

Abstract
Background
Heart failure (HF) is a progressive disorder characterized by reduced cardiac output and increased peripheral resistance, ultimately leading to tissue perfusion deficits and devastating consequences for several organs including the brain. We previously described a tumor necrosis factor-α (TNF-α)–dependent enhancement of posterior cerebral artery tone and concomitant reduced cerebral blood flow in a mouse model of early HF in which blood pressure remains minimally affected. HF is often associated with cognitive impairments such as memory deficits, even before any overt changes in brain structure and function occur. The pathophysiology underlying the development of cognitive impairments in HF is unknown, and appropriate treatment strategies are lacking.
Methods and Results
We used a well-established mouse model in which HF was induced by experimental myocardial infarction produced by permanent surgical ligation of the left anterior descending coronary artery (infarct size ≈25% of the left ventricular wall). Ligated mice developed enlarged hearts, congested lungs, and reduced cardiac output and blood pressure, with elevated peripheral resistance within 6 to 8 weeks after ligation. In this study, we demonstrated the significance of the proinflammatory cytokine TNF-α during HF-mediated neuroinflammation and associated impaired hippocampus-independent nonspatial episodic memory function. Augmented cerebral TNF-α expression and microglial activation in HF mice, indicative of brain inflammation, were accompanied by morphological changes and significant reduction of cortical dendritic spines (61.39±8.61% for basal and 61.04±9.18% for apical spines [P<0.001]). The significance of TNF-α signaling during the observed HF-mediated neurodegenerative processes is supported by evidence showing that sequestration or genetic deletion of TNF-α ameliorates the observed reduction of cortical dendritic spines (33.51±7.63% for basal and 30.13±6.98% for apical spines in wild-type mice treated with etanercept; 17.09±6.81% for basal and 17.21±7.29% for apical spines in TNF-α−/−). Moreover, our data suggest that alterations in cerebral serum and glucocorticoid-inducible kinase 1 (SgK1) expression and phosphorylation during HF may be TNF-α dependent and that an increase of SgK1 phosphorylation potentially plays a role in the HF-associated reduction of dendritic spine density.
Conclusions
Our findings demonstrate that TNF-α plays a pivotal role in HF-mediated neuroinflammation and associated alterations of cortical dendritic spine density and has the potential to reveal novel treatment strategies for HF-associated memory deficits.
Keywords: heart failure, inflammation, nervous system
Congestive heart failure (HF) is a progressive disorder characterized by reduced cardiac output and increased peripheral resistance, ultimately leading to tissue perfusion deficits.1 As the heart fails, severe dysfunction of liver, kidney, and lung contributes to mortality in HF2; however, a concomitant decline in cognitive function also significantly reduces patients’ quality of life. HF-associated cognitive impairments (CIs), such as memory deficits, inability to make sound decisions, attention deficits, impaired visual perception, and difficulties in problem solving, negatively affect many aspects of daily life and may impair patients’ capacity to recognize worsening HF symptoms, thereby lowering self-care.3
Neurological deficits in HF are reported to result from structural changes in the cortex, alterations in white matter, cerebral hypoperfusion, and impaired blood flow regulation in small vessels.4 Hemodynamic abnormalities, specifically cerebral hypoperfusion and impaired cerebral vascular function, are hallmarks of a variety of cerebrovascular conditions including hemodynamic stroke, microangiopathy, and vascular dementia. HF has emerged as a model for the effects of whole-brain hypoperfusion on cognitive dysfunction that affects complex cognitive tasks such as memory, attention, and executive skills more than the focal deficits that result from embolic stroke types mediated by cardiac thromboemboli.5 As HF progresses to more severe states, reduced cerebral blood flow correlates with declining neurological function.6,7 Moreover, some pathophysiological links between cerebral hypoperfusion and structural changes, including white matter hyperintensities, have already been established.8 To date, it is unknown whether impairments in dendritic spine morphology, which is associated with most neurological disorders that are characterized by alterations in cognition and memory loss, also occur in HF.
Dendritic spines are specialized structures with distinct functions and are vital elements of synaptic function and plasticity.9 As dynamic structures they undergo morphological remodeling in development, in adaptation to sensory stimuli, or in learning and memory. Changes in both the number and morphology of spines govern alterations in synaptic plasticity, which, in turn, influence learning and memory.9 Spines may be regulated by extracellular factors including neurotransmitters, growth factors, and hormones.10 Importantly, cytokines released from glial cells can also alter spine morphology and affect synaptic function.11 Increased levels of proinflammatory cytokines, resulting from microglial cell proliferation, were found to be associated with alterations in dendritic spine morphology and density in animal models of neurodegenerative disorders.12
Following a myocardial infarction (MI), the immune system is activated and levels of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukins 6 and 1β are elevated.13 Augmented circulating levels of these cytokines have been shown to correlate with the degree of HF and increased mortality with this condition14 and have been associated with the occurrence of devastating cerebral complications such as stroke.15,16 In addition to the elevation of proinflammatory cytokines in the periphery,14 their levels in the brain are increased.17 This elevation likely results from local cytokine production.18 Within the brain, glial cells are a major source of many of the resident cytokines in the central nervous system.19 When microglia are activated, they secrete TNF-α,20 contributing to local elevation of proinflammatory cytokines reported in neurodegenerative diseases such as Alzheimer disease and Parkinson disease.21 Microglial activation and increases in TNF-α have been found in various models of peripheral inflammation.22 Whether a similar proinflammatory response also occurs following MI is currently unknown.
TNF-α and its receptors activate the nuclear factor-κB (NF-kB) pathway, a pivotal transcriptional factor essential for controlling the expression of several genes involved in inflammation, adhesion, and memory function.23 NF-kB is activated by a diverse array of stimuli including proinflammatory cytokines, free radicals, and growth factors. Its functions in the brain have yet to be fully elucidated. Evidence demonstrates that NF-kB can be activated by serum and glucocorticoid-inducible kinase 1 (SgK1),24 which is known to regulate a variety of cellular functions including neuronal excitability.25 By activating I-κB kinase and p300, SgK1 increases NF-kB activity and regulates neuronal plasticity by enhancing N-methyl-D-aspartate receptor (NMDAR) NR2A and NR2B expression.24 Compelling evidence suggests a role for SgKs in the pathophysiology of brain disease,25,26 although the precise mechanisms by which SgKs may be involved in the deterioration or maintenance of neuronal function remain to be elucidated.
In the present study, we demonstrated an HF-mediated loss of cortical dendritic spines and parallel hippocampus-independent mild CI. We described an important role for TNF-α in these observations because blocking TNF-α, either pharmacologically by etanercept (Etn) or genetically (TNF-α−/−), significantly attenuated the HF-associated loss of dendritic spine density and memory deficits. Furthermore, our results suggest that an increase of SgK1 phosphorylation may contribute to the HF-associated reduction of dendritic spine density and CI.
Methods
Reagents
Etn was purchased from Amgen. All chemical reagents and solutions were purchased from Sigma-Aldrich unless otherwise stated. Commercially available primary antibodies against TNF-α and CD11b (Abcam); NMDAR2A (Chemicon); PSD-95 (postsynaptic density protein 95; Life Technologies); NF-kB/p65, phosphorylated SgK (Ser 422), and GAPDH (Santa Cruz Biotechnology); and SgK1 (Millipore) were used for Western blotting. These were conjugated with either horseradish peroxidase–conjugated anti-mouse or anti-rabbit IgG (Sigma-Aldrich) or peroxidase-labeled sheep anti-mouse/rabbit IgG antibody (GE Healthcare) and detected with ECL Plus enhanced chemiluminescence substrates (GE Healthcare) using an Odyssey fluorescence imager or x-ray film. For immunohistochemistry, commercially available primary antibody against ionized calcium-binding adapter molecule 1 (Iba1; Wako Chemicals) was conjugated with ImmPRESS anti-Rabbit IgG kit (Vector Laboratories). Primers for quantitative polymerase chain reaction (PCR) were purchased from Applied Biosystems. A detailed list of primer sequences can be found in Table 1.
Table 1.
RT and qPCR Primers
| Gene | Primer Sequence |
|---|---|
| SGK1 (qPCR) | Forward: 5′-GATGGGCCTGAACGATTTTA-3′ |
| Reverse: 5′-GTTCATAAGCTCCGGCTCCT-3′ | |
| TNF-α (qPCR) | Forward: 5′-GGGACAGTGACCTGGACTGT-3′ |
| Reverse: 5′-CTCCCTTTGCAGAACTCAGG-3′ | |
| TNFR1 (RT) | Forward: 5′-GGGCACCTTTACGGCTTCC-3′ |
| Reverse: 5′-GGTTCTCCTTACAGCCACACA-3′ | |
| TNFR2 (RT) | Forward: 5′-CAGGTTGTCTTGACACCCTAC-3′ |
| Reverse: 5′-GCACAGCACATCTGAGCCT-3′ | |
| β-actin (qPCR) | Forward: 5′-GGCACCACACCTTCTACAATG-3′ |
| Reverse: 5′-TGGATGGCTACGTACATGGCTG-3′ | |
| GAPDH (RT) | Forward: 5′-TTCACCACCATGGAGAAGG-3′ |
| Reverse: 5′-CTCGTGGTTCACACCCATC-3′ |
SGK1 indicates serum and glucocorticoid-inducible kinase 1; qPCR, quantitative polymerase chain reaction; RT, reverse transcription; TNF-α, tumor necrosis factor-α.
Animals
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH; publication No. 85-23, revised 1996). All animal care and experimental protocols were approved by the institutional animal care and use committee at the University of Toronto and were conducted in accordance with Canadian animal protection laws. Commercially available wild-type mice (2 to 3 months; C57BL/6N) were purchased from Charles River Laboratories (Montreal, Canada). Tumor necrosis factor (TNF-α−/−) knockout mice27 were purchased commercially from Taconic Laboratories (Hudson, NY, USA). All mice were housed under a standard 12:12-hour light–dark cycle, fed normal chow, and had access to water ad libitum.
