

Abstract
Background
Cystathionine γ-lyase, cystathionine β-synthase, and 3-mercaptopyruvate sulfurtransferase are endogenous enzymatic sources of hydrogen sulfide (H2S). Functions of H2S are mediated by several targets including ion channels and signaling proteins. Nuclear factor-erythroid 2-related factor 2 is responsible for the expression of antioxidant response element–regulated genes and is known to be upregulated by H2S. We examined the levels of H2S, H2S-producing enzymes, and nuclear factor-erythroid 2-related factor 2 activation status in skeletal muscle obtained from critical limb ischemia (CLI) patients.
Methods and Results
Gastrocnemius tissues were attained postamputation from human CLI and healthy control patients. We found mRNA and protein levels of cystathionine γ-lyase, cystathionine β-synthase, and 3-mercaptopyruvate sulfurtransferase were significantly decreased in skeletal muscle of CLI patients as compared to control. H2S and sulfane sulfur levels were significantly decreased in skeletal muscle of CLI patients. We also observed significant reductions in nuclear factor-erythroid 2-related factor 2 activation as well as antioxidant proteins, such as Cu, Zn-superoxide dismutase, catalase, and glutathione peroxidase in skeletal muscle of CLI patients. Biomarkers of oxidative stress, such as malondialdehyde and protein carbonyl formation, were significantly increased in skeletal muscle of CLI patients as compared to healthy controls.
Conclusions
The data demonstrate that H2S bioavailability and nuclear factor-erythroid 2-related factor 2 activation are both attenuated in CLI tissues concomitant with significantly increased oxidative stress. Reductions in the activity of H2S-producing enzymes may contribute to the pathogenesis of CLI.
Keywords: antioxidant proteins, critical limb ischemia, hydrogen sulfide, NRF2, oxidative stress
Critical limb ischemia (CLI) is a manifestation of peripheral arterial disease (PAD), affecting nearly 2 million people in United States.1–3 During PAD, arteries that supply blood to the legs are narrowed, typically due to atherosclerosis, and results in a number of symptoms called intermittent claudication. Patients with CLI have a 6-month risk of major amputation of 25% to 40% and an annual mortality of 20%.4 Patients with CLI share the same traditional risk factors as patients with atherosclerosis in other vascular territories. Patients with CLI usually have concomitant severe cardiac (20%) and cerebrovascular disease (50% to 75%).5–7 With an aging population and the rising incidence of diabetes and chronic kidney disease, the prevalence of CLI is likely to increase. Early recognition of PAD and aggressive risk factor modification is likely to ameliorate the severity of PAD presentation and will hopefully reduce the incidence of CLI.8
Hydrogen sulfide (H2S), historically viewed as an environmental toxic gas,9–11 is now known to be an endogenous gaseous signaling molecule12–16 that is required for the maintenance of normal vascular function. The production of H2S in mammalian systems has been attributed to at least 3 endogenous enzymes: cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), 3-mercaptopyruvate sulfur transferase (3-MST).17–25 The rate of H2S production in tissue homogenates is in the range of 1 to 10 pmol/s per mg of protein, resulting in low micromolar extracellular concentrations.26,27 At this physiological concentration, H2S is cytoprotective in various models of cellular injury.28,29 As a gaseous signaling molecule, H2S diffuses across cell membranes in a receptor-independent manner and activates various cellular targets. This distinct ability makes H2S an attractive pharmacological agent for the treatment of cardiovascular disease. Recent evidence suggests that H2S is a mediator of many physiological and pathological processes.13,14,16,27,30–39 While the exact mechanism of action of H2S is still under investigation, some chemical and biochemical catabolic reactions of H2S have been disclosed that may be responsible for its biological functions. This molecule provides significant protection in cardiac, neurologic, and hepatic models of ischemia reperfusion injury when delivered before an ischemic insult.40–47 H2S dilates most mammalian blood vessels; the possibility that a deficiency in the biosynthesis of this gas contributes to or predisposes to cardiovascular diseases is appealing. Over the past few years, numerous other cardiovascular effects of H2S have been noted. These include proangiogenesis in vitro and in vivo,48 positive vascular remodeling (ie, reduced vascular hyperplasia) in blood vessels of hypertensive rats,49,50 anti-atherosclerotic activity,51 and reduced52 binding of neutrophils to the blood vessel wall.
