

Abstract
Melanoma is the least common form of skin cancer, but it is responsible for the majority of skin cancer deaths. Traditional therapeutics and immunomodulatory agents have not shown much efficacy against metastatic melanoma. Agents that target the RAS/RAF/MEK/ERK (MAPK) signaling pathway—the BRAF inhibitors vemurafenib and dabrafenib, and the MEK1/2 inhibitor trametinib—have increased survival in patients with metastatic melanoma. Further, the combination of dabrafenib and trametinib has been shown to be superior to single agent therapy for the treatment of metastatic melanoma. However, resistance to these agents develops rapidly. Studies of additional agents and combinations targeting the MAPK, PI3K/AKT/mTOR (PI3K), c-kit, and other signaling pathways are currently underway. Furthermore, studies of phytochemicals have yielded promising results against proliferation, survival, invasion, and metastasis by targeting signaling pathways with established roles in melanomagenesis. The relatively low toxicities of phytochemicals make their adjuvant use an attractive treatment option. The need for improved efficacy of current melanoma treatments calls for further investigation of each of these strategies. In this review, we will discuss synthetic small molecule inhibitors, combined therapies and current progress in the development of phytochemical therapies.
Keywords: Melanoma, signaling pathways, targeted therapy, phytochemicals
Introduction
Melanoma is a malignant tumor of melanocytes, cells responsible for producing the skin pigment melanin. Of all skin cancers, melanoma is the least common, but because of its metastatic potential, it is responsible for approximately 80% of deaths related to skin cancer.1 The American Cancer Society estimates that the lifetime probability of Caucasian men being diagnosed with melanoma is 1/35, while for Caucasian women it is 1/54.2 Furthermore, it is projected that there will be ~76,710 new cases of melanoma in the United States in 2014, and ~9,710 of those cases will result in death.2 Established risk factors for the development of melanoma include fair features (light skin, hair and eye color) and ultraviolet exposure. In particular, blistering sunburns early in life have been shown to play a causal role.3 For cutaneous melanomas of low thickness (Breslow depths of up to 1.0 mm) surgery is curative for the majority patients.4 Rates of survival drop precipitously with increased tumor thickness due to the increased risk of metastasis.4 This transition from a mostly benign disease to one with a more serious prognosis occurs as melanoma progresses through the radial and vertical growth phases. The prognosis for metastatic melanoma is grim: 5-year survival ranges from 12 to 28%, depending on the location of the metastasis.4
Traditional cytotoxic therapy and immunomodulatory agents have failed to demonstrate significant efficacy, with fewer than 5% of patients having complete responses at 5 years.5 Fortunately, the last decade has been an exciting time for melanoma research, with advances in oncogene related therapies as well as immunotherapies. Immunotherapies that block inhibitory checkpoint molecules, CTLA-4 and PD-1, have been shown to improve survival for patients with metastatic melanoma and have gained FDA approval.6,7,8 Likewise, pivotal advances in oncogene directed therapies have led to improvements in patient survival, resulting in FDA approval of agents that target the RAS/RAF/MEK/ERK (MAPK) pathway, such as vemurafenib, dabrafenib and trametinib. Yet, in spite of these advancements, the extension of life offered by these agents is only a matter of months due to the rapid development of resistance. Also, they only target a fraction of the oncogenic signaling that leads to melanoma. Current research in this area is focused on the discovery of additional inhibitors of the MAPK pathway and inhibitors of other pathways that play key roles in melanomagenesis and resistance, such as PI3K/AKT/mTOR (PI3K) and c-kit signaling. The study of combination therapy with existing agents and the further elucidation of mechanisms of resistance are also underway. Furthermore, preclinical studies of phytochemicals, both alone and in combination with traditional cytotoxic and targeted therapies, have recently yielded promising results. The relatively low toxicities of these substances make the adjuvant use of natural agents an attractive treatment option for metastatic melanoma. In summary, the need for improved efficacy of current melanoma treatments calls for innovative strategies, such as the elucidation of combination therapies, continued discovery of novel therapeutic targets, and preclinical investigation of natural agents as adjuvant therapy. In this review, we will discuss progress in targeting MAPK, PI3K, and c-kit signaling pathways, preclinical studies of phytochemicals, and combined oncogene directed therapies (Tables 1, 2, 3; Figures 1, 2, 3).
Table 1.
Selected clinical trials of synthetic small molecule inhibitors in metastatic melanoma
| Inhibitor (code and common name) | Target(s) | Mechanism of action | Phase of clinical trial | Treatment effect or trial status | Mutations selected in patient population | Reference or NCT trial number (if not completed) |
|---|---|---|---|---|---|---|
| Tipifarnib (R115777) | RAS | Inhibition of posttranslation farnesylation of RAS | Phase II | No evidence of clinical activity | None selected | 24 |
| Sorafenib (BAY 43-9006; Nexavar) | BRAF CRAF VEGF PFGF |
Multikinase inhibitor | Phase III | No significant improvement in PFS, OS or RR | None selected | 45 |
| Vermurafenib (PLX4032; Zelboraf) | BRAFV600E BRAFV600K |
Selective BRAF kinase inhibitor | Phase III | Significant improvement in PFS and OS | BRAFV600E BRAFV600K |
52 |
| Dabrafenib (GSK 2118436) | BRAFV600E BRAFV600K |
Selective BRAF kinase inhibitor | Phase III | Significant improvement in PFS | BRAFV600E BRAFV600K |
55 |
| Encorafenib (LGX818) | BRAFV600 | Selective BRAF kinase inhibitor | Phase I | In progress | BRAFV600 | NCT01436656 |
| RAF265 (CHIR-265) | BRAFV600E VEGFR2 |
Multikinase inhibitor | Phase II | Completed results pending | None selected | NCT00304525 |
| Cobimetinib (GDC-0973; XL518) | MEK1/2 | MEK1/2 inhibitor | Phase I | In progress | None selected | NCT01271803 |
| Selumetinib (ARRY-142886; AZD6244) | MEK1/2 | MEK1/2 inhibitor | Phase II | Significantly improved PFS when combined with dacarbazine but no significant improvement in OS | BRAFV600 | 78 |
| Trametinib (GSK1120212; JTP-74057; Mekinist) | MEK1/2 | MEK1/2 inhibitor | Phase III | Significantly improved PFS and OS | BRAFV600E BRAFV600K |
82 |
| MEK162 (ARRY-438162; PD0325901) | MEK1/2 | MEK1/2 inhibitor | Phase II | Partial responses observed | NRAS BRAFV600 |
34 |
| Phase III | In progress | NRAS | NCT01763164 | |||
| Pimasertib (AS703026; MSC1936369B) | MEK1/2 | MEK1/2 inhibitor | Phase II | In progress | NRAS | NCT01693068 |
| Temsirolimus (CCI-779; Torisel) | mTOR | Rapamycin analog, inhibitor of mTORC1 | Phase II | No significant improvement in OS or PFS | None selected | 118 |
| Phase II | In progress | mTOR | NCT01960829 | |||
| Imatinib (CGP 57148; CGP57148B; STI-571; Gleevec) | c-kit | Tyrosine kinase inhibitor | Phase II | Objective responses observed | c-kit | 148, 149 |
| Phase II | In progress | c-kit | NCT00470470 | |||
| Dasatinib (BMS-354825; Sprycel) | c-kit | Tyrosine kinase inhibitor | Phase II | No significant improvement in PFS or RR | None selected | 151 |
| Phase II | In progress | c-kit | NCT00700882 | |||
| Nilotinib AMN 107; Tasigna) | c-kit | Tyrosine kinase inhibitor | Phase II | Preliminary results showed objectives responses | c-kit | 152, NCT01099514 |
| Phase II | In progress | c-kit | NCT01028222 |
OS – Overall survival, PFS – Progression free survival, RR – Response Rate
Table 2.
Selected clinical trials of synthetic small molecule inhibitor combinations in metastatic melanoma
| Inhibitor combination | Target(s) | Phase of clinical trial | Treatment effect or trial status | Mutations selected in patient population | Reference or NCT trial number (if not completed) |
|---|---|---|---|---|---|
| Dabrafenib and trametinib compared to dabrafenib monotherapy | BRAF + MEK1/2 | Phase II Phase III |
No significant improvement in PFS In progress |
BRAFV600 BRAFV600 |
89 NCT01584648 |
| Dabrafenib and trametinib compared to verumafenib monotherapy | BRAF + MEK1/2 | Phase III | In progress | BRAFV600E BRAFV600K |
NCT01597908 |
| Sorafenib with temsirolimus or Sorafenib with tipifarnib |
BRAF + mTOR or BRAF + RAS |
Phase II | No significant improvement in PFS | None selected | 120 |
| LGX818 plus MEK162 and LGX818 monotherapy to verumafenib monotherapy | BRAF + MEK1/2 | Phase III | In progress | BRAFV600 | NCT01909453 |
| Cobimetinib in combination with vermurafenib | BRAF + MEK1/2 | Phase Ib | Anti-tumor activity | BRAFV600 | 91 |
| Cobimetinib in combination with vemurafenib vs vemurafenib alone | BRAF + MEK1/2 | Phase III | In progress | brafV600 | NCT01689519 |
OS – Overall survival, PFS – Progression free survival, RR – Response Rate
Table 3.