Myocardial Infarction
Equal numbers of male and female mice underwent ligation of the left anterior descending coronary artery or sham operation at 9 to 12 weeks of age, according to a protocol approved by the University of Toronto committee on animal care. Animals were anesthetized with 1.5% isoflurane in 30% O2 and 70% N2O, intubated, and ventilated using a pressure-control ventilator (Kent Scientific). The thorax and pericardium were opened, and the heart was exposed. With the use of a 7-0 silk suture (Deknatel), the left anterior descending coronary artery was ligated, the chest was closed, and the animal was allowed to recover. In sham-operated control animals, the thorax and pericardium were opened, but the left anterior descending coronary artery was not ligated. Following either procedure, the chest was closed, and the mice were extubated on spontaneous respiration. As described previously, HF mice (infarct size ≈25% of the left ventricular wall) develop enlarged hearts, congested lungs, and reduced cardiac output and blood pressure, with elevated peripheral resistance. All of these parameters remained normal in sham-operated controls.28 At 6 weeks after MI, cohorts of wild-type HF mice were treated with either the TNF-α scavenger Etn (1 mg/mL subcutaneously; n=12) or saline (n=12) twice weekly for 2 weeks, as described previously.29 Sham-operated animals served as control (n=12). In TNF-α−/− mice, neither the HF group (n=10) nor the sham group (n=10) received any drug treatment. Dissected brains were used for Golgi staining and subsequent dendritic spine analysis (n=4) or for immunohistochemistry experiments (n=3), or the 2 hemispheres were separated and equally distributed to protein or RNA isolation (n=5).
Locomotor Activity Assessment
Spontaneous locomotor activity was assessed using an automated movement-detection system for 60 minutes (AM1053 activity monitors; Linton Instrumentation). This apparatus comprises a Perspex box (20×30 cm) surrounded by a frame (45×25 cm) containing an array of 24 infrared beams, forming a grid across 2 levels. As the animal moves, a beam is broken, and an activity count is registered. Several behavioral parameters are measured using this system, depending on the location and succession of the activity counts. The following behavioral parameters were measured: activity, rearing, center rearing, front to back count, active time, mobile time, rearing time, slow activity, slow rearing, slow center rearing, mobile counts, and slow mobile counts.
Novel Object Recognition
Wild-type mice (n=10 for each group) were habituated to the testing arena for 15 minutes over a period of 7 days. Habituation and object memory testing were conducted, as described previously.30 On the test day, each mouse was exposed to a LEGO construct (LEGO Group) and a Hot Wheels car (Mattel Inc) for 10 minutes. Five minutes later, mice were reexposed to 1 object from the original test pair and to a novel object. The novel object was selected from a larger pool of objects that mice were demonstrated to spend an equal time exploring, as described previously.31
A delay interval of 5 minutes was chosen to test short-term retention of object familiarity, which is dependent on rhinal cortical function.30,32,33 With delay intervals >15 minutes, retention becomes dependent on reactivation of memory traces in the hippocampus.34 The placement of the new object was counterbalanced between mice for either the left or right side of the cage to address any confounding influences of handedness. In between testing states, the objects and the cage were wiped with Virox 5 (Diversey Inc). The movements of the animal were video tracked with the computer software AnyMaze (Stoelting Co). A discrimination index was calculated as DI=(Tn−Tf)/(Tn+Tf), in which DI was the discrimination index, Tn was the time spent exploring the novel object, and Tf was the time spent exploring the familiar object. This result could vary between +1 and −1; a positive score indicated more time spent with the novel object, a negative score indicated more time spent with the familiar object, and a zero score indicated a null preference.35
Dendritic Spine Morphology Analysis and Quantification
Brains were removed and processed for Golgi histology based on Glaser and Van der Loos’ modified Golgi stain,36 using a commercially available Rapid GolgiStain Kit (Neurodigitech Inc). Freshly dissected tissue was immersed in osmium dichromate at room temperature in the dark. Following 7 days incubation, tissue was rinsed 3 times in distilled water and then in 0.75% silver nitrate. Tissue was incubated in 0.75% silver nitrate in the dark at room temperature for 24 hours and rinsed in distilled water 3 times before dehydration in 70% ethanol for 2 hours. Tissue was then embedded in paraffin blocks using an automated tissue processor.
Paraffin-embedded Golgi-stained brains were sectioned at a sagittal plane (25- to 30-μm thickness) using a sliding microtome (Leica Microsystems) and mounted on slides. Slides were incubated at 60°C for 1 hour to remove excess wax and enforce adherence to the slide. Slides were then cooled to room temperature for 1 hour before deparafinization. The slides were processed with 2× xylene solution and coverslipped with DPX mounting medium (EMS). The slides were air dried at room temperature for ≈2 to 3 weeks before quantitative analysis.
Analysis was performed as described previously by Wong et al.37 Regions included in analysis encompassed both apical and basal dendrites of cortical neurons from both frontal and parietal cortical regions. Dendritic segments were analyzed using stereology-based software (MBF Bioscience) coupled to a Zeiss Axioplan 2 image microscope with motorized X, Y, and Z-focus for high-resolution image acquisition and digital quantitation. All analysis was carried out by an experimenter who was blinded to the experimental condition of the sample.
Synaptosome Preparation and RNA Extraction
Intact synaptosomes were prepared from 3 sham-operated mice and 3 congestive HF–operated mice 5 months after surgery. Synaptosome preparation was performed, as described previously.1 Briefly, freshly dissected whole brains were homogenized in ice-cold buffer A (320 mmol/L sucrose, 1 mmol/L EGTA, and 5 mmol/L HEPES; pH 7.4) and centrifuged (1000g, 10 minutes). The supernatant was centrifuged (24 000g, 10 minutes), and the resulting pellet P2 was resuspended in buffer A. The P2 fraction was loaded onto a discontinuous Ficoll gradient (13%, 9%, and 5% in buffer A) and centrifuged (35 000g, 35 minutes). The interface from 13% to 9%, containing intact synaptosomes, was resuspended in buffer B (140 mmol/L NaCl, 5 mmol/L KCl, 20 mmol/L HEPES, 5 mmol/L NaHCO3, 1.2 mmol/L Na2HPO4, 1 mmol/L MgCl2, 1 mmol/L EGTA, and 10 mmol/L glucose) and centrifuged (24 000g, 10 minutes). Synaptosomal RNA was extracted and purified according the manufacturer’s instructions using an Absolutely RNA nanoprep kit (Stratagene) with in-column DNA digestion. RNA quantity was determined using a NanoDrop 1000 spectrophotometer, and quality was determined using an Agilent 2100 Bioanalyzer.
Gene Expression Profiling
For gene expression profiling, 1 ng of extracted RNA from each brain homogenate (Stratagene or Agilent) was amplified using the WT-Ovation Pico RNA amplification system version 1.0 (NuGEN), and cDNA was run on a bioanalyzer (Agilent) for quality control. All samples had an RNA integrity number of >7.8. Subsequently, 3.5 μg cDNA was biotin labeled (NuGEN Illumina), and 1.5 μg of cDNA was hybridized to the Illumina Mouse WG-6 v2.0 Expression BeadChip containing 45 821 probes. BeadChips were incubated at 48°C with a rotation speed of 5 for 18.0 hours of hybridization. BeadChips were then washed and stained, according to Illumina’s protocol, and scanned on the iScan (Illumina). Data files were quantified in GenomeStudio version 2010.2 (Illumina). All samples passed Illumina’s sample-dependent and -independent quality control metrics. Data were further checked for overall quality using R (version 2.14.1; R Foundation) with the Bioconductor framework and the lumi package installed. There were no discernible outliers.
Data was imported into Genespring v12.0 (Agilent) for analysis and normalized with a quantile normalization followed by a median centering. All data analysis and visualization were performed on log2 transformed data. There were only 2 groups, (CHF and sham) therefore standard t-test style statistics were used. A total of 45821 probes are represented on the Illumina WG6 V2R2 array. Data was first filtered to remove the confounding effect probes that show no signal may have on subsequent analysis. Only probes that were in the upper 80th percentile of the distribution of intensities in 100% of any of the 1 of 2 above categories were allowed to pass through this filtering. The final set for analysis contained 33252 probes.
A t test was performed using a Benjamini and Hochberg false discovery rate multiple-testing correction threshold of P<0.05. With the multiple testing correction in place, no probes were found to be different among groups. An uncorrected t test yielded a list of 2052 probes that significantly differed among treatment groups. A cutoff of >1.5-fold change was applied, yielding a final list of 6 genes.
Quantitative Real-Time PCR
RNA was isolated using a Qiagen RNeasy Kit, according to instructions using a Superscript III kit (Invitrogen Life Technologies), and RNA was reverse transcribed with random hexamer primers. The resulting cDNA was diluted and subsequently used as a template for PCR. Quantitative PCR was performed in duplicate using Promega GoTaq PCR mastermix, according to the manufacturer’s instructions. Each reaction comprised 1 ng cDNA, 12.5 μL GoTaq, and 0.2 μmol/L final concentration of each primer. Cycling and detection were carried out using an Applied Biosystems 7500 Real Time PCR System, and data were quantified using Sequence Detection Software version 1.4 (Applied Biosystems). PCR was performed for a total of 40 cycles (95° 15 seconds, 60° 60 seconds) followed by a dissociation stage. All data were normalized to actin, and quantification was carried out by the absolute method using standard curves generated from pooled cDNA representative of each sample to be analyzed. Details of primer sequences used are provided in Table 1.
