

SUMMARY
During viral RNA synthesis, Ebola virus (EBOV) nucleoprotein (NP) alternates between an RNA template-bound form and a template-free form to provide the viral polymerase access to the RNA template. In addition, newly synthesized NP must be prevented from indiscriminately binding to noncognate RNAs. Here we investigate the molecular bases for these critical processes. We identify an intrinsically disordered peptide derived from EBOV VP35 (NPBP) that binds NP with high affinity and specificity, inhibits NP oligomerization, and releases RNA from NP-RNA complexes in vitro. The structure of the NPBP/ΔNPNTD complex, solved to 3.7 Å resolution, reveals how NPBP peptide occludes a large surface area that is important for NP-NP and NP-RNA interactions and for viral RNA synthesis. Together, our results identify a highly conserved viral interface that is important for EBOV replication and can be targeted for therapeutic development.
Keywords: Ebola virus, RNA virus replication, nucleocapsid protein, antiviral peptides
Graphical Abstract
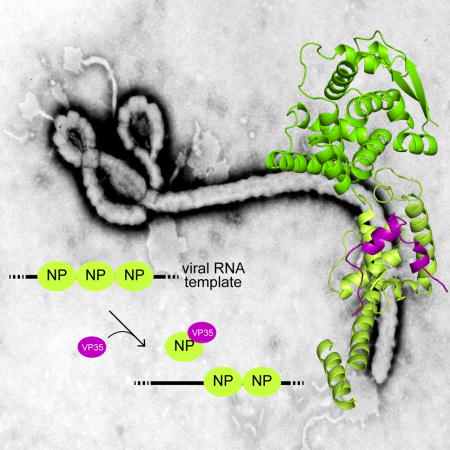
INTRODUCTION
Ebolaviruses and marburgviruses are nonsegmented negative sense RNA viruses (NNSVs) that cause severe hemorrhagic fever (Sanchez et al., 2006). Because of the severity of filovirus disease, which is associated with case fatality rates approaching 90% during some outbreaks, ebolaviruses and marburgviruses remain significant threats to global human health. The recent epidemic in Western Africa caused by Zaire ebolavirus (EBOV) is the largest filovirus outbreak on record, and the subsequent import of EBOV to non-African countries highlights the public health impact of these zoonotic pathogens.
Like other non-segmented negative strand virus (NNSV) family members, EBOV has a single stranded RNA genome that undergoes transcription upon entry into the host cytosol prior to the generation of viral proteins (Muhlberger, 2007; Sanchez et al., 2006). The roughly 19 kb ebolavirus genome has seven genes that encode for at least 8 distinct translation products. (Sanchez and Kiley, 1987; Sanchez et al., 1993; Sanchez et al., 2007; Shabman et al., 2014). Viral genome replication and transcription of individual genes into distinct 5′-capped, 3′-polyadenylated monocistronic mRNAs are carried out by the viral RNA-dependent RNA polymerase complex (RdRp) (Muhlberger, 2007). This complex consists of the enzymatic component of the RDRP, the large protein (L), along with the viral nucleoprotein (NP), viral protein (VP)35 and VP30 and acts upon the nucleocapsid, which consists of single-stranded (ss) viral genomic and antigenomic RNAs (Muhlberger, 2007) that are encapsidated by NP. The EBOV core nucleocapsid protects ssRNAs from degradation, similar to that observed for paramyxoviruses and rhabdoviruses (Masters and Banerjee, 1988).
Based on sequence homology, the filovirus NP has a paramyxovirus/filovirus conserved region spanning residues 1 to 450 and a region unique to filoviruses from 451 to 739 (Shi et al., 2008; Watanabe et al., 2006) (Figure 1A and S1A). The N-terminal region mediates NP self-association and forms helical tube-like structures in vitro (Watanabe et al., 2006) and in cells (Huang et al., 2002). Mapping studies indicate that the NP C-terminus contributes to NP-VP40 interaction and NP incorporation into ebolavirus-like particles (Noda et al., 2007). Recently, a crystal structure of the EBOV C-terminal residues encompassing 641–739 was described (Dziubanska et al., 2014), but how this structure contributes to EBOV infection is unknown.
Figure 1. The first 25 residues of the EBOV NP are important for oligomerization and replication, related to Figure S1.

(A) Domain organization of EBOV NP protein, including regions identified in this study. Size exclusion chromatography results for NPNTD (residue 1–457, blue) and ΔNPNTD (residue 25–457, green) in (B) 150 mM NaCl and (C) 500 mM NaCl buffer. (D) Representative chromatograms for NP 20–550 wildtype (cyan) and Tyr21Ala/His22Ala double mutant (red). NP 25–550 (black) is shown as a comparison (500 mM NaCl). The arrow marks the void volume in panels B–D. (E–F) Minigenome activity assays using NP truncation and point mutations as indicated in the figure. (E) N-terminal residues, including Tyr21 and His22, are important for oligomerization and mutation of these residues result in loss of MG activity. (F) Mutants lacking only the C-terminus inhibit MG activity of full-length NP.
Viral polymerase cofactor VP35 is a suppressor of innate immune signaling and is also critical for viral RNA synthesis (Feng et al., 2007; Luthra et al., 2013; Prins et al., 2009; Schumann et al., 2009). Its role in RNA synthesis is thought to be functionally analogous to the P protein in other NNSVs (Sanchez et al., 2006). VP35 has an N-terminal oligomerization domain and a C-terminal interferon inhibitory domain (IID), which binds dsRNA and is critical for innate immune inhibition (Basler et al., 2003; Hartman et al., 2004; Leung et al., 2009; Leung et al., 2010b). The N-terminal oligomerization domain is thought to participate in protein-protein interactions, including VP35 self-association (Moller et al., 2005; Reid et al., 2005) as well as association with the viral polymerase L (Trunschke et al., 2013). VP35 oligomerization is also important for RNA synthesis (Moller et al., 2005), but appears to be dispensable for VP35/NP co-localization (Moller et al., 2005).
Despite previous efforts, it is unclear how VP35-NP interactions modulate EBOV RNA synthesis. Here, we describe an interaction between the EBOV VP35 and NP proteins that critically impacts NP function and EBOV RNA synthesis.
RESULTS
The N-terminus of NP is important for oligomerization
Previous deletion analysis of EBOV NP revealed that three truncations (residues 2–150, 151–300, and 301–450) resulted in loss of NP function in the viral RDRP complex (Watanabe et al., 2006). In order to identify specific residues involved in NP-NP association, we generated a series of truncation constructs (Figure S1A), which were evaluated in both low (150 mM NaCl) and high (500 mM NaCl) salt buffers. All constructs tested, including NP 1–457 and 25–457, formed large homo-oligomeric complexes as NP elutes in the void volume in buffer containing 150 mM NaCl (Figure 1B). In buffer with 500 mM NaCl, many constructs displayed behavior similar to those in 150 mM NaCl except for NP 25–457 (ΔNPNTD), where some protein eluted in the included volume and some near the void volume (V0), representing a multimeric population that is likely an aggregate. (Figure 1C). Moreover, we observed >60% monodispersed (well-behaved) ΔNPNTD protein in solution at concentrations >10 mg/mL based on multiangle light scattering (MALS) studies. Results from finer truncations (i.e. 2–3 residue deletions) within the first 24 residues suggested a critical role for residues 21–22 as proteins starting at residue 23 eluted in the included volume. In order to further evaluate the impact of Y21 and H22, we generated single mutants (Y21A or H22A) and a double mutant (Y21A/H22A) of NP 1–457. Of these NP mutants, only the Y21A/H22A double mutant eluted in the included volume and the peak displayed low levels of polydispersity (Figure 1D). These results further support an important role for the NP paramyxovirus/filovirus conserved region (NP residues 1–457) in NP-NP self-association.