A Summary of the molecular mechanisms(s)/cellular targets of phytochemicals in the treatment of melanoma
| Phytochemical | Dietary Source | Category | In vitro Studies | In vivo Studies | Molecular Mechanism(s)/Cellular Targets | References |
|---|---|---|---|---|---|---|
| Fisetin | Strawberries, mangoes, apples, grapes, peaches, persimmons, onions, tomatoes and cucumbers | Flavonoid | A375 cells, 3-D melanoma skin constructs of A375 cells | 451Lu melanoma xenografts | Inhibited cell growth and cell cycle progression Inhibited mTOR and p70S6K through direct binding and phosphorylation of AKT |
159 |
| A375, SK-MEL-28, RPMI-7951, SK-MEL-119 and Hs294T cells, 3-D melanoma skin constructs of A375 cells | Reduced cell invasion Inhibited phosphorylation of MEK1/2 and ERK1/2 Inhibited NFκB activation And EMT |
158 | ||||
| 451Lu cells | Inhibited cell growth, cell cycle progression Wnt/β-catenin signaling, MITF and c-myc | 157 | ||||
| EGCG | Green Tea | Catechins | A375 and Hs294T cells | Inhibited cell growth Induced cell cycle arrest and apoptosis by reducing Cyclin D1 and cdk2, p16INK4a, p21WAF1/CIP1 and p27KIP1 Modulated expression of Bcl-2 family proteins |
177 | |
| 1205Lu and Hs294T cells | Inhibited cell growth Decreased IL-1β secretion Down regulated NLRP1 Reduced caspase-1 activation Inhibited NFκB activities |
178 | ||||
| B16 cells | Inhibited B16 cells adhesion to laminin | 179 | ||||
| B16-F10 | Increased cell stiffness and inhibited cell migration | 180 | ||||
| A375 and Hs294T cells | Inhibited cell migration and invasion Reduced COX-2 and PGE2 receptors Inhibited NFκB activation and EMT |
181 | ||||
| B16-F3m cells | B16-F3m cells injected intraperitoneally in to Balb/c mice | Inhibited cell spreading on extracellular matrix and cell-cell interactions Inhibited phosphorylation of FAK Inhibited MMP-9 activity Reduced lung metastasis in mice Increased the survival of melanoma bearing mice |
182 | |||
| A375 and SK-MEL-28 cells | Inhibited cell growth Induced apoptosis Induced phosphorylation of ERK1/2 in A375, but not in SK-MEL-28 cells No effect on phosphorylation of p38 or JNK MAP kinases in either cell line |
162 | ||||
| Resveratrol | Grapes | Stilbene | B16 cells | Induced autophagy through ceramide accumulation Inhibited AKT/mTOR pathway |
163 | |
| B16 cells | Inhibited growth of an established B16/DOX melanoma and prolonged survival | Inhibited cell growth Induced cell cycle arrest and apoptosis Down regulated cyclin D1/cdk4 Increased p53 expression |
164 | |||
| Autologous human melanoma cell line (weakly metastatic Line IV clone 3 and on autologous, highly metastatic Line IV clone) originally established from a primary malignant melanoma lesion | Inhibited cell growth and colony formation Induced cell cycle arrest Increased expression of p53 |
165 | ||||
| Lu1205 cells | Reduced cell invasion Decreased AP-1/JunD, MMP-1, Bcl-2, and iNOS protein levels |
166 | ||||
| B16 cells | Inhibited cell growth and invasion Increased expression of α -MSH signaling-related molecules, including β-catenin, c-kit, and MITF Inhibited MMP-9 expression |
167 | ||||
| B16-F10 and B16-BL6 cells | B16-F10 cells injected in tail vein of C57BL/6 mice | Reduced tumor volume and metastasis; reduced AKT expression | 168 | |||
| YUZAZ6 and M14 cells | Reduced melanoma induced angiogenesis Increased expression of p53, matrix protein TSP1, Inhibition HIF-1α and VEGF production |
169 | ||||
| K1735 and B16-F10 cells | C57BL/6 mice | Enhanced cisplatin cytotoxicity Increased Connexin 43 | 170 | |||
| WM3211, c81-46A and c83-2c cells | Enhanced dacarbazine cytotoxicity Inhibition of Ref-1-activated AP-1 DNA-binding and endonuclease activities |
171 | ||||
| SK-MEL-5 and SK-MEL-28 cells | Inhibited cell growth Induced cell cycle arrest Inhibited kinase activity MEK1/2 and ribosomal S6 kinase-2 Reduced phosphorylation of ERK1/2 Reduced activation of NFκB, AP-1, and STAT3 |
173 | ||||
| Silymarin | Milk Thistle | Flavonoid | A375, Hs294T, Mel 1241 and Mel 1011 cells | Inhibited cell migration and invasion Enhanced the levels of casein kinase 1α and GSK-3β Inhibited β-catenin signaling Reduced MMP-2 and MMP-9 levels |
174 | |
| A375-S2 cells | Enhanced pro-apoptotic actions of anti-Fas agonistic antibody CH11 Increased the expression of Fas-associated proteins with death domain Increased cleavage of procaspose-3 Increased digestion of the inhibitor of caspase-activated DNase |
175 | ||||
| B16 and B16-BL6 cells | Spontaneous metastatic tumor model of C57BL/6J mice | Inhibited cell invasion to the draining lymph nodes Inhibited Src and STAT3 phosphorylation through PRL-3 down-regulation |
183 | |||
| Curcumin | Turmeric | Polyphenol | MMAN, MMRU, RPEP, PMWK, SK-MEL-2, SK-MEL-5, SK-MEL-28 and MeWo cells | Induced apoptosis independent of p53 Activated caspases-3 and -8 Inhibited NFκB Induced Fas receptor aggregation in FasL-independent manner Suppressed the apoptotic ihibitor, XIAP |
184 | |
| B78H1, SK-MEL-28 and MeWo cells | C57BL/6 mice | miRNA expression signature in tumors was substantially altered by curcumin intake with mmu-miR-205-5p over 100 times higher expressed when compared to controls Induced apoptosis Down regulated Bcl-2 and PCNA |
186 | |||
| B16–F10 cells | Induced apoptosis, ER stress, p23 cleavage Down regulated Mcl-1 protein | 187 | ||||
| 4046T cells | Induced apoptosis Suppressed NFκB, COX-2 and cyclin D1 |
189 | ||||
| C32, G-361, and WM 266-4 cells | Induced apoptosis Suppressed NFκB activation independent of BRAF/MEK/ERK and AKT pathway |
190 | ||||
| A375 and MeWo cells | Induced cell cycle arrest and apoptosis Down regulated NFκB activation, iNOS Up regulated p53, p21(Cip1), p27(Kip1) and checkpoint kinase 2 |
188 | ||||
| B16 and WM-115 cells | Induced ROS production and apoptosis Induced MST1 and JNK activation, Foxo3a nuclear translocation |
191 | ||||
| B16-F10 cells | Inhibited cell growth Induced cell cycle arrest Enhanced expression of cyclin-dependent kinase inhibitors (cyclin A, p21 and p27) Decreased expression of DNMT1 Inhibition of cyclic nucleotide phosphodiesterase 1–5 activities |
192 | ||||
| B16-2F2 cells | Induced differentiation | 198, 199 | ||||
| Lupeol | White cabbages, green peppers, strawberries, olives, mangoes and grapes | Triterpene | B16-2F2 cells | Induced differentiation Increased expression of melanosome transport proteins: tyrosinase, Rab27a, and myosin-Va |
200 | |
| B16-2F2 cells | Inducted differentiation Activation of p38 MAPK, |
201 | ||||
| B16-2F2 cells | Suppressed migration; Disassembly of the actin cytoskeleton | 202 | ||||
| Mel 928, Mel 1011 and Mel 1241 cells | Mel 928 and Mel 1011 xenograft | Inhibited cell growth Inducted apoptosis Decreased β-catenin transcriptional activity Decrease in the expression of Wnt target genes Suppressed c-myc, cyclin D1, PCNA, Ki-67 and osteopontin expression |
195 | |||
| B16-2F2 | C57BL/6 mice subcutaneously injected into with B16 2F2 cells | Suppressed tumor growth Induced cell cycle arrest Decreased Ki-67 and PCNA expression |
197 | |||
| WM35 and 451Lu cells | 451Lu xenograft | Inhibited cell growth Induced cell cycle arrest Decreased expression of Cyclins and cdk2 Modulated Bcl-2 family proteins Reduced 451Lu tumor growth, proliferation and cell cycle regulatory proteins |
196 | |||
| SK-MEL2 and MeWo cells | Inhibited cell growth Increased cytosolic cytochrome c, caspase activity, and mitochondrial depolarization |
204 | ||||
| Honokiol | Magnolia | Biphenol | B16-F10 cells | Inhibited cell growth Inhibited AKT/mTOR and Notch signaling |
205 | |
| CHL-1 and WM266-4 cells | Induced apoptosis due to ER stress from an interaction with glucose regulated protein 78 | 206 |
Figure 1.
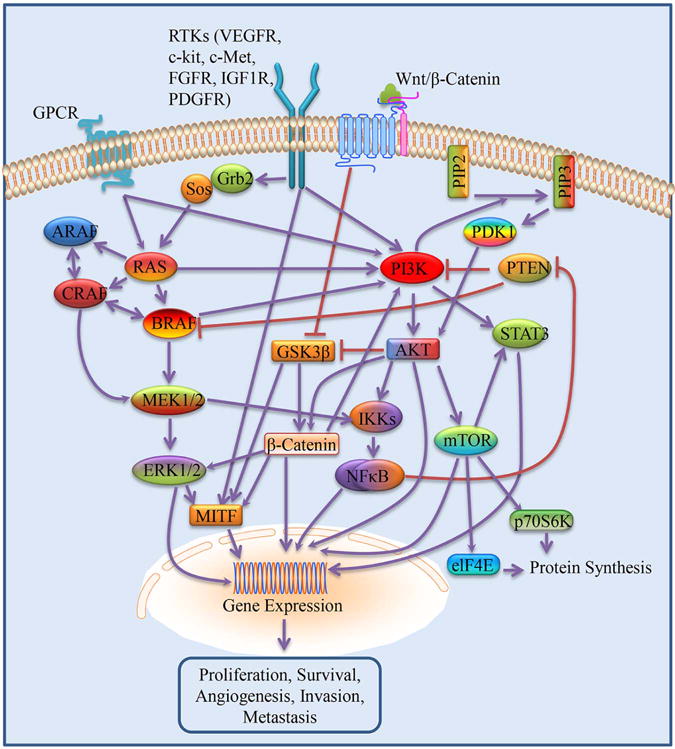
Signaling pathways activated in melanoma
Figure 2.
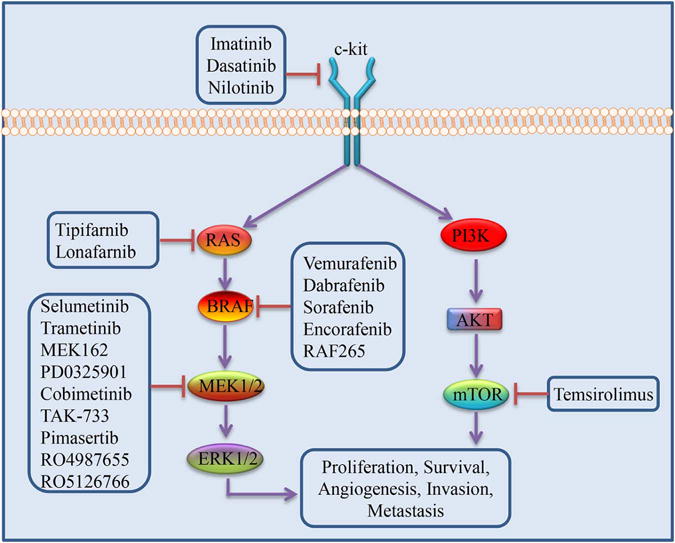
Signaling pathways targeted by synthetic small molecule inhibitors
Figure 3.
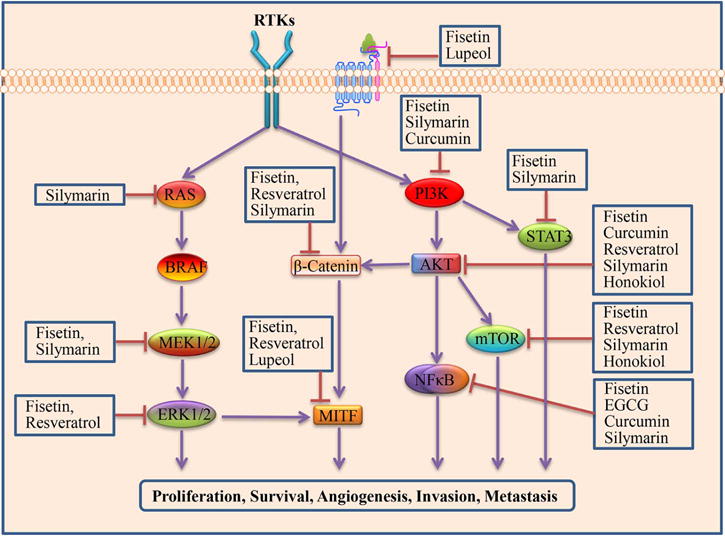
Signaling pathways targeted by phytochemicals
RAS/RAF/MEK/ERK (MAPK) Signaling in Melanoma
Activation of the MAPK has been described in roughly 90% of melanomas.9 Activation of the MAPK pathway occurs when RAS-GTP causes RAF kinase dimerization.9 An important target of activated RAF kinases is MEK1/2, which catalyzes the phosphorylation of ERK1/2.10,11 ERKs can translocate into the nucleus and regulate numerous cellular processes, including proliferation, differentiation, survival, motility, and angiogenesis.12
RAS
Some of the first oncogenes described in humans were RAS proteins. Through cellular stimuli, such as receptor tyrosine kinases (RTKs), RAS transmits extracellular signals to intracellular effector pathways, which include the RAS/RAF/MEK/ERK (MAPK) and the PI3K/AKT/mTOR (PI3K) signaling pathways.13 RAS signaling regulates a multitude of functions, including cell cycle progression, apoptosis, and differentiation.14,15 The conversion between inactive RAS-GDP and active RAS-GTP is regulated by guanine nucleotide exchange factors (GEFs) and by GTPase-activating proteins (GAPs). GEFs promote the exchange of GDP for GTP leading to RAS activation. GAPs accelerate RAS-mediated GTP hydrolysis and lead to inactivation of RAS.16 There are three main RAS isotypes: HRAS, KRAS, and NRAS.16 The most common RAS gene mutation in melanoma is NRAS, which is mutated in 15–20% of all melanomas.17 In accordance with the importance of NRAS mutations in maintaining melanoma cell growth, inactivation of NRAS in melanoma cell lines by RNA interference leads to induction of apoptosis.18 The most common NRAS mutation is at codon 61; this prevents RAS GTP hydrolysis, causing the NRAS protein to be constitutively active.19,20 Less common mutations at codon 12 and 13 prevent the association of GAP proteins with the NRAS complex.21
Association with the inner face of the plasma membrane is necessary for RAS function. Inhibition of post-translational farnesylation blocks RAS activation by impeding translocation of RAS to the plasma membrane. In mouse models, farnesyltransferase inhibitors (FTIs) were shown to have powerful anti-tumor activity and caused minimal toxicity to normal tissue in various cancer cell lines.22,23 Despite this evidence of the efficacy of RAS inhibition by FTIs, results from clinical trials were disappointing. A phase II clinical trial examining tipifarnib (a FTI) as a single agent in advanced melanoma was curtailed due to a lack of initial response to treatment, thus halting further clinical investigations.24 It is now believed that the RAS proteins can escape FTI through prenylation by a geranylgeranyl transferase that results in the transfer of an alternate isoprenoid group to RAS and allows continued activity.25,26 Despite unfavorable results as a monotherapy, there is still hope for the utility of FTIs when combined with other agents. It has been shown that combination treatment with cisplatin and lonafarnib (SCH66336), another FTI, amplified cisplatin-induced apoptosis in human and mouse melanoma cell lines.27,28 In melanoma cells, lonafarnib has also been shown to block mammalian targets of rapamycin (mTOR) signaling and enhance sorafenib-induced apoptosis.29 A phase I clinical trial combining tipfarnib and sorafenib (a BRAF inhibitor) showed stable disease in patients with various cancers, including one with metastatic melanoma.30 Further, an in vitro study showed that the combination of lonafarnib and sorafenib led to significant enhancement of sorafenib-induced apoptosis and complete suppression of melanoma cell invasion in raft culture.29 Blockade of NRAS signaling through inhibition of BRAF with vemurafenib has also been attempted, but was unsuccessful due to paradoxical hyperactivation of MEK-ERK signaling, causing activation of CRAF and induction of growth in cells with mutated RAS.31,32 In contrast to the results obtained with BRAF inhibitors, a recent study using NRAS mutant, patient-derived melanoma cell cultures showed that MEK inhibition reduced ERK1/2 phosphorylation and induced apoptosis.33 Promisingly, results of a phase II clinical trial of the MEK inhibitor, MEK162, exhibited objective responses in patients with NRAS mutations, providing support for the clinical use of MEK inhibitors for NRAS mutant metastatic melanoma treatment.34 There is a phase III study currently underway to compare the efficacy of MEK162 to dacarbazine in patients with NRAS mutations, along with a phase II trial of another MEK inhibitor, pimasertib, in patients with NRAS mutant melanoma (NCT01763164, NCT01693068).