Western Blotting
Dissected brain tissue was homogenized in 10× volume homogenization buffer (100 mmol/L NaCl, 50 mmol/L Tris, 1 mmol/L EDTA, 1% NP-40, and protease inhibitor cocktail). After 20 minutes of incubation at 4°C, homogenates were centrifuged at 20 000g for 5 minutes. Protein concentration was determined by bicinchoninic acid assay (Thermo Scientific). Western blotting was carried out according to standard protocols. Briefly, samples were heated for 10 minutes at 70°C in 25% NuPage LDS sample buffer (Invitrogen). Proteins were then separated on 4% to 12% bis-Tris NuPage Novex Mini gels (Invitrogen) and transferred onto nitrocellulose membranes (Life Sciences) to probe with primary antibodies to TNF-α (1:1000), phosphorylated SgK (Ser 422, 1:500), SgK1 (1:800), and PSD-95 (1:100). Bound horseradish peroxidase–conjugated anti-mouse or anti-rabbit IgG were revealed by chemiluminescence using ECL Plus and detected with an Odyssey fluorescent imager. For immunostaining of CD11b, NMDAR2, and NF-kB, proteins were separated electrophoretically and transferred onto PVDF membranes (Millipore). The membranes were blocked for 60 minutes in 5% nonfat skim milk (in PBST [PBS containing 1% Tween 20]; 137 mmol/L NaCl, 2.7 mmol/L KCl, 10 mmol/L Na2HPO4, 1.76 mmol/L K2HPO4; pH 7.4) and sequentially incubated with the primary and secondary antibodies. Dilution of 1:1000 for primary antibodies and 1:40 000 for the peroxidase-labeled secondary antibody (all diluted in 2% BSA in PBST) were used. A standard chemiluminescence procedure was used to expose x-ray film; developed films were evaluated densitometrically using Image J software (NIH).
Immunohistochemistry
Whole hemispheres of brain were fixed in 4% paraformaldehyde and then embedded in paraffin. Paraffin-embedded tissue was sectioned at a thickness of 5 μm in a sagittal plane and mounted on glass slides. After removal of paraffin in xylene and rehydration in a graded series of alcohols (100%, 100%, 95%), sections were digested in 1% pepsin in 0.01 N HCl at 37°C for 15 minutes and blocked in 2.5% normal horse serum. Sections were incubated in primary antibody to Iba1 (1:800) overnight at room temperature. Sections were washed in 1× Tris-buffered saline, and staining was completed using the ImmPRESS anti-rabbit Ig kit. Sections were washed in 1× Tris-buffered saline and developed with freshly prepared DAB solution (Dako) before dehydration in graded alcohols, clearing in xylene, and mounting in Permount (Fisher Scientific).
Iba1 Semiquantification
Immunostained sections were scanned at ×40 using a ScanScope digital slide scanner (Aperio Technologies, Inc). Image files were viewed with ImageScope (Aperio Technologies, Inc). Positive pixel analysis of tissue stained with Iba1 was carried out using a custom-designed algorithm in ImageScope, as described previously.38 An experimenter blinded to the treatment group of the tissue sample performed the analysis. The mean number of Iba1-positive pixels was calculated in 10 microglia per section, providing a semiquantitative measure of microglial size.
Assessment of myocardial infarct size
A standard procedure for determining infarct size was followed.28,39 Following euthanization, hearts were removed, fixed in 10% formaldehyde, and paraffin embedded. Slices (10 μm) were stained with hematoxylin and eosin. Infarct size was calculated by comparing the circumferential length of the infarct to the total myocardial length, resulting in a percentage-based index in infarct size.
Magnetic resonance imaging–based measurement of brain perfusion
The FAIR (flow-sensitive alternating inversion recovery) technique was used to evaluate brain perfusion with a 7-T micro–magnetic resonance imaging system (BioSpec 70/30 USR; Bruker BioSpin), including the B-GA12 gradient insert, 72-mm inner diameter linear volume resonator for radio frequency transmission, and anteriorly placed head coil for radio frequency reception. FAIR isolates perfusion as an accelerated T1 signal relaxation following slice-selective versus nonselective inversion preparation, based on the following equation, where T1 refers to spin-lattice relaxation: CBF=λ (1/T1, ss−1/T1, ns) (mL/(100 g×minute)). CBF indicates cerebral blood flow, ss and ns denote slice-selective and nonselective measurements, and λ is the blood–brain partition coefficient, defined as the ratio between water concentration per gram of brain tissue and per milliliter of blood. This coefficient is ≈90 mL/100 g in mice.4
FAIR optimization used in our study was a single-shot echo-planar imaging technique with preceding adiabatic inversion. Parameters included echo time of 11 milliseconds, repetition time of 17 seconds, 18 inversion times ranging from 25 to 6825 milliseconds in 400-millisecond increments, 3-mm slice-selective inversion slab, 16.8×16.8-mm field of view with a 64×64 matrix for 263-μm in-plane resolution, 1-mm slice thickness, and 10 minutes and 12 seconds of data acquisition time. Acquisitions were repeated in fore-, middle-, and hind-brain vertical sections, corresponding to anterior, mixed, and posterior circulation.
FAIR images were evaluated by manual prescription (MIPAV application, NIH; mipav.cit.nih.gov) of subhemispheric regions of interest, termed global and local regions of interest, corresponding to cortical and subcortical parenchyma in fore-brain sections, cortical and paraventricular parenchyma in middle sections, and cortical and midbrain parenchyma in hind-brain sections. Regions of interest were drawn directly on T1-weighted signal images to enable manual correction for intrascan motion. Regions of interest were registered with parametric cerebral blood flow maps to verify absence of bias from high-perfusion vessels and meninges. T1 regressions and cerebral blood flow calculations were performed using Matlab (MathWorks). Significance was defined by 1-tailed Student t test (P<0.05).
Statistical Analysis
All data are expressed as mean±SEM. For comparison of multiple independent groups, a nonparametric 1-way ANOVA (Kruskal–Wallis) was used, followed by a Bonferroni test with exact P value computation as a post hoc test for comparisons. For comparison of 2 groups, a 2-tailed unpaired t test was used. Differences were considered significant at error probabilities of P<0.05.
Results
Heart Failure Attenuates Hippocampus-Independent Nonspatial Episodic Memory Function
We assessed cortical components of memory using a nonspatial novel object recognition task with a short 5-minute retention interval, as described previously.30 Figure 1A and 1B illustrate that HF animals spent less time exploring the novel object (18.12±2.37 seconds) compared with sham control mice (24.25±3.86 seconds) and showed a clear preference for the familiar object (for all tasks: sham n=9, HF n=10, P<0.05) (Figure 1A and 1B). Deficits in exploration time for the novel objects were shown not to be related to locomotor activity because no difference in any aspect of locomotor activity was apparent between sham and HF mice (Figure 1C). Western blot analyses of whole-brain protein expression of NMDAR2A and PSD-95 revealed no difference between control and HF groups (NMDAR2A: 0.28±0.04 versus 0.31±0.06, n=7, P=0.6494; PSD-95: 0.71±0.04 versus 0.72±0.03, n=6, P=0.9074) (Figure 1D and1E).
Figure 1.
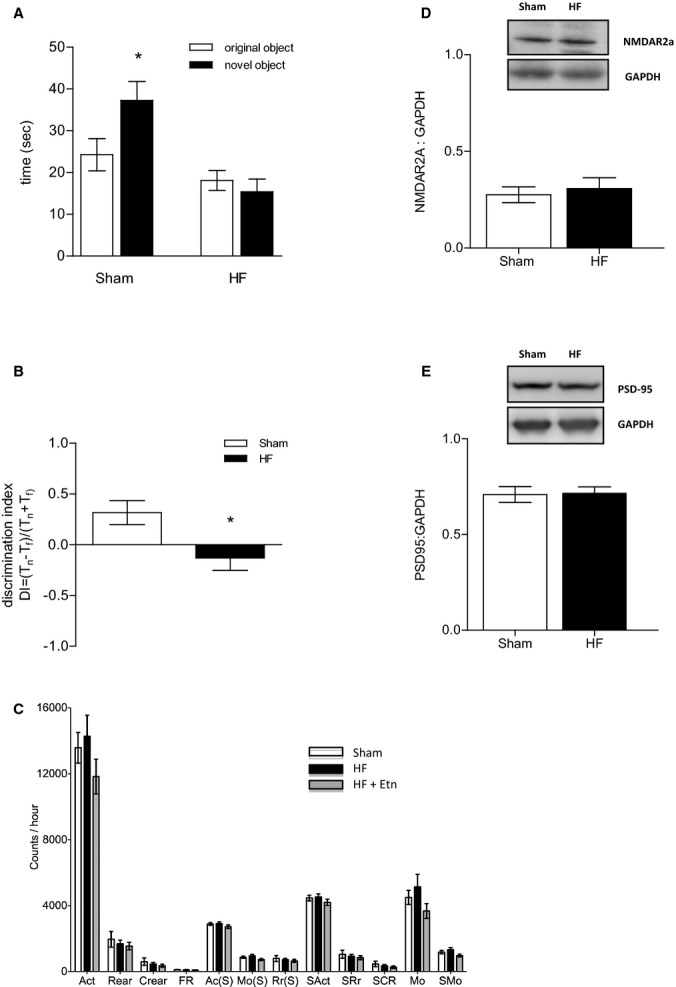
HF attenuates hippocampus-independent, short-term memory. A, HF attenuated the rodents’ short-term retention of object familiarity in a nonspatial novel object recognition task with a 5-minute retention interval. B, The calculated DI revealed a clear preference for the familiar rather than the novel object in HF mice. C, All aspects of locomotor activity were shown not to be significantly different between sham and HF mice. For all tasks: sham n=9, HF n=10,*P<0.05. D, Cerebral protein expression of NMDAR2A (n=7, P=0.6494) and (E) PSD-95 (n=6, P=0.9074) was not affected during HF. Ac(S) indicates active time; Act, activity; Crear, center rearing; DI, discrimination index; Etn, etanercept; FR, front to back count; HF, heart failure; Mo(S), mobile time; MO, mobile counts; NMDAR, N-methyl-D-aspartate receptor; PSD-95, postsynaptic density protein 95; Rear, rearing; Rr(S), rearing time; SAct, slow activity; SCR, slow center rearing; SMO, slow mobile counts; SRr, slow rearing.