The N- and C-termini of NP are required for viral RNA synthesis
In order to further test the role of NP-NP self-association in viral RNA synthesis, we tested N- and C-terminal truncations of NP using a minigenome (MG) assay that uses a model Ebola virus genomic RNA encoding Renilla luciferase as a template for transcription and replication by a viral polymerase complex reconstituted by expression of NP, VP35, VP30 and L (Muhlberger, 2007; Muhlberger et al., 1999). In these assays, mutation of Y21A/H22A in the context of the full length NP and the 20–739 NP construct resulted in a complete loss of function (Figure 1E and S1B). Deletion of the first 19 residues of NP (NP 20–739) results in a partial loss in activity (about 60%), suggesting that these residues are important, but not essential for activity. In contrast, C-terminal deletion mutants, such as NP 1–550 or NP 1–601, were severely impaired (<10% of wild type activity) in the MG assay (Figure 1E).
We tested three additional EBOV NP constructs, NP 25–739, NP 25–550, and NP 1–550, for their ability to support MG activity in the presence and absence of WT NP (Figure 1F and Figure S1C). Expression of N-terminal NP truncations, such as NP 25–739 and NP 25–550, did not affect the MG function of NP. However, a C-terminal truncation construct that retained the entire N-terminus (NP 1–550) and the ability to oligomerize displayed dominant negative activity, suggesting that interactions through the N-terminus limit the MG activity of the WT NP (1–739). Collectively these results support a role of both the NP N- and C-termini during viral RNA synthesis.
NPBP peptide from VP35 is necessary and sufficient to bind NP
EBOV NP associates with EBOV VP35 in the context of the replication complex, and this interaction is thought to play a major role during viral RNA synthesis (Elliott et al., 1993; Huang et al., 2002). Previous analyses of EBOV VP35 identified several regions that are highly conserved among filoviruses, including an N-terminal oligomerization domain and a C-terminal IID (Figure 2A) (Hartman et al., 2004; Leung et al., 2010a; Reid et al., 2005). However, sequence analysis of ebolaviruses revealed an additional, highly conserved region N-terminal to the previously predicted oligomerization domain (Figure S1D) (Reid et al., 2005). To determine whether this region in VP35 is required to support viral RNA synthesis, we generated a series of VP35 truncated constructs, where we systematically deleted 10-residue segments from the N-terminus and evaluated the role of these residues in viral RNA synthesis using the MG assay. The resulting data revealed that the first 19 residues are largely dispensable for MG activity (Figure 2B). In contrast, residues 20–52 are critical for EBOV MG activity as N-terminal truncations that extend into this conserved region, including truncations such as in the 30–340 construct, resulted in >60% loss of activity relative to WT VP35 (residues 1–340). Deletion of the entire NPBP region plus the first 19 amino acids results in near-complete loss of MG activity, suggesting that the entire NPBP is required for activity.
Figure 2. A peptide derived from EBOV polymerase co-factor VP35 (NPBP) binds ΔNPNTD with high affinity and specificity, related to Figure S1D, S2, S3, S4 and Table S1.

(A) Domain organization of EBOV VP35, including an oligomerization and IID domains. There is a highly conserved region N-terminal to the oligomerization domain, termed NPBP. (B) Impact of the VP35 N-terminus was evaluated by the Ebola minigenome assay. Ability of either wildtype or mutant VP35 proteins lacking various N-terminal sequences were tested and plotted as normalized minigenome activity, where wildtype VP35 activity was set to 100%. Below representative western blots show similar levels of VP35 protein expression. β-tubulin was used as loading control. (C) Summary of binding measurements by ITC between ΔNPNTD and VP35 N-terminal truncation mutants reveal that residues 20–48 are necessary and sufficient for high affinity binding. n.d., not determined. (D) The structure of the dual-octameric ring of ΔNPNTD in complex with VP35 NPBP peptide in a ribbon representation (left) and in a 90° rotated configuration (right). Each NPBP/ΔNPNTD complex in the asymmetric unit is colored yellow/orange (mol A/mol E), magenta/blue (mol B/mol F), green/purple (mol C/mol G), and cyan/pink (mol D/mol H).
In order to determine the molecular basis for this observation, we next tested interactions between VP35 and NP using in vitro binding assays and various VP35 and NP truncations. These results revealed that EBOV ΔNPNTD was sufficient for high affinity binding to the EBOV VP35 N-terminus. These studies also revealed that VP35-NP association is coordinated, at least in part, through the VP35 N-terminus as VP35 1–168 binds NP whereas VP35 58–340 does not (Figure 2C, S2A). In order to further characterize this interaction, we generated a series of peptides spanning VP35 residues 20–52. Peptides were subjected to quantitative analysis by isothermal titration calorimetry (ITC) and a summary of the results is shown in Figure 2C. These results revealed that VP35 residues 20–48 (i.e. NPBP) comprise the most critical region for high affinity ΔNPNTD binding (KD=18.0 ± 6.0 nM at 500 mM NaCl, Figure 2C and S2B; and KD=28.9 ± 6.4 nM at 150 mM NaCl, Figure S2C). VP35 peptides 1–45 and 27–48 resulted in 1.5- and 6.5-fold reduction in binding affinity relative to that of NPBP for ΔNPNTD protein, which support the importance of the core NPBP peptide. In contrast to these observations, VP35 peptides 30–60 and 20–41 displayed significantly weaker binding to ΔNPNTD (Figure 2C and Figure S2C). Further analysis of the ITC data and MALS results indicates that NPBP binds to ΔNPNTD with a 1:1 stoichiometry (Figure S2B) with a complex molecular weight corresponding to one molecule of ΔNPNTD and one molecule of NPBP (Figure S2D). We also assessed the secondary structure of the NPBP peptide by circular dichroism (CD) spectroscopy, which revealed that NPBP and truncations of NPBP are intrinsically disordered in solution. However, the characteristic random coil sequence of NPBP can also form helical structure as the addition of trifluoroethanol (TFE) resulted in CD spectra with higher helical characteristics (Figure S2E). However, helical propensity alone is not sufficient to bind NP protein as some peptides (for example VP35 20–43) do not bind despite being helical in TFE containing buffers (Figure S2C and S2E). Together, these results reveal that the residues encompassed by VP35 NPBP peptide are intrinsically disordered in solution but have a tendency to form helical structure, which may be critical for high affinity binding to NP in the context of the full length VP35 protein.
Structure of the VP35 NPBP/ΔNPNTD complex
We solved the crystal structure of the VP35 NPBP/ΔNPNTD complex to 3.7 Å resolution by selenomethionine single wavelength anomalous dispersion (Se-SAD) and observed four NPBP/ΔNPNTD heterodimers in the asymmetric unit (termed molA, molB, molC, and molD for ΔNPNTD and molE, molF, molG, and molH for NPBP) (Figure 2D, Figure S3A and Table S1). Analysis of crystal packing revealed that sixteen copies of the NPBP/ΔNPNTD complex form a back-to-back double ring with eight ΔNPNTD molecules arranged in each ring and an approximate 6 nm inner and 20 nm outer diameters (Figure 2D and S3B). The overall structure of ΔNPNTD contains two identifiable lobes, a head lobe spanning NP residues 38 to 240 and a foot lobe from 244 to 383 that is connected together by a flexible hinge located within the head lobe (Figure S4). The head lobe contains 12 alpha helices, with two parallel beta strands (β1 and β2), including the first beta strand located at the beginning of the structured region of NP.
The first 37 residues at the N-terminus are unstructured. The foot lobe contains 10 alpha helices with two short antiparallel beta strands (β3 and β4). The last two helices (α21 and α22) of the foot lobe make structural contacts that are unique to Ebola virus NP (Figure S3B–C) when compared to other NNSV nucleoproteins (N). Specifically, these helices make extensive non-bonded contacts with helices α21 and α22 from n-1 and n+1 NP molecules, where n is one of the eight molecules that make up the ring (Figure S3B–C). The α21 and α22 alpha helices extend into the middle of the ring structure formed by symmetry related molecules. From the structure, the function of the ring configuration is unclear. However, previous data for other nucleoprotein molecules from NNSVs and previous electron microscopy studies of EBOV suggest that the ring configuration we observe here, where NP-NP interactions stabilize ring formation, may provide the intermolecular interactions to maintain NP-NP contacts in the Ebola virus nucleocapsid (Bharat et al., 2012).