H2S is known to increase intracellular reduced glutathione concentrations and to suppress oxidative stress in mitochondria.44 Coupled with the finding that mitochondria contain an H2S-producing enzyme, 3-MST is particularly interesting. The antioxidant activity of H2S may explain a number of the reported biological effects of this gas, including protection against (1) heart,45 liver,28 and intestinal53 damage following ischemia/reperfusion injury; (2) H2O2-induced damage in rat gastric epithelial cells; (3) myocardial54 and renal55 injury due to hyperhomocysteinemia in rats; (4) methionine-induced56 and homocysteine-induced57 oxidative stress; and (5) hemin-mediated oxidation of low-density lipoprotein.58 H2S is known to increase heme oxygenase 1 levels.4,59 Based on these facts, it is clear that H2S is a potent reducing agent and is likely to be consumed by endogenous oxidant species, such as peroxynitrite,60 superoxide,54 and H2O2.61 Treatment with exogenous H2S or modulation of the endogenous production of H2S protects against acute myocardial ischemia/reperfusion injury and heart failure by attenuating oxidative stress, inhibiting apoptosis, and reducing inflammation.4,62 Recently, we have demonstrated that 1 mechanism by which H2S exerts cytoprotective actions is via upregulation of cellular antioxidants in a nuclear factor-erythroid-2-related factor 2 (NRF2)-dependent manner.4 NRF2 regulates the gene expression of a number of enzymes that serve to detoxify pro-oxidative stressors,63 by binding to the antioxidant response element (ARE) found in the gene-promoter region.4 It is important to note here that most reactive oxygen species (ROS), such as superoxide, H2O2, lipid peroxides, and any other lipid radicals may not directly react with H2S but are presumably scavenged by either Cu, Zn-superoxide dismutase or catalase, and/or GPx1 or by their combined actions. On the other hand, H2S is known to activate NRF2 signaling, which is responsible for the upregulation of these antioxidant proteins.4,63
The present study was undertaken to investigate the effects of CLI on endogenous levels of H2S, the expression of H2S-producing enzymes, and NRF2 and related downstream signaling pathways.
Materials and Methods
Collection of Skeletal Muscle From Human CLI and Healthy Controls
Gastrocnemius tissues were attained from human CLI patients and otherwise healthy individuals (non-CLI) who underwent leg amputations. All patients were consented to the use of their amputated tissue for research purposes and signed a HIPPA release for use of the medical record. Patients were segregated into normal perfusion or CLI according to pulse examination and objective imaging criteria.
Institutional Review Board approval with consent was obtained.
Immunoblot Analysis
Protein samples obtained from skeletal muscle of healthy controls and CLI patients were analyzed by immunoblotting64 using specific antibodies to CBS (Santa Cruz), CSE (Abcam), 3-MST, thioredoxin-1 and -2 and thioredoxin reductase 1 and 2 (Santa Cruz), catalase (Santa Cruz), glutathione peroxidase-1 (Santa Cruz), NRF2 (Santa Cruz), fibrillarin (Cell Signaling), and GAPDH (Santa Cruz).
