

Abstract
Background
Identified genetic variants are insufficient to explain all cases of inherited arrhythmia. We tested whether the integration of whole exome sequencing with well-established clinical, translational, and basic science platforms could provide rapid and novel insight into human arrhythmia pathophysiology and disease treatment.
Methods and Results
We report a proband with recurrent ventricular fibrillation, resistant to standard therapeutic interventions. Using whole-exome sequencing, we identified a variant in a previously unidentified exon of the dipeptidyl aminopeptidase-like protein-6 (DPP6) gene. This variant is the first identified coding mutation in DPP6 and augments cardiac repolarizing current (Ito) causing pathological changes in Ito and action potential morphology. We designed a therapeutic regimen incorporating dalfampridine to target Ito. Dalfampridine, approved for multiple sclerosis, normalized the ECG and reduced arrhythmia burden in the proband by >90-fold. This was combined with cilostazol to accelerate the heart rate to minimize the reverse-rate dependence of augmented Ito.
Conclusions
We describe a novel arrhythmia mechanism and therapeutic approach to ameliorate the disease. Specifically, we identify the first coding variant of DPP6 in human ventricular fibrillation. These findings illustrate the power of genetic approaches for the elucidation and treatment of disease when carefully integrated with clinical and basic/translational research teams.
Keywords: arrhythmia, genetics, ion channel
The transient-outward K+ current (Ito) initiates the early repolarization phase of the cardiac action potential1 and demonstrates marked transmural variability.2 Ito is conducted by a pore-forming α subunit (Kv4.3) and β subunits, including dipeptidyl aminopeptidase-like protein-6 (DPP6).3,4 DPP6 associates with5 and modulates the trafficking, kinetics, and pharmacology of Kv4 channels in brain and heart.4,6–8 Here we describe a proband with recurrent ventricular fibrillation (VF) despite multiple pharmacological and ablation interventions. Whole exome sequencing (WES) in the patient identified an inherited variant (H332R) in DPP6-T, a previously unreported but dominant human isoform of DPP6. Notably, the human DPP6-T variant displays increased binding activity for Kv4.3 compared with canonical DPP6-T. Further, the DPP6-T human variant results in increased association with Kv4.3 and a proarrhythmic augmentation of Ito. Treatment of the patient with dalfampridine, an Ito antagonist used in the treatment of multiple sclerosis, dramatically decreased arrhythmic events.
Methods
Statistics
Data are presented as mean±SEM. For our study, a value of P<0.05 was considered statistically significant. For the comparison of 2 groups, we performed Wilcoxon–Mann–Whitney U tests. For the comparison of >2 groups, we applied a Kruskal–Wallis test. When we obtained a significant P value, we continued with pairwise comparisons by using Wilcoxon–Mann–Whitney U tests according to the closed testing principle. Incidence of arrhythmia was analyzed by using χ2 test. The null hypothesis was rejected for P<0.05.
Clinical Genetic Testing
Genetic testing for both Brugada and long QT syndromes were sent after the proband's initial presentation (GeneDx). Both clinical panels were negative for mutations in SCN5A,GPD1L,CACNA1C,CACNB2,SCN1B,SCN4B,ANK2,KCNQ1, KCNH2, KCNE1, KCNE2, KCNJ2, CAV3, AKAP9, and SNTA1.
Human Tissue
Cardiac tissue was obtained from explanted hearts of patients undergoing heart transplantation through the Cooperative Human Tissue Network: Midwestern Division at Ohio State University. Approval for use of human subjects was obtained from the Institutional Review Board of Ohio State University. Tissue from healthy donor hearts not suitable for transplantation (subclinical atherosclerosis, age, no matching recipients) was obtained through Lifeline of Ohio. The investigation conforms to the principles outlined in the Declaration of Helsinki. Age and sex were the only identifying data acquired from tissue providers.
Genetic Research Studies
This research study was approved by the OSU Institutional Review Board, and the proband and family members provided written informed consent. Extracted DNA was sent to the Baylor College of Medicine Whole Genome Laboratory for WES. WES and bioinformatics analysis of the sequence data were performed at Baylor's Human Genome Sequencing Center. Genomic DNA was fragmented via sonication, ligated to Illumina multiplexing paired-end adapters, amplified by using a polymerase chain reaction (PCR) assay with the use of primers with sequencing barcodes (indexes), and hybridized to biotin-labeled VCRome, version 2.1, for target enrichment/exome capture. The postcapture library DNA underwent massively parallel sequencing on the Illumina HiSeq 2000 platform. Variants deemed clinically significant were confirmed by Sanger sequencing in the proband and other family members to determine whether the variants were inherited or de novo.
Computational Modeling
A well-validated mathematical model of the human ventricular action potential with modifications to account for differences in ion channel expression in endocardial and epicardial cells was used for computer simulations.9 To account for defects in Ito due to the H332R variant, Ito density was increased 7-fold in the model. Difference in action potential duration (ΔAPD measured at 90% repolarization) between the endocardial and epicardial cell after steady-state pacing at a cycle length of 1000 ms was determined for the wild-type and H332R variant. Therapeutic intervention was simulated by reducing max Ito conductance and increasing the pacing cycle length.
Molecular Biology
Human DPP6 isoform 1 (NM_130797) and Kv4.3 was acquired from Origene and used as a template to generate PCR fragments for cloning into pcDNA3.1+, pDsRed-Monomer-N1, and pcDNA6/myc-HisB. DPP6-T was generated by using a human heart cDNA library and isoform 1 to generate 2 products that were subsequently ligated to produce the DPP6-T isoform. The H332R mutation was generated by using unique primers and site-directed mutagenesis. All sequences were verified prior to use to confirm sequence and frame.
Tissue Preparation
For immunoblotting and coimmunoprecipitation analysis, cardiac tissues (whole heart [control, ischemic heart failure, and nonischemic heart failure], right atria, right ventricle, left ventricle, left atria) were harvested and flash frozen with liquid nitrogen.10,11 The frozen tissues were resuspended in 2 volumes of ice-cold homogenization/IP buffer (0.025 mol/L Tris, 0.15 mol/L NaCl, 0.001 mol/L EDTA, 1% NP-40, 5% glycerol, pH 7.4, 1 mmol/L PMSF, 1 mmol/L AEBSF, 10 μg/mL leupeptin, and 10 μg/mL pepstatin) and homogenized by using a handheld homogenizer. The homogenate was centrifuged at 1000g at 4°C to remove nuclei. The lysate was pelleted at high speed for 15 minutes at 4°C. The resulting supernatant was quantitated by bicinchoninic acid assay (BCA; Thermo-Scientific) before analysis.
Biochemistry
Coimmunoprecipitations were performed by using the Pierce Co-Immunoprecipitation Kit and the manufacturer's instructions. Briefly, 5 μg of Ig were conjugated to beads and incubated with 100 μg lysate overnight at 4°C in homogenization/IP buffer.12 Beads were washed 5 times with Dulbecco's PBS, and the proteins were eluted, electrophoresed, and transferred to nitrocellulose. Immunoblotting was carried out in a vehicle of 5% nonfat dry milk/Tris-buffered saline+Tween 20. For pull-downs, 100 μg of whole heart lysate were incubated with GST or GST-Kv4.3 NT beads overnight in binding buffer. Beads were washed 3 times in wash buffer (500 mmol/L NaCl) and eluted. Proteins were separated via electrophoresis on a 4% to 15% gel (BioRad), and the proteins transferred to nitrocellulose and immunoblotted.