NPBP binds the foot lobe of ΔNPNTD
All four ΔNPNTD molecules are present in the structure. However, the head lobes of molecules B and D show a high degree of disorder with corresponding high average and individual B-factors (Table S1 and Figure S3D). The B-factors indicate that the conformations of head lobes in symmetrically-equivalent copies of these molecules across the crystal lattice differ in their orientation by 1–2 Å. In contrast, molecules A and C are well ordered (Figure 3A and Figure S3D). Thus, in the analysis of intermolecular interactions between NPBP and ΔNPNTD, we use the coordinates of molG (NPBP) and molC (ΔNPNTD), respectively (Figure 3). In the structure of molG/molC, NPBP interacts exclusively with the foot lobe of ΔNPNTD. NPBP forms two orthogonal helices (VP35 residues 26–36 form an alpha helix and residues 40–42 form a turn of a 310 helix) and makes multiple hydrogen bonds and non-bonded contacts with NP (Figure 3B–E). In an effort to validate the presence of NPBP peptide in our structure, we obtained additional NPBP/ΔNPNTD complex crystals where the peptide was labeled at positions Met20 and Met34 with selenomethionine (SeMet) and the ΔNPNTD was also SeMet labeled. Analysis of the diffraction data for one of these crystals revealed anomalous signal at the side chains of Met20 and Met34, which confirms that the peptide observed in the structure results from the VP35 NPBP (Figure 3C).
Figure 3. The complex structure of ΔNPNTD and NPBP reveals extensive intermolecular interactions, related to Figure S2 and S4.

(A) The 1:1 complex of NPBP/ΔNPNTD (mol C/mol G). The ΔNPNTD head lobe (residues 37–146, dark green) also contains a flexible hinge region (residues 147–239) that connects to the foot lobe (residues 240–285, light green). The VP35 NPBP interacts with the binding surface formed exclusively by the ΔNPNTD foot lobe. (B) Surface representation of ΔNPNTD with a 1.0 sigma electron density (sigmaA weighted Fo–Fc) mesh shown for NPBP. (C) An anomalous difference map was calculated from data collected at the selenium edge on a crystal of SeMet derivatized ΔNPNTD bound to NPBP synthesized with Se-Met instead of Met. Relative orientation of the figure as shown is 180° rotated from (B). The anomalous map (pink) is contoured at 3.0 sigma to show the position of selenium atoms in the methionine residues within the NPBP structure (Cα backbone shown in stick representation). Residues SeMet 20 and SeMet 34 are shown in stick representation. (D) Analysis of the NPBP/ΔNPNTD complex shows surface complementarity and extensive hydrophobic interactions among residues at the interface. Electrostatic surface representations of the NPBP/ΔNPNTD complex with (left) and without NPBP (right). Red, white, and blue colors represent negative, neutral, and positive electrostatic potential, respectively (−10 to +10 kBT e−1). Arrowhead denotes potential RNA binding site. A dotted outline highlights the region where the VP35 NPBP binds to ΔNPNTD. (E) Residue specific contacts between ΔNPNTD and NPBP are highlighted by the amino acid sidechains shown within 5 Å of NPBP.
Next, we evaluated the interactions between NPBP and ΔNPNTD at the molecular level by assessing surface electrostatics, hydrogen bonding and non-bonded contacts. Evaluation of the NPBP and ΔNPNTD in the complex suggest that there is limited charge complementarity (Figure 3D). Closer examination of the residue specific binding interactions shows that nearly all the residues in the peptide (from residues 20–47) are within 4.5 Å of NP residues, which is consistent with the formation of a large binding interface with many important contacts that drive specificity and affinity (Figure 3E and 4A). The complementary binding interface from Ebola virus NP consists of helix α13, α14, and α15 and β3 and β4 beta strands, where NPBP wraps around the β3 and β4 beta strands (Figure 3E). All residues of NP that contribute to the binding interface are conserved across different Ebola viruses (Figure S1A). This interface contains approximately 2600 Å2 of shared buried surface area, of which approximately 1200 Å2 is contributed by NPBP. We also observe several important hydrogen bonds. For example NPBP residues Pro21, Gly22, Glu24, Ser26, Ser30, Glu31 and Phe44 are engaged in intermolecular H-bonds with NP residues (Figure 4A). Interestingly, Arg37 from NPBP also makes H-bond with Glu292 from NP and the molecular interface formed by these bonds also further occlude the NPBP alpha helix from the solvent (Figure 4B). Collectively, these observations further support the specificity of the NPBP/NP interactions.
Figure 4. Mutational analysis validates the NPBP binding site on NP.

(A) Non-bonded contacts drive NPBP/ΔNPNTD binding. LigPlot+ diagram showing extensive hydrophobic and hydrogen bond interactions between ΔNPNTD and NPBP. Protein side chains are shown as ball and sticks. Hydrogen bonds are shown as orange dotted lines. Non-bonded contacts are shown as spoked arcs. (B) Surface and ribbon representation highlighting the interaction between NPBP (purple) and the foot lobe of ΔNPNTD (light green). VP35 NPBP residues Glu24 and Arg37 and NP residues Lys257 and Glu292 are shown in stick representation. (C) Summary of ITC binding measurements between ΔNPNTD mutants and NPBP reveal that residues R240, K248, and D252 are involved in NPBP/ΔNPNTD complex interactions. n.d., not determined. (D) Impact of the NP mutants was evaluated by the MG assay. Ability of either wildtype or mutant NP proteins to promote MG activity were tested and plotted as MG activity relative to no NP control. The p values are determined by student t-test. ** p< 0.001. Representative western blots to show similar levels of NP protein expression. (E) Representative western blots for NP proteins used in (D) at 250 and 500 ng plasmid transfections.
To further define the NPBP/ΔNPNTD interface, NP residues were mutated to alanine and binding to VP35 NPBP in vitro and cell-based MG activity were assessed. Arg240 and Asp252, were important for VP35 NPBP interactions as the observed ITC-based binding affinities for the mutants are substantially lower than wildtype NP binding to NPBP (Figure 4C). Specific interactions between NPBP and NP also significantly depend on Lys248 as mutation of this residue to alanine results in near-complete loss of binding. We also tested the importance of these residues in the MG in order to confirm that Arg240, Lys248, and Asp252 are critical for viral RNA synthesis. Resulting data shown in Figure 4D reveals that mutations altering NPBP and NP binding impact Ebola virus RNA synthesis in the MG assay. Since these mutations are at the interface, a potential explanation is that these residues enhance the interaction between VP35 and NP.
ΔNPNTD binds ssRNA and the ssRNA binding is inhibited by NPBP
The ability of NP to bind RNA is well-established (Bharat et al., 2012; Huang et al., 2002; Muhlberger et al., 1999; Noda et al., 2010; Noda et al., 2011; Watanabe et al., 2006). Therefore, we assessed if ΔNPNTD can bind single stranded RNA (ssRNA) and double stranded RNA (dsRNA). Using a dot blot assay, we find that NP selectively binds ssRNA with high affinity (KD = 620 ± 230 nM), whereas NP does not bind dsRNA (Figure 5A). Analysis of the NPBP/ΔNPNTD structure identified several highly basic regions (Figure 3D). Using surface charge as a guide we identified K160, K171, and K174 as potential ssRNA binding sites (Figure 5B). Mutation of these residues to alanine demonstrates their role in ssRNA binding (Figure 5C). In contrast, NP residues that are critical for NPBP binding and for EBOV MG activity, such as R240, K248, and D252, are not important for ssRNA binding. Next we conducted competitive binding assays between ssRNA and NPBP, which revealed that VP35 NPBP and ssRNA can compete for binding to ΔNPNTD. In these experiments, NPBP was titrated into the reaction while ssRNA and ΔNPNTD concentrations were held constant. Increasing NPBP concentration resulted in a proportional loss of ssRNA from the ssRNA-NP complex (Figure 5D), yielding an IC50 of 4 μM. These results indicate that ssRNA and NPBP binding to NP are mutually exclusive.