Memory Deficits of HF Mice Are Accompanied by Increased Cerebral TNF-α Levels and Activated Microglia
We showed the significant upregulation of CD11b, a marker of microglial activation,40 in whole-brain lysates of HF mice (0.48±0.14 versus 1.08±0.14, n=5, P<0.05) (Figure 2A), and that was accompanied by visually observed morphological alterations in microglia of enlarged cytoplasm and reduced, thickened processes in tissue sections from HF animals (Figure 2B). The large cells represent neurons exhibiting lipofuscin, whereas the blue stain is the counterstain with toluidine blue. Semiquantitative analysis revealed that the mean number of positive and strongly positive pixels per microglia was significantly higher in HF animals (181 716±19 445) versus sham animals (104 475±10 838) (n=10; P<0.05).
Figure 2.
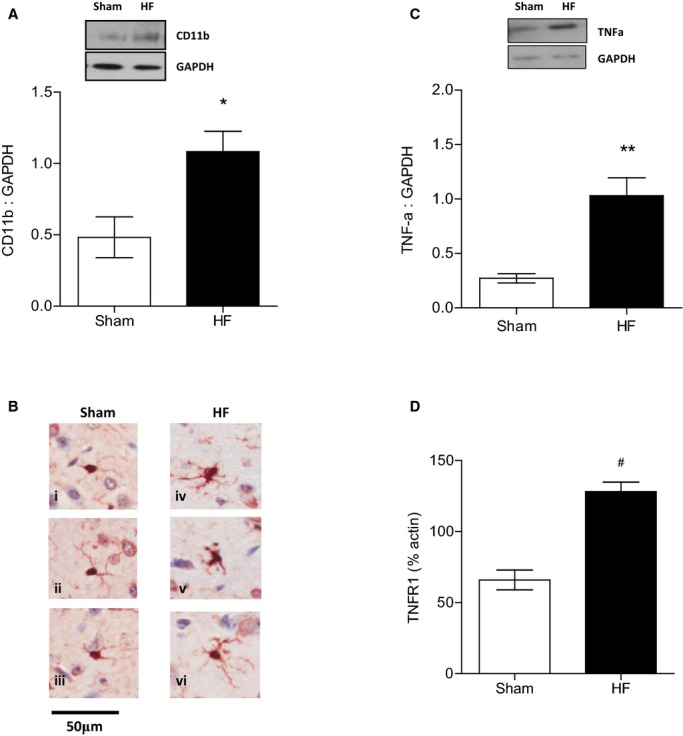
Memory deficits of HF mice are accompanied by increased cerebral TNF-α levels and activated microglia. A, Increased expression of CD11b, the β-integrin marker of microglia, represents microglial activation during neurodegenerative inflammation during HF (n=5, P<0.05). B, Cerebral tissue sections of HF mice show evidence of microglial activation with microglia exhibiting visibly enlarged cytoplasm and reduced, thickened processes (iv through vi) compared with sham-operated animals (i through iii). C, TNF-α protein levels in the brain were significantly increased in mice 8 to 10 weeks after myocardial infarction compared with sham controls (n=5, P<0.01). D, TNF-α receptor type 1 mRNA was significantly upregulated during HF compared with sham controls (n=6, P<0.001). *P<0.05, **P<0.01 and #P<0.001. HF indicates heart failure; TNF-α, tumor necrosis factor-α.
We also found that during HF, brain TNF-α levels were significantly elevated compared with sham control levels (1.12±0.17 versus 0.47±0.01; n=5; P<0.05) (Figure 2C). Interestingly, TNFR1 mRNA was also found to be significantly upregulated during HF compared with sham controls (65.9±6.9 versus 128.2±6.7, n=6, P<0.001) (Figure 2D), whereas TNFR2 mRNA expression remained unaltered (data not shown).
HF Induces Morphological Changes and a Reduction in Number of Dendritic Spines
Unbiased morphological analyses of dendritic spines revealed significant morphological alterations in cortical neurons of HF mice. The total dendritic spine density was reduced in the frontal cortex of HF mice by 61.39±8.61% for basal and 61.04±9.18% for apical spines (P<0.001) (Figure 3B). Breakdown analysis (dendrograms) further supported a significant loss of spine density in both basal and apical dendrites of HF mice compared with the sham group (n=6, P<0.001) (Figure 3C and 3D).
Figure 3.
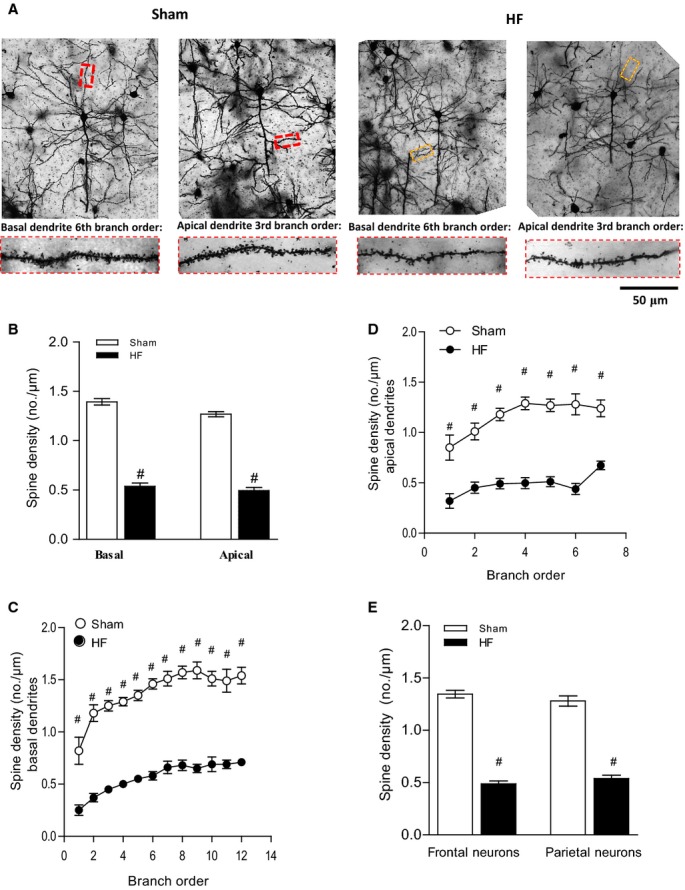
HF induces morphological changes and a reduction in number of dendritic spines. The sample population was composed of pyramidal cells chosen rostrocaudally from motor, sensory, and visual cortical regions (4 to 6 cells per animal). A, Depicts representative Golgi-stained images of cortical neurons of sham and HF wild-type mice and illustrates the differences between the basal and apical neurons of sham mice, which had significant more dendritic branches and thicker branch diameters than those of HF mice. The magnification shows details of both basal and apical dendrite morphology. B, Spine density of both basal and apical dendrites was reduced in the frontal cortex in HF mice. Breakdown analysis (dendrograms) further revealed a significant loss of spines or spine density in both (C) basal and (D) apical dendrites of HF compared with sham. E, Spine density of frontal neurons and parietal neurons is significantly reduced during HF. For all tasks: sham n=18, HF n=20, #P<0.001. HF indicates heart failure.
Pyramidal neurons consist of frontal neurons associated with attention, short-term memory, and motivation and parietal neurons associated with the integration of sensory information in short-term memory.32,33,41 The differential analysis revealed a significant reduction of spine density in both frontal and parietal neurons during HF compared with sham control (1.34±0.04 versus 0.49±0.03 for frontal neurons and 1.28±0.05 versus 0.54±0.03 for parietal neurons) (Figure 3E).
HF-Induced Effects in the Brain Are Partially Rescued by the TNF-α Scavenger Etn
We showed that by antagonizing TNF-α signaling, the effect of HF on dendritic spine density was significantly ameliorated in the frontal cortex of HF animals treated with Etn; however, HF mice treated with Etn displayed significantly reduced spine density (0.91±0.13/μm in basal dendrites and 0.79±0.16/μm in apical dendrites) compared with sham control animals (P<0.001) (Figure 4A). Breakdown analysis (dendrograms) further supported a significantly higher spine density in both basal and apical dendrites of Etn-treated HF mice compared with the untreated HF group (P<0.001) (Figure 4B and 4C). Surprisingly, the differential analysis of frontal and parietal neurons revealed a protective effect of Etn on frontal but not parietal neurons (Figure 4D).
Figure 4.