In Vitro Translation/Binding
DPP6-T and DPP6-T H332R constructs were in vitro translated by using rabbit reticulocyte lysate, [35S]methionine (20 μCi Redivue l-[35S]methionine; GE Healthcare), T7 polymerase, and 1 μg plasmid DNA (TNT Coupled Rabbit Reticulocyte Lysate System; Promega). For binding experiments, in vitro translated products were incubated with immobilized GST or immobilized GST-Kv4.3 NT constructs overnight at 4°C in binding buffer (PBS+150 mmol/L NaCl, 0.1% Triton X-100). Reactions were washed 3 times in wash buffer (150 mmol/L, 500 mmol/L, and 1 mol/L NaCl), eluted, and separated by using SDS-PAGE. Gels were stained with Coomassie to show the presence of GST proteins before drying the gel in a gel dryer (Bio-Rad Laboratories). Radiolabeled proteins were detected by using standard autoradiography.
Patient Sequencing for p.H332R Variant
Genomic DNA from whole blood was extracted by using the Qiagen DNAeasy kit and the manufacturer's instructions. Primers were designed to amplify a fragment that was gel excised/purified and sequenced.
GST-Fusion Proteins
cDNA for Kv4.3 NT was PCR generated, cloned into pGEX6P-1 (Amersham). BL21(DE3)pLysS cells (Agilent) were transformed with the pGEX6P-1 constructs and grown overnight at 37°C in LB supplemented with ampicillin (50 μg/mL). Overnight cultures were subcultured for large-scale expression. Cells were grown to an optical density of 0.6 to 0.8 and induced with 1 mmol/L isopropyl 1-thio-α-d-galactopyranoside (IPTG) for 4 hours at 37°C. Cells were centrifuged at 6000g, resuspended in lysis buffer (1 mmol/L DTT, 1 mmol/L EDTA, 40 μg/mL AEBSF, 10 μg/mL leupeptin, 40 μg/mL benzamidine, 10 μg/mL pepstatin), and frozen at −20°C.13,14 Cells were lysed via thawing and homogenized via sonication.15,16 Cell debris was removed via centrifugation at 4000g at 4°C. Supernatants were added to 1 mL equilibrated glutathione-agarose (Amersham) and incubated overnight at 4°C. The glutathione-agarose solutions were washed with PBS containing 1% Triton X-100 (3×), PBS containing 500 mmol/L NaCl (3×), and stored in PBS containing 1 mmol/L NaN3. Protein purification and sizes were verified with SDS-PAGE followed by Coomassie Blue staining.
Reagents
Antibodies included Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Fitzgerald Industries), DPP6 (Sigma Aldrich), and Kv4.3 (Covance Immunology Services) Igs. A rabbit polyclonal antibody specific for the novel truncated DPP6-T isoform was generated by Covance Immunology Services and purified in-house. Specifically, a KLH-conjugated peptide, KVKSRKLTLPHSKSC, was used to inoculate 2 rabbits. The final bleed was validated against in vitro translated DPP6-T and lysates from HEK293 cells transfected with myc-tagged DPP6 and either DPP6-T-myc or myc-tagged DPP6-T H332R cDNA. Transfected cells were lysed and coimmunoprecipitated by using anti-myc Ig. Associated proteins were separated via SDS-PAGE, transferred to nitrocellulose and immunoblotted with the DPP6-T antibody. Additional validation was performed by in vitro translating DPP6-T protein (TNT Coupled Rabbit Reticulocyte Lysate System; Promega) followed by SDS-PAGE/immunoblotting.
Electrophysiology
Electrophysiological experiments were performed as described previously.10–12,17,18
Additional Patient Case Information
Previous medical history was not significant for cardiac disease. The proband was treated for seizures as a child with phenytoin; the seizures resolved and phenytoin treatment was discontinued during preadolescence. At presentation, the proband was undergoing treatment for a mood disorder and restless leg syndrome. Home medications at initial presentation included lamotrigine, ropinirole, and lithium. Admission lab values were notable for a serum potassium of 3.3 mEq/L. After the initiation of mexiletine, the patient returned with symptoms of dizziness, palpitations, and nausea on home medications of mexiletine, oxcarbazepine, escitalopram, pramipexole, and lamotrigine. Mexiletine dose reduction failed to relieve these adverse effects, and it was discontinued. Escitalopram was discontinued due to potential proarrhythmia concerns and replaced by sertraline. During the hospital stay, when the first ablation occurred, intermittent hypokalemia was again noted and treated with potassium supplements. A follow-up evaluation by nephrology consult did not identify a renal cause for the hypokalemia. The patient had 2 episodes of atrial fibrillation documented during hospitalizations for ventricular tachycardia (VT)/VF storms.
Pharmacologic Management of the Patient
At the time of initial presentation, home medications at presentation included lamotrigine, lithium, and ropinirole. Ropinirole was discontinued due to concerns about potential proarrhythmia.19 Lithium therapy has been associated with the appearance of a Brugada-type ECG pattern, at therapeutic and supratherapeutic lithium concentrations. The association between lithium and BrS is limited to case reports, and discontinuation may either normalize the ECG or convert the pattern from a type 1 to a type 2 or 3 Brugada pattern.20–25 Notably, in this patient, discontinuation of lithium did not result in resolution or modification of the J-point amplitude.
Oxcarbazepine
There is a case report of oxcarbazepine-associated ST elevation in leads V1 and V2 and associated VF.26 This may be attributable to oxcarbazepine-mediated effects on cardiac sodium channels.27 A trial off of oxcarbazepine was not successful, and this therapy has been resumed as a mood stabilizer.
Quinidine
Quinidine is a recommended therapy for Brugada syndrome,28 and while the patient did not fulfill the Brugada diagnostic criteria, he was empirically started on quinidine. He apparently did well clinically, albeit for a short duration, on therapeutic doses of quinidine and had a serum trough concentration of 1.9 µg/mL on 648 mg every 12 hours. However, the patient was unable to tolerate this dosage due to gastrointestinal intolerance. Due to patient preference, the dose was reduced after the initial hospitalization, and adherence to quinidine therapy has been intermittent. The patient has continued on a low dose (324 mg every 8 hours), which is subtherapeutic. Because quinidine is known to block Ito29,30 in addition to multiple other cardiac ion channels, it is possible that the combination of quinidine with dalfampridine is beneficial even at a subtherapeutic concentration.
Cilostazol
The high-output implantable cardioverter-defibrillator (ICD) implanted in this patient (external and internal defibrillation has been required in the past) does not have atrial pacing capability. Cilostazol is a phosphodiesterase inhibitor used in the treatment of intermittent claudication. An off-target effect of cilostazol is increased heart rate, and this increase in heart rate has been reported to be potentially and variably beneficial in patients with Brugada or early repolarization syndromes.31–34 Once a putative increase in Ito was linked to this patient's arrhythmia syndrome, acceleration of the heart rate was identified as an additional therapeutic strategy. Ito has a relatively slow recovery from inactivation35; therefore, acceleration of the heart rate can be used to reduce the current amplitude. Notably, in this patient, the increase in heart rate appears to be sustained with long-term cilostazol therapy. However, patient adherence has been intermittent with cilostazol therapy.