Figure 5. EBOV VP35 NPBP binding can compete with NP-ssRNA binding.

(A) Measurement of the fraction of 32P-end labeled ssRNA (20 base, black) and dsRNA (20 nucleotide basepair, red) bound to ΔNPNTD. The ratio of 32P captured on nitrocellulose (from protein bound RNA) or nylon membranes (free RNA) is shown normalized to the fraction bound. (B) Ribbon representation of ΔNPNTD from the ΔNPNTD/NPBP complex structure (inset) highlighting several residues (stick representation) in one of the major basic patches that is important for ssRNA binding. (C) Dot blot binding results. Relative comparison of mutant NP proteins binding to 20 nt ssRNA compared to WT NP. (D) ΔNPNTD binding to 5 nM ssRNA a function of VP35 NPBP concentration. Shown are representative data from at least 3 independent experiments.
EBOV NP alone forms oligomers and a putative function of VP35 is to maintain EBOV NP in an RNA-free non-oligomeric form. Initial cryo-electron microscopy (cryoEM) studies revealed that NPNTD (1–457) and WT NP (1–739) can form ring structures. Representative images for NPNTD reveal that these protein oligomers can assemble into near-uniform rings, with an outer diameter of approximately 40 nm (Figure 6A). Individual images of different ring structures reveal the top and bottom conformation of these rings and provide sideviews. While the relevance of the double-ring conformation observed in the crystal structure is currently unclear (Figure 2D), our observation of double rings under distinct sample conditions by cryo-EM suggests that this conformation may be functionally important. However, in our crystal structure, crystal packing may impact the ring formation since ΔNPNTD/NPBP is a monomer in solution. Addition of NPBP to the cryo-EM sample results in loss of the ring conformation, presumably due to the formation of 1:1 heterodimers between NPBP and NPNTD (Figure 6B). Taken together, our cryoEM and X-ray results show that NPBP can cause morphological differences between the oligomeric ring structures formed by NPNTD and NPNTD/NPBP complexes. Next, we evaluated a variety of NP constructs, including ΔNPNTD construct and NPNTD using size exclusion chromatography as a measure of the hydrodynamic behavior of NP upon binding to NPBP under low (150 mM) and high (500 mM) NaCl buffer conditions. In contrast to previous results for the peptide-free NP proteins (Figure 1B–C), which eluted at the V0, all NPBP-bound NP proteins eluted at a volume consistent with a well-behaved heterodimer (one molecule of NP and one NPBP molecule) (Figure 6C), indicating that NPBP interaction also prevents oligomerization.
Figure 6. EBOV VP35 NPBP binding can dismantle oligomeric ring structures formed by NPNTD.

(A) Representative cryoEM images of NPNTD forming double ring structures, observed from top and side views. Scale bar: 100 nm (up), 20 nm (down). (B) Ring-like structures formed by NPNTD disappears upon addition of NPBP peptide just prior to cryoEM grid preparation. Scale bar: 100 nm. (C) Representative chromatograms of NPBP complex of NPNTD in 150 mM NaCl (blue dotted line) and 500 mM NaCl buffers (blue solid line), ΔNPNTD in 150 mM NaCl (green dotted line) and 500 mM NaCl (green solid line) from size exclusion chromatography. NPBP peptide alone is shown in 150 mM NaCl (purple dotted line) and 500 mM NaCl (purple solid line) buffer conditions. Location of the void volume for the Superdex 200 column is indicated with an arrow.
VP35 NPBP inhibits Ebola RNA synthesis
In order to determine whether VP35 NPBP can function in trans to complement a VP35 protein lacking the NPBP region and support RNA synthesis, we tested various VP35 N-terminal truncation mutants together with plasmids expressing GFP-NPBP or using a cell-penetrating peptide fused to the NPBP sequence in the MG assay. Specifically, we fused the NPBP with an N-terminal TAT peptide (sequence YGRKKRRQRRR). In this assay, we used VP35 lacking the N-terminal 51 residues and assessed how addition of different regions of EBOV VP35 impact MG activity. Resulting data shows that GFP-NPBP or other VP35 truncations (1–218, 219–340 or 52–340) were nonfunctional in this assay, suggesting that the function of NPBP is required in cis with the rest of the VP35 polypeptide sequence (Figure 7A and S5A).
Figure 7. VP35 NPBP inhibits EBOV replication, related to Figure S5 and S6.

(A) NPBP in the context of VP35, but not as an isolated peptide can support RNA synthesis. MG assay performed with VP35 52–340 with additional VP35 truncations as indicated in the figure, failed to support RNA synthesis. 500 ng of GFP-NPBP and 100 μM of TAT-NPBP was used. Representative results from MG assay with either (B) GFP control plasmid or GFP-NPBP peptide expressing plasmid with three different doses (125, 250 and 500 ng; *p=0.04, **p=0.001). (C) Control TAT-peptide or TAT-NPBP (1, 10 and 100 μM). Activity in A–C reported as a percent of the average activity recorded for wild type VP35 (*p=0.01, *p=0.002). (D) VP35 NPBP inhibits EBOV infection in HeLa cells. Cells were transfected with either plasmid encoding GFP-NPBP peptide fusion (top) or GFP expressing plasmid (bottom) and then challenged with Zaire Ebola virus at an MOI of 0.05 48 hours post plasmid transfection. Representative images show cell nuclei stained with Hoechst 33342 dye (blue), plasmid expression (green) and virus replication (red, stained with anti-Ebola NP mAb). (E) Quantitative reverse transcription-PCR (qRT-PCR) analysis for GFP or GFP-NPBP (VP35 residues 20–48) expressing cells that were infected with Ebola virus. At 15h and 24h post-infection supernatant was collected, TRIZOL treated, and subjected to reverse transcription using a primer a complementary to the negative-sense viral RNA (vRNA) followed by qRT-PCR using primers directed to EBOV NP (*p=0.06, **p=0.006).
Since NPBP occupies a functionally critical site on NP and is required for RNA synthesis, we next tested the ability of NPBP to inhibit Ebola virus RDRP activity. We used plasmid expressing GFP-NPBP and observed dose dependent inhibition of MG activity by NPBP (GFP-NPBP) with an IC50 = 33 μM relative to a GFP peptide control (Figure 7B and S5B). We then tested how TAT-NPBP would function in a similar assay. These peptides were applied to cells prior to transfection with MG assay plasmids. Similar to the GFP-NPBP peptide, we observed dose dependent inhibition of MG activity by Tat-NPBP, but not by the control peptide (Figure 7C and S5C).
To further test activity of NPBP in the context of EBOV infections, HeLa cells were pretransfected with the GFP-NPBP or GFP-control peptide constructs and then challenged with EBOV. Representative images of these infections show that GFP-NPBP, but not the GFP control plasmid was able to inhibit EBOV replication (Figure 7D). Consistent with the immunofluorescence results and with the MG assays, quantitative reverse transcription-PCR (qRT-PCR) results show that the presence of GFP-VP35 NPBP, but not the GFP-control, results in lower viral RNA in the cell supernatants (Figure 7E). Collectively, these results suggest that NPBP can functionally interact with the NP in the viral RDRP complex and has the potential to inhibit viral RNA synthesis.
DISCUSSION
Here we describe the identification and first characterization of the core regulatory elements from EBOV NP and VP35 proteins and their impact on viral RNA synthesis. NPBP, a peptide derived from EBOV VP35, is necessary and sufficient to maintain NP in a non-oligomeric and RNA-free state (NP0), but NPBP cannot support viral RNA synthesis in trans. Our results support a model where Ebola VP35 NPBP binding to NP prevents NP from associating with noncognate RNA (i.e. cellular and non-genomic viral RNA). In addition, NPBP supports viral RNA synthesis in the context of full length VP35, but in isolation NPBP inhibits viral RNA synthesis.