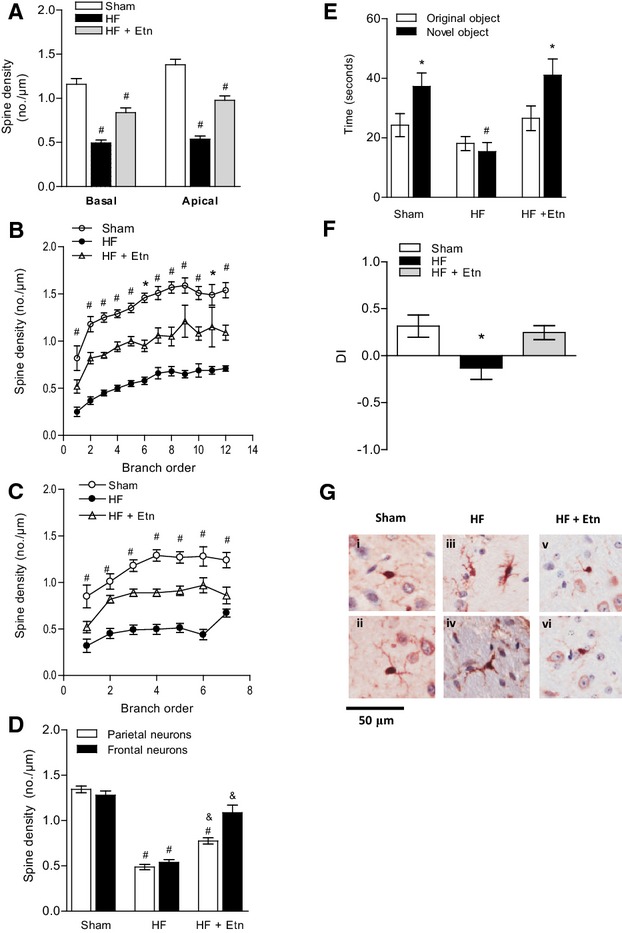
TNF-α scavenger etanercept partially rescues HF-induced effects in the brain. A, Dendritic spine density was found to be reduced in the frontal cortex in HF mice treated with Etn compared with sham controls but significantly increased compared with untreated HF mice. Breakdown analysis (dendrograms) further revealed significant differences of spines or spine density in both (B) basal and (C) apical dendrites of Etn-treated HF compared with HF mice. D, Spine density of frontal and parietal neurons is significantly reduced during HF. Etn fully prevented HF-mediated reduction in the frontal neurons but only partially prevented it in the parietal neurons. For all tasks: sham n=18, HFn=20, HFplus Etn n=13. #P<0.001 between control and HF, &P<0.001 between HF and HF plus Etn. E, In a nonspatial novel object recognition task with a 5-minute retention period, HF animals treated with Etn spent more time exploring the novel object and (F) showed a clear preference for novelty compared with untreated mice. For all tasks: sham n=9, HF n=10, HFplus Etn n=10. *P<0.05 and #P<0.001. G, Cerebral tissue sections of HF mice treated with Etn show, similar to control mice (i and ii), no evidence of microglial activation (v and vi) compared with untreated HF animals, which exhibit microglia with visibly enlarged cytoplasm and reduced, thickened processes (iii and iv) (n=3). DI indicates discrimination index; Etn, etanercept; HF, heart failure; TNF-α, tumor necrosis factor-α.
Although dendritic spine density was still reduced in Etn-treated HF mice, performance in the novel object recognition task was fully restored after treatment with Etn (40.09±5.57 seconds for HF plus Etn, 39.97±6.51 seconds for sham, and 17.1±3.24 seconds for HF; n=10, P<0.05) (Figure 4E). The calculated discrimination index illustrated that HF mice treated with Etn showed a clear preference for the novel object compared with untreated HF animals, which spent more time exploring the familiar rather than the novel object (0.32±0.12 versus −0.13±0.12, n=10, P<0.05) (Figure 4F).
We also showed that microglial activation following HF (as noted by visually observed enlarged cytoplasm and reduced, thickened processes) appeared to be ameliorated by Etn, with microglial morphology in Etn-treated animals appearing similar to that in sham animals (Figure 4G). Semiquantitative analysis revealed that the mean number of positive and strongly positive pixels per microglia was significantly lower in HF animals treated with Etn (96 269±20 561) versus HF animals treated with saline (181 716±19 445) (n=10; P<0.01).
HF-Induced Effects in the Brain Are Ameliorated in TNF-α−/−Animals
In accordance with our Etn results, TNF-α−/− mice show no signs of cerebral inflammation during HF because there was no notable increase of CD11b levels in HF brain compared with sham control (data not shown).
Remarkably, genetic deletion of TNF-α failed to fully prevent the HF-associated reduction in spine density in this mouse model. Figure 5A represents images of dendritic spines of pyramidal cells from TNF-α−/− mice. Whereas the general morphology of dendritic structures did not differ significantly, the total spine number was markedly reduced in HF mice compared with sham controls, with 17.09±6.81% for basal and 17.21±7.29% for apical spines (P<0.001) (Figure 5B). Breakdown analysis of spine density between sham and HF TNF-a−/− mice revealed a significant reduction in spine density of basal and apical dendrites spanning several branch orders (P<0.05) (Figure 5C and 5D).
Figure 5.
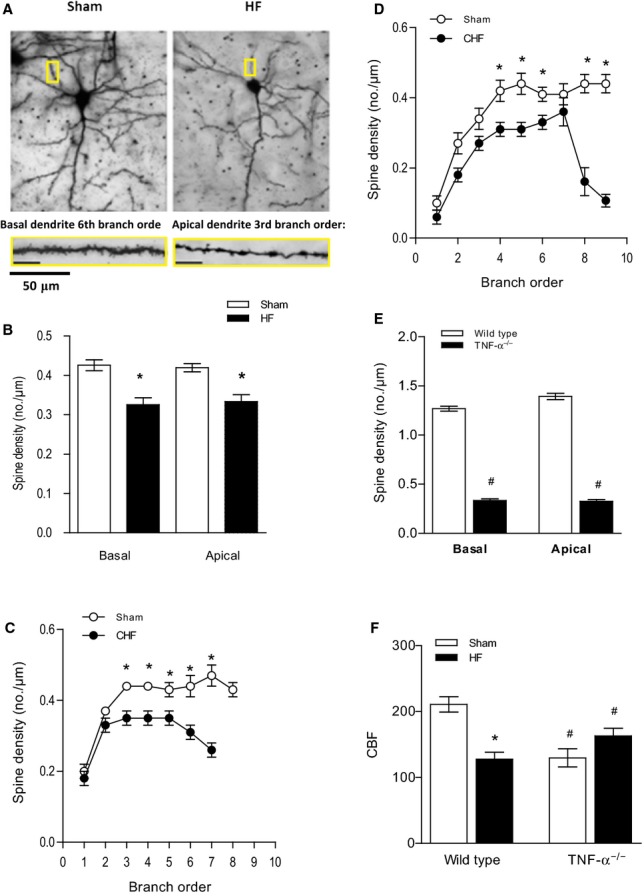
HF-induced effects in the brain are ameliorated in TNF-α−/− mice. A, Representative images of dendritic spines of pyramidal cells from TNF-α−/− mice. The magnification shows details of both basal and apical dendrite morphology (5 to 7 cells per animal). B, Total spine density of basal and apical dendrites was significantly reduced in HF mice (4 to 6 cells per animal). Breakdown analysis of spine density found that significant reductions occurred across several branch orders of (C) basal dendrites and (D) apical dendrites between sham and HF mice. D, The average spine density of TNF-α−/− sham mice was significantly lower than that of normal wild-type animals in both basal and apical dendrites. For all tasks: sham n=24, HF n=24. *P<0.05, #P<0.001. E, HF had no effect on cerebral blood flow in TNF-α−/− mice (n=5, P=0.9069) compared with TNF-α−/− sham-operated mice. F, CBF, shown as mL/(100 g×minute), was significantly reduced compared with wild-type control animals (n=5, P<0.001). *P<0.05 within a group, #P<0.05 between wild-type and TNF-α−/− mice. CBF indicates cerebral blood flow; CHF, congestive heart failure; HF, heart failure; TNF-α, tumor necrosis factor-α.
Interestingly, the average spine density of healthy TNF-α−/− mice was found to be significantly lower than in normal wild-type animals for both basal and apical dendrites (1.27±0.03 versus 0.49±0.03 for basal and 1.39±0.03 versus 0.54±0.03 for apical dendrites, n=5, P<0.001) (Figure 4E); that could be explained by the reduced cerebral blood flow measured in TNF-α−/− mice compared with wild-type control animals (129.6±13.8 versus 210.8±11.8, n=6, P<0.001) (Figure 4F). In accordance with our previous findings showing that HF failed to augment myogenic tone in cerebral arteries isolated from TNF-α−/− mice,42 cerebral blood flow remained unaltered during HF in these animals (129.6±13.8 versus 127.5±10.8, n=6, P=0.9069) (Figure 4F).
SgK1 Protein Expression and Phosphorylation Status Is Altered in a Mouse Model of Congestive HF
To further investigate long-term brain alterations following HF, in a separate group of animals, we carried out gene expression analysis on RNA isolated from whole-brain synaptosomes 5 months after MI. Six genes were found to have a >1.5-fold change in the HF mice versus sham-operated animals (Table 2). SgK1, which is implicated in increasing NF-kB activity stimulating dendrite growth, was also found to be significantly upregulated ≈2-fold during HF. Quantitative PCR confirmed that cerebral SgK1 mRNA levels were significantly upregulated in synaptosomal RNA 5 months after MI (0.8±0.03 in sham versus 1.28±0.1 in HF mice, n=5, P<0.01) (data not shown).
Table 2.
Gene Expression Profiling of Whole-Brain Synaptosomal RNA Isolated From HF Mice 5 Months After Myocardial Infarction and Respective Sham Controls
| Gene | Probe ID | Fold Change (HF vs Control) | Regulation (HF vs Control) | P Value |
|---|---|---|---|---|
| Sgk1 | 4900519 | 1.96 | Up | 0.001 |
| Cd59a | 1440019 | 1.70 | Up | 0.001 |
| Tsc22d3 | 6840382 | 1.64 | Up | 0.017 |
| Ddit4 | 130634 | 1.62 | Up | 0.013 |
| LOC331139 | 4070072 | 1.60 | Down | 0.028 |
| Mt2 | 2190196 | 1.55 | Up | 0.012 |
HF indicates heart failure.
No obvious relationship could be established with the other modulated genes, although future information regarding the function of these genes may reveal a role in HF-mediated effects in the brain.