Dalfampridine
Dalfampridine is the US adopted name for 4-aminopyridine, and in 2010 an extended-release oral formulation of the drug (Ampyra) was approved to improve walking in patients with multiple sclerosis.36 4-Aminopyridine blocks Ito and the ultrarapid delayed rectifier current (IKur) at relatively low concentrations.37 Other repolarizing potassium currents such as IKr (hERG) or IKs are relatively insensitive and not blocked by low concentrations of 4-aminopyridine,18,38 and safety assessments have not found QT interval prolongation.36 Epileptogenic adverse effects have been reported with dalfampridine39 but are dose related (more common at doses >10 mg twice daily36) or occur with immediate-release formulations. Long-term use with the extended-release formulation does not appear to confer an increased risk of seizures,36 and no such adverse events have been noted in this patient.
Atrial fibrillation often occurs in patients with heritable forms of lethal ventricular arrhythmias.40 Atrial repolarization is strongly influenced by Ito, and Kv1.5-encoded IKur is also an important modulator of atrial repolarization. We have reported that increased atrial Ito in a canine model was associated with increased inducibility of atrial fibrillation.41 Thus, the inhibition of Ito with dalfampridine (as well as quinidine) not only may be beneficial for this patient's VF but also may mitigate atrial fibrillation via prolongation of the atrial action potential.
Results
Abnormal Repolarization and VF in Proband Negative for Identified Arrhythmia Gene Variants
A 37-year-old white man was transferred from a community hospital after resuscitation from a witnessed cardiac arrest. He was found to be in VF and treated with CPR, defibrillation, hypothermia, and amiodarone. The presenting ECG showed partial right bundle-branch block and J-point elevation in leads V1 and V2 but was nondiagnostic for Brugada syndrome or early repolarization (Figure 1).42 Previous cardiac history was negative; family history was significant for sudden death in the proband's mother. Cardiac magnetic resonance imaging revealed left ventricular ejection fraction of 46%, with no evidence of arrhythmogenic right ventricular cardiomyopathy or infiltrative disease. An electrophysiology study was negative for accessory pathways, and procainamide challenge was nondiagnostic for a Brugada pattern (Figure 2, Table 1).43 A subcutaneous ICD was placed, and the patient was discharged.
Figure 1.
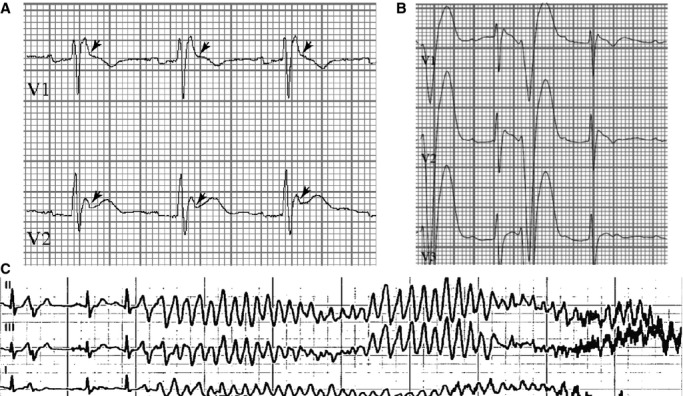
Abnormal repolarization and ventricular fibrillation. A, Presenting ECG with right bundle-branch block; J-point elevation (arrows) <2 mV in leads V1 to V3; thus, not consistent with Brugada pattern. B, Ectopy with short-coupled PVCs. C, Short-coupled PVC initiates VF requiring ICD shock in the presence of quinidine. Over 16 months, proband had 168 appropriate ICD discharges for VF (≈8.6 per month). ICD indicates implantable cardioverter-defibrillator; VF, ventricular fibrillation; PVC, premature ventricular contraction.
Figure 2.

Negative response to procainamide challenge. Leads V1 through V3 are shown at baseline and every 5 minutes after procainamide treatment. Note the absence of dynamic changes in morphology, lack of transition from saddleback to coved type morphology in lead V2, and no elevation of the J point.
Table 1.
ECG Intervals in Proband on Various Treatment Regimens
| Months After Presentation | Antiarrhythmic Treatment | Heart Rate (bpm) | PR (ms) | QRS (ms) | QT (ms) | QTc (ms) |
|---|---|---|---|---|---|---|
| 0 | None | 86 | 162 | 124 | 366 | 431 |
| 4 | Qunidine 648 mg every 12 h | 59 | 180 | 112 | 442 | 438 |
| 15 | Mexiletine 300 mg every 8 h | 73 | 176 | 118 | 368 | 406 |
| 17 | Quinidine 648 mg every 12 h | 74 | 186 | 118 | 406 | 451 |
| 17 | Ablation 1 | |||||
| 20 | Quinidine 324 mg every 8 h | 65 | 208 | 114 | 434 | 451 |
| 25 | Quinidine 324 mg every 12 h | 81 | 168 | 114 | 346 | 402 |
| 32 | Ablation 2 | |||||
| 33 | Quinidine 324 mg every 8 h | 78 | 158 | 100 | 414 | 472 |
| 33 | Quinidine 324 mg every 8 h; cilostazol 50 mg 2 times/d | 92 | 148 | 102 | 342 | 423 |
| 33 | Quinidine 324 mg every 8 h; cilostazol 50 mg 2 times/d; after first dose of dalfampridine 10 mg | 91 | 176 | 98 | 342 | 421 |
| 33 | Quinidine 324 mg every 8 h; cilostazol 50 mg 2 times/d; dalfampridine 10 mg every 12 h | 83 | 160 | 106 | 382 | 449 |
| 39 | Quinidine 324 mg every 8 to 12 h; cilostazol 50 mg 2 times/d; dalfampridine 10 mg every 12 h | 75 | 184 | 112 | 422 | 471 |
| 44 | Quinidine 324 mg every 8 to 12 h; cilostazol 50 mg QD (once daily); dalfampridine 10 mg every 12 h | 68 | 162 | 108 | 394 | 419 |
Note that ECGs were obtained as part of clinical care and thus were not taken with consistent timing after drug dosing, and the apparent pharmacological response to quinidine may have varied as a function of time from previous doses and/or patient adherence. The QT/QTc was never significantly prolonged on any ECG, by any drug regimen, although the J point was modified as shown in Figure 8.