In the structure of the ΔNPNTD/NPBP complex, we observe several characteristics that are unique to the EBOV NP protein. First, the location of the VP35 peptide binding site is highly conserved in filoviruses. Second, NP makes important contacts with NPBP, such as H-bonds with Glu292, because the NPBP peptide forms an unusual kink facilitated by Thr35 and Gly36, which allows for a 310 helical turn (Figure 3E). Third, the NP-NP association in the C-terminal lobe involves a helix-helix interaction that is not observed in any of the NNSV nucleoprotein structures to date (Figure S7). Finally, we do not observe electron density for approximately 75 residues at the C-terminus of Ebola virus NP. We note that the sequence in this region (i.e. residues 380–450) has low sequence complexity and therefore may be in largely random coil conformation. This observation coupled with sequence alignments with other NNSVs suggest that the Ebola virus NP N-terminal domain may be smaller than previously recognized (Watanabe et al., 2006).
As Ebola virus NP protein is an integral part of the viral nucleocapsid and is intimately associated with the viral RNA template, its oligomeric state is of particular importance for maintaining template integrity and to provide template access to the viral RDRP. Our data show that deletion of the N-terminal 24 residues or mutation of highly conserved Tyr21 and His22 to alanine in Ebola virus NP results in loss of oligomerization and corresponding loss of function in the MG assays. However, in addition to Tyr21 and His22, other residues within the first 40 amino acids of NP may be involved in facilitating NP-NP interactions.
The NP C-terminus is dispensable for oligomerization but not for viral RNA synthesis as deletions C-terminal to residue 551 result in loss of Ebola RNA binding. Our observations are also consistent with previous studies, where a 150 residue deletion at the N-terminus resulted in loss of NP-NP association (Watanabe et al., 2006). However, such large deletions can result in the loss of structural integrity, thus it is difficult to attribute these results to a role for oligomerization. Our results also suggest that the NP-NP self-association is regulated by the region that binds NPBP (Figure 2A). Importantly, this binding and resulting change in the NP oligomeric state does not depend on the oligomeric state of VP35 (Moller et al., 2005; Reid et al., 2005) as we observe similar affinities between NP-VP35 and NP-NPBP (See Figure 2C and S2).
VP35 prevents high affinity NP-RNA interactions until nascent NP reaches the site of viral RNA synthesis, where VP35 facilitates transfer of a monomeric and template free NP (NP0) to the viral template RNA, as our data show that VP35 NPBP binding releases RNA from NP. Upon binding to template RNA to form the NP-RNA complex, NP is released from interactions with VP35 NPBP through a yet unknown mechanism. Release from NPBP and subsequent binding to viral RNA likely induces NP oligomerization, which can further stabilize the RNA-bound NP proteins. Our results, particularly data that show how NPBP can out-compete ssRNA binding, are consistent with a model where NPBP overrides NP-NP and NP-RNA interactions in order to maintain Ebola NP in the NP0 state. Not surprisingly, deletion of the NPBP sequence from Ebola VP35 results in loss of viral RNA synthesis likely due to the lack of NP0 to support viral RNA synthesis. However, when a truncated VP35 lacking NPBP (residues 52–340) was co-expressed with the VP35 NPBP peptide and other components in the MG assay, NPBP failed to support viral RNA synthesis in trans. Our results suggest that NPBP-binds NP with high affinity to form NPBP-NP complexes. In addition, proper viral RNA synthesis may require NPBP in the context of the full length VP35 to bind NP to generate NP0. Among the possible reasons for this outcome is a need for NPBP, which is located in the VP35 N-terminus, to function together with VP35 C-terminal IID region. Our previous studies revealed that VP35 IID is also important for virus RNA synthesis. Specifically, we were able to show that conserved basic residues within the alpha-helical subdomain of VP35 IID were critical for VP35-NP interactions (Leung et al., 2009; Prins et al., 2010). Another possibility is that the NPBP binding to NP also provides a means to correctly localize the NP0 proteins to the viral RDRP at a site near the viral nucleocapsid (NP-RNA). In such an arrangement, full length VP35 can also provide template access to the polymerase L.
NNSV P proteins are essential for viral RNA synthesis despite the lack of any sequence similarity among P proteins. In NNSVs, the P protein, which is functionally equivalent to Ebola virus VP35, is thought to maintain newly synthesized nucleoprotein molecules in a non-oligomeric and RNA-free state (N0). Studies on vesicular stomatitis virus (VSV) and Nipah virus (NiV) have shown that an N-terminal region of each respective P protein is sufficient to bind N0. In addition, P peptide binding (N0-P) presumably blocks RNA access to a groove formed between the N-terminal and C-terminal lobes of nucleoprotein based upon structural alignments of the N0-P and N-RNA complex structures. Comparison of the NP structure from our Ebola virus ΔNPNTD/NPBP complex with corresponding structures of VSV (Leyrat et al., 2011a; Leyrat et al., 2011b) and (Yabukarski et al., 2014) NiV N in complex with the P-derived peptide (N0-P), analogous to the Ebola VP35 NPBP, reveals several differences. The P peptide binding site and the secondary structure of the P peptide are different from Ebola virus VP35 NPBP bound to NP (Figure S7). In addition, there are intra-peptide interactions which stabilize the NPBP-bound conformation that are absent in the corresponding VSV and NiV N bound peptides. Moreover, in VSV and NiV, the N-N contacts are different from those observed for Ebola virus NP-NP. Specifically, we observe inter-NP interactions (see Figure S3B–C) that occur via helices in the foot lobe whereas the inter-N interactions in the VSV and NiV occur mainly via loop-loop interactions. Despite these structural differences, we observe several similarities. VSV and NiV N proteins and Ebola virus NP have similar folds and display similar domain arrangements in the first ~350–400 residues of each protein. In addition, our ΔNPNTD/NPBP structure along with ssRNA binding studies show that NPBP binding is incompatible with RNA binding. Comparison of the ΔNPNTD/NPBP structure from the current study with ssRNA bound structures of VSV and RSV nucleoproteins suggest that the highly basic region highlighted in Figure 3D (left) may recognize and bind ssRNA in Ebola NP. Our results also show that the VP35 NPBP binding relieves NP oligomerization and NP-RNA binding by interacting with two distinct regions within NP that are important for viral RNA synthesis. These sites on EBOV NP do not appear to overlap since mutations of individual residues important for ssRNA binding and for NPBP binding appear to function independently. In contrast, structural alignments of the N0-P and N-RNA structures suggest that P peptide binding to N protein in VSV and NiV may also limit N-RNA interactions by steric hindrance (Leyrat et al., 2011a; Yabukarski et al., 2014). Collectively, these results suggest that NNSVs may use variations of a common mechanism to control viral RNA synthesis.
Our results here show for the first time how EBOV VP35 uses the NPBP region to control viral RNA synthesis and highlight NNSV-common and filoviral-specific mechanisms by which EBOV VP35 regulates NP-NP and NP-RNA template interactions. These results also provide the framework to develop anti-viral therapeutics that target filoviruses.
EXPERIMENTAL PROCEDURES
Cloning and purification
Ebola virus NP and VP35 constructs were subcloned into a modified pET15b (Novagen) vector containing a maltose binding protein fusion tag and TEV protease site and expressed in BL21(DE3) Escherichia coli cells grown at 37 °C. Cells were induced for 12–14 hours at 18 °C with 0.5 mM IPTG and harvested at 8,000 x g for 10 minutes, resuspended in buffer A (20 mM Tris pH 7.5, 1 M or 150 mM NaCl for NP and VP35, respectively), 20 mM imidazole pH 7, 5 mM β-mercaptoethanol, and a protease inhibitors. Cells were lysed using an Avestin C3 homogenizer, clarified by centrifugation at 47,000 x g for 30 minutes, and supernatant was purified by affinity and ion exchange chromatography. Fusion tag removed by TEV protease and purified by ion exchange columns prior to final size exclusion column. VP35 peptides were purchased from GenScript. Selenomethionine labeled protein: Proteins were expressed in M9-minimal media, supplemented with appropriately labeled metabolites, and purified in a manner similar to native protein described above.