HF mice 10 to 12 weeks after MI also showed significantly increased mRNA levels of SgK1 in the brain (0.72±0.089 in sham versus 1.14±0.08 in HF mice; n=5, P<0.05) (Figure 6A). Interestingly, scavenging TNF-α by Etn did not reverse this effect; quantitative PCR experiments revealed no detectable difference in SgK1 mRNA levels between Etn-treated (0.89±0.08) and untreated HF mice (n=6, P=0.2) (Figure 6A). Augmented SgK1 activity has been reported in severe Alzheimer disease, in which its phosphorylation status was found to be important for survival signals.43 Consequently, we determined both SgK1 protein levels and activity and found increased phosphorylation in whole-brain lysates of HF animals compared with sham controls but, remarkably, a clear reduction of SgK1 phosphorylation in the Etn-treated HF group compared with untreated HF mice (1.38±0.09 for sham versus 2.12±0.21 for HF and 1.51±0.04 for HF plus Etn; n=5, P<0.05) (Figure 6B). HF did not affect SgK1 mRNA levels, protein expression, or phosphorylation in TNF-α−/− mice (Figure 6C and 6D), further supporting an important role of TNF-a signaling in HF-mediated cerebral complications. Because TNF-α and its receptors as well as SgK1 activate the NF-kB pathway, it seems surprising that cerebral NF-kB (p65) is activated during HF in TNF-α–deficient mice (0.79±0.13 versus 1.79±0.26; n=5, P<0.01) (Figure 6E), suggesting an alternative activation route independent of TNF-α and SgK1 in these animals. In contrast, in wild-type animals, cerebral NF-kB (p65) activation is significantly enhanced during HF (0.42±0.07 versus 0.83±0.14; n=5, P<0.05) (Figure 6F), possibly mediated through TNFR1, which has mRNA levels increased in the brain under HF conditions (Figure 2D), and enhanced SgK1 activity, which has been implicated in the regulation of neuronal plasticity via NF-kB (p65) activation.24
Figure 6.
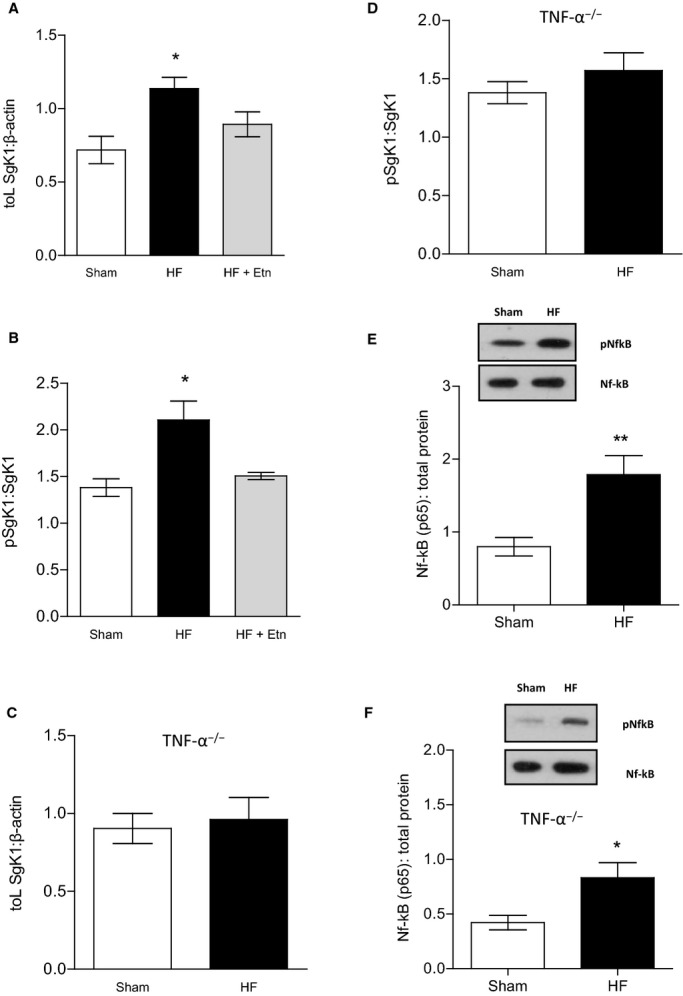
SgK1 mRNA levels, protein expression, and phosphorylation status are altered during HF. A, During HF, SgK1 mRNA levels were upregulated compared with sham mice (n=6, P<0.05). Etn-treated HF mice showed SgK1 mRNA expression similar to that of untreated HF mice (n=6, P=0.2). B, HF induced an increase in SgK1 protein phosphorylation, which is significantly reduced after treating HF mice with Etn (n=5, P<0.05). HF had no effect on (C) SgK1 mRNA levels in TNF-α−/− mice (n=5, P=0.74) or (D) on SgK1 protein phosphorylation (n=5, P=0.38). Cerebral NF-kB (p65) was found to be activated during HF in (E) wild-type mice (n=5, versus <0.01) and (F) TNF-α deficient mice (n=5, P<0.01). *P<0.05 and **P<0.01. Etn indicates etanercept; HF, heart failure; NF-kB, nuclear factor-κB; p, phosphorylated; SgK1, serum and glucocorticoid-inducible kinase 1; TNF-α, tumor necrosis factor-α.
Discussion
The key finding of the present study suggests that cerebral inflammation, marked by an increase in TNF-α levels, may be the underlying cause of alterations in number and morphology of dendritic spines in an animal model of HF. Furthermore, we described evidence that TNF-α represents a key player in SgK1 signaling and altered dendritic spine morphology, which may contribute to the cortical memory impairment observed in this mouse model of HF. These findings may have implications for the mechanisms underlying the CI reported in human HF and, most important, for development of novel clinical treatment strategies.
Various studies have described increased cerebral levels of the proinflammatory cytokine TNF-α, especially after acute brain injury or ischemia but also during neurodegenerative diseases21 and after systemic inflammatory challenge.22 To our knowledge, this study provides the first demonstration of an elevation of cerebral TNF-α in a mouse model of HF. TNF-α is produced mainly by cells of the macrophage lineage, which includes microglia in the central nervous system.20 An increase of TNF-α in the brain occurs (1) when circulating TNF-α enters the brain through regions that lack or have a compromised blood–brain barrier, (2) when activated microglia locally produce the proinflammatory cytokine, or (3) a combination of both. Previous studies from our laboratory have already suggested that the smooth muscle layer of cerebral arteries contributes to local TNF-α production during HF42; however, the present data suggest that activated microglia may also contribute to the proinflammatory process in HF brain.
The decline in cognitive function in HF includes memory deficits, inability to make sound decisions, attention deficits, impaired visual perception, and difficulties in problem solving.3 Such neurological deficits are reported to result from structural changes in the cortex, alterations in white matter, cerebral hypoperfusion, and impaired blood flow regulation in small vessels4 but also can result from alterations of dendritic spine morphology and number. The presently observed alterations in number and morphology of dendritic spines in the frontal cortex in HF mice likely result from the above-described cerebral inflammation because the sequestration of TNF-α by Etn, a soluble fusion protein that binds TNF-α and decreases its role in disorders involving excess inflammation in humans and other animals,44 ameliorates the HF-mediated reduction in dendritic spine number. Furthermore, several in vitro studies using mixed cultures have reported involvement of TNF-α in neuronal loss, neurite outgrowth, and apoptosis.45,46 Proinflammatory cytokines were also shown to contribute to demyelination and axonal damage in a cerebellar culture model of neuroinflammation.47 The question remains whether this is a consequence of the impaired brain blood flow previously reported in this mouse model of HF, which is also restored by TNF-α sequestration with Etn,29,42 or whether alternative TNF-α signaling mechanisms play a role. Although less pronounced, HF still reduced the number of dendritic spines in TNF-α–depleted mice, in which brain blood flow was not altered during HF, suggesting the existence of alternative mechanisms. Interestingly, sham-operated TNF-α−/− mice demonstrated significantly reduced dendritic spine numbers compared with their littermate controls. This observation might be explained by the significantly reduced basal brain blood flow measured in these animals compared with wild-type mice, or other mechanisms may contribute. A recent study, for example, demonstrated that another member of the TNF superfamily, RANKL (receptor activator of NF-kB ligand, TNFSF11), inhibited neurotrophin-promoted neurite growth in the developing mouse peripheral nervous system, suggesting a direct role for members of the TNF superfamily in neurite development.48 The absence of TNF-α has been correlated previously with poor cognitive functioning.49 These authors concluded from their results that the deletion of both TNF-α receptors reduced cognitive functioning and that low levels of TNF-α appear essential for normal cognitive function. These data, taken together with our findings of reduced cerebral blood flow and a marked structural impairment in TNF-α–deficient mice, are supportive of an important role for TNF-α in cognitive function in health and disease.
TNF-α also serves as a potent activator of glucocorticoids, which are a known and powerful stress response and have been reported to induce structural plasticity in the brain including changes in dendritic spines.50 SgK1 has been reported to be upregulated after ischemic brain injury in rodents and humans26 and is described as a potent player in neuronal excitability,25 neurite outgrowth, long-term potentiation, and learning.51 Furthermore, recent evidence demonstrating a role for SgK1 in stress-induced changes in oligodendrocyte morphology in a mouse model provides support for our present evidence suggestive of a role of SgK1 in dendritic morphology during HF.52 In this study, we showed that HF also leads to a significant increase in cerebral SgK1 protein expression and phosphorylation, which has been reported to be important for survival signals in Alzheimer disease.43 Genetic deletion of TNF-α prevented both upregulation and phosphorylation of SgK1, whereas a treatment with Etn only reversed the HF-associated SgK1 phosphorylation. Sgk1 activation was reported to indirectly affect cognitive processes by upregulating the NMDA receptor subunits NR2A and NR2B5 via activating NF-kB.53 Although HF mediated an increase of cerebral NF-kB protein activity, no significant differences in NMDAR and PSD-95 protein expression were observed. Marchetti et al provided strong support that TNF-α receptor type 2, but not type 1, induces persistent NF-kB activation via the phosphoinositide 3-kinase/protein kinase B (Akt) pathway with the opposite effect on NMDAR signaling.54 In our model of HF, mRNA levels of cortical TNFR1 were significantly upregulated, whereas levels of cortical TNFR2 remained unaltered. The potential involvement of this upregulation in the positive feedback loop was reportedly involved in the prolonged activation of microglia through TNFR1-dependent self-induced TNF-α production by microglia,55 whether TNFR2 may also play a role in NF-kB activation is currently unknown and certainly worthy of future study.