Four months later, he experienced syncope and VF ×4; quinidine was initiated (648 mg every 12 hours; trough plasma concentration 1.9 μg/mL) based on modest J-point elevation, and he was discharged on quinidine. The patient later discontinued quinidine due to gastrointestinal intolerance. Ten months later, the patient had unifocal premature ventricular contraction (PVC)-triggered recurrent VF and mexiletine was initiated to suppress the triggering PVCs. One month later, he was admitted to an outside hospital with a VT/VF storm (91 ICD discharges) requiring intubation; atrial fibrillation was also noted. Amiodarone was initiated, the ICD was replaced, and he was transferred to our institution. A second electrophysiology study with planned PVC ablation was undertaken. Cardiac mapping was done, but no PVCs were elicited during the electrophysiology study even with isoproterenol infusion. Thus, no site for PVCs was identified, although previous ECGs were suggestive of a right ventricular apical site. There were areas of electrical fractionation in the right ventricular free wall, and these sites were ablated; initiation of VF during delivery of radiofrequency energy was noted. Postablation, he was discharged on amiodarone and mexiletine, but he subsequently experienced multiple episodes of VF requiring internal and external defibrillation. He was acutely treated with lidocaine and amiodarone and then oral mexiletine and amiodarone. After a second opinion was obtained, amiodarone and mexiletine were discontinued, and low-dose quinidine therapy was reinitiated. After he developed recurrent VF, quinidine was increased to 324 mg 3 times daily. At patient's request, he was transferred to another facility for electrophysiology study and evaluation for a second ablation. At this third electrophysiology study, VF was induced and epicardial radiofrequency ablation was done over the right ventricular outflow tract, targeting areas of electrogram fragmentation. This approach was undertaken based on what appeared to be a “Brugada-like” ECG pattern (although never fully meeting the diagnostic criteria for BrS), a history of previous unsuccessful endocardial ablation, and the continuing low burden of PVCs. After ablation, the ST segment was lower in leads V1 to V3, and VF was noninducible. Quinidine, potassium and spironolactone were discontinued, and verapamil was initiated.
At 3 weeks post epicardial ablation, the patient was admitted after 2 ICD discharges (VF). This was followed by multiple episodes of VT/VF with recurrent shocks resulting in intubation for 7 days. Quinidine and lidocaine were started; after electrolyte repletion and extubation, therapy was transitioned to oral quinidine and cilostazol to accelerate heart rate in an attempt to minimize PVC-triggered VF. His course was complicated by pneumonia and sepsis. Three days later, he developed atrial fibrillation/flutter with rapid ventricular response that was treated with rate control, anticoagulation, and cardioversion.
The proband's mother died at age 64, with cause of death specified as malignant VF without coronary atherosclerosis. Based on family history and clinical presentation, the proband underwent clinical genetic testing for Brugada and long QT syndromes: no pathogenic mutations were identified. Additional genetic evaluation was performed, and WES identified multiple nonsynonymous, nonsense, splice site, and frameshift variants. A heterozygous A>G variant on chromosome 7 (7:154379727) within a noncoding region of the Kv4.3-associated subunit DPP6 was identified (Figure 3A through 3D). This variant was of interest because DPP6 modulates Kv4.3-encoded Ito, a contributor to cardiac repolarization. This locus was previously associated via genome-wide haplotype-sharing analysis as the only validated gene in a 1.5-Mb size risk haplotype associated with familial idiopathic VF.44 Sanger sequencing confirmed the variant; both his father and brother were negative, but a maternal uncle (twin of proband's mother) was positive, defining his mother as an obligate carrier. Minor allele frequency is 0.0047 in the NHLBI Exome Sequencing Project and 0.0010 in 1000 Genomes. Despite past association with idiopathic VF, the variant was initially dismissed based on its presumed location in an intron hundreds of kilobases from identified genomic regulatory elements and minor allele frequency.
Figure 3.

Identification of novel splice form of DPP6 (DPP6-T). A, Human DPP6 (red, novel exon 5B). B and C, Domain organization of canonical DPP6 and truncated DPP6-T (red=novel residues). D, DPP6-T A>G variant. E, Confirmation of DPP6-T transcript from human heart (primers denoted by arrows in Panel C). F, DPP6-T expression in human heart by using DPP6-T Ig (F409 in Figure 2C). G, Relative DPP6 and DPP6-T expression in human heart chambers (n=4; *P<0.05 compared with LV). DPP6 indicates dipeptidyl aminopeptidase-like protein-6; LA, left atrium; LV, left ventricle; MW, molecular weight; RA, right atrium; RV, right ventricle.
Identification of a Novel Human Splice Form of DPP6
Subsequent analysis determined that 7:154379727 is located in a putative novel coding exon of DPP6 (Figure 3B). The novel exon (DPP6-5B) was predicted to encode 144 residues with a termination codon, ultimately encoding a truncated version of DPP6 (DPP6-T). The DPP6-T cDNA was confirmed by RT-PCR of human heart (Figure 3E). Further, an antibody raised against the truncated isoform confirmed the presence of the novel polypeptide in human heart (Figures 3F and 4). Notably, compared with the canonical DPP6 that is expressed similarly in all 4 human heart chambers, we observed elevated DPP6-T expression in both human left atria and right ventricle compared with left ventricle and right atria (Figure 3G; P<0.05).
Figure 4.

Validation of DPP6-T–specific affinity-purified antibody. A polyclonal DPP6-T antibody was designed and generated. A, In vitro translated DPP6-T, but not DPP6, was recognized by anti–DPP6-T Ig. B, HEK cells were transfected with myc-tagged DPP6 and either myc-tagged DPP6-T or myc-tagged DPP6-T H332R. Cell lysates were immunoprecipitated by using anti-myc Ig and blotted with the DPP6-T Ig. The DPP6-T Ig specifically recognizes the DPP6-T and DPP6-T H332R with no reactivity against canonical DPP6. DPP6 indicates dipeptidyl aminopeptidase-like protein-6.
Human DPP6-T p.H332R Variant Displays Increased Binding Activity for Kv4.3
We performed biochemical experiments from human left ventricular lysates as a first step to define the molecular role of the new DPP6-T isoform in heart. We first performed coimmunoprecipitations from detergent-soluble lysates from the human left ventricle by using DPP6-T Ig. DPP6-T Ig coimmunoprecipitates the Kv4.3 α-subunit from human heart (Figure 5A). Conversely, Kv4.3 Ig coimmunoprecipitates DPP6-T in parallel biochemical assays (Figure 5B). Based on coimmunoprecipitation experiments, DPP6-T may directly or indirectly associate with Kv4.3. We therefore tested the ability of the 2 molecules to associate using purified GST-Kv4.3. As shown in Figure 5C, GST-Kv4.3 but not GST alone associated with DPP6-T in pull-down assays. Finally, we tested direct association by using purified GST-Kv4.3 and radiolabeled DPP6-T. We observed association of GST-Kv4.3 but not GST with radiolabeled DPP6-T (Figure 5D). By using identical assays, we next measured relative binding activity of DPP6-T versus the human DPP6-T H332R variant. Radiolabeled DPP6-T p.H332R showed nearly a 2-fold increase in binding activity for purified Kv4.3 relative to DPP6-T (Figure 5D and 5E). Together, our data support that DPP6-T is expressed in heart and associates directly with Kv4.3. Further, our biochemical data illustrate that the human DPP6-T variant (p.H332R) linked with arrhythmia displays significantly increased binding activity for Kv4.3 compared with DPP6-T.
Figure 5.