RAS-driven melanomas represent a high percentage of metastatic melanomas.17 Despite the well-established role of NRAS in melanomagenesis, the development of effective therapies for patients with NRAS-driven melanoma remains elusive. Direct inhibition of RAS, thus far, has not been effective and RAS inhibition through blockade of BRAF has been shown to be ineffective.24,35,36 However, despite the failure of FTIs in monotherapy, these agents may support modulation of RAS signaling when used in combination with other treatment regimens. Moreover, MEK inhibition has shown promise as a therapy for NRAS mutant melanoma.33,34 These treatment strategies and other means of RAS inhibition are actively being pursued.
BRAF
The RAF isoforms include ARAF, BRAF, and CRAF/RAF-1.37 BRAF mutations are found in approximately 60% of all melanomas, and the oncogenic contribution of BRAF in melanoma has been validated in numerous cell and animal models.38,39,40 The BRAFV600E mutation accounts for nearly 90% of all such mutations found in melanoma.38 A substitution of valine for glutamic acid at position 600 results in the BRAFV600E mutation, causing the protein to remain in the active conformation permanently. Less common mutations (V600D, V600K, V600R) contribute another 5–6%, and are due to alternative point mutations at the same position.38 Of note, BRAF mutations are also found in many benign nevi.41 In fact, BRAF expression in human melanocytes has been shown to cause cell cycle arrest.42 Based on this evidence, BRAF is believed to induce the cancer sequence and with additional mutations, namely in tumor suppressor genes, transformation to melanoma ensues.41
The development of agents targeted at BRAF mutations, specifically the BRAFV600E mutation, is responsible for much-needed advancement in the treatment of metastatic melanoma. The first targeted agent to be tested in clinical trials for BRAF mutant melanoma was sorafenib.43 Sorafenib is a nonspecific kinase inhibitor, and has been shown to inhibit BRAF, CRAF, and the vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and various other RTK.43 However, a phase II clinical trial of sorafenib monotherapy showed a lack of response in patients with metastatic melanoma.44 Further trials evaluated the effectiveness of sorafenib in combination with cytotoxic agents. Unfortunately, a phase III clinical trials of sorafenib with carboplatin and paclitaxel similarly failed to shown a significant survival benefit.45,46 It is believed that due to sorafenib’s BRAF-independent cellular effects, therapeutic doses could not be achieved because of significant toxicity.47 The development and use of second-generation BRAF inhibitors with greater selectivity has been met with great success. Vemurafenib binds selectively to the ATP-binding site of the BRAFV600E mutation, resulting in reduced proliferation and downstream inhibition of ERK phosphorylation.48 Preclinical studies showed vemurafenib-induced RAF inhibition reduced the proliferation of BRAF mutant melanoma cell lines, but did not inhibit melanoma cell lines without BRAF mutations.49 Phase I and II clinical trials showed tumor shrinkage and vemurafenib-induced clinical responses in more than half the patients treated and showed improvement in rates of overall survival (OS) and progression-free survival (PFS) in patients with BRAFV600E mutant metastatic melanoma.50,51 A pivotal phase III study (BRIM-3) validated vemurafenib’s superiority to cytotoxic therapy in patients with the BRAFV600E mutation and also in patients with the BRAFV600K mutation. In patients with the BRAFV600E mutation, the estimated median PFS in the vemurafenib group was 6.9 months compared to 1.6 months for the dacarbazine group. For the patients with the BRAFV600K mutation, median PFS in the vemurafenib and dacarbazine group was 5.9 months and 1.7 months, respectively.52 These landmark results led to the FDA approval of vemurafenib for treatment of patients with BRAFV600E metastatic melanoma in 2011 along with a BRAFV600 mutation test. Another agent targeting BRAF mutant melanoma, dabrafenib, has recently received FDA approval. Phase I and II clinical trials of dabrafenib showed PFS ranging from 5.5–6.3 months for BRAFV600E melanoma and 4.5–5.6 months for BRAFV600K melanoma.53,54 A phase III trial comparing dabrafenib to dacarbazine showed a median PFS of 5.1 months for dabrafenib and 2.7 months for dacarbazine.55
Despite this exciting progress, 10% of patients with BRAF inhibitor-responsive melanoma show tumor progression early in the course of therapy and a majority of patients relapse in less than a year.50,56 The aberrations that result in resistance are numerous, but most reactivate the MAPK pathway by bypassing BRAF inhibition and restoring ERK activation. Resistance mechanisms that restore ERK activation include, but are not limited to, elevated expression of RAF kinases,57,58 activating mutations in NRAS or MEK1,59,60 stimulation of receptor tyrosine kinases59,61 and a splice variant of BRAFV600E.62 Moreover, the use of BRAF inhibitors comes with many troubling side effects, including development of keratoaconthomas and invasive squamous cell carcinoma.52,55 These proliferations occur in BRAF wild-type cells with mutated RAS and are due to up-regulation of BRAF-CRAF leading to ERK1/2 hyperactivation.63,32 This paradoxical ERK1/2 hyperactivation has been shown to lead to the development of RAS-driven squamous cell carcinomas and keratoaconthomas while patients are on vemurafenib and other BRAF inhibitors and, as mentioned above, is thought to be responsible for the inefficacy of BRAF inhibitors in NRAS mutant melanoma.64
Other BRAF inhibitors are currently in development or in clinical trials for metastatic melanoma (NCT01436656, NCT00304525).65 RAF265 is a dual inhibitor of mutant BRAFV600E and VEGFR2; a phase II clinical trial in patients with metastatic melanoma has been recently completed with pending results (NCT00304525). Preclinical studies with BRAF inhibitors lacking paradoxical ERK1/2 hyperactivation in cell lines with wild-type BRAF are also currently underway. For example, a non-paradox inducing BRAF inhibitor, PLX7904, was shown recently to reduce ERK1/2 phosphorylation and growth of NRAS mutant, vemurafenib-resistant melanoma cell lines.65
BRAF inhibitors have produced exciting and much-needed progress in the treatment of metastatic melanoma, but unfortunately, they have only been shown to inhibit the RAF/MEK/ERK pathway in cell lines that harbor BRAFV600E or BRAFV600K mutations. Of note, a clinical report on patients with the BRAFV600R mutation who were treated with vemurafenib or dabrafenib has shown promise; however, the activity of BRAF inhibitors in other, more rare, BRAF mutations remains unknown.66 It is clear that progress has been made for patients with certain BRAF mutations, but unfortunately, this only represents about half of the patients with metastatic melanoma. Responsive melanomas also develop resistance to BRAF inhibition quickly, and the mechanisms of resistance to dabrafenib and vemurafenib have been shown to be similar.67 The numerous mechanisms of resistance to BRAF inhibitors support the need for combined therapies of BRAF inhibitors with agents that target other locations in the RAF/MEK/ERK or alternative pathways. Most recently, the combination of MEK and BRAF inhibitors has shown great promise.68
MEK
BRAF mutated cells have been shown to possess enhanced sensitivity to MEK inhibition.69 MEK inhibitors are believed to function by inducing apoptosis through suppression of Mcl-1, a member of the anti-apoptotic B-cell CLL/lymphoma 2 (Bcl-2) family.70 There are two major classes of MEK inhibitors, ATP non-competitive and ATP competitive inhibitors.71 Currently, most MEK inhibitors are noncompetitive, indicating that they do not compete for the ATP-binding site and instead bind to an adjacent allosteric site, which explains their high specificity.71 Selumetinib, a selective, noncompetitive inhibitor of MEK1/2, has been shown to reduce the growth of melanoma cells by inducing G1-phase cell cycle arrest.72,73,74 Furthermore, cell growth inhibition was demonstrated in melanoma lines possessing the BRAFV600E mutation.75,76 A phase I clinical trial of selumetinib resulted in disease stabilization and tumor biopsies demonstrated reduced ERK phosphorylation in patients with metastatic melanoma.72 In a phase II clinical trial, patients with metastatic melanoma and an unknown NRAS/BRAF status received therapy with selumetinib or temozolomide.77 Results showed no significant difference in PFS. However, it was later found that five of the six patients who showed a partial response to selumetinib had BRAF mutant tumors.77 Therefore, more recent studies with MEK inhibitors have selected patients with BRAF mutant melanoma. Of note, a randomized phase II trial combining selumetinib and dacarbazine showed an improved median PFS of 5.6 months compared to 3.0 months with dacarbazine monotherapy; however, no significant change was seen in OS.78
The second generation ATP noncompetitive MEK1/2 inhibitors have shown improved effectiveness and the MEK1/2 inhibitor, trametinib (GSK1120212) has recently gained FDA approval for the treatment of metastatic melanoma. Preclinical trials with BRAFV600E melanoma cell lines and xenografts showed trametinib to be a reversible allosteric inhibitor of both MEK1/2 activation and kinase activity.79 A phase I trial of trametinib showed a response rate of 33% in patients with BRAF mutant melanoma and 10% in patients with wild-type BRAF, confirming the importance of selecting for mutational status.80 A phase II clinical trial of trametinib showed a median PFS of 4.0 months in BRAF mutant melanoma that had not been treated previously with a BRAF inhibitor, however no efficacy was seen in patients who had been treated previously with a BRAF inhibitor.81 The pivotal phase III trial, METRIC, enrolled patients with metastatic melanoma and BRAFV600E or BRAFV600K mutations that had not been previously treated with a BRAF or MEK inhibitor. Results showed significant improvements in OS and PFS, with a median PFS of 4.8 months in the trametinib group and 1.5 months in the chemotherapy group.82 Because of these results, the FDA approved trametinib in May 2013 for the treatment of patients with metastatic melanoma and BRAFV600E or BRAFV600K mutations who had not formerly received BRAF inhibitor treatment.