RNA Isolation and Reverse Transcriptase Real Time q-Polymerase Chain Reaction
RNA was isolated from the skeletal muscle of CLI patients and healthy controls. One microgram of RNA was transcribed using an I-script cDNA synthesis kit from Bio-Rad. TaqMan primers for CBS, CSE, 3-MST, NRF2, TXN1 and 2, superoxide dismutase 1 and 2, catalase, and glutathione peroxidase-1 (GPx-1) from Life Technology were used to amplify q-polymerase chain reaction. For real-time q-polymerase chain reaction experiments, 18s was used as a housekeeping gene and values were corrected to 18s. 2ΔΔCT was used for the data analysis of all q-polymerase chain reaction.64
Determination of Protein Carbonyl Content
Protein carbonyl (CO) contents in skeletal muscle from CLI patients and healthy controls were measured as described previously.65
Measurement of MDA Levels
Malondialdehyde (MDA) levels in skeletal muscle from CLI patients and healthy controls were assayed as described previously.65
Measurement of H2S and Sulfane Sulfur
H2S and sulfane sulfur levels were measured in protein extracts from skeletal muscle of healthy controls and CLI patients by gas chromatography chemiluminescence.66
Electrophoretic Mobility Shift Assay
Nuclear extracts (NE) were prepared from skeletal muscle of healthy controls and CLI patients. DNA-protein interaction was determined by utilizing electrophoretic mobility shift assay as described previously.64
Statistical Analysis
All data in this study are expressed as the mean±SEM. Differences in data between the groups were compared using Prism 6 (GraphPad Software) with nonparametric test (Wilcoxon rank sum test). A P value of <0.05 was considered statistically significant.
Results
Decreased Levels of H2S and Sulfane Sulfur in Skeletal Muscle of CLI Patients
H2S and sulfane levels were measured in samples obtained from skeletal muscle of CLI and healthy controls. As can be seen in Figure 1, both H2S (A) and sulfane sulfur (B) were significantly decreased in CLI patients as compared to control. Reduction in H2S (≈3-fold decrease) and sulfane sulfur may indicate reduced levels of H2S-producing enzymes such as CBS, CSE, and 3-MST in skeletal muscle of CLI patients.
Figure 1.
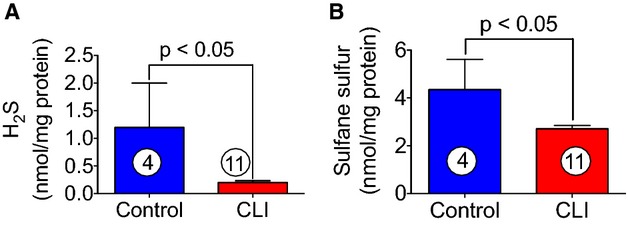
Levels of free H2S and sulfane sulfur in skeletal muscle of healthy controls and CLI patients. H2S and sulfane sulfur were measured in protein extracts obtained from skeletal muscle of CLI patients and aged-matched controls as described in Materials and Methods. H2S and sulfane sulfur are represented by (A) and (B), respectively. Circle with number inside bar denotes number of patients per group. CLI indicates critical limb ischemia; H2S, hydrogen sulfide.
Reduction in the Levels of CBS, CSE, and 3-MST in Skeletal Muscle of CLI Patients
Due to the decreased of levels H2S and sulfane sulfur in CLI patients, we next measured tissue levels of CBS, CSE, and 3-MST in skeletal muscle of CLI patients. Figures2A through 2C and 3A through 3D show that both mRNA and protein levels of all 3 enzymes were significantly decreased in skeletal muscle of CLI patients, and it was clearly observed that the expression levels of CBS and CSE in CLI were reduced to about 2-fold or more as compared to control subjects. It is well appreciated that H2S increases cellular antioxidant levels.
Figure 2.
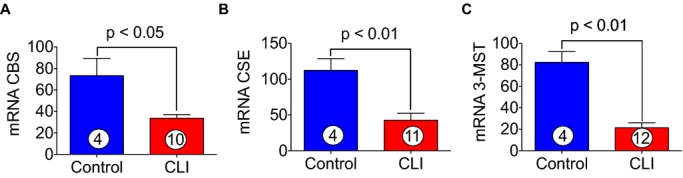
Expression of CBS, CSE, and 3-MST in skeletal muscle of healthy controls and CLI patients. A through C, represent the mRNA expression of CBS, CSE, and 3-MST genes, respectively, in the skeletal muscle of healthy controls and CLI patients. Circle with number inside bar denotes number of patients per group. CBS indicates cystathionine β-synthase; CLI, critical limb ischemia; CSE, cystathionine γ-lyase; 3-MST, 3-mercaptopyruvate sulfurtransferase.
