E, Razdan S, Gauda E, Martin L. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab. 2011;31:178–189. doi: 10.1038/jcbfm.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pirooznia SK, Dawson VL, Dawson TM. Motor neuron death in ALS: programmed by astrocytes? Neuron. 2014;81:961–963. doi: 10.1016/j.neuron.2014.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Re LV, Yu A, Politi P, Ikiz H, Koolen N, Papadimitriou N, Mitsumoto K, Wichterle H, Przedborski necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81:1001–1008. doi: 10.1016/j.neuron.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenbaum D, Degterev A, David J, Rosenbaum P, Roth S, Grotta J, Cuny G, Yuan J, Savitz S. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88:1569–1576. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tristão V, Gonçalves P, Dalboni M, Batista M, Durão M, Martins J. Nec-1 protects against nonapoptotic cell death in cisplatin-induced kidney injury. Ren Fail. 2012;34:373–377. doi: 10.3109/0886022X.2011.647343. [DOI] [PubMed] [Google Scholar]
- 43.MacVicar BA, Thompson RJ. Non-junction functions of pannexin-1 channels. Trends Neurosci. 2010;33(2):93–102. doi: 10.1016/j.tins.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 44.Laird M, Wakade C, Alleyne J, Dhandapani K. Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free Radic Biol Med. 2008;45:1103–1114. doi: 10.1016/j.freeradbiomed.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 45.Simenc L-S. Staurosporine induces different cell death forms in cultured rat astrocytes. Radiol Oncol. 2012;46:312–320. doi: 10.2478/v10019-012-0036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S, Dayani L, Rosenberg P, Li J. RIP1 kinase mediates arachidonic acid-induced oxidative death of oligodendrocyte precursors. Int J Physiol Pathophysiol Pharmacol. 2010;2:137–147. [PMC free article] [PubMed] [Google Scholar]