As discussed above, the MEK1/2 inhibitor, MEK162, has also recently shown promise for treatment of patients with NRAS-mutated melanoma in both preclinical and clinical trials.33,34 Of interest, it has been shown that variances in the activation state of MEK exist in KRAS versus BRAF mutant tumors.83 Specifically, inhibition of the RAF/MEK/ERK pathway in BRAF mutated melanoma occurs through inhibition of active, phosphorylated MEK, whereas inhibitors that block feedback phosphorylation by wild-type BRAF may be more effective for KRAS inhibition.83,84 Inhibitors that target the unique activation states of MEK are in clinical trials (NCT01689519, NCT01271803).83 Other MEK1/2 inhibitors (PD-0325901, TAK733, pimasertib, and RO4987655) are also currently in clinical development.85,86,12,87 Clinical Trials of targeted therapies in the treatment of metastatic melanoma are summarized in Table 1.
MEK and BRAF combined therapy
Numerous preclinical and clinical studies have demonstrated that BRAF and MEK co-inhibition is a successful treatment strategy for metastatic melanoma. Preclinical studies have shown that the combination of a BRAF and MEK inhibitor reduces tumor growth and delays onset of resistance when compared to monotherapy.88,68 In a phase II trial dabrafenib, either as a monotherapy or in combination with trametinib showed a significant improvement in PFS.89 The median PFS was 9.4 months in the combination group and 5.8 months in the monotherapy group.89 Moreover, the addition of trametinib was shown to reduce the appearance of cutaneous squamous cell carcinomas.64,89 These results led to the accelerated approval of the combination of dabrafenib and trametinib for the treatment of patients with metastatic melanomas that carry the BRAFV600E or BRAFV600K mutation in January 2014. Recent results of a phase III trial comparing the combination of dabrafenib and trametinib to dabrafenib monotherapy in patients with BRAF-mutant melanoma likewise showed improvements in PFS, with a median PFS of 9.3 months in the dabrafenib and trametinib group and 8.8 months in the dabrafenib monotherapy group.90 The rate of cutaneous SCC was also lower in the dabrafenib trametinib combination group compared to the dabrafenib-only group.90 A phase III clinical trial comparing dabrafenib and trametinib to vemurafenib monotherapy (COMBI-v) is also in progress (NCT01597908). Other BRAF and MEK inhibitor combinations have also shown promise and are in phase III trials (NCT01909453, NCT01689519).91,89 Unfortunately, it has been shown that resistance to MEK inhibition may cross over from BRAF inhibitor resistance.60 However, the combination of BRAF and MEK inhibition seems to delay this resistance,60,92 and a recent study has shown benefit for patients treated with a BRAF inhibitor after MEK inhibitor failure, suggesting that the mechanisms of resistance for MEK inhibition may be different.93 Moreover, a dual BRAF/MEK inhibitor, RO5126766, has recently shown activity in patients with metastatic melanoma, suggesting tandem inhibition of BRAF and MEK may one day be a possibility.94 Clinical Trials of synthetic small molecule inhibitor combinations in metastatic melanoma are summarized in Table 2.
PI3K/AKT/mTOR (PI3K) Signaling in Melanoma
The PI3K/AKT/mTOR (PI3K) pathway promotes cell survival and proliferation, and is hyperactivated in most malignancies, including melanoma.95,96 Stimulation of the PI3K pathway arises via GTP binding of RAS proteins and stimulation of RTK.96 Activation of the pathway generates phosphoinositide 3-kinase regulating subunit, allowing the catalytic subunit to phosphorylate membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) converting it to phosphatidylinositol 3,4,5-trisphosphate (PIP3), which is a key propagator of intracellular signaling. The tumor suppressor phosphatase and tensin homolog (PTEN) is a key regulator of the PI3K pathway. Lack of PTEN antagonism leads to association of phosphoinositide-dependent protein kinase 1 to the cell membrane and phosphorylation of AKT, a widely known oncogene.97,98 The numerous substrates of AKT include cellular regulators of insulin signaling, proliferation, and survival.99,100
AKT induces protein synthesis and cell proliferation by causing abrogation of TSC1/2, which leads to activation of the mTORC1 complex.101,102 Both PI3K and AKT participate in activation of mTOR,103,104 and the full activation of AKT requires phosphorylation of another of its kinase domains via mTOR complex 2 (mTORC2).105 These two distinct complexes of mTOR, mTORC1 and mTORC2, are believed to have differing functions, but their regulation overlaps in important ways. A primary role of mTORC2 is regulation of the actin cytoskeleton.106 However, as stated above, activation of mTORC2 leads directly to phosphorylation of AKT, linking this complex to the activation of the mTORC1 pathway.107,98 Enhanced protein translation is the result of mTORC1 activation and occurs through targeting of eukaryotic translation initiation factor (eIF4E) and p70S6 kinase.108
Elevated AKT phosphorylation and/or activated mTOR functioning arises in 70% of malignant melanomas.109,110 Elevated phospho-AKT levels are linked with reduced survival in melanoma patients111 and promote motility, invasion, and angiogenesis.112 The major mechanisms of PI3K pathway activation in melanoma are loss of PTEN and NRAS mutations, as discussed above. Loss of PTEN is seen in 10–30% of melanoma cell lines, with most exhibiting concurrent BRAFV600E mutations.113,114 In a mouse melanoma model, the BRAFV600E mutation alone led to benign melanocytic hyperplasias, and metastasis was induced with the concurrent loss of PTEN, suggesting a model for melanomagenesis.114
Agents that inhibit mTOR have demonstrated anti-proliferative effects against many human cancers.115 Most studies of PI3K signaling blockade in melanoma have used the first-generation agent rapamycin and the second-generation agents, everolimus (RAD001) and temsirolimus (CCI-779), which allosterically inhibit the mTORC1 complex.116,117 These agents have long been used as immunosuppressants in patients with organ transplants and are tolerated reasonably well. However, clinical trials in melanoma have shown a lack of objective responses to mTORC1 inhibitors as single agents or in combination with BRAF inhibitors.118,119,120 The reason for this seems to be interruption of negative feedback loops mediated by mTORC1, causing activation of PI3K, AKT and ERK.121,122 Specifically, compensatory PI3K pathway activation occurs due to interruption of baseline mTORC1 mediated inhibition of insulin receptor substrate 1, a second messenger of the insulin-like growth factor 1 RTK.123,122
The efficacy of mTORC1 inhibitors is limited by dysregulation of negative feedback loops and a lack of mTORC2 inhibition.117,124 Promisingly, a preclinical study evaluating the efficacy of dual mTORC1/2 inhibition showed blockade of compensatory AKT hyperactivation in sensitive cell lines.125 Dual PI3K-mTOR inhibitors are also being examined as a means to block compensatory activation of PI3K signaling.126 In melanoma, dual PI3K-mTOR inhibitors have shown impressive antiproliferative activity and durable suppression of AKT phosphorylation in both in vitro and in vivo studies.127,128 Furthermore, previous trials of mTOR inhibitors have not been performed in patients with mTOR mutations.118 There is currently a phase II clinical trial underway in patients selected for mTOR mutations (NCT01960829).
Combined therapy with PI3K inhibitors
Resistance to BRAF inhibitors and MEK inhibitors underscores the need to target alternative pathways.129,130 In particular, acquired resistance after BRAF therapy has been shown to occur through activation of PI3K signaling and can be overcome by MEK inhibitor therapy.131,61 Crosstalk between the MAPK and PI3K pathway is known to be a major cause of resistance, and inhibition of the PI3K pathway is being examined as a means of combating BRAF and MEK inhibitor resistance.61,129 In vivo models showing that activation of PI3K signaling with mutant BRAF enhances melanomagenesis more than either mutation alone supports the use of PI3K and MEK inhibitor combinations in vemurafenib-resistant, BRAF-mutant melanoma.114,132 Moreover, vemurafenib and selumetinib co-resistant BRAF-mutant melanoma cell lines have shown dependence on AKT induction for survival.130,133 In one study, the addition of an AKT inhibitor overcame acquired resistance to both vemurafenib and selumetinib, with the only exception being vemurafenib-resistant cell lines that secondarily acquired a NRAS mutation.130 In another study, BRAFV600E mutant melanoma cell lines harboring MEK or NRAS mutations also showed resistance to dabrafenib and trametinib monotherapy. However, when these cell lines were treated with a combination of a BRAF inhibitor and the PI3K inhibitor GSK2126458, they showed improved cell growth inhibition.129 Furthermore, in a RAS-driven, Ink4a/Arf-deficient mouse model of melanoma, the combination of BEZ235, a dual PI3K-mTOR inhibitor, with the MEK inhibitor AZD6244, produced significant tumor regression and improved survival.134 A recent preclinical study has shown superior growth inhibition and a delay in resistance in a melanoma cell line with homozygous PTEN loss when treated with the triple therapy of dabrafenib, trametinib and the AKT inhibitor GSK2141795B.135 Currently, a phase I study is examining the efficacy of selumetinib alone and in combination with the AKT inhibitor MK2206 (NCT01021748).
c-kit Signaling in Melanoma
The receptor tyrosine kinase c-kit is universally expressed in mature melanocytes and promotes proliferation and survival. Binding of c-kit to its ligand, stem cell factor, results in receptor dimerization, autophosphorylation, and stimulation of several signaling pathways, which include MAPK, PI3K, and janus kinase-signal transducer and activator of transcription (JAK-STAT) pathways. A recent preclinical study demonstrated that c-kit-driven PI3K activation led to MAPK pathway activation and increased melanocyte proliferation and melanoma survival.136 In addition, common variants at the c-kit locus have been shown to affect nevus number and increase the risk of melanoma.137 In melanomas that harbor c-kit mutations, point mutations resulting in substitutions at L576P and K642E have been shown to account for 55% of c-kit mutated melanomas.138 Recently, it has been shown that tumors retaining c-kit overexpression number fewer than 3% in unselected melanomas.139 In contrast to the overall scarcity of c-kit mutations, melanomas that occur in mucosal, acral, or chronically sun-damaged skin have been reported to have high rates of c-kit-activating mutations or amplifications (~28–39%).139,140
Imatinib mesylate is an ATP-competitive inhibitor of several tyrosine kinases, including c-kit. Interest in imatinib as a treatment for melanoma began with two separate case reports that showed striking responses to imatinib in metastatic melanomas with c-kit mutations.141,142 More recently, another case report has shown similar results.143 Preclinical evidence has also shown that, when treated with the tyrosine kinase inhibitor imatinib, melanoma cell lines with c-kit mutations exhibit reduced rates of melanoma cell proliferation and increased apoptosis, along with suppression of the MAPK, PI3K, JAK-STAT, and anti-apoptotic pathways.144 As imatinib is known to exhibit activity at several targets and to have efficacy in other tumor types, initial phase II studies in metastatic melanoma did not select for specific c-kit aberrations. As a probable result of this lack of clinical selectivity, these early trials did not show statistically significant results.145,146 A third phase II trial also failed to demonstrate clinical efficacy; however, it showed a dramatic response in one patient with a known c-kit mutation.147 Therefore, most recent trials of imatinib have selected for patients with c-kit-mutated metastatic melanoma.148,149 With this selected patient population, two recent phase II trials have shown clinically significant results. In the first study, 23.4% of the cases demonstrated c-kit mutations and/or amplifications, with a median PFS of 12.0 weeks and an OS of 46.3 weeks.148 This study also found that certain c-kit variants achieved greater responses; specifically, either K642E or L576P substitutions were present in all responses observed.148 In the second phase II trial of imatinib, all patients harbored c-kit aberrations, which resulted in a median PFS of 3.5 months and an OS of 14 months. Conversely, correlations between response and kit mutations were not seen in this study.149 Another phase II trial of imatinib in patients with c-kit aberrations and mucosal or acral metastatic melanoma is in progress (NCT00470470).
Other RTK inhibitors are currently being tested in clinical trials (NCT00700882, NCT01099514). In one study, two metastatic melanoma patients with the c-kitL576P mutation demonstrated responses to another RTK inhibitor, dasatinib.150 Like imatinib, further trials with dasatinib have shown a lack of efficacy in unselected melanoma patients.151 Preliminary results of a phase II clinical trial of another c-kit inhibitor, nilotinib, resulted in durable responses of 8.4 months and 10.0+ months, respectively, in two patients with c-kit mutations (NCT01099514).152 A randomized phase II trial (NCT01028222) comparing the efficacy of nilotinib vs. dacarbazine in patients with c-kit mutations and mucosal or acral metastatic melanoma is currently in progress.