Figure 3.
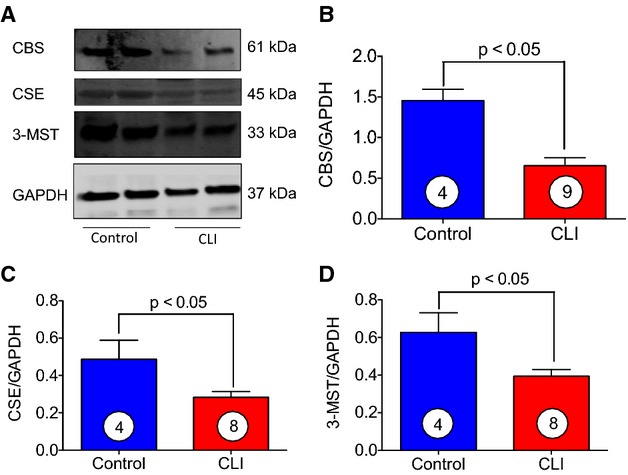
Levels of CBS, CSE, and 3-MST in skeletal muscle of control and CLI patients. A, Representative immunoblots of CBS, CSE, and 3-MST from either skeletal muscle of CLI patients or healthy controls. B through D, Represent relative intensity of immunoblots in (A). Circle with number inside bar denotes number of patients per group. CBS indicates cystathionine β-synthase; CLI, critical limb ischemia; CSE, cystathionine γ-lyase; 3-MST, 3-mercaptopyruvate sulfurtransferase.
Skeletal Muscle of CLI Patients Shows Decreased NRF2 Activity
NRF2 is a nuclear factor that binds to the ARE site in a specific promoter and thereby enhances the transcription of antioxidant protein genes. It was of next interest to determine the NRF2 activity in skeletal muscle of CLI patients. Figure 4A shows mRNA of NRF2 was markedly decreased in skeletal muscle of CLI patients as compared to control subjects. Nuclear level of NRF2 was also found to be decreased significantly (≈2-fold) in CLI patients compared to control subjects (Figure 4B and 4C). DNA binding activity of NRF2 to consensus ARE site was analyzed using electrophoretic mobility shift assay. Figure 4D shows that NRF2-DNA binding activity was markedly decreased in nuclear extracts of skeletal muscle of CLI patients as compared to control subjects. Figure 4E represents the binding specificity of NRF2 to ARE site.
Figure 4.
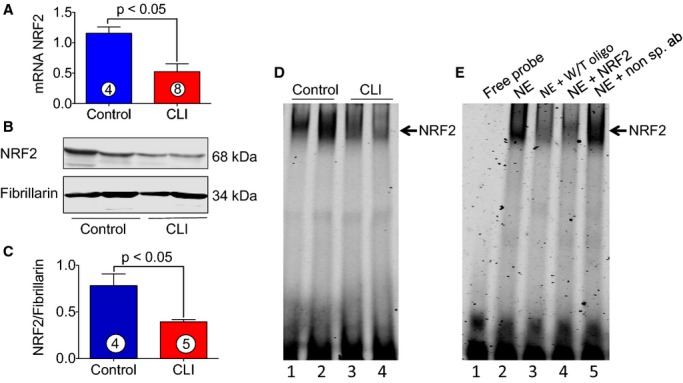
Activity of NRF2 in skeletal muscle of CLI patients. A, Represents mRNA expression of NRF2 in the skeletal muscle of control and CLI patients. B, Represents immunoblots of nuclear levels of NRF2 and fibrillarin in the skeletal muscle of control and CLI patients. C, Quantitation of immunoblots in (B). D, NRF2-DNA binding activity as judged by EMSA; lanes 1 and 2, control; lanes 3 and 4, CLI. E, The binding specificity of NRF2; lane 1, free probe; lane 2, NE; lane 3, NE+W/T oligo (250× excess); lane 4, NE+NRF2 antibody; and lane 5, NE+nonspecific antibody (non sp. ab). Circle with number inside bar denotes number of patients per group. CLI indicates critical limb ischemia; EMSA, electrophoretic mobility shift assay; NE, nuclear extracts; NRF2, nuclear factor-erythroid 2-related factor 2; W/T, wild type.