DPP6-T H332R displays increased Kv4.3 binding relative to DPP6-T. A and B, Coimmunoprecipitations of Kv4.3 and DPP6-T from detergent-soluble human left ventricle lysates. C, Purified GST-Kv4.3, but not GST alone associates with DPP6-T in pull-down experiments from detergent-soluble human left ventricular lysates. D and E, Purified GST-Kv4.3, but not GST, associates with radiolabeled DPP6-T and DPP6-T H332R. However, GST-Kv4.3 shows >2-fold increase in relative binding activity for DPP6-T H332R vs DPP6-T when corrected for input and protein loading (n=3; *P<0.05 compared to wild-type). Input lanes in top panel of (D) were taken from same gel at different exposure times. DPP6 indicates dipeptidyl aminopeptidase-like protein-6; WT, wild-type.
Human DPP6-T p.H332R Variant Increases Kv4.3-Dependent Ito
To define the molecular mechanism of DPP6-T function in heart, we evaluated DPP6 and DPP6-T for the regulation of Ito. Compared with Kv4.3 alone, we observed increased Ito in cells coexpressing Kv4.3 plus DPP6. Notably, coexpression of DPP6-T with Kv4.3 increased Ito nearly 2-fold compared with canonical DPP6 (Figure 6A and 6B). We then compared DPP6-T with the proband's DPP6-T p.H332R variant. As noted earlier, DPP6-T p.H332R demonstrated increased binding affinity for Kv4.3 compared with DPP6-T (Figure 5D and 5E). Consistent with these data, we observed ≈7-fold increase in Ito for DPP6-T p.H332R compared with DPP6-T (Figure 6A and 6B). In summary, our data support a role for DPP6-T in increasing Kv4.3-dependent Ito in heart relative to canonical DPP6. Further, our data demonstrate that the DPP6-T p.H332R variant significantly augments Ito compared with DPP6-T.
Figure 6.

DPP6-T+ Kv4.3 displays augmented Ito compared with DPP6+ Kv4.3. A and B, DPP6-T H332R +Kv4.3 shows ≈7-fold increase in peak Ito compared with WT DPP6-T (n=6/condition; *P<0.05 compared with Kv4.3; #P<0.05 compared with Kv4.3+DPP6-T). DPP6 indicates dipeptidyl aminopeptidase-like protein-6; WT, wild-type.
DPP6-T p.H332R Associated Ito Displays Proarrhythmic Phenotypes
Computational modeling of observed H332R Ito in a human myocyte model revealed significant arrhythmogenic phenotypes consistent with the proband's clinical presentation (Figure 7A through 7D). Specifically, incorporation of H332R-related defects in Ito into mathematical models of human endocardial and epicardial (relatively large Ito at baseline, Figure 7A) action potentials demonstrated dramatic premature repolarization in the epicardial but not endocardial cell (Figure 7B). The resulting dispersion of repolarization (estimated as difference in APD between endocardial/epicardial cells [ΔAPD], Figure 7D) promotes a substrate for arrhythmia. These phenotypes were eliminated by directly reducing Ito or by increasing the basal heart rate (Ito slowly recovers from inactivation). Thus, our combined data support increased Ito as the molecular mechanism underlying the human DPP6-T H332R variant. A combined approach of modest Ito reduction and modest increase in rate was sufficient to restore normal repolarization in the epicardial cell and decrease dispersion of repolarization (Figure 7C and 7D).
Figure 7.

Ito-directed therapies are predicted to reduce repolarization dispersion and arrhythmia burden. A and B, Computer-simulated human endocardial (Endo) and epicardial (Epi) action potentials associated with DPP6-T and DPP6-T H332R. C, Simulated therapeutic intervention involving modest Ito block combined with modest increase in pacing rate. D, Simulated difference in endocardial and epicardial ΔAPD. Mathematical modeling predicts that Ito reduction combined with pacing reduces proarrhythmic dispersion in repolarization for H332R. DPP6 indicates dipeptidyl aminopeptidase-like protein-6; WT, wild-type; ΔAPD, action potential duration.
Ito-Based Therapies Correct ECG and Arrhythmia Phenotypes in DPP6-T p.332R Proband
Based on the preceding analysis and suboptimal response to conventional therapies, the proband was admitted for initiation of dalfampridine therapy (10 mg every 12 hours) to directly inhibit Ito and cilostazol (100 mg twice daily) to increase heart rate. Secondary therapies included quinidine, enoxaparin, warfarin, and potassium. The patient has remained on a regimen of dalfampridine, cilostazol, and low-dose quinidine (1.1 µg/mL) for 25.5 months. During this time, no PVCs were seen on repeated ambulatory ECG recordings (Figure 8). ECGs were notable for a lower J point in leads V1 to V3 (compare Figures 1A and 8A). The patient's ICD has appropriately delivered electrical therapy during this time period (Figure 8B); however, both events occurred when the patient was nonadherent with the dalfampridine/cilostazol regimen. In summary, even with the inclusion of these 2 events, initiation of Ito-directed therapy reduced VF (defined by appropriate ICD therapies) in this proband by ≈95.6-fold (8.31 to 0.086 event per month; Figure 8B and 8C).
Figure 8.

Ito-directed therapies reduce repolarization dispersion and arrhythmia burden. A, Proband ECG following dalfampridine and cilostazol. Note altered terminal portion of QRS complex and lower J point vs Figure 1A. B and C, ICD discharge events following initial event. Ablations noted with arrows. VF events reduced >90-fold since initiation of Ito-targeted therapy (*P<0.05 compared to approach lacking dalfampridine/cilostazol). The patient's ICD has fired twice following the new therapeutic approach; both events occurred when patient was nonadherent with the dalfampridine/cilostazol regimen. ICD indicates implantable cardioverter-defibrillator; VF, ventricular fibrillation.
Discussion
While the DPP6 locus has been previously associated with idiopathic ventricular fibrillation (IVF), DPP6 coding variants have not been reported. Wilde and colleagues previously linked 7q36 with IVF.44 While a coding variant was not identified, a putative DPP6 promoter variant was identified and 7q36 variant haplotype carriers showed ≈20-fold increase in DPP6 mRNA.44 Similar to our proband, the presenting VF event occurred postadolescence.44 However, unlike our proband, members of this kindred did not show J-wave alterations. Subsequently, Xiao et al reported alterations in Ito phenotypes in these patients that resolved through ablation.6 In our proband, ablation initially appeared to be beneficial but paroxysmal VF storms recurred. The emergence of VF in an adult suggests that age-dependent increases in ectopy may have been required for arrhythmia initiation; notably, the short coupled ectopy suggests an “R-on-T” phenomenon as the PVCs occur during the T wave.
Dalfampridine (4-aminopyridine) is a potent and relatively specific blocker of Ito without evidence of QT interval prolongation.36 Further, cilastozol also regulates Ito activity.45,46 After >25 months, no significant adverse events have been noted in this patient. Our data support the link of the DPP6-T variant with the disease phenotype. However, there may be other variants and/or environmental triggers associated with the VF. The integrated clinical, genetic, and experimental approach outlined here may serve as a model for the diagnosis and treatment of rare but potentially significant forms of human disease. It should be noted that 4-aminopyridine, while relatively specific for Ito, blocks other potassium channels including IKur that has been implicated in increased susceptibility to AF (also observed in proband).47,48 Furthermore, cilastozol has been linked with direct Ito block,45,46,49 albeit at concentrations significantly higher than expected with clinically used doses.46 These data highlight the complexity for channel-targeted antiarrhythmia therapies.