Isothermal titration calorimetry
NP and VP35 binding was measured by VP-ITC microcalorimeter (MicroCal/GE Healthcare). Samples were dialyzed overnight at 25 °C against 1 L of buffer B (20 mM Tris pH 7.5, 500 mM NaCl, and 2 mM TCEP). Raw ITC data were processed using Origin software and data fit by non-linear least squares analysis to yield KD (equilibrium binding constant) and n (number of binding sites). Representative of 2–4 independent experiments is shown.
SEC-MALS
SEC-MALS experiments were performed using a DAWN-HELEOS II detector (Wyatt Technologies) coupled to a Superdex SD200 column (GE Healthcare) in buffer C (20 mM Tris pH 7.5, 250 mM NaCl, and 2 mM TCEP). 2 mg/mL sample were injected and raw data were analyzed using ASTRA 6 software (Wyatt Technologies) to determine the weight averaged molecular mass (MW). Protein concentrations were determined using the refractive index measured by an Optilab T-rEX (Wyatt Technologies) and a dn/dc = 0.185 mL · g−1.
Minigenome assays
BSRT7 cells were co-transfected by using Lipofectamine 2000 (Invitrogen) with T7-driven expression plasmids encoding the EBOV NP, L, VP30, and VP35 proteins along with plasmid expressing a T7 promoter-driven EBOV minigenome RNA which encodes a Renilla luciferase reporter gene and constitutively expressing firefly luciferase expression plasmid that served as a transfection control. At 24 h posttransfection, cells were lysed with passive lysis buffer (Promega) and reporter activities were determined using the Dual Luciferase assay kit (Promega). Renilla luciferase activity was normalized to firefly luciferase activity. Minigenome reporter activation was expressed as percent minigenome activity setting activity of WT VP35 as 100%. Error bars represent the standard deviation from three independent replicates.
Crystallization, data collection, and structure solution
ΔNPNTD and VP35 NPBP were incubated together in 1:1 molar ratio prior to loading onto a Superdex SD200 column (GE Healthcare) with buffer containing 20 mM Tris pH 7, 250 mM NaCl, and 2 mM TCEP. NPBP/ΔNPNTD complex crystals were generated using hanging drop vapor diffusion method and streak seeding in well solution containing 100 mM Tris pH 7.2, 50 mM Hepes pH 7, and 23% PEG400. Crystals were cryoprotected in stabilization solution containing 100 mM Tris pH 7.2, 50 mM Hepes pH 7, 50 mM NaCl, 2 mM TCEP, 15% PEG400, and 15% PEG3350 followed by vitrification in liquid nitrogen.
X-ray data were collected at the Structural Biology Center 19ID at the Advanced Photon Source (Argonne, IL), Datasets from two crystals of SeMet derivatized NP protein bound to native VP35 NPBP were collected at low remote energy to minimize radiation damage with an oscillation angle of 0.8 and a crystal-to-detector distance of 500 mm. Dataset from one crystal of SeMet derivitized NP bound to SeMet derivatized VP35 NPBP was collected at the Se absorption peak to maximize the anomalous signal with an oscillation angle of 0.8 and a crystal-to-detector distance of 450 mm.
The structure of Ebola virus VP35 NPBP/DNPNTD complex was solved using the diffraction data obtained from three crystals. The partial model was built using phases obtained from the averaging datasets of these crystals. The data averaging took the differences in the levels of anomalous signal into account. HKL3000 was used to process diffraction data sets for all crystals (Minor et al., 2006; Otwinowski and Minor, 1997). Computational corrections for absorption in a crystal and imprecise calculations of the Lorentz factor resulting from a minor misalignment of the goniostat were applied (Borek et al., 2003; Otwinowski et al., 2003). Anisotropic diffraction was corrected to adjust the error model and to compensate for the phasing signal for radiation-induced increase of non-isomorphism within the crystal (Borek et al., 2010; Borek et al., 2013; Borek et al., 2007). The data statistics are presented in Table S1. The crystals diffracted anisotropically to a resolution of 3.7 Å in the best direction, and 4.0 and 4.2 Å in the other two directions. The estimated level of anomalous signal was 3.6% of the native intensity. We performed a search for heavy atom positions to a resolution of 7.0 Å with ShelxD (Sheldrick, 2008), which identified 28 Se positions with correlations coefficients: CCAll = 54.79% and CCWeak = 28.18%. The handedness of the solution was determined with ShelxE by analyzing the connectivity and contrast of the electron density maps. 26 Se positions were refined anisotropically to 4.4 Å with MLPHARE (Otwinowski, 1991), with the final FOM reaching 0.205. 4-fold NCS was identified by Resolve (Terwilliger, 2004; Terwilliger, 2003). NCS-averaging and solvent flattening was performed by DM (Cowtan and Main, 1998). The resulting electron density map was used for model building, which consisted of several cycles of BUCCANEER (Cowtan, 2006). Intermediate models from different cycles of BUCCANEER were combined manually into a more complete model for one of the four chains, which had the lowest thermal motions. This model was subsequently propagated by applying the NCS operators, and then iterative model building was repeated. The resulting oligomer was then manually rebuilt and corrected by iterative application of Coot, Refmac (Emsley and Cowtan, 2004; Murshudov et al., 1997; Murshudov et al., 1999) along with the local NCS restraints, jelly body refinement, and in the later cycles with TLS refinement (Winn et al., 2003).
The ΔNPNTD chains in the asymmetric unit have different levels of order, with average B-factors for chains A, B, C and D: 122, 187, 88, and 233 Å2, respectively. Although the N-terminal domains for chains B and D could not be accurately modeled, the order of chains A and C was sufficient to build the entire peptide chain that consists of N-terminal head lobe and C-terminal foot lobe. SeMet positions for the ΔNPNTD protein together with the SeMet derivatized NPBP data set analyzed separately provided unambiguous tracing for the ΔNPNTD chains and NPBP chains. This model was propagated through an NCS symmetry operator and provides a credible indication as to where the N-terminal domains of chains B and D are located. However, due to their large thermal motions (equivalent to 1–2 Å shift of subdomains) the validation statistics, which analyze how this region fits to the electron density, are not meaningful. The model was refined with these domains, because the electron density and packing interactions indicate where the domains are located, albeit the individual secondary structure elements can barely be fitted. All atomic interactions are analyzed based on the chains C and G.
CryoEM specimen preparation and data acquisition
The 400 mesh R1.2/1.3 holey carbon Quantifoil grid (Quantifoil Micro Tools GmbH) was cleaned with acetone (Sigma-Aldrich) for 12 hours and glow discharged before use. 3.0 μl aliquot of purified Ebola NP oligomer at ~2 mg/ml in the presence or absence of 0.5 μl disruption peptide was applied to the grid, blotted for 8 seconds, and immediately frozen in liquid ethane using a Leica EM GP (Leica Microsystems, Inc.) with temperature held constantly at 10 °C and 99% humidity during the process. The grid was stored in liquid nitrogen until imaging. All grids were examined on a JEM 2100 (LaB6 gun) cryo-electron microscope (JEOL) operated at 200 kV, spot size of 2, condenser aperture of 70 μm, and objective aperture of 60 μm. Images were recorded under low-dose condition on a Gatan 4k × 4k CCD camera (Gatan, Inc.) at ×50,000 microscope magnification (corresponding to a calibrated sampling of 2.16 Å/pixel) and a dose of 50–60 electrons/Å2 with defocus ranging from 3–5 μm. Particle images were manually boxed and extracted using EMAN2 (Tang et al., 2007) program with a box size of 288 × 288 pixels.