Induction of Oxidative Stress in Skeletal Muscle of CLI Patients
One of the pathological consequences of chronic tissue ischemia is increased oxidative stress and cellular injury. Therefore, it was of interest to determine the levels of oxidative stress in the protein samples obtained from skeletal muscle of CLI patients. Oxidative stress was measured by determining the levels of protein CO contents and MDA formation. As can be seen in Figure 5A and 5B, both protein CO contents and MDA were significantly increased in skeletal muscle of CLI patients as compared to control subjects. Protein CO contents and MDA are considered biomarkers of oxidative stress; therefore, based on the induction of protein CO (≈2-fold) and MDA (≈2-fold) formation in skeletal muscle of CLI patients, these data suggest that the cellular oxidative stress is increased in CLI patients.
Figure 5.
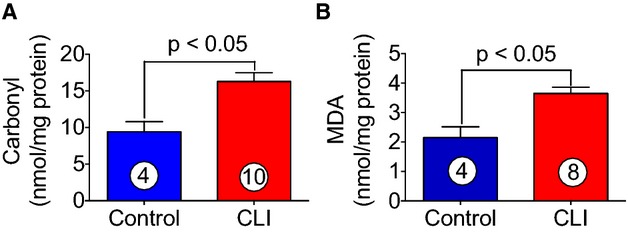
Induction of oxidative stress in skeletal muscle of CLI patients. Biomarkers of oxidative stress such as protein CO contents (A) and MDA (B) formation were significantly increased in skeletal muscle of CLI patients as compared with healthy controls. Circle with number inside bar denotes number of patients per group. CLI indicates critical limb ischemia; CO, carbonyl; MDA, malondialdehyde.
Skeletal Muscle of CLI Patients Exhibits Reduced Levels of Antioxidant Proteins
We next determined the levels of antioxidant proteins in skeletal muscle of CLI patients. We determined both mRNA and protein levels of antioxidant protein in skeletal muscle of CLI patients as well as in control subjects. As can be seen in Figure 6, significant reductions (≈2-fold) in mRNA expression of catalase (A), glutathione peroxidase-1 (GPx-1) (B), and Cu, Zn-superoxide dismutase (SOD1) (C) were observed in skeletal muscle of CLI patients as compared to control subjects. Immunoblot analysis demonstrated significant reduction in protein levels of GPx-1 and superoxide dismutase in skeletal muscle of CLI patients as compared to control (Figure 7A, 7C, and 7D). These data clearly demonstrate reduced expression of antioxidant enzymes results in CLI patients in addition to increased levels of oxidative stress.
Figure 6.
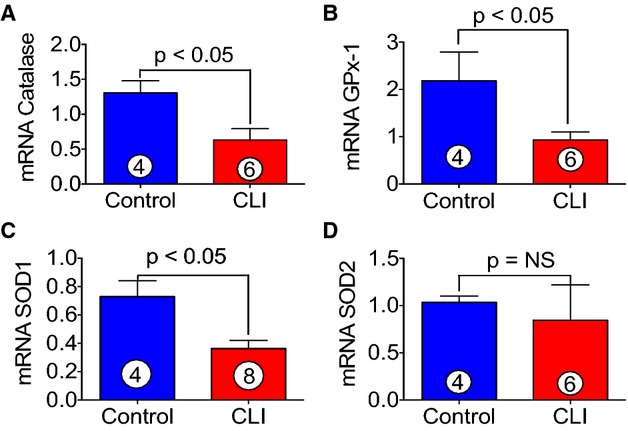
Decreased expression of antioxidant gene in skeletal muscle of CLI patients. A through D, Represent the mRNA expression of catalase, GPx-1, SOD1, and SOD2, respectively, in the skeletal muscle of control and CLI patients. Circle with number inside bar denotes number of patients per group. CLI indicates critical limb ischemia; GPx-1, glutathione peroxidase-1; NS, not significant; SOD1, Cu, Zn-superoxide dismutase.