We previously observed no differences between mice with HF and sham-operated controls in the Morris water maze, a spatial memory task that is heavily dependent on hippocampal function.42 Baker and Kim56 blocked long-term potentiation with an NMDAR antagonist in the dorsal hippocampus of rats and reported selective impairment of object memory when tested with a 3-hour delay but not with a 5-minute delay interval. We used a nonspatial object memory task with a 5-minute retention interval and found significant memory impairment in mice with HF. We observed no difference in NMDAR2A and PSD-95 protein expression between HF and control mice, suggesting that the memory impairment we observed during HF may be attributed to the reduction of dendritic spine density in the cortex. Indeed, the frontal cortex has been implicated in short-term memory, attention, and problem solving.57 Parietal lobe function involves sensation and perception and the integration of sensory input, primarily with the visual system.58 Damage to the parietal lobes often induces striking deficits, such as abnormalities in body image and spatial relations.59 It has been reported that in primates, prefrontal neuronal responses carry information necessary for long-term visual recognition memory.60 Scavenging TNF-α restored memory function in HF mice by fully rescuing the spine density of frontal neurons and partially rescuing the spine density of parietal neurons, suggesting an important role for TNF-α signaling in short-term memory and recognition.
Although our data provide compelling evidence for the involvement of TNF-α and SgK1 signaling in cortical memory impairment in a mouse model of HF, it is important to recognize that limitations in translating these findings to CI in HF in humans. A study comparing the postsynaptic proteome of excitatory synapses in the human and mouse found that although there was >70% overlap in postsynaptic density proteins, there were significant interspecies differences in the abundance of several key synaptic proteins, including a group of interacting proteins implicated in dendritic spine morphology and plasticity.61 This study illustrates an important caveat that must be considered when translating findings regarding synaptic proteins from murine models to human brain disease. Future studies examining interspecies diversity in the cortical presynaptic proteome may enhance our ability to harness information from murine models to better understand the complexity of alterations in brain-signaling mechanisms contributing to CI in HF.
In conclusion, using a well-characterized mouse model of HF, we provided evidence that inflammatory processes following MI lead to pronounced neuroinflammation. We found evidence of increased TNF-α levels and activated microglia in the brain of HF mice, with corresponding alterations in the protein phosphorylation of SgK1, reduction in dendritic spine density, and concomitant cortical memory impairment. Our findings are suggestive of a major role for TNF-α because blocking TNF-α (by either Etn, a soluble TNF-α receptor, or genetic deletion) attenuated the HF-associated loss of cortical dendritic spine density and associated cognitive deficits. Future studies, for example, using cortical neuron cultures to study the molecular mechanisms involved in the TNF-α–mediated morphological alterations of dendrites and associated neuronal loss, will investigate the potential for intervening in these proinflammatory pathways as a potential treatment for CI in HF. Although both the Randomized Etanercept North American Strategy to Study Antagonism of Cytokines (RENAISSANCE) and Research Into Etanercept Cytokine Antagonism in Ventricular Dysfunction (RECOVER) clinical trials were stopped prematurely due to lack of beneficial effects on the end points of clinical composite score, HF hospitalization, and mortality,62 these trials did not evaluate cerebral perfusion or neurological function. We believe that TNF-α therapy may ameliorate key components driving organ dysfunction in our mouse model of HF and that these findings may reveal specific therapeutic targets to potentially treat and prevent cognitive decline in patients with cardiovascular diseases like HF.
Sources of Funding
We gratefully acknowledge the following funding support: Operating and Infrastructure Grants from the Heart and Stroke Foundation of Ontario (HSFO; Bolz, G13-0002610); the Canadian Foundation for Innovation and Ontario Research Fund (Bolz, 11810); and Research Support from the University of Toronto (Bolz). This work is also supported by HSFO Career Investigator Awards (Bolz CI-7432), a Marie Curie Action of the 7th Framework Program of the European Commission (Meissner) and the August Pi i Sunyer Biomedical Research Institute (IDIBAPS) (Meissner).
Disclosures
None.
References
- Malik AS, Giamouzis G, Georgiopoulou VV, Fike LV, Kalogeropoulos AP, Norton CR, Sorescu D, Azim S, Laskar SR, Smith AL, Dunbar SB, Butler J. Patient perception versus medical record entry of health-related conditions among patients with heart failure. Am J Cardiol. 2011;107:569–572. doi: 10.1016/j.amjcard.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havranek EP, Dwinnell B, Smith KS. Multiorgan failure after cardiac arrest in a 20-year-old man. Circulation. 1995;92:3139–3143. doi: 10.1161/01.cir.92.10.3139. [DOI] [PubMed] [Google Scholar]
- Bennett SJ, Oldridge NB, Eckert GJ, Embree JL, Browning S, Hou N, Chui M, Deer M, Murray MD. Comparison of quality of life measures in heart failure. Nurs Res. 2003;52:207–216. doi: 10.1097/00006199-200307000-00001. [DOI] [PubMed] [Google Scholar]
- Vogels RL, Oosterman JM, van Harten B, Gouw AA, Schroeder-Tanka JM, Scheltens P, van der Flier WM, Weinstein HC. Neuroimaging and correlates of cognitive function among patients with heart failure. Dement Geriatr Cogn Disord. 2007;24:418–423. doi: 10.1159/000109811. [DOI] [PubMed] [Google Scholar]
- Marshall RS, Lazar RM. Pumps, aqueducts, and drought management: vascular physiology in vascular cognitive impairment. Stroke. 2011;42:221–226. doi: 10.1161/STROKEAHA.110.595645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson OB, Jarden JO, Godtfredsen J, Vorstrup S. Cerebral blood flow in patients with congestive heart failure treated with captopril. Am J Med. 1984;76:91–95. doi: 10.1016/0002-9343(84)90892-1. [DOI] [PubMed] [Google Scholar]
- Choi BR, Kim JS, Yang YJ, Park KM, Lee CW, Kim YH, Hong MK, Song JK, Park SW, Park SJ, Kim JJ. Factors associated with decreased cerebral blood flow in congestive heart failure secondary to idiopathic dilated cardiomyopathy. Am J Cardiol. 2006;97:1365–1369. doi: 10.1016/j.amjcard.2005.11.059. [DOI] [PubMed] [Google Scholar]
- Pantoni L, Garcia JH. Pathogenesis of leukoaraiosis: a review. Stroke. 1997;28:652–659. doi: 10.1161/01.str.28.3.652. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Chen JR, Yan YT, Wang TJ, Chen LJ, Wang YJ, Tseng GF. Gonadal hormones modulate the dendritic spine densities of primary cortical pyramidal neurons in adult female rat. Cereb Cortex. 2009;19:2719–2727. doi: 10.1093/cercor/bhp048. [DOI] [PubMed] [Google Scholar]
- Bitzer-Quintero OK, González-Burgos I. Immune system in the brain: a modulatory role on dendritic spine morphophysiology? Neural Plast. 2012;2012:348642. doi: 10.1155/2012/348642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Kohsaka S, Okabe S. Long-term changes of spine dynamics and microglia after transient peripheral immune response triggered by LPS in vivo. Mol Brain. 2011;4:27. doi: 10.1186/1756-6606-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the vesnarinone trial (VEST) Circulation. 2001;103:2055–2059. doi: 10.1161/01.cir.103.16.2055. [DOI] [PubMed] [Google Scholar]
- Pinto A, Tuttolomondo A, Casuccio A, Di Raimondo D, Di Sciacca R, Arnao V, Licata G. Immuno-inflammatory predictors of stroke at follow-up in patients with chronic non-valvular atrial fibrillation (NVAF) Clin Sci (Lond) 2009;116:781–789. doi: 10.1042/CS20080372. [DOI] [PubMed] [Google Scholar]
- Tuttolomondo A, Di Raimondo D, Pecoraro R, Arnao V, Pinto A, Licata G. Inflammation in ischemic stroke subtypes. Curr Pharm Des. 2012;18:4289–4310. doi: 10.2174/138161212802481200. [DOI] [PubMed] [Google Scholar]
- Kang YM, Zhang ZH, Johnson RF, Yu Y, Beltz T, Johnson AK, Weiss RM, Felder RB. Novel effect of mineralocorticoid receptor antagonism to reduce proinflammatory cytokines and hypothalamic activation in rats with ischemia-induced heart failure. Circ Res. 2006;99:758–766. doi: 10.1161/01.RES.0000244092.95152.86. [DOI] [PubMed] [Google Scholar]
- Francis J, Chu Y, Johnson AK, Weiss RM, Felder RB. Acute myocardial infarction induces hypothalamic cytokine synthesis. Am J Physiol Heart Circ Physiol. 2004;286:H2264–H2271. doi: 10.1152/ajpheart.01072.2003. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Lee SC, Liu W, Dickson DW, Brosnan CF, Berman JW. Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta. J Immunol. 1993;150:2659–2667. [PubMed] [Google Scholar]
- Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87:10–20. doi: 10.1016/j.brainresbull.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazi K, Galic MA, Kuzmiski JB, Ho W, Sharkey KA, Pittman QJ. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci USA. 2008;105:17151–17156. doi: 10.1073/pnas.0806682105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–1078. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- Tai DJ, Su CC, Ma YL, Lee EH. Sgk1 phosphorylation of IkappaB kinase alpha and p300 up-regulates NF-kappaB activity and increases N-methyl-D-aspartate receptor NR2A and NR2B expression. J Biol Chem. 2009;284:4073–4089. doi: 10.1074/jbc.M805055200. [DOI] [PubMed] [Google Scholar]