While this report provides a framework for the identification of a potential disease mechanism and treatment, there are limitations to this work. Incomplete penetrance is well known in the field of familial arrhythmias. Incomplete penetrance is more common than not for the cardiac channelopathies, with only ≈40% penetrance observed for the congenital LQTS50 and ≈16% for small BrS kindreds.51 Procainamide challenge and multiple ECG evaluations in this patient did not fulfill the diagnostic criteria for BrS; procainamide was used as the intravenous provocative agent based on availability in the United States, with the ECG leads in the standard, rather than the alternative superior, position, which may have affected the sensitivity of the text.40 Collectively, the clinical presentation is suggestive of a variant of BrS. Here, we observed phenotypic diversity between the proband and his mother (both severe forms of the disease) and the uncle of the proband. Further, while not common, this variant is present in the general population (minor allele frequency 0.0047 in NHLBI ESP; 0.0010 in 1000 Genomes). However, as recently shown, disease-causing variants are found with significantly higher prevalence rates in the population than predicted.52 Now that this new DPP6 coding exon is identified, it will be important to define the prevalence of variants in this region in larger phenotype-positive/genotype-negative arrhythmia cohorts. As with many variants, it is possible that this variant represents a modifier allele with additional factors required for disease susceptibility. In fact, as shown by McNally and colleagues for cardiomyopathy, and by Bezzina et al with Brugada syndrome, modifier genes are likely central in defining genotype/phenotype relationships, as genetic risk variants are more prevalent in the population than predicted.53,54 Given the age of onset in the proband and his mother, a second critical direction will be to define the cause of the underlying arrhythmia trigger(s). We cannot rule out the possible contribution of the Purkinje system to the initiating ectopy in this patient6,55; however, it is notable that the cardiac tissue had increased expression of DPP6-T in the right ventricle relative to the left. This combined with the computational data suggests a possible role for phase 2 reentry in the right ventricle as an initiator of the ectopy, although the potential contribution of the Purkinje system cannot be ruled out. Interpretation of the proband's response to therapy is complicated by the altered state of the cardiac substrate after ablation and ICD discharges. Collectively, when considering these substrate issues and the nonselective pharmacology of dalfampridine that inhibits IKur in addition to Ito, further investigation is warranted to elucidate these atrial and ventricular arrhythmia mechanisms. Finally, it will be important to perform a full characterization of the role of the novel isoform of DPP6, as well as the underlying mechanism of the arrhythmia variant. While we observed increased Ito density, it will be important to further characterize DPP6-T for altered expression, targeting, and turnover phenotypes related to Kv4.3 in disease. As a first step toward examining these mechanisms, we evaluated DPP6 and DPP6-T in left ventricular samples from nonfailing hearts, as well as hearts from patients displaying ischemic and nonischemic heart failure. These disease samples, particularly nonischemic heart failure, were selected as they have previously been linked with reduced Ito and Kv4.3 expression.56 As expected, we observed reduced Kv4.3 expression in nonischemic heart failure samples compared with nonfailing samples (Figure 9). Relevant for this study, we observed a decreased trend for DPP6 expression in both ischemic and nonischemic samples compared with nonfailing tissue (Figure 9). In contrast, DPP6-T showed a trend for increased expression in both disease states compared with nonfailing samples (Figure 9). The relative change in expression between DPP6 and DPP6-T in both disease states was significant (Figure 9). Based on these findings, while we propose that the 2 molecules may be differentially regulated to tune Ito and cardiac repolarization, additional experiments will be necessary to better define the role of these molecules in vivo and in disease.
Figure 9.

DPP6-T expression in human ischemic (IHF) and nonischemic heart failure (NIHF). Figure shows primary immunoblot data (A) for nonfailing heart samples and IHF and NIHF samples. B, Levels were normalized to Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) for protein loading and expressed as relative to levels in nonfailing samples (n=5; *P<0.05 compared to non-failing heart tissue). DPP6 indicates dipeptidyl aminopeptidase-like protein-6.
Sources of Funding
Supported by National Institutes of Health (HL084583, HL083422, HL114383 to Dr Mohler; HL114893 to Dr Hund; HL113084 to Dr Janssen); James S. McDonnell Foundation (to Dr Hund), American Heart Association (to Dr Mohler), and Fondation LeDucq (to Drs Priori, Hund, and Mohler).
Disclosures
None.
References
- Antzelevitch C. Molecular basis for the transmural distribution of the transient outward current. J Physiol. 2001;533:1. doi: 10.1111/j.1469-7793.2001.0001b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J Mol Cell Cardiol. 2010;48:12–25. doi: 10.1016/j.yjmcc.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourrier M, Schram G, Nattel S. Properties, expression and potential roles of cardiac K+ channel accessory subunits: MinK, MiRPs, KChiP, and KChAP. J Membr Biol. 2003;194:141–152. doi: 10.1007/s00232-003-2034-8. [DOI] [PubMed] [Google Scholar]
- Radicke S, Cotella D, Graf EM, Ravens U, Wettwer E. Expression and function of dipeptidyl-aminopeptidase-like protein 6 as a putative beta-subunit of human cardiac transient outward current encoded by Kv4.3. J Physiol. 2005;565:751–756. doi: 10.1113/jphysiol.2005.087312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh H, Goldstein SA. I SA channel complexes include four subunits each of DPP6 and Kv4.2. J Biol Chem. 2008;283:15072–15077. doi: 10.1074/jbc.M706964200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Koopmann TT, Ordog B, Postema PG, Verkerk AO, Iyer V, Sampson KJ, Boink GJ, Mamarbachi MA, Varro A, Jordaens L, Res J, Kass RS, Wilde AA, Bezzina CR, Nattel S. Unique cardiac Purkinje fiber transient outward current beta-subunit composition: a potential molecular link to idiopathic ventricular fibrillation. Circ Res. 2013;112:1310–1322. doi: 10.1161/CIRCRESAHA.112.300227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguy A, Le Bouter S, Comtois P, Chartier D, Villeneuve L, Wakili R, Nishida K, Nattel S. Ion channel subunit expression changes in cardiac Purkinje fibers: a potential role in conduction abnormalities associated with congestive heart failure. Circ Res. 2009;104:1113–1122. doi: 10.1161/CIRCRESAHA.108.191809. [DOI] [PubMed] [Google Scholar]