Figure 7.
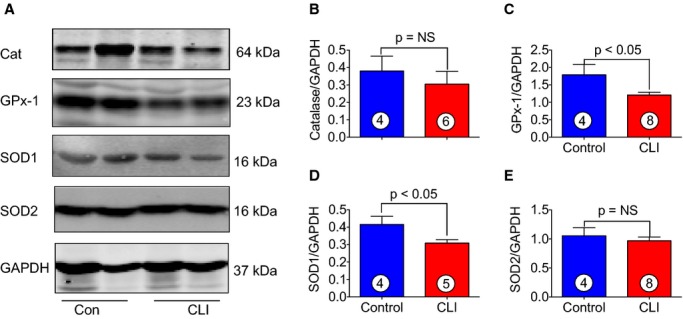
Antioxidant protein levels in skeletal muscle of healthy controls and CLI patients. A, Immunoblots for the levels of Cat, GPx-1, SOD1 and SOD2 of the skeletal muscle of CLI patients and healthy controls. B through E, Quantitation of immunoblots in (A). Circle with number inside bar denotes number of patients per group. Cat indicates catalase; CLI, critical limb ischemia; GPx-1, glutathione peroxidase-1; NS, not significant; SOD1, Cu, Zn-superoxide dismutase; SOD2, Mn-superoxide dismutase.
Decreased Levels of Thioredoxins and Thioredoxin Reductases in Skeletal Muscle of CLI Patients
Thioredoxins are proteins that act as antioxidants by facilitating the reduction of other proteins by cysteine thiol-disulfide exchange. Thioredoxin plays a central role in humans and is increasingly linked to medicine through their response to ROS. In order to define the roles of thioredoxins in CLI patients, both mRNA and protein levels of thioredoxin 1 and 2 were analyzed. As can be seen in Figure 8A through 8E, both mRNA and protein levels of thioredoxin 1 and 2 significantly decreased in CLI patients as compared to healthy control subjects. Furthermore, an experiment was also performed to analyze the expression of thioredoxin reductase 1 and 2. Thioredoxin reductases catalyze the NADPH-dependent reduction of thioredoxin 1 and 2 and other oxidized cellular components and thereby prevent cellular oxidative damage. Figure 9A and 9B shows that mRNA expression of thioredoxin reductases 1 and 2 were significantly decreased in skeletal muscle of CLI patients as compared to control subjects.
Figure 8.
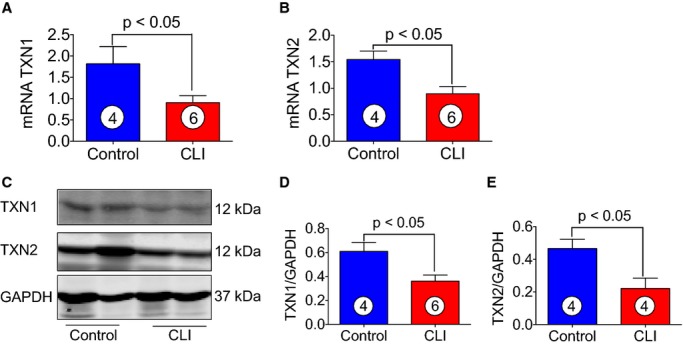
Levels of TXN1 and 2 in skeletal muscle of healthy controls and CLI patients. A and B, Represent the mRNA expression of TXN1 and TXN2, respectively, in the skeletal muscle of control and CLI patients. C, Immunoblots of TXN1 and TXN2 for the protein samples obtained from skeletal muscle tissues from CLI patients and controls. D and E, Quantitation of immunoblots in (C). Circle with number inside bar denotes number of patients per group. CLI indicates critical limb ischemia; TXN, thioredoxin.