- Lang F, Bohmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86:1151–1178. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- Mitsios N, Saka M, Krupinski J, Pennucci R, Sanfeliu C, Wang Q, Rubio F, Gaffney J, Kumar P, Kumar S, Sullivan M, Slevin M. A microarray study of gene and protein regulation in human and rat brain following middle cerebral artery occlusion. BMC Neurosci. 2007;8:93. doi: 10.1186/1471-2202-8-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino MW, Dunn A, Grail D, Inglese M, Noguchi Y, Richards E, Jungbluth A, Wada H, Moore M, Williamson B, Basu S, Old LJ. Characterization of tumor necrosis factor-deficient mice. Proc Natl Acad Sci USA. 1997;94:8093–8098. doi: 10.1073/pnas.94.15.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefer J, Azam MA, Kroetsch JT, Leong-Poi H, Momen MA, Voigtlaender-Bolz J, Scherer EQ, Meissner A, Bolz SS, Husain M. Sphingosine-1-phosphate-dependent activation of p38 MAPK maintains elevated peripheral resistance in heart failure through increased myogenic vasoconstriction. Circ Res. 2010;107:923–933. doi: 10.1161/CIRCRESAHA.110.226464. [DOI] [PubMed] [Google Scholar]
- Meissner A, Yang J, Kroetsch JT, Sauve M, Dax H, Momen A, Noyan-Ashraf MH, Heximer S, Husain M, Lidington D, Bolz SS. Tumor necrosis factor-alpha-mediated downregulation of the cystic fibrosis transmembrane conductance regulator drives pathological sphingosine-1-phosphate signaling in a mouse model of heart failure. Circulation. 2012;125:2739–2750. doi: 10.1161/CIRCULATIONAHA.111.047316. [DOI] [PubMed] [Google Scholar]
- Francis BM, Kim J, Barakat ME, Fraenkl S, Yücel YH, Peng S, Michalski B, Fahnestock M, McLaurin J, Mount HT. Object recognition memory and BDNF expression are reduced in young TgCRND8 mice. Neurobiol Aging. 2012;33:555–563. doi: 10.1016/j.neurobiolaging.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaucher E, Fluit P, Chishti MA, Westaway D, Mount HT, Kar S. Object recognition memory and cholinergic parameters in mice expressing human presenilin 1 transgenes. Exp Neurol. 2002;175:398–406. doi: 10.1006/exnr.2002.7915. [DOI] [PubMed] [Google Scholar]
- Mumby DG, Pinel JP. Rhinal cortex lesions and object recognition in rats. Behav Neurosci. 1994;108:11–18. doi: 10.1037//0735-7044.108.1.11. [DOI] [PubMed] [Google Scholar]
- Steckler T, Drinkenburg WH, Sahgal A, Aggleton JP. Recognition memory in rats—II. Neuroanatomical substrates. Prog Neurobiol. 1998;54:313–332. doi: 10.1016/s0301-0082(97)00061-0. [DOI] [PubMed] [Google Scholar]
- Hammond RS, Tull LE, Stackman RW. On the delay-dependent involvement of the hippocampus in object recognition memory. Neurobiol Learn Mem. 2004;82:26–34. doi: 10.1016/j.nlm.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Aggleton JP, Albasser MM, Aggleton DJ, Poirier GL, Pearce JM. Lesions of the rat perirhinal cortex spare the acquisition of a complex configural visual discrimination yet impair object recognition. Behav Neurosci. 2010;124:55–68. doi: 10.1037/a0018320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser EM, Van der Loos H. Analysis of thick brain sections by obverse-reverse computer microscopy: application of a new, high clarity Golgi-Nissl stain. J Neurosci Methods. 1981;4:117–125. doi: 10.1016/0165-0270(81)90045-5. [DOI] [PubMed] [Google Scholar]
- Wong JC, Visanji NP, Dabek MK, Laposa RR, Hazrati LN. Dendritic spine density is altered in a mouse model of Cockayne syndrome. Neuropathol Appl Neurobiol. 2013;39:437–440. doi: 10.1111/j.1365-2990.2012.01305.x. [DOI] [PubMed] [Google Scholar]
- Visanji NP, Collingwood JF, Finnegan ME, Tandon A, House E, Hazrati LN. Iron deficiency in parkinsonism: region-specific iron dysregulation in parkinson’s disease and multiple system atrophy. J Parkinsons Dis. 2013;3:523–537. doi: 10.3233/JPD-130197. [DOI] [PubMed] [Google Scholar]
- Hochman JS, Choo H. Limitation of myocardial infarct expansion by reperfusion independent of myocardial salvage. Circulation. 1987;75:299–306. doi: 10.1161/01.cir.75.1.299. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Berryhill ME, Phuong L, Picasso L, Cabeza R, Olson IR. Parietal lobe and episodic memory: bilateral damage causes impaired free recall of autobiographical memory. J Neurosci. 2007;27:14415–14423. doi: 10.1523/JNEUROSCI.4163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Noyan-Ashraf MH, Meissner A, Voigtlaender-Bolz J, Kroetsch JT, Foltz W, Jaffray D, Kapoor A, Momen A, Heximer SP, Zhang H, van Eede M, Henkelman RM, Matthews SG, Lidington D, Husain M, Bolz SS. Proximal cerebral arteries develop myogenic responsiveness in heart failure via tumor necrosis factor-alpha-dependent activation of sphingosine-1-phosphate signaling. Circulation. 2012;126:196–206. doi: 10.1161/CIRCULATIONAHA.111.039644. [DOI] [PubMed] [Google Scholar]
- Sahin P, McCaig C, Jeevahan J, Murray JT, Hainsworth AH. The cell survival kinase SGK1 and its targets FOXO3a and NDRG1 in aged human brain. Neuropathol Appl Neurobiol. 2013;39:623–633. doi: 10.1111/nan.12023. [DOI] [PubMed] [Google Scholar]
- Peppel K, Crawford D, Beutler B. A tumor necrosis factor (TNF) receptor-IgG heavy chain chimeric protein as a bivalent antagonist of TNF activity. J Exp Med. 1991;174:1483–1489. doi: 10.1084/jem.174.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Huang Y, Zhao L, Li Y, Sun L, Zhou Y, Qian G, Zheng JC. IL-1beta and TNF-alpha induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem. 2013;125:897–908. doi: 10.1111/jnc.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neniskyte U, Vilalta A, Brown GC. Tumour necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett. 2014;588:2952–2956. doi: 10.1016/j.febslet.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Penta A, Moreno B, Reix S, Fernandez-Diez B, Villanueva M, Errea O, Escala N, Vandenbroeck K, Comella JX, Villoslada P. Oxidative stress and proinflammatory cytokines contribute to demyelination and axonal damage in a cerebellar culture model of neuroinflammation. PLoS One. 2013;8:e54722. doi: 10.1371/journal.pone.0054722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez H, Kisiswa L, O’Keeffe GW, Smithen MJ, Wyatt S, Davies AM. Regulation of neurite growth by tumour necrosis superfamily member RANKL. Open Biol. 2013;3:120150. doi: 10.1098/rsob.120150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baune BT, Wiede F, Braun A, Golledge J, Arolt V, Koerner H. Cognitive dysfunction in mice deficient for TNF- and its receptors. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1056–1064. doi: 10.1002/ajmg.b.30712. [DOI] [PubMed] [Google Scholar]
- Tanokashira D, Morita T, Hayashi K, Mayanagi T, Fukumoto K, Kubota Y, Yamashita T, Sobue K. Glucocorticoid suppresses dendritic spine development mediated by down-regulation of caldesmon expression. J Neurosci. 2012;32:14583–14591. doi: 10.1523/JNEUROSCI.2380-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KJ, Chen SK, Ma YL, Hsu WL, Lee EH. Sgk, a primary glucocorticoid-induced gene, facilitates memory consolidation of spatial learning in rats. Proc Natl Acad Sci USA. 2002;99:3990–3995. doi: 10.1073/pnas.062405399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata S, Koyama Y, Takemoto K, Yoshikawa K, Ishikawa T, Taniguchi M, Inoue K, Aoki M, Hori O, Katayama T, Tohyama M. Plasma corticosterone activates SGK1 and induces morphological changes in oligodendrocytes in corpus callosum. PLoS One. 2011;6:e19859. doi: 10.1371/journal.pone.0019859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Hagan JJ, Rawlins JN. Allocentric spatial learning by hippocampectomised rats: a further test of the “spatial mapping” and “working memory” theories of hippocampal function. Q J Exp Psychol B. 1986;38:365–395. [PubMed] [Google Scholar]
- Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel UL. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem. 2004;279:32869–32881. doi: 10.1074/jbc.M311766200. [DOI] [PubMed] [Google Scholar]
- Kuno R, Wang J, Kawanokuchi J, Takeuchi H, Mizuno T, Suzumura A. Autocrine activation of microglia by tumor necrosis factor-alpha. J Neuroimmunol. 2005;162:89–96. doi: 10.1016/j.jneuroim.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Baker KB, Kim JJ. Effects of stress and hippocampal NMDA receptor antagonism on recognition memory in rats. Learn Mem. 2002;9:58–65. doi: 10.1101/lm.46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Raine A. Prefrontal structural and functional brain imaging findings in antisocial, violent, and psychopathic individuals: a meta-analysis. Psychiatry Res. 2009;174:81–88. doi: 10.1016/j.pscychresns.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb J, Snyder LH. Spatial and non-spatial functions of the parietal cortex. Curr Opin Neurobiol. 2010;20:731–740. doi: 10.1016/j.conb.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabeza R, Ciaramelli E, Olson IR, Moscovitch M. The parietal cortex and episodic memory: an attentional account. Nat Rev Neurosci. 2008;9:613–625. doi: 10.1038/nrn2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang JZ, Brown MW. Neuronal responses related to long-term recognition memory processes in prefrontal cortex. Neuron. 2004;42:817–829. doi: 10.1016/j.neuron.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Jiwa NS, Garrard P, Hainsworth AH. Experimental models of vascular dementia and vascular cognitive impairment: a systematic review. J Neurochem. 2010;115:814–828. doi: 10.1111/j.1471-4159.2010.06958.x. [DOI] [PubMed] [Google Scholar]
- Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L, Krum H, Liu P, Nieminen M, Tavazzi L, van Veldhuisen DJ, Waldenstrom A, Warren M, Westheim A, Zannad F, Fleming T. Targeted anticytokine therapy in patients with chronic heart failure: results of the randomized etanercept worldwide evaluation (RENEWAL) Circulation. 2004;109:1594–1602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]