Structural figure generation and analysis
Surface area and surface complementarity were calculated using AREAIMOL and Sc, respectively as implemented in CCP4 program suite (COLLABORATIVE COMPUTATIONAL PROJECT, 1994). Structure figures were prepared using PyMOL (DeLano, 2002). Protein-protein interactions were analyzed using LigPlot+ (Laskowski and Swindells, 2011). Topology diagrams were generated by PDBSum (Laskowski, 2007) and TopDraw (Bond, 2003). Sequence alignment was performed using ClustalW (Thompson et al., 2002) and prepared using ESPript 3.0 (Gouet et al., 2003).
RNA binding and competition assays
20 nt ssRNA was labeled at the RNA 5′ using 32P-γ-ATP. 25 μM of ΔNPNTD was serially diluted and incubated with 32P-20 nt ssRNA at 5 nM for 15 min prior to loading onto a dot blot apparatus (Bio-Rad). For peptide-competition experiments, 20 μM of VP35 NPBP was serially diluted and incubated with ΔNPNTD at 4 μM for 30 min prior to the addition of 32P-20 nt ssRNA (at 5 nM). The protein-RNA complex was then passed through nitrocellulose (NC) and nylon (NY) membranes. The membranes were scanned using a Typhoon 9410 Variable Model Imager and the amount of 32P-20 nt ssRNA bound to NC and NY membranes was quantified from the radioactivity detected. Data analyzed by Origin and the fraction ssRNA bound to ΔNPNTD proteins was calculated using the equation shown below:
Virus infection
HeLa cells were transfected with either a plasmid encoding GFP-NPBP peptide fusion or a GFP control expressing plasmid. 24 hours post plasmid transfection, cells were transferred to the BSL-4 laboratory and challenged with Zaire Ebola virus at an MOI of 0.05 and were fixed by immersing in neutral buffered formalin overnight. Fixed cells were washed three times with PBS and then stained with anti-Ebola-GP antibody followed by an anti-Mouse-546 (Alexa) secondary antibody, nuclei were stained with Hoechst-33342 (Life-Technologies - 1:50,000 dilution). Cells were imaged on a Nikon Ti Eclipse microscope. Total cells (blue), transfected cells (green; GFP) and Ebola infected cells (red; GP specific mAb) were counted using CellProfiler software (Broad Institute) using customized pipelines which are available from R. Davey upon request.
qRT-PCR
293T cells were transfected with GFP or VP35 NPBP and infected with Zaire Ebola virus. At 15 and 24 hours postinfection, supernatant and cells were collected for RNA isolation using TRIZOL. For reverse transcription, either an oligo(dT) primer (for detection of mRNA) or a primer complementary to the negative-sense viral RNA (vRNA) was used, followed by quantitative real-time PCR using primers directed against NP. In parallel, quantitative RT-PCR directed against β-actin mRNA was performed, to control for differences in the amount of total input RNA. The relative copy number of target RNA was calculated from the change in threshold cycle (CT) value between β-actin mRNA and target RNA. mRNA was reverse transcribed using a vRNA specific primer (NP), followed by quantitative real-time PCR using NP-specific primers. Shown are the means and standard errors from two independent experiments.
Supplementary Material
Acknowledgments
We thank: Drs. T. Ellenberger, T. Brett, N. Tolia, W. Li, and J. Payton for discussions; members of the Amarasinghe/Leung, Basler and Davey laboratories for experimental support; Drs. S. Ginell, N. Duke, and J. Lazarz at Advanced Photon Source (APS) Sector 19 for beamline access and assistance during data collection. Use of Structural Biology Center beamlines at the APS is supported by the U.S. D.O.E. under contract DE-AC02-06CH11357. Use of the National Magnetic Resonance Facility at Madison is supported by NIH grant P41GM103399 (NIGMS). This work was supported in part by a contract to the Center for Structural Genomics of Infectious Diseases (CSGID) from the NIAID/NIH/DHS (contract number HHSN272201200026C, Anderson-PI) to ZO, by DOD grants DTRA 1-21-1-0002 to RD, DTRA-HDTRA1-12-1-0051 and DTRA HDTRA1-14-1-0013 to CFB and GKA, and by NIH grants (R01AI107056 to DWL; R01GM053163 to ZO, R01AI077519 to RD, R01AI059536 to CFB; U19AI109945 (Basler-PI) to CFB, DWL, and GKA and U19AI109664 (Basler-PI) to DWL, DMB, CFB and GKA; U19 AI070489 (Holtzman-PI), P41GM103832 and P50 GM1003297 to WC, and R01AI081914 to GKA).
Footnotes
Author contributions: DWL, CFB and GKA conceived and designed the overall study. DWL defined VP35 NPBP. DWL, JMB and GKA developed NP purification methods. JP purified proteins for crystallization. DWL identified and optimized crystallization conditions and acquired crystallographic data, which was used by DMB and ZO to solve the structure, build and refined the model. DWL, CFB, and GKA conducted analysis of the structure and designed validation assays. DWL, GL, and GKA designed and conducted RNA binding studies. IH conducted peptide analysis. PL, RSS, and CFB designed functional validation assays. PL and AE-F conducted cell-based studies and analyzed results. ZS and WC conducted cryoEM studies. MA and RD designed and executed BSL4 studies. DWL, DMB, CFB, and GKA wrote manuscript with input from all co-authors. All authors analyzed results and approved the submission.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, Klenk HD, Palese P, Garcia-Sastre A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol. 2003;77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat TA, Noda T, Riches JD, Kraehling V, Kolesnikova L, Becker S, Kawaoka Y, Briggs JA. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc Natl Acad Sci U S A. 2012;109:4275–4280. doi: 10.1073/pnas.1120453109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond CS. TopDraw: a sketchpad for protein structure topology cartoons. Bioinformatics. 2003;19:311–312. doi: 10.1093/bioinformatics/19.2.311. [DOI] [PubMed] [Google Scholar]
- Borek D, Cymborowski M, Machius M, Minor W, Otwinowski Z. Diffraction data analysis in the presence of radiation damage. Acta crystallographica Section D, Biological crystallography. 2010;66:426–436. doi: 10.1107/S0907444909040177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borek D, Dauter Z, Otwinowski Z. Identification of patterns in diffraction intensities affected by radiation exposure. Journal of synchrotron radiation. 2013;20:37–48. doi: 10.1107/S0909049512048807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borek D, Ginell SL, Cymborowski M, Minor W, Otwinowski Z. The many faces of radiation-induced changes. Journal of synchrotron radiation. 2007;14:24–33. doi: 10.1107/S0909049506046589. [DOI] [PubMed] [Google Scholar]
- Borek D, Minor W, Otwinowski Z. Measurement errors and their consequences in protein crystallography. Acta Crystallogr D. 2003;59:2031–2038. doi: 10.1107/s0907444903020924. [DOI] [PubMed] [Google Scholar]
- COLLABORATIVE COMPUTATIONAL PROJECT, N. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta crystallographica Section D, Biological crystallography. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- Cowtan K, Main P. Miscellaneous algorithms for density modification. Acta crystallographica Section D, Biological crystallography. 1998;54:487–493. doi: 10.1107/s0907444997011980. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific; 2002. [Google Scholar]
- Dziubanska PJ, Derewenda U, Ellena JF, Engel DA, Derewenda ZS. The structure of the C-terminal domain of the Zaire ebolavirus nucleoprotein. Acta crystallographica Section D, Biological crystallography. 2014;70:2420–2429. doi: 10.1107/S1399004714014710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott LH, Sanchez A, Holloway BP, Kiley MP, McCormick JB. Ebola protein analyses for the determination of genetic organization. Arch Virol. 1993;133:423–436. doi: 10.1007/BF01313780. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Feng Z, Cerveny M, Yan Z, He B. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J Virol. 2007;81:182–192. doi: 10.1128/JVI.01006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouet P, Robert X, Courcelle E. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman AL, Towner JS, Nichol ST. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology. 2004;328:177–184. doi: 10.1016/j.virol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Huang Y, Xu L, Sun Y, Nabel GJ. The assembly of Ebola virus nucleocapsid requires virion-associated proteins 35 and 24 and posttranslational modification of nucleoprotein. Mol Cell. 2002;10:307–316. doi: 10.1016/s1097-2765(02)00588-9. [DOI] [PubMed] [Google Scholar]