Figure 9.
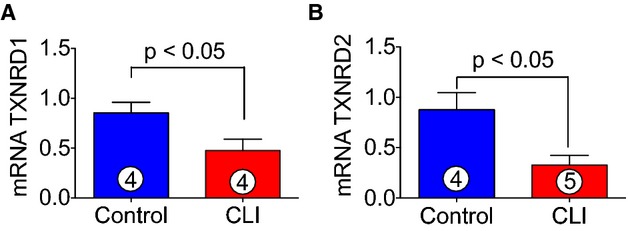
Expression of TXNRD1 and TXNRD2 in CLI patients. A and B, Represent the mRNA expression of TXNRD1 and TXNRD2 in CLI patients and healthy controls. Circle with number inside bar denotes number of patients per group. CLI indicates critical limb ischemia; TXNRD, thioredoxin reductase.
Discussion
In the present study, we provide novel insights into the biochemical and molecular derangements in isolated skeletal muscle in the setting of CLI. The major finding of this study is that the pathological conditions occurring in chronic CLI decrease H2S bioavailability and expression of H2S-producing enzymes in CLI patients. Furthermore, we have determined that NRF2 activity is also significantly attenuated in CLI patients as compared to control subjects. Uncontrolled oxidative stress resulting in decreased H2S bioavailability and attenuated NRF2 activity may significantly contribute to the pathogenesis of CLI. Therapies aimed at restoring redox balance, thereby increasing H2S, may prove beneficial for the treatment of CLI.
H2S is synthesized in tissues throughout the body by enzymes located either in the cytosol (CBS, CSE) or in mitochondria (3-MST). Because H2S is a highly diffusible gas, upon formation it is likely to be either sequestered or catabolized rapidly. The molecular targets for H2S are largely unknown, but recent experimental studies suggest that H2S impacts on intracellular proteins, enzymes, transcription factors, as well as an array of membrane ion channels. H2S produces anti-apoptotic, anti-inflammatory, antihypertrophic, cardioprotective, and anti-oxidant effects, which ultimately can lead to protection from cellular injury or oxidative damage. H2S is also known to have protean effects, including the regulation of inflammation, vascular permeability and tone, and cellular metabolism. H2S also activates prosurvival signaling cascades such as the reperfusion injury salvage kinases, which include PI3-kinase/Akt and extracellular signal-regulated kinase, among others.2,67 Previous studies clearly demonstrate that H2S-releasing drugs significantly limit myocardial ischemia/reperfusion injury and myocardial injury in heart failure.2,4,68,69
A major mechanism by which H2S exerts cytoprotective actions is via upregulation of cellular antioxidants in an NRF2-dependent manner.4 In addition, H2S markedly reduces cellular ROS levels and suppresses oxidative stress in mitochondria via increasing intracellular reduced glutathione concentration.44 Furthermore, mitochondria are able to synthesize H2S via 3-MST and thereby regulate redox balance and oxidant stress within the mitochondria.44 Significant increases in oxidative stress that overwhelm endogenous antioxidant defenses result in potentially cytotoxic reactions with membrane phospholipids, proteins, nucleic acids, and other cellular components, and ultimately impair cell structure and function, leading to cell damage. In the present study, we determined that H2S bioavailability was significantly reduced in the skeletal muscle of CLI patients. We also observed significant reductions in the protein expression of the H2S-producing enzymes in CLI skeletal tissue. It is well appreciated that H2S leads to NRF2 activation,70,71 which attenuates oxidative damage by increasing the levels of antioxidant proteins. Previous reports have indicated that NRF2 deficiency is associated with enhanced oxidative stress and cell death.72,73 NRF2 is a family of nuclear basic leucine zipper transcription factors that regulate the gene expression of a number of antioxidant enzymes that serve to detoxify pro-oxidative stressors63 including GPx-1 and heme oxygenase 1.4 NRF2 is widely expressed and is thought to translocate to the nucleus after treatment with xenobiotics and antioxidants, which stimulate its release from a repressor protein Kelch-like ECH (enoyl-CoA hydratase)-associated protein 1. Following translocation into the nucleus, NRF2 binds the ARE site in the promoter region of a gene and enhances the transcription of antioxidant proteins, which results in suppression of an oxidative stress response. Therefore, NRF2 controls the basal and inducible expression of antioxidant genes and other cytoprotective phase II detoxifying enzymes that are ubiquitously expressed throughout the cardiovascular system. Emerging evidence has revealed that NRF2 and its target genes are critical regulators of cardiovascular homeostasis via the suppression of oxidative stress/ROS production.74 Similarly, oxidative stress/reactive oxygen species may also potentiate development of CLI.