Phytochemicals
Metastatic melanoma treatment has made great strides in recent years; however, these new signal transduction inhibitors have notable adverse side effects and the development of resistance progresses rapidly. It is apparent that new strategies and novel agents are needed to complement current therapies. Naturally occurring chemical compounds are referred to collectively as phytochemicals. Phytochemicals have gained attention as promising therapeutics due to studies demonstrating their ability to prevent the development of cutaneous malignancies.153,154 There are several classes of phytochemicals that have been studied, including polyphenols, flavonoids, isoflavonoids, phytoalexins, phenols and carotenoids. Here, we discuss and summarize recent research on classes of phytochemicals in the treatment of melanoma (Table 3, Figure 3).
Flavonoids are polyphenolic compounds that are ubiquitous in plants. Flavonoids have been shown to modulate various cancer signaling pathways, including proliferation, progression and metastasis.155 Several preclinical studies have shown that the flavonoid fisetin is active against melanoma. Specifically, fisetin is a flavone sub-class found in the Rhus family, which includes strawberries, mangoes and other plants.156 Studies have demonstrated that fisetin reduces melanoma cell proliferation, invasion and tumor growth by inhibiting β-catenin/Microphthalmia-associated transcription factor (MITF),157 MAPK,158 NFκB,158 and PI3K signaling159. In one study, inhibition of AKT and mTOR was linked with a significant reduction in the transition from the radial to the vertical growth phase in cell and xenograft models.159 It has similarly been shown that fisetin suppresses melanoma cell invasion by promoting mesenchymal to epithelial transition, which occurred through inhibition of the MAPK and NFκB signaling pathways.158 Fisetin has also been shown to cause G1 cell cycle arrest and to decrease β-catenin/MITF signaling, leading to inhibition of melanoma cell proliferation and progression.157
The stilbenoid resveratrol is a polyphenol that is found in peanuts, red wine, grape skins, and mulberries.160 In plants, resveratrol is synthesized in response to stess.161 Resveratrol has been shown to induce apoptosis in both A375 and SK-MEL-28 melanoma cell lines.162 Ceramide accumulation and AKT/mTOR pathway inhibition has been shown to be another possible mechanism of apoptosis induction.163 Furthermore, resveratrol has been shown to reduce proliferation and induce apoptosis of melanoma cells by causing down regulation of cyclin D1/cdk4 and increased p53 expression.164,165 In one study, a reduction in invasion was associated with a significant reduction in activator protein-1 (AP-1)/JunD, MMP-1, Bcl-2, and iNOS protein levels.166 Resveratrol treatment has also been shown to suppress invasion and expression of alpha-melanocyte-stimulation hormone (α-MSH) signaling-related molecules, including β-catenin, c-kit, and MITF.167 A syngeneic mouse model of melanoma showed that treatment with resveratrol reduced tumor volume and metastasis, which was thought to occur through reduced AKT expression.168 Evidence also suggests that resveratrol reduces melanoma-induced angiogenesis. In this study, the growth inhibition of vascular endothelial cells in co-culture with melanoma cells was associated with amplified melanoma cell expression of p53, matrix protein TSP1, and inhibition of VEGF production.169 Resveratrol has also shown promise as an adjuvant chemotherapeutic agent. Resveratrol treatment enhanced cisplatin cytotoxicity in a mouse melanoma model, which was believed to occur through increased Connexin 43, a ubiquitous gap junction protein.170 In another study, exposure of melanoma cells to resveratrol inhibited Ref-1-activated AP-1 DNA-binding and endonuclease activities, rendering melanoma cells more sensitive to dacarbazine treatment.171
The polyphenolic flavonoid silymarin is found in milk thistle.172 Silymarin has been shown to inhibit melanoma cell proliferation through suppression of MEK- and RSK-mediated signaling pathways that results in decreased activation of NFκB, AP-1, and STAT3.173 In another study, human melanoma cell lines treated with silymarin showed decreased melanoma cell migration via β-catenin inactivation and reduced MMP-2 and MMP-9 levels.174 Silymarin was also shown to amplify the pro-apoptotic actions of anti-Fas agonistic antibody CH11 in A375-S2 melanoma cells.175
Catechins, which are extracted from green tea leaves, have well-established anti-carcinogenic activity.176 Epigallocatechin gallate (EGCG) is a polyphenol flavonoid that is the most abundant green tea catechin.176 EGCG has been shown to cause significant reduction in melanoma cell growth and increased apoptosis through alterations in the cki-cyclin-cdk network and Bcl-2 family proteins.177 In one study, EGCG resulted in NFκB inhibition, which was associated with reduced melanoma cell interleukin-1beta (IL-1β) secretion, downregulation of the inflammasome component, nuclear localization leucine-rich-repeat protein 1 (NLRP1), and decreased caspase-1 activation.178 Further, both in vitro and in vivo evidence supports the anti-invasive and anti-metastatic actions of EGCG; specifically, it has been shown to reduce cell adhesion179 and decrease cell motility.180 EGCG has also been shown to suppress melanoma cell invasion/migration by targeting epithelial to mesenchymal transition via inhibition of endogenous expression of COX-2 and PGE(2) receptors.181 In an in vivo study, EGCG treatment showed reduced cell spreading, cell-extracellular matrix, and cell-cell interactions, along with inhibition of MMP-9 and focal adhesion kinase (FAK) activities.182 In this same study, the combination of EGCG and dacarbazine inhibited melanoma growth and metastasis significantly when compared to monotherapy.
Curcumin is a polyphenol found in turmeric, a widely used spice. In melanoma, curcumin has been shown to activate apoptosis through p53-independent pathways.183,184 In one study, curcumin treatment resulted in growth inhibition of B16BL6 melanoma cells and p53 independent down regulation of phosphatase of regenerating liver-3.183 Similarly, curcumin was shown not to induce p53, but to suppress the NFκB cell survival pathway and the apoptotic inhibitor, X-linked inhibitor of apoptosis protein (XIAP).184 Other mechanisms that have been shown to induce apoptosis in melanoma cell lines after curcumin treatment include down regulation of anti-apoptotic signaling molecules, including Bcl-2,185,186 proliferating cell nuclear antigen (PCNA),186 and induced myeloid leukemia cell differentiation protein (Mcl-1) protein.187 Suppression of NFκB signaling has also been demonstrated in several cell lines.188,189,190,184 Macrophage stimulating 1/hepatocyte growth factor-like (MST1) activation is also believed to play a role.191 The antiproliferative effects of curcumin have been shown to occur through blockade of cyclic nucleotide phosphodiesterases.192 One preclinical study showed that the combination of tamoxifen and curcumin increased phosphatidyl serine flipping, mitochondria depolarization, and reactive oxygen species generation in A375 and G361 melanoma cell lines.193
Lupeol is a phytosterol and triterpene that is found in white cabbages, green peppers, strawberries, olives, mangoes and grapes.194 Tumor growth inhibition after lupeol treatment has also been associated with suppression of Wnt target genes (c-myc and cyclin D1, proliferation markers proliferating cell nuclear antigen and Ki-67) and the invasion marker osteopontin.195 In mouse melanoma models, lupeol was shown to decrease melanoma tumor growth and promote cell cycle arrest.196,197 Lupeol has been shown to induce melanoma cell differentiation in B162F2 melanoma cells.198,199,200 This induction of differentiation has been suggested to occur via activation of the MAPK pathway.201 Lupeol has also been shown to suppress the migration of human melanoma cells by promoting disassembly of the actin cytoskeleton.202
Honokiol is a biphenolic compound from a species of magnolia native to China. It has long been used in traditional Chinese and Japanese medicine.203 Honokiol has been shown to inhibit proliferation in melanoma.204,205 This has been suggested to occur through attenuation of AKT/mTOR and Notch signaling.205 Further, honokiol has been shown to induce apoptosis through interaction with glucose-regulated protein 78, a sensor of endoplasmic reticulum stress.206
Phytochemicals have demonstrated their potential utility in the treatment of metastatic melanoma, and several studies have already shown their potential as adjuvant therapies.170,193 Moreover, phytochemicals are remarkably nontoxic. The proposed mechanisms of these phytochemicals are yet to be fully elucidated, however, it has been proposed that due to their ubiquity in nature, through evolution, phytochemicals inherently possess diverse mechanisms of action.207 Furthermore, development of phytochemical analogs with more specific spectra of activity is also underway. For some of these agents, the ability to achieve physiologically relevant concentrations has also been a challenge. Therefore, further studies are needed to delineate their target molecules, create novel vehicles to improve bioavailability, and/or develop effective analogs.
Conclusions and future directions
The elucidation of melanoma cell signaling pathways and development of cell signaling inhibitors represent a momentous accomplishment in the treatment of metastatic melanoma, and have led to much-needed new treatments. The combination of dabrafenib and trametinib is now the current treatment of choice for metastatic melanoma with BRAF mutations and offers improved survival for these patients. Despite these accomplishments, however, a cure for patients with metastatic melanoma remains a distant goal. Current research is focused on discovering even more effective combined regimens that will lead to inhibition of coexistent melanoma signaling pathways in the hope of stalling or preventing resistance. The discovery of agents, or agent combinations, with superior toxicity profiles is also a major concern. Development of therapies for patients with other mutational aberrations, such as NRAS and c-kit, is also underway. Moreover, phytochemical therapies are on the horizon and have shown promise in preclinical studies; they also show low toxicity to non-neoplastic cells. The clinical utility of these agents will be determined by efforts to characterize their mechanisms, improve bioavailability, and/or develop effective analogs.
Highlights.
Agents targeting members of the MAPK pathway have increased survival in patients with metastatic melanoma
Combined therapy with agents that target members of the MAPK pathway has been shown to be superior to single agent therapy in clinical trials
Studies of agents that target MAPK, PI3K, c-kit, and other signaling pathways are currently being pursued both alone and in combination
Phytochemicals have been shown to inhibit signaling pathways with well-established roles in cell proliferation, survival, invasion and melanomagenesis
Acknowledgments
This work was supported by NIH Grant 1R21CA173043-01A1 to FA.