- Nadal MS, Ozaita A, Amarillo Y, Vega-Saenz de Miera E, Ma Y, Mo W, Goldberg EM, Misumi Y, Ikehara Y, Neubert TA, Rudy B. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron. 2003;37:449–461. doi: 10.1016/s0896-6273(02)01185-6. [DOI] [PubMed] [Google Scholar]
- Grandi E, Pasqualini FS, Bers DM. A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol. 2010;48:112–121. doi: 10.1016/j.yjmcc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, Leymaster ND, Dun W, Wright PJ, Cardona N, Qian L, Mitchell CC, Boyden PA, Binkley PF, Li C, Anderson ME, Mohler PJ, Hund TJ. Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation. 2012;126:2084–2094. doi: 10.1161/CIRCULATIONAHA.112.105320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Scouarnec S, Bhasin N, Vieyres C, Hund TJ, Cunha SR, Koval O, Marionneau C, Chen B, Wu Y, Demolombe S, Song LS, Le Marec H, Probst V, Schott JJ, Anderson ME, Mohler PJ. Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc Natl Acad Sci USA. 2008;105:15617–15622. doi: 10.1073/pnas.0805500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran J, Makara MA, Little SC, Musa H, Liu B, Wu X, Polina I, Alecusan JS, Wright P, Li J, Billman GE, Boyden PA, Gyorke S, Band H, Hund TJ, Mohler PJ. EHD3-dependent endosome pathway regulates cardiac membrane excitability and physiology. Circ Res. 2014;115:68–78. doi: 10.1161/CIRCRESAHA.115.304149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdi KM, Mohler PJ, Davis JQ, Bennett V. Isoform specificity of ankyrin-B: a site in the divergent C-terminal domain is required for intramolecular association. J Biol Chem. 2006;281:5741–5749. doi: 10.1074/jbc.M506697200. [DOI] [PubMed] [Google Scholar]
- Mohler PJ, Hoffman JA, Davis JQ, Abdi KM, Kim CR, Jones SK, Davis LH, Roberts KF, Bennett V. Isoform specificity among ankyrins. An amphipathic alpha-helix in the divergent regulatory domain of ankyrin-b interacts with the molecular co-chaperone Hdj1/Hsp40. J Biol Chem. 2004;279:25798–25804. doi: 10.1074/jbc.M401296200. [DOI] [PubMed] [Google Scholar]
- Gudmundsson H, Curran J, Kashef F, Snyder JS, Smith SA, Vargas-Pinto P, Bonilla IM, Weiss RM, Anderson ME, Binkley P, Felder RB, Carnes CA, Band H, Hund TJ, Mohler PJ. Differential regulation of EHD3 in human and mammalian heart failure. J Mol Cell Cardiol. 2012;52:1183–1190. doi: 10.1016/j.yjmcc.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudmundsson H, Hund TJ, Wright PJ, Kline CF, Snyder JS, Qian L, Koval OM, Cunha SR, George M, Rainey MA, Kashef FE, Dun W, Boyden PA, Anderson ME, Band H, Mohler PJ. EH domain proteins regulate cardiac membrane protein targeting. Circ Res. 2010;107:84–95. doi: 10.1161/CIRCRESAHA.110.216713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline CF, Wright PJ, Koval OM, Zmuda EJ, Johnson BL, Anderson ME, Hai T, Hund TJ, Mohler PJ. Betaiv-spectrin and CaMKII facilitate Kir6.2 regulation in pancreatic beta cells. Proc Natl Acad Sci USA. 2013;110:17576–17581. doi: 10.1073/pnas.1314195110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar A, da Cunha DN, Lacombe VA, Zhou Q, Fox JJ, Hamlin RL, Carnes CA. The plateau outward current in canine ventricle, sensitive to 4-aminopyridine, is a constitutive contributor to ventricular repolarization. Br J Pharmacol. 2007;152:870–879. doi: 10.1038/sj.bjp.0707403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simko J, Szentandrassy N, Harmati G, Barandi L, Horvath B, Magyar J, Banyasz T, Lorincz I, Nanasi PP. Effects of ropinirole on action potential characteristics and the underlying ion currents in canine ventricular myocytes. Naunyn Schmiedebergs Arch Pharmacol. 2010;382:213–220. doi: 10.1007/s00210-010-0538-1. [DOI] [PubMed] [Google Scholar]
- Chandra PA, Chandra AB. Brugada syndrome unmasked by lithium. South Med J. 2009;102:1263–1265. doi: 10.1097/SMJ.0b013e3181bfae69. [DOI] [PubMed] [Google Scholar]
- Darbar D, Yang T, Churchwell K, Wilde AA, Roden DM. Unmasking of Brugada syndrome by lithium. Circulation. 2005;112:1527–1531. doi: 10.1161/CIRCULATIONAHA.105.548487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laske C, Soekadar SR, Laszlo R, Plewnia C. Brugada syndrome in a patient treated with lithium. Am J Psychiatry. 2007;164:1440–1441. doi: 10.1176/appi.ajp.2007.07020226. [DOI] [PubMed] [Google Scholar]
- Pirotte MJ, Mueller JG, Poprawski T. A case report of Brugada-type electrocardiographic changes in a patient taking lithium. Am J Emerg Med. 2008;26:113.e111–113.e113. doi: 10.1016/j.ajem.2007.06.028. [DOI] [PubMed] [Google Scholar]
- Roberts-Thomson KC, Teo KS, Young GD. Drug-induced Brugada syndrome with ST-T wave alternans and long QT. Intern Med J. 2007;37:199–200. doi: 10.1111/j.1445-5994.2006.01286.x. [DOI] [PubMed] [Google Scholar]
- Wright D, Salehian O. Brugada-type electrocardiographic changes induced by long-term lithium use. Circulation. 2010;122:e418–e419. doi: 10.1161/CIRCULATIONAHA.110.951244. [DOI] [PubMed] [Google Scholar]
- El-Menyar A, Khan M, Al Suwaidi J, Eljerjawy E, Asaad N. Oxcarbazepine-induced resistant ventricular fibrillation in an apparently healthy young man. Am J Emerg Med. 2011;29:693.e691–693.e693. doi: 10.1016/j.ajem.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Ambrosio AF, Soares-Da-Silva P, Carvalho CM, Carvalho AP. Mechanisms of action of carbamazepine and its derivatives, oxcarbazepine, BIA 2-093, and BIA 2-024. Neurochem Res. 2002;27:121–130. doi: 10.1023/a:1014814924965. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze-Bahr E, Tan H, Wilde A. Brugada syndrome: report of the second consensus conference. Heart Rhythm. 2005;2:429–440. doi: 10.1016/j.hrthm.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Imaizumi Y, Giles WR. Quinidine-induced inhibition of transient outward current in cardiac muscle. Am J Physiol. 1987;253:H704–H708. doi: 10.1152/ajpheart.1987.253.3.H704. [DOI] [PubMed] [Google Scholar]
- Clark RB, Sanchez-Chapula J, Salinas-Stefanon E, Duff HJ, Giles WR. Quinidine-induced open channel block of K+ current in rat ventricle. Br J Pharmacol. 1995;115:335–343. doi: 10.1111/j.1476-5381.1995.tb15882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi K, Noda T, Kamakura S, Shimizu W. Beneficial effects of cilostazol in a patient with recurrent ventricular fibrillation associated with early repolarization syndrome. Heart Rhythm. 2013;10:604–606. doi: 10.1016/j.hrthm.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Tsuchiya T, Ashikaga K, Honda T, Arita M. Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 2002;13:698–701. doi: 10.1046/j.1540-8167.2002.00698.x. [DOI] [PubMed] [Google Scholar]