- Laskowski RA. Enhancing the functional annotation of PDB structures in PDBsum using key figures extracted from the literature. Bioinformatics. 2007;23:1824–1827. doi: 10.1093/bioinformatics/btm085. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Leung DW, Ginder ND, Fulton DB, Nix J, Basler CF, Honzatko RB, Amarasinghe GK. Structure of the Ebola VP35 interferon inhibitory domain. Proc Natl Acad Sci U S A. 2009;106:411–416. doi: 10.1073/pnas.0807854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung DW, Prins KC, Basler CF, Amarasinghe GK. Ebolavirus VP35 is a multifunctional virulence factor. Virulence. 2010a;1:526–531. doi: 10.4161/viru.1.6.12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung DW, Prins KC, Borek DM, Farahbakhsh M, Tufariello JM, Ramanan P, Nix JC, Helgeson LA, Otwinowski Z, Honzatko RB, et al. Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nat Struct Mol Biol. 2010b;17:165–172. doi: 10.1038/nsmb.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyrat C, Jensen MR, Ribeiro EA, Jr, Gerard FC, Ruigrok RW, Blackledge M, Jamin M. The N(0)-binding region of the vesicular stomatitis virus phosphoprotein is globally disordered but contains transient alpha-helices. Protein science : a publication of the Protein Society. 2011a;20:542–556. doi: 10.1002/pro.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyrat C, Yabukarski F, Tarbouriech N, Ribeiro EA, Jr, Jensen MR, Blackledge M, Ruigrok RW, Jamin M. Structure of the vesicular stomatitis virus N(0)-P complex. PLoS pathogens. 2011b;7:e1002248. doi: 10.1371/journal.ppat.1002248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra P, Ramanan P, Mire CE, Weisend C, Tsuda Y, Yen B, Liu G, Leung DW, Geisbert TW, Ebihara H, et al. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe. 2013;14:74–84. doi: 10.1016/j.chom.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters PS, Banerjee AK. Complex formation with vesicular stomatitis virus phosphoprotein NS prevents binding of nucleocapsid protein N to nonspecific RNA. J Virol. 1988;62:2658–2664. doi: 10.1128/jvi.62.8.2658-2664.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution - from diffraction images to an initial model in minutes. Acta Crystallogr D. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Moller P, Pariente N, Klenk HD, Becker S. Homo-oligomerization of Marburgvirus VP35 is essential for its function in replication and transcription. J Virol. 2005;79:14876–14886. doi: 10.1128/JVI.79.23.14876-14886.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlberger E. Filovirus replication and transcription. Future virology. 2007;2:205–215. doi: 10.2217/17460794.2.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J Virol. 1999;73:2333–2342. doi: 10.1128/jvi.73.3.2333-2342.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta crystallographica Section D, Biological crystallography. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- Noda T, Hagiwara K, Sagara H, Kawaoka Y. Characterization of the Ebola virus nucleoprotein-RNA complex. The Journal of general virology. 2010;91:1478–1483. doi: 10.1099/vir.0.019794-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Halfmann P, Sagara H, Kawaoka Y. Regions in Ebola Virus VP24 That Are Important for Nucleocapsid Formation. J Infect Dis. 2007;196(Suppl 2):S247–250. doi: 10.1086/520596. [DOI] [PubMed] [Google Scholar]
- Noda T, Kolesnikova L, Becker S, Kawaoka Y. The importance of the NP: VP35 ratio in Ebola virus nucleocapsid formation. The Journal of infectious diseases. 2011;204(Suppl 3):S878–883. doi: 10.1093/infdis/jir310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. Maximum likelihood refinement of heavy atom parameters. Paper presented at: CCP4 Study Weekend (Science and Engineering Research Council).1991. [Google Scholar]
- Otwinowski Z, Borek D, Majewski W, Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallographica Section A. 2003;59:228–234. doi: 10.1107/s0108767303005488. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Prins KC, Binning JM, Shabman RS, Leung DW, Amarasinghe GK, Basler CF. Basic residues within the ebolavirus VP35 protein are required for its viral polymerase cofactor function. J Virol. 2010;84:10581–10591. doi: 10.1128/JVI.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins KC, Cardenas WB, Basler CF. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol. 2009;83:3069–3077. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid SP, Cardenas WB, Basler CF. Homo-oligomerization facilitates the interferon-antagonist activity of the ebolavirus VP35 protein. Virology. 2005;341:179–189. doi: 10.1016/j.virol.2005.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A, Geisbert TW, Feldmann H. Filoviridae:Marburg and Ebola Viruses. In: Knipe DM, Howley PM, Griffin RA, Martin MA, Roizman B, Straus SE, editors. Fields Virology. Philadenphia, PA: Lippincott Williams & Wilkins; 2006. pp. 1409–1448. [Google Scholar]
- Sanchez A, Kiley MP. Identification and analysis of Ebola virus messenger RNA. Virology. 1987;157:414–420. doi: 10.1016/0042-6822(87)90283-2. [DOI] [PubMed] [Google Scholar]
- Sanchez A, Kiley MP, Holloway BP, Auperin DD. Sequence analysis of the Ebola virus genome: organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res. 1993;29:215–240. doi: 10.1016/0168-1702(93)90063-s. [DOI] [PubMed] [Google Scholar]
- Sanchez A, Wagoner KE, Rollin PE. Sequence-based human leukocyte antigen-B typing of patients infected with ebola virus in Uganda in 2000: identification of alleles associated with fatal and nonfatal disease outcomes. J Infect Dis. 2007;196(Suppl 2):S329–336. doi: 10.1086/520588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann M, Gantke T, Muhlberger E. Ebola virus VP35 antagonizes PKR activity through its C-terminal interferon inhibitory domain. J Virol. 2009;83:8993–8997. doi: 10.1128/JVI.00523-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabman RS, Jabado OJ, Mire CE, Stockwell TB, Edwards M, Mahajan M, Geisbert TW, Basler CF. Deep Sequencing Identifies Noncanonical Editing of Ebola and Marburg Virus RNAs in Infected Cells. mBio. 2014;5 doi: 10.1128/mBio.02011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick GM. A short history of SHELX. Acta Crystallographica Section A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Shi W, Huang Y, Sutton-Smith M, Tissot B, Panico M, Morris HR, Dell A, Haslam SM, Boyington J, Graham BS, et al. A filovirus-unique region of Ebola virus nucleoprotein confers aberrant migration and mediates its incorporation into virions. J Virol. 2008;82:6190–6199. doi: 10.1128/JVI.02731-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Terwilliger T. SOLVE and RESOLVE: automated structure solution, density modification and model building. Journal of synchrotron radiation. 2004;11:49–52. doi: 10.1107/s0909049503023938. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC. SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2002;Chapter 2(Unit 2 3) doi: 10.1002/0471250953.bi0203s00. [DOI] [PubMed] [Google Scholar]
- Trunschke M, Conrad D, Enterlein S, Olejnik J, Brauburger K, Muhlberger E. The L-VP35 and L-L interaction domains reside in the amino terminus of the Ebola virus L protein and are potential targets for antivirals. Virology. 2013;441:135–145. doi: 10.1016/j.virol.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Noda T, Kawaoka Y. Functional mapping of the nucleoprotein of Ebola virus. J Virol. 2006;80:3743–3751. doi: 10.1128/JVI.80.8.3743-3751.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- Yabukarski F, Lawrence P, Tarbouriech N, Bourhis JM, Delaforge E, Jensen MR, Ruigrok RW, Blackledge M, Volchkov V, Jamin M. Structure of Nipah virus unassembled nucleoprotein in complex with its viral chaperone. Nature structural & molecular biology. 2014;21:754–759. doi: 10.1038/nsmb.2868. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.