Abbreviations
- OS
Overall survival
- MAPK
mitogen-activated protein kinase
- RTKs
receptor tyrosine kinases
- PI3K
phosphatidylinositide 3-kinases
- GEFs
guanine nucleotide exchange factors
- GAPs
GTPase-activating proteins
- FTIs
farnesyltransferase inhibitors
- mTOR
mammalian targets of rapamycin
- VEGF
vascular endothelial growth factor
- PDGF
platelet-derived growth factor
- PFS
progression-free survival
- PIP2
phospholipid phosphatidylinositol 4,5-bisphosphate
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- PTEN
phosphatase and tensin homolog
- eIF4E
eukaryotic translation initiation factor
- JAK-STAT
janus kinase-signal transducer and activator of transcription
- MITF
β-catenin/Microphthalmia-associated transcription factor
- NFκB
nuclear factor kappa B
- MMP
matrix metalloproteinase
- AP-1
activator protein-1
- α-MSH
alpha-melanocyte-stimulation hormone
- EGCG
Epigallocatechin gallate
- IL-1β
interleukin-1beta
- NLRP1
nuclear localization leucine-rich-repeat protein 1
- FAK
focal adhesion kinase
- XIAP
X-linked inhibitor of apoptosis protein
- Bcl-2
B-cell CLL/lymphoma 2
- PCNA
proliferating cell nuclear antigen
- Mcl-1
induced myeloid leukemia cell differentiation protein
- MST1
Macrophage stimulating 1/hepatocyte growth factor-like
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: No potential conflicts of interest
References
- 1.Jerant AF, Johnson JT, Sheridan CD, Caffrey TJ. Early detection and treatment of skin cancer. Am Fam Physician. 2000;62:357–368. [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Garibyan L, Fisher DE. How sunlight causes melanoma. Curr Oncol Rep. 2010;12:319–326. doi: 10.1007/s11912-010-0119-y. [DOI] [PubMed] [Google Scholar]
- 4.Balch CM, et al. Final Version of 2009 AJCC Melanoma Staging and Classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flaherty KT. Chemotherapy and targeted therapy combinations in advanced melanoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2006;12:2366s–2370s. doi: 10.1158/1078-0432.CCR-05-2505. [DOI] [PubMed] [Google Scholar]
- 6.Robert C, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109–1117. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 7.Hodi FS, et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Topalian SL, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol Off J Am Soc Clin Oncol. 2014;32:1020–1030. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nikolaou VA, Stratigos AJ, Flaherty KT, Tsao H. Melanoma: new insights and new therapies. J Invest Dermatol. 2012;132:854–863. doi: 10.1038/jid.2011.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sacks DB. The role of scaffold proteins in MEK/ERK signalling. Biochem Soc Trans. 2006;34:833–836. doi: 10.1042/BST0340833. [DOI] [PubMed] [Google Scholar]
- 11.Liang H, Liu T, Chen F, Liu Z, Liu S. A full-length 3D structure for MAPK/ERK kinase 2 (MEK2) Sci China Life Sci. 2011;54:336–341. doi: 10.1007/s11427-011-4156-z. [DOI] [PubMed] [Google Scholar]
- 12.Akinleye A, Furqan M, Mukhi N, Ravella P, Liu D. MEK and the inhibitors: from bench to bedside. J Hematol Oncol J Hematol Oncol. 2013;6:27. doi: 10.1186/1756-8722-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hocker TL, Singh MK, Tsao H. Melanoma genetics and therapeutic approaches in the 21st century: moving from the benchside to the bedside. J Invest Dermatol. 2008;128:2575–2595. doi: 10.1038/jid.2008.226. [DOI] [PubMed] [Google Scholar]
- 14.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J Off Publ Fed Am Soc Exp Biol. 1995;9:726–735. [PubMed] [Google Scholar]
- 15.Steelman LS, et al. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189–218. doi: 10.1038/sj.leu.2403241. [DOI] [PubMed] [Google Scholar]
- 16.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van ’t Veer LJ, et al. N-ras mutations in human cutaneous melanoma from sun-exposed body sites. Mol Cell Biol. 1989;9:3114–3116. doi: 10.1128/mcb.9.7.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eskandarpour M, et al. Suppression of oncogenic NRAS by RNA interference induces apoptosis of human melanoma cells. Int J Cancer J Int Cancer. 2005;115:65–73. doi: 10.1002/ijc.20873. [DOI] [PubMed] [Google Scholar]
- 19.Brose MS, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. [PubMed] [Google Scholar]
- 20.Dahl C, Guldberg P. The genome and epigenome of malignant melanoma. APMIS Acta Pathol Microbiol Immunol Scand. 2007;115:1161–1176. doi: 10.1111/j.1600-0463.2007.apm_855.xml.x. [DOI] [PubMed] [Google Scholar]
- 21.Posch C, Ortiz-Urda S. NRAS mutant melanoma–undrugable? Oncotarget. 2013;4:494–495. doi: 10.18632/oncotarget.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cox AD, Der CJ. Farnesyltransferase inhibitors and cancer treatment: targeting simply Ras? Biochim Biophys Acta. 1997;1333:F51–71. doi: 10.1016/s0304-419x(97)00011-5. [DOI] [PubMed] [Google Scholar]
- 23.Wlodarczyk N, et al. In vitro and in vivo evaluation of two rational-designed nonpeptidic farnesyltransferase inhibitors on HT29 human colon cancer cell lines. Oncol Res. 2005;16:107–118. doi: 10.3727/000000006783981170. [DOI] [PubMed] [Google Scholar]
- 24.Gajewski TF, et al. Phase II study of the farnesyltransferase inhibitor R115777 in advanced melanoma (CALGB 500104) J Transl Med. 2012;10:246. doi: 10.1186/1479-5876-10-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sebti SM, Der CJ. Opinion: Searching for the elusive targets of farnesyltransferase inhibitors. Nat Rev Cancer. 2003;3:945–951. doi: 10.1038/nrc1234. [DOI] [PubMed] [Google Scholar]
- 26.Whyte DB, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 27.Smalley KSM, Eisen TG. Farnesyl transferase inhibitor SCH66336 is cytostatic, pro-apoptotic and enhances chemosensitivity to cisplatin in melanoma cells. Int J Cancer J Int Cancer. 2003;105:165–175. doi: 10.1002/ijc.11064. [DOI] [PubMed] [Google Scholar]
- 28.Morgillo F, Lee HY. Lonafarnib in cancer therapy. Expert Opin Investig Drugs. 2006;15:709–719. doi: 10.1517/13543784.15.6.709. [DOI] [PubMed] [Google Scholar]
- 29.Niessner H, et al. The farnesyl transferase inhibitor lonafarnib inhibits mTOR signaling and enforces sorafenib-induced apoptosis in melanoma cells. J Invest Dermatol. 2011;131:468–479. doi: 10.1038/jid.2010.297. [DOI] [PubMed] [Google Scholar]
- 30.Hong DS, et al. Phase I trial of a combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in advanced malignancies. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15:7061–7068. doi: 10.1158/1078-0432.CCR-09-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hatzivassiliou G, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 32.Heidorn SJ, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thumar J, Shahbazian D, Aziz SA, Jilaveanu LB, Kluger HM. MEK targeting in N-RAS mutated metastatic melanoma. Mol Cancer. 2014;13:45. doi: 10.1186/1476-4598-13-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ascierto PA, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14:249–256. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- 35.Kaplan FM, Shao Y, Mayberry MM, Aplin AE. Hyperactivation of MEK-ERK1/2 signaling and resistance to apoptosis induced by the oncogenic B-RAF inhibitor, PLX4720, in mutant N-RAS melanoma cells. Oncogene. 2011;30:366–371. doi: 10.1038/onc.2010.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halaban R, et al. PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment Cell Melanoma Res. 2010;23:190–200. doi: 10.1111/j.1755-148X.2010.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roskoski R. RAF protein-serine/threonine kinases: structure and regulation. Biochem Biophys Res Commun. 2010;399:313–317. doi: 10.1016/j.bbrc.2010.07.092. [DOI] [PubMed] [Google Scholar]
- 38.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 39.Karasarides M, et al. B-RAF is a therapeutic target in melanoma. Oncogene. 2004;23:6292–6298. doi: 10.1038/sj.onc.1207785. [DOI] [PubMed] [Google Scholar]
- 40.Hoeflich KP, et al. Oncogenic BRAF is required for tumor growth and maintenance in melanoma models. Cancer Res. 2006;66:999–1006. doi: 10.1158/0008-5472.CAN-05-2720. [DOI] [PubMed] [Google Scholar]
- 41.Pollock PM, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 42.Sharpless NE, DePinho RA. Cancer: crime and punishment. Nature. 2005;436:636–637. doi: 10.1038/436636a. [DOI] [PubMed] [Google Scholar]
- 43.Wilhelm SM, et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 44.Eisen T, et al. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer. 2006;95:581–586. doi: 10.1038/sj.bjc.6603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hauschild A, et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J Clin Oncol Off J Am Soc Clin Oncol. 2009;27:2823–2830. doi: 10.1200/JCO.2007.15.7636. [DOI] [PubMed] [Google Scholar]
- 46.Flaherty KT, et al. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31:373–379. doi: 10.1200/JCO.2012.42.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whittaker S, et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci Transl Med. 2010;2:35ra41. doi: 10.1126/scitranslmed.3000758. [DOI] [PubMed] [Google Scholar]
- 48.Tsai J, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joseph EW, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci U S A. 2010;107:14903–14908. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flaherty KT, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sosman JA, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McArthur GA, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323–332. doi: 10.1016/S1470-2045(14)70012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Falchook GS, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379:1893–1901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ascierto PA, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31:3205–3211. doi: 10.1200/JCO.2013.49.8691. [DOI] [PubMed] [Google Scholar]
- 55.Hauschild A, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 56.Salama AKS, Flaherty KT. BRAF in melanoma: current strategies and future directions. Clin Cancer Res Off J Am Assoc Cancer Res. 2013;19:4326–4334. doi: 10.1158/1078-0432.CCR-13-0779. [DOI] [PubMed] [Google Scholar]
- 57.Montagut C, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–4861. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corcoran RB, et al. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3:ra84. doi: 10.1126/scisignal.2001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nazarian R, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wagle N, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol Off J Am Soc Clin Oncol. 2011;29:3085–3096. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Villanueva J, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poulikakos PI, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Poulikakos PI, Zhang C, Bollag G, Shokat KM. Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Su F, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–215. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Le K, Blomain ES, Rodeck U, Aplin AE. Selective RAF inhibitor impairs ERK1/2 phosphorylation and growth in mutant NRAS, vemurafenib-resistant melanoma cells. Pigment Cell Melanoma Res. 2013;26:509–517. doi: 10.1111/pcmr.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klein O, et al. BRAF inhibitor activity in V600R metastatic melanoma. Eur J Cancer Oxf Engl 1990. 2013;49:1073–1079. doi: 10.1016/j.ejca.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 67.Shi H, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Paraiso KHT, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102:1724–1730. doi: 10.1038/sj.bjc.6605714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Solit DB, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang YF, et al. Apoptosis induction in human melanoma cells by inhibition of MEK is caspase-independent and mediated by the Bcl-2 family members PUMA, Bim, and Mcl-1. Clin Cancer Res Off J Am Assoc Cancer Res. 2007;13:4934–4942. doi: 10.1158/1078-0432.CCR-07-0665. [DOI] [PubMed] [Google Scholar]
- 71.Wallace EM, Lyssikatos JP, Yeh T, Winkler JD, Koch K. Progress towards therapeutic small molecule MEK inhibitors for use in cancer therapy. Curr Top Med Chem. 2005;5:215–229. doi: 10.2174/1568026053507723. [DOI] [PubMed] [Google Scholar]
- 72.Adjei AA, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol Off J Am Soc Clin Oncol. 2008;26:2139–2146. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yeh TC, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res Off J Am Assoc Cancer Res. 2007;13:1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 74.Haass NK, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res Off J Am Assoc Cancer Res. 2008;14:230–239. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 75.Friday BB, et al. BRAF V600E disrupts AZD6244-induced abrogation of negative feedback pathways between extracellular signal-regulated kinase and Raf proteins. Cancer Res. 2008;68:6145–6153. doi: 10.1158/0008-5472.CAN-08-1430. [DOI] [PubMed] [Google Scholar]