- Abud A, Bagattin D, Goyeneche R, Becker C. Failure of cilostazol in the prevention of ventricular fibrillation in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 2006;17:210–212. doi: 10.1111/j.1540-8167.2005.00290.x. [DOI] [PubMed] [Google Scholar]
- Kanlop N, Chattipakorn S, Chattipakorn N. Effects of cilostazol in the heart. J Cardiovasc Med (Hagerstown) 2011;12:88–95. doi: 10.2459/JCM.0b013e3283439746. [DOI] [PubMed] [Google Scholar]
- Patel SP, Campbell DL. Transient outward potassium current, ‘Ito’, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J Physiol. 2005;569:7–39. doi: 10.1113/jphysiol.2005.086223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornblath DR, Bienen EJ, Blight AR. The safety profile of dalfampridine extended release in multiple sclerosis clinical trials. Clin Ther. 2012;34:1056–1069. doi: 10.1016/j.clinthera.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Nattel S, Matthews C, De Blasio E, Han W, Li D, Yue L. Dose-dependence of 4-aminopyridine plasma concentrations and electrophysiological effects in dogs: potential relevance to ionic mechanisms in vivo. Circulation. 2000;101:1179–1184. doi: 10.1161/01.cir.101.10.1179. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- Marquez MF, Salica G, Hermosillo AG, Pastelin G, Gomez-Flores J, Nava S, Cardenas M. Ionic basis of pharmacological therapy in Brugada syndrome. J Cardiovasc Electrophysiol. 2007;18:234–240. doi: 10.1111/j.1540-8167.2006.00681.x. [DOI] [PubMed] [Google Scholar]
- Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C, Ackerman M, Belhassen B, Estes NA, III, Fatkin D, Kalman J, Kaufman E, Kirchhof P, Schulze-Bahr E, Wolpert C, Vohra J, Refaat M, Etheridge SP, Campbell RM, Martin ET, Quek SC. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace. 2013;15:1389–1406. doi: 10.1093/europace/eut272. [DOI] [PubMed] [Google Scholar]
- Sridhar A, Nishijima Y, Terentyev D, Khan M, Terentyeva R, Hamlin RL, Nakayama T, Gyorke S, Cardounel AJ, Carnes CA. Chronic heart failure and the substrate for atrial fibrillation. Cardiovasc Res. 2009;84:227–236. doi: 10.1093/cvr/cvp216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–1963. doi: 10.1016/j.hrthm.2013.05.014. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze-Bahr E, Tan H, Wilde A. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–670. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- Alders M, Koopmann TT, Christiaans I, Postema PG, Beekman L, Tanck MW, Zeppenfeld K, Loh P, Koch KT, Demolombe S, Mannens MM, Bezzina CR, Wilde AA. Haplotype-sharing analysis implicates chromosome 7q36 harboring DPP6 in familial idiopathic ventricular fibrillation. Am J Hum Genet. 2009;84:468–476. doi: 10.1016/j.ajhg.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szel T, Koncz I, Antzelevitch C. Cellular mechanisms underlying the effects of milrinone and cilostazol to suppress arrhythmogenesis associated with Brugada syndrome. Heart Rhythm. 2013;10:1720–1727. doi: 10.1016/j.hrthm.2013.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao GS, Liao YH. Effect of cilostazol on transient outward potassium current in human atrial myocytes. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2004;20:238–241. [PubMed] [Google Scholar]
- Wettwer E, Hala O, Christ T, Heubach JF, Dobrev D, Knaut M, Varro A, Ravens U. Role of IKur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation. 2004;110:2299–2306. doi: 10.1161/01.CIR.0000145155.60288.71. [DOI] [PubMed] [Google Scholar]
- Burashnikov A, Antzelevitch C. Can inhibition of IKur promote atrial fibrillation? Heart Rhythm. 2008;5:1304–1309. doi: 10.1016/j.hrthm.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurabi Z, Koncz I, Patocskai B, Nesterenko VV, Antzelevitch C. Cellular mechanism underlying hypothermia-induced ventricular tachycardia/ventricular fibrillation in the setting of early repolarization and the protective effect of quinidine, cilostazol, and milrinone. Circ Arrhythm Electrophysiol. 2014;7:134–142. doi: 10.1161/CIRCEP.113.000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge KE, Haugaa KH, Fruh A, Anfinsen OG, Gjesdal K, Siem G, Oyen N, Greve G, Carlsson A, Rognum TO, Hallerud M, Kongsgard E, Amlie JP, Leren TP. Molecular genetic analysis of long QT syndrome in Norway indicating a high prevalence of heterozygous mutation carriers. Scand J Clin Lab Invest. 2008;68:362–368. doi: 10.1080/00365510701765643. [DOI] [PubMed] [Google Scholar]
- Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Brignole M, Giordano U, Giovannini T, Menozzi C, Bloise R, Crotti L, Terreni L, Schwartz PJ. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: a prospective evaluation of 52 families. Circulation. 2000;102:2509–2515. doi: 10.1161/01.cir.102.20.2509. [DOI] [PubMed] [Google Scholar]
- Pan S, Caleshu CA, Dunn KE, Foti MJ, Moran MK, Soyinka O, Ashley EA. Cardiac structural and sarcomere genes associated with cardiomyopathy exhibit marked intolerance of genetic variation. Circ Cardiovasc Genet. 2012;5:602–610. doi: 10.1161/CIRCGENETICS.112.963421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave-Castillo LM, Wolfgeher D, McNally EM. Population-based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5:391–399. doi: 10.1161/CIRCGENETICS.112.962928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi F, Guicheney P, Fressart V, Leenhardt A, Antzelevitch C, Bartkowiak S, Borggrefe M, Schimpf R, Schulze-Bahr E, Zumhagen S, Behr ER, Bastiaenen R, Tfelt-Hansen J, Olesen MS, Kaab S, Beckmann BM, Weeke P, Watanabe H, Endo N, Minamino T, Horie M, Ohno S, Hasegawa K, Makita N, Nogami A, Shimizu W, Aiba T, Froguel P, Balkau B, Lantieri O, Torchio M, Wiese C, Weber D, Wolswinkel R, Coronel R, Boukens BJ, Bezieau S, Charpentier E, Chatel S, Despres A, Gros F, Kyndt F, Lecointe S, Lindenbaum P, Portero V, Violleau J, Gessler M, Tan HL, Roden DM, Christoffels VM, Le Marec H, Wilde AA, Probst V, Schott JJ, Dina C, Redon R. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45:1044–1049. doi: 10.1038/ng.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haissaguerre M, Shoda M, Jais P, Nogami A, Shah DC, Kautzner J, Arentz T, Kalushe D, Lamaison D, Griffith M, Cruz F, de Paola A, Gaita F, Hocini M, Garrigue S, Macle L, Weerasooriya R, Clementy J. Mapping and ablation of idiopathic ventricular fibrillation. Circulation. 2002;106:962–967. doi: 10.1161/01.cir.0000027564.55739.b1. [DOI] [PubMed] [Google Scholar]
- Zicha S, Xiao L, Stafford S, Cha TJ, Han W, Varro A, Nattel S. Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J Physiol. 2004;561:735–748. doi: 10.1113/jphysiol.2004.075861. [DOI] [PMC free article] [PubMed] [Google Scholar]