- 76.Madhunapantula SV, Robertson GP. Is B-Raf a good therapeutic target for melanoma and other malignancies? Cancer Res. 2008;68:5–8. doi: 10.1158/0008-5472.CAN-07-2038. [DOI] [PubMed] [Google Scholar]
- 77.Kirkwood JM, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2012;18:555–567. doi: 10.1158/1078-0432.CCR-11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Robert C, et al. Selumetinib plus dacarbazine versus placebo plus dacarbazine as first-line treatment for BRAF-mutant metastatic melanoma: a phase 2 double-blind randomised study. Lancet Oncol. 2013;14:733–740. doi: 10.1016/S1470-2045(13)70237-7. [DOI] [PubMed] [Google Scholar]
- 79.Gilmartin AG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res Off J Am Assoc Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 80.Falchook GS, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–789. doi: 10.1016/S1470-2045(12)70269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim KB, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31:482–489. doi: 10.1200/JCO.2012.43.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Flaherty KT, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 83.Hatzivassiliou G, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501:232–236. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- 84.Lito P, et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25:697–710. doi: 10.1016/j.ccr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.LoRusso PM, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res Off J Am Assoc Cancer Res. 2010;16:1924–1937. doi: 10.1158/1078-0432.CCR-09-1883. [DOI] [PubMed] [Google Scholar]
- 86.Von Euw E, et al. Antitumor effects of the investigational selective MEK inhibitor TAK733 against cutaneous and uveal melanoma cell lines. Mol Cancer. 2012;11:22. doi: 10.1186/1476-4598-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zimmer L, et al. Phase I expansion and pharmacodynamic study of the oral MEK inhibitor RO4987655 (CH4987655) in selected patients with advanced cancer with RAS-RAF mutations. Clin Cancer Res Off J Am Assoc Cancer Res. 2014;20:4251–4261. doi: 10.1158/1078-0432.CCR-14-0341. [DOI] [PubMed] [Google Scholar]
- 88.King AJ, et al. Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions. PloS One. 2013;8:e67583. doi: 10.1371/journal.pone.0067583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Flaherty KT, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Long GV, et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N Engl J Med. 2014 doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 91.Ribas A, et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAF(V600)-mutated melanoma: a phase 1b study. Lancet Oncol. 2014;15:954–965. doi: 10.1016/S1470-2045(14)70301-8. [DOI] [PubMed] [Google Scholar]
- 92.Gowrishankar K, et al. Acquired resistance to BRAF inhibition can confer cross-resistance to combined BRAF/MEK inhibition. J Invest Dermatol. 2012;132:1850–1859. doi: 10.1038/jid.2012.63. [DOI] [PubMed] [Google Scholar]
- 93.Goldinger SM, et al. Upstream mitogen-activated protein kinase (MAPK) pathway inhibition: MEK inhibitor followed by a BRAF inhibitor in advanced melanoma patients. Eur J Cancer Oxf Engl 1990. 2014;50:406–410. doi: 10.1016/j.ejca.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 94.Honda K, et al. Phase I and pharmacokinetic/pharmacodynamic study of RO5126766, a first-in-class dual Raf/MEK inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;72:577–584. doi: 10.1007/s00280-013-2228-4. [DOI] [PubMed] [Google Scholar]
- 95.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 96.Davies MA. The role of the PI3K-AKT pathway in melanoma. Cancer J Sudbury Mass. 2012;18:142–147. doi: 10.1097/PPO.0b013e31824d448c. [DOI] [PubMed] [Google Scholar]
- 97.Chudnovsky Y, Adams AE, Robbins PB, Lin Q, Khavari PA. Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nat Genet. 2005;37:745–749. doi: 10.1038/ng1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 99.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Luo J, et al. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab. 2006;3:355–366. doi: 10.1016/j.cmet.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 101.Brunet A, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 102.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998;12:502–513. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Scott PH, Brunn GJ, Kohn AD, Roth RA, Lawrence JC. Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1998;95:7772–7777. doi: 10.1073/pnas.95.13.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bellacosa A, et al. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 106.Jacinto E, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 107.Laplante M, Sabatini D. M mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 109.Slipicevic A, et al. Expression of activated Akt and PTEN in malignant melanomas: relationship with clinical outcome. Am J Clin Pathol. 2005;124:528–536. doi: 10.1309/YT58WWMTA6YR1PRV. [DOI] [PubMed] [Google Scholar]
- 110.Karbowniczek M, Spittle CS, Morrison T, Wu H, Henske E. P mTOR is activated in the majority of malignant melanomas. J Invest Dermatol. 2008;128:980–987. doi: 10.1038/sj.jid.5701074. [DOI] [PubMed] [Google Scholar]
- 111.Dai DL, Martinka M, Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J Clin Oncol Off J Am Soc Clin Oncol. 2005;23:1473–1482. doi: 10.1200/JCO.2005.07.168. [DOI] [PubMed] [Google Scholar]
- 112.Restuccia DF, Hemmings BA. From man to mouse and back again: advances in defining tumor AKTivities in vivo. Dis Model Mech. 2010;3:705–720. doi: 10.1242/dmm.004671. [DOI] [PubMed] [Google Scholar]
- 113.Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004;122:337–341. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dankort D, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rowinsky EK. Targeting the molecular target of rapamycin (mTOR) Curr Opin Oncol. 2004;16:564–575. doi: 10.1097/01.cco.0000143964.74936.d1. [DOI] [PubMed] [Google Scholar]
- 116.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway–beyond rapalogs. Oncotarget. 2010;1:530–543. doi: 10.18632/oncotarget.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Margolin K, et al. CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer. 2005;104:1045–1048. doi: 10.1002/cncr.21265. [DOI] [PubMed] [Google Scholar]
- 119.Davies MA, et al. Phase I study of the combination of sorafenib and temsirolimus in patients with metastatic melanoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2012;18:1120–1128. doi: 10.1158/1078-0432.CCR-11-2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Margolin KA, et al. Randomized phase II trial of sorafenib with temsirolimus or tipifarnib in untreated metastatic melanoma (S0438) Clin Cancer Res Off J Am Assoc Cancer Res. 2012;18:1129–1137. doi: 10.1158/1078-0432.CCR-11-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Carracedo A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.O’Reilly KE, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther. 2005;4:1533–1540. doi: 10.1158/1535-7163.MCT-05-0068. [DOI] [PubMed] [Google Scholar]
- 124.Ogita S, Lorusso P. Targeting phosphatidylinositol 3 kinase (PI3K)-Akt beyond rapalogs. Target Oncol. 2011;6:103–117. doi: 10.1007/s11523-011-0176-7. [DOI] [PubMed] [Google Scholar]
- 125.Gopal YNV, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010;70:8736–8747. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Maira SM, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 127.Marone R, et al. Targeting melanoma with dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitors. Mol Cancer Res MCR. 2009;7:601–613. doi: 10.1158/1541-7786.MCR-08-0366. [DOI] [PubMed] [Google Scholar]
- 128.Aziz SA, et al. Vertical targeting of the phosphatidylinositol-3 kinase pathway as a strategy for treating melanoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2010;16:6029–6039. doi: 10.1158/1078-0432.CCR-10-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Greger JG, et al. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012;11:909–920. doi: 10.1158/1535-7163.MCT-11-0989. [DOI] [PubMed] [Google Scholar]
- 130.Atefi M, et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PloS One. 2011;6:e28973. doi: 10.1371/journal.pone.0028973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Paraiso KHT, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–2760. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cheung M, Sharma A, Madhunapantula SV, Robertson GP. Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res. 2008;68:3429–3439. doi: 10.1158/0008-5472.CAN-07-5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jiang CC, et al. MEK-independent survival of B-RAFV600E melanoma cells selected for resistance to apoptosis induced by the RAF inhibitor PLX4720. Clin Cancer Res Off J Am Assoc Cancer Res. 2011;17:721–730. doi: 10.1158/1078-0432.CCR-10-2225. [DOI] [PubMed] [Google Scholar]
- 134.Roberts PJ, et al. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin Cancer Res Off J Am Assoc Cancer Res. 2012;18:5290–5303. doi: 10.1158/1078-0432.CCR-12-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lassen A, et al. Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Mol Cancer. 2014;13:83. doi: 10.1186/1476-4598-13-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Todd JR, Scurr LL, Becker TM, Kefford RF, Rizos H. The MAPK pathway functions as a redundant survival signal that reinforces the PI3K cascade in c-Kit mutant melanoma. Oncogene. 2014;33:236–245. doi: 10.1038/onc.2012.562. [DOI] [PubMed] [Google Scholar]
- 137.Bourillon A, et al. Genetic variation at KIT locus may predispose to melanoma. Pigment Cell Melanoma Res. 2013;26:88–96. doi: 10.1111/pcmr.12032. [DOI] [PubMed] [Google Scholar]
- 138.Beadling C, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res Off J Am Assoc Cancer Res. 2008;14:6821–6828. doi: 10.1158/1078-0432.CCR-08-0575. [DOI] [PubMed] [Google Scholar]
- 139.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol Off J Am Soc Clin Oncol. 2006;24:4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 140.Curtin JA, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 141.Hodi FS, et al. Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol Off J Am Soc Clin Oncol. 2008;26:2046–2051. doi: 10.1200/JCO.2007.14.0707. [DOI] [PubMed] [Google Scholar]
- 142.Lutzky J, Bauer J, Bastian BC. Dose-dependent, complete response to imatinib of a metastatic mucosal melanoma with a K642E KIT mutation. Pigment Cell Melanoma Res. 2008;21:492–493. doi: 10.1111/j.1755-148X.2008.00475.x. [DOI] [PubMed] [Google Scholar]
- 143.Brown MC, Casasola RJ. Complete response in a melanoma patient treated with imatinib. J Laryngol Otol. 2012;126:638–640. doi: 10.1017/S0022215112000527. [DOI] [PubMed] [Google Scholar]
- 144.Jiang X, et al. Imatinib targeting of KIT-mutant oncoprotein in melanoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2008;14:7726–7732. doi: 10.1158/1078-0432.CCR-08-1144. [DOI] [PubMed] [Google Scholar]
- 145.Wyman K, et al. Multicenter Phase II trial of high-dose imatinib mesylate in metastatic melanoma: significant toxicity with no clinical efficacy. Cancer. 2006;106:2005–2011. doi: 10.1002/cncr.21834. [DOI] [PubMed] [Google Scholar]
- 146.Ugurel S, et al. Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer. 2005;92:1398–1405. doi: 10.1038/sj.bjc.6602529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kim KB, et al. Phase II trial of imatinib mesylate in patients with metastatic melanoma. Br J Cancer. 2008;99:734–740. doi: 10.1038/sj.bjc.6604482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Carvajal RD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA J Am Med Assoc. 2011;305:2327–2334. doi: 10.1001/jama.2011.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Guo J, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol Off J Am Soc Clin Oncol. 2011;29:2904–2909. doi: 10.1200/JCO.2010.33.9275. [DOI] [PubMed] [Google Scholar]
- 150.Woodman SE, et al. Activity of dasatinib against L576P KIT mutant melanoma: molecular, cellular, and clinical correlates. Mol Cancer Ther. 2009;8:2079–2085. doi: 10.1158/1535-7163.MCT-09-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kluger HM, et al. A phase 2 trial of dasatinib in advanced melanoma. Cancer. 2011;117:2202–2208. doi: 10.1002/cncr.25766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cho JH, Kim KM, Kwon M, Kim JH, Lee J. Nilotinib in patients with metastatic melanoma harboring KIT gene aberration. Invest New Drugs. 2012;30:2008–2014. doi: 10.1007/s10637-011-9763-9. [DOI] [PubMed] [Google Scholar]
- 153.Afaq F, Adhami VM, Mukhtar H. Photochemoprevention of ultraviolet B signaling and photocarcinogenesis. Mutat Res. 2005;571:153–173. doi: 10.1016/j.mrfmmm.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 154.Surh Y-J. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 155.Moon YJ, Wang X, Morris ME. Dietary flavonoids: effects on xenobiotic and carcinogen metabolism. Toxicol Vitro Int J Publ Assoc BIBRA. 2006;20:187–210. doi: 10.1016/j.tiv.2005.06.048. [DOI] [PubMed] [Google Scholar]
- 156.Khan N, Syed DN, Ahmad N, Mukhtar H. Fisetin: a dietary antioxidant for health promotion. Antioxid Redox Signal. 2013;19:151–162. doi: 10.1089/ars.2012.4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Syed DN, et al. Inhibition of human melanoma cell growth by the dietary flavonoid fisetin is associated with disruption of Wnt/β-catenin signaling and decreased Mitf levels. J Invest Dermatol. 2011;131:1291–1299. doi: 10.1038/jid.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pal HC, et al. Fisetin inhibits human melanoma cell invasion through promotion of mesenchymal to epithelial transition and by targeting MAPK and NFκB signaling pathways. PloS One. 2014;9:e86338. doi: 10.1371/journal.pone.0086338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Syed DN, et al. Fisetin inhibits human melanoma cell growth through direct binding to p70S6K and mTOR: findings from 3-D melanoma skin equivalents and computational modeling. Biochem Pharmacol. 2014;89:349–360. doi: 10.1016/j.bcp.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zheng YY, Viswanathan B, Kesarwani P, Mehrotra S. Dietary agents in cancer prevention: an immunological perspective. Photochem Photobiol. 2012;88:1083–1098. doi: 10.1111/j.1751-1097.2012.01128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Soleas GJ, Diamandis EP, Goldberg D. M Resveratrol: a molecule whose time has come? And gone? Clin Biochem. 1997;30:91–113. doi: 10.1016/s0009-9120(96)00155-5. [DOI] [PubMed] [Google Scholar]
- 162.Niles RM, et al. Resveratrol is a potent inducer of apoptosis in human melanoma cells. Cancer Lett. 2003;190:157–163. doi: 10.1016/s0304-3835(02)00676-6. [DOI] [PubMed] [Google Scholar]
- 163.Wang M, et al. Resveratrol triggers protective autophagy through the ceramide/Akt/mTOR pathway in melanoma B16 cells. Nutr Cancer. 2014;66:435–440. doi: 10.1080/01635581.2013.878738. [DOI] [PubMed] [Google Scholar]
- 164.Gatouillat G, Balasse E, Joseph-Pietras D, Morjani H, Madoulet C. Resveratrol induces cell-cycle disruption and apoptosis in chemoresistant B16 melanoma. J Cell Biochem. 2010;110:893–902. doi: 10.1002/jcb.22601. [DOI] [PubMed] [Google Scholar]
- 165.Hsieh T, Wang Z, Hamby CV, Wu JM. Inhibition of melanoma cell proliferation by resveratrol is correlated with upregulation of quinone reductase 2 and p53. Biochem Biophys Res Commun. 2005;334:223–230. doi: 10.1016/j.bbrc.2005.06.073. [DOI] [PubMed] [Google Scholar]
- 166.Yang Z, et al. Nitric oxide initiates progression of human melanoma via a feedback loop mediated by apurinic/apyrimidinic endonuclease-1/redox factor-1, which is inhibited by resveratrol. Mol Cancer Ther. 2008;7:3751–3760. doi: 10.1158/1535-7163.MCT-08-0562. [DOI] [PubMed] [Google Scholar]
- 167.Chen Y-J, Chen Y-Y, Lin Y-F, Hu H-Y, Liao H-F. Resveratrol inhibits alpha-melanocyte-stimulating hormone signaling, viability, and invasiveness in melanoma cells. Evid-Based Complement Altern Med ECAM. 2013;2013:632121. doi: 10.1155/2013/632121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bhattacharya S, Darjatmoko SR, Polans AS. Resveratrol modulates the malignant properties of cutaneous melanoma through changes in the activation and attenuation of the antiapoptotic protooncogenic protein Akt/PKB. Melanoma Res. 2011;21:180–187. doi: 10.1097/CMR.0b013e3283456dfc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Trapp V, Parmakhtiar B, Papazian V, Willmott L, Fruehauf JP. Anti-angiogenic effects of resveratrol mediated by decreased VEGF and increased TSP1 expression in melanoma-endothelial cell co-culture. Angiogenesis. 2010;13:305–315. doi: 10.1007/s10456-010-9187-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cheng Y-J, et al. Resveratrol enhances chemosensitivity in mouse melanoma model through connexin 43 upregulation. Environ Toxicol. 2014 doi: 10.1002/tox.21952. [DOI] [PubMed] [Google Scholar]
- 171.Yang S, Irani K, Heffron SE, Jurnak F, Meyskens FL. Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE/Ref-1) in human melanoma and identification of the therapeutic potential of resveratrol as an APE/Ref-1 inhibitor. Mol Cancer Ther. 2005;4:1923–1935. doi: 10.1158/1535-7163.MCT-05-0229. [DOI] [PubMed] [Google Scholar]
- 172.Milić N, Milosević N, Suvajdzić L, Zarkov M, Abenavoli L. New therapeutic potentials of milk thistle (Silybum marianum) Nat Prod Commun. 2013;8:1801–1810. [PubMed] [Google Scholar]
- 173.Lee M-H, et al. Direct targeting of MEK1/2 and RSK2 by silybin induces cell-cycle arrest and inhibits melanoma cell growth. Cancer Prev Res Phila Pa. 2013;6:455–465. doi: 10.1158/1940-6207.CAPR-12-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Vaid M, Prasad R, Sun Q, Katiyar S. K Silymarin targets β-catenin signaling in blocking migration/invasion of human melanoma cells. PloS One. 2011;6:e23000. doi: 10.1371/journal.pone.0023000. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 175.Li L-H, et al. Silymarin enhanced cytotoxic effect of anti-Fas agonistic antibody CH11 on A375-S2 cells. J Asian Nat Prod Res. 2007;9:593–602. doi: 10.1080/10286020600882502. [DOI] [PubMed] [Google Scholar]
- 176.Yang CS, Wang X, Lu G, Picinich SC. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat Rev Cancer. 2009;9:429–439. doi: 10.1038/nrc2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nihal M, Ahmad N, Mukhtar H, Wood GS. Anti-proliferative and proapoptotic effects of (−)-epigallocatechin-3-gallate on human melanoma: possible implications for the chemoprevention of melanoma. Int J Cancer J Int Cancer. 2005;114:513–521. doi: 10.1002/ijc.20785. [DOI] [PubMed] [Google Scholar]
- 178.Ellis LZ, et al. Green tea polyphenol epigallocatechin-3-gallate suppresses melanoma growth by inhibiting inflammasome and IL-1β secretion. Biochem Biophys Res Commun. 2011;414:551–556. doi: 10.1016/j.bbrc.2011.09.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Suzuki Y, Isemura M. Inhibitory effect of epigallocatechin gallate on adhesion of murine melanoma cells to laminin. Cancer Lett. 2001;173:15–20. doi: 10.1016/s0304-3835(01)00685-1. [DOI] [PubMed] [Google Scholar]
- 180.Watanabe T, et al. Higher cell stiffness indicating lower metastatic potential in B16 melanoma cell variants and in (−)-epigallocatechin gallate-treated cells. J Cancer Res Clin Oncol. 2012 doi: 10.1007/s00432-012-1159-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Singh T, Katiyar SK. Green tea catechins reduce invasive potential of human melanoma cells by targeting COX-2, PGE2 receptors and epithelial-to-mesenchymal transition. PloS One. 2011;6:e25224. doi: 10.1371/journal.pone.0025224. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 182.Liu JD, Chen SH, Lin CL, Tsai SH, Liang YC. Inhibition of melanoma growth and metastasis by combination with (−)-epigallocatechin-3-gallate and dacarbazine in mice. J Cell Biochem. 2001;83:631–642. doi: 10.1002/jcb.1261. [DOI] [PubMed] [Google Scholar]
- 183.Wang L, et al. An anticancer effect of curcumin mediated by down-regulating phosphatase of regenerating liver-3 expression on highly metastatic melanoma cells. Mol Pharmacol. 2009;76:1238–1245. doi: 10.1124/mol.109.059105. [DOI] [PubMed] [Google Scholar]
- 184.Bush JA, Cheung KJ, Li G. Curcumin induces apoptosis in human melanoma cells through a Fas receptor/caspase-8 pathway independent of p53. Exp Cell Res. 2001;271:305–314. doi: 10.1006/excr.2001.5381. [DOI] [PubMed] [Google Scholar]
- 185.Bill MA, et al. Curcumin induces proapoptotic effects against human melanoma cells and modulates the cellular response to immunotherapeutic cytokines. Mol Cancer Ther. 2009;8:2726–2735. doi: 10.1158/1535-7163.MCT-09-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Dahmke IN, et al. Curcumin intake affects miRNA signature in murine melanoma with mmu-miR-205-5p most significantly altered. PloS One. 2013;8:e81122. doi: 10.1371/journal.pone.0081122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bakhshi J, et al. Coupling endoplasmic reticulum stress to the cell death program in mouse melanoma cells: effect of curcumin. Apoptosis Int J Program Cell Death. 2008;13:904–914. doi: 10.1007/s10495-008-0221-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zheng M, Ekmekcioglu S, Walch ET, Tang CH, Grimm EA. Inhibition of nuclear factor-kappaB and nitric oxide by curcumin induces G2/M cell cycle arrest and apoptosis in human melanoma cells. Melanoma Res. 2004;14:165–171. doi: 10.1097/01.cmr.0000129374.76399.19. [DOI] [PubMed] [Google Scholar]
- 189.Marín YE, et al. Curcumin downregulates the constitutive activity of NF-kappaB and induces apoptosis in novel mouse melanoma cells. Melanoma Res. 2007;17:274–283. doi: 10.1097/CMR.0b013e3282ed3d0e. [DOI] [PubMed] [Google Scholar]
- 190.Siwak DR, Shishodia S, Aggarwal BB, Kurzrock R. Curcumin-induced antiproliferative and proapoptotic effects in melanoma cells are associated with suppression of IkappaB kinase and nuclear factor kappaB activity and are independent of the B-Raf/mitogen-activated/extracellular signal-regulated protein kinase pathway and the Akt pathway. Cancer. 2005;104:879–890. doi: 10.1002/cncr.21216. [DOI] [PubMed] [Google Scholar]
- 191.Yu T, Ji J, Guo Y. MST1 activation by curcumin mediates JNK activation, Foxo3a nuclear translocation and apoptosis in melanoma cells. Biochem Biophys Res Commun. 2013;441:53–58. doi: 10.1016/j.bbrc.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 192.Abusnina A, Keravis T, Yougbaré I, Bronner C, Lugnier C. Anti-proliferative effect of curcumin on melanoma cells is mediated by PDE1A inhibition that regulates the epigenetic integrator UHRF1. Mol Nutr Food Res. 2011;55:1677–1689. doi: 10.1002/mnfr.201100307. [DOI] [PubMed] [Google Scholar]
- 193.Chatterjee SJ, Pandey S. Chemo-resistant melanoma sensitized by tamoxifen to low dose curcumin treatment through induction of apoptosis and autophagy. Cancer Biol Ther. 2011;11:216–228. doi: 10.4161/cbt.11.2.13798. [DOI] [PubMed] [Google Scholar]
- 194.Saleem M. Lupeol, a novel anti-inflammatory and anti-cancer dietary triterpene. Cancer Lett. 2009;285:109–115. doi: 10.1016/j.canlet.2009.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tarapore RS, et al. Specific targeting of Wnt/β-catenin signaling in human melanoma cells by a dietary triterpene lupeol. Carcinogenesis. 2010;31:1844–1853. doi: 10.1093/carcin/bgq169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Saleem M, et al. Lupeol inhibits growth of highly aggressive human metastatic melanoma cells in vitro and in vivo by inducing apoptosis. Clin Cancer Res Off J Am Assoc Cancer Res. 2008;14:2119–2127. doi: 10.1158/1078-0432.CCR-07-4413. [DOI] [PubMed] [Google Scholar]
- 197.Nitta M, et al. Systemic and local injections of lupeol inhibit tumor growth in a melanoma-bearing mouse model. Biomed Rep. 2013;1:641–645. doi: 10.3892/br.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hata K, Ishikawa K, Hori K, Konishi T. Differentiation-inducing activity of lupeol, a lupane-type triterpene from Chinese dandelion root (Hokouei-kon), on a mouse melanoma cell line. Biol Pharm Bull. 2000;23:962–967. doi: 10.1248/bpb.23.962. [DOI] [PubMed] [Google Scholar]
- 199.Hata K, et al. Differentiation-inducing activity of lupane triterpenes on a mouse melanoma cell line. Cytotechnology. 2006;52:151–158. doi: 10.1007/s10616-007-9069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ogiwara K, Hata K. Melanoma cell differentiation induced by lupeol separates into two stages: morphological and functional changes. J Nat Med. 2009;63:323–326. doi: 10.1007/s11418-009-0319-7. [DOI] [PubMed] [Google Scholar]
- 201.Hata K, Hori K, Takahashi S. Role of p38 MAPK in lupeol-induced B16 2F2 mouse melanoma cell differentiation. J Biochem (Tokyo) 2003;134:441–445. doi: 10.1093/jb/mvg162. [DOI] [PubMed] [Google Scholar]
- 202.Hata K, Hori K, Murata J, Takahashi S. Remodeling of actin cytoskeleton in lupeol-induced B16 2F2 cell differentiation. J Biochem (Tokyo) 2005;138:467–472. doi: 10.1093/jb/mvi151. [DOI] [PubMed] [Google Scholar]
- 203.Ponnurangam S, et al. Honokiol in combination with radiation targets notch signaling to inhibit colon cancer stem cells. Mol Cancer Ther. 2012;11:963–972. doi: 10.1158/1535-7163.MCT-11-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mannal PW, Schneider J, Tangada A, McDonald D, McFadden DW. Honokiol produces anti-neoplastic effects on melanoma cells in vitro. J Surg Oncol. 2011;104:260–264. doi: 10.1002/jso.21936. [DOI] [PubMed] [Google Scholar]
- 205.Kaushik G, et al. Honokiol induces cytotoxic and cytostatic effects in malignant melanoma cancer cells. Am J Surg. 2012;204:868–873. doi: 10.1016/j.amjsurg.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Martin S, et al. Inducing apoptosis of cancer cells using small-molecule plant compounds that bind to GRP78. Br J Cancer. 2013;109:433–443. doi: 10.1038/bjc.2013.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Arbiser JL, Fisher DE. Fisetin: a natural fist against melanoma? J Invest Dermatol. 2011;131:1187–1189. doi: 10.1038/jid.2011.39. [DOI] [PubMed] [Google Scholar]