In the present study, we found a significant induction of oxidative stress in skeletal muscle of CLI patients compared to controls, as judged by measuring the levels of protein CO contents and MDA formation (as protein CO contents and MDA formation are considered biomarkers of oxidative stress). Under basal conditions, the ability of NRF2 to induce endogenous antioxidants is tightly regulated when anchored in the cytoplasm, and is targeted for ubiquitination and proteasome degradation.75 Only when there is a disruption in the binding of NRF2 to the cytosolic anchoring proteins (such as Kelch-like ECH-associated protein 1) is NRF2 able to translocate to the nucleus and protect the cells from oxidative damage through enhancing the transcription of antioxidant genes. Previous studies have indicated that H2S exerts cytoprotective actions through upregulation of cellular antioxidants via the NRF2 signaling pathway.4
Medical management of CLI entails pain control, risk factors modification, proper wound care, and infection management. The management of this complex patient population often warrants a multidisciplinary approach with collaboration with an endovascular interventionist, vascular surgeons, podiatrist, infectious disease specialist, and wound-care specialist. Although data are limited, with regard to pharmacological interventions in CLI patients, most of the pharmacological measures are of proven benefit in reducing the risk of death, myocardial infarction, heart failure, and stroke in patients with atherosclerosis, and therefore the use of such therapy, despite the lack of evidence in the setting of CLI, is justified to limit adverse cardiovascular events.76–82 Various reports have demonstrated that the use of “cardioprotective” medications such as statins, angiotensin-converting enzyme inhibitors and angiotensin receptor blockers, and antiplatelet agents is associated with a decreased cardiovascular event rate in patients with PAD.77 However, little is known about the effectiveness of these drugs in patients with CLI. Effective new treatments for CLI are critically important, given the high incidence of major amputation and mortality seen in this population.83 Increasing blood flow, tissue perfusion, and oxygen and nutrient delivery via new vessel growth (ie, angiogenesis) or collateralization could promote tissue regeneration, and/or delay or even prevent tissue necrosis and the development of conditions such as gangrene.
In the present study, we observed that levels of H2S and H2S-producing enzymes were significantly decreased in skeletal muscle of CLI patients as compared to healthy controls. We also found significant reductions in NRF2 activation and antioxidant proteins concomitant with increased skeletal muscle oxidative stress in CLI patients. In conclusion, the present study clearly demonstrates that induction of oxidative stress and attenuated NRF2 signaling appears to be involved in the pathogenesis of CLI and is associated with significant reductions in skeletal muscle H2S bioavailability. Given the recent elucidation of the potent cytoprotective, antioxidant, and pro-angiogenic properties of physiological or pharmacological levels of H2S, it is possible that H2S-based therapies or H2S-induced NRF2 activation may prove beneficial for the treatment of CLI. In the present study, we examined a small sample of control tissues, which represents a limitation of this research. However, it is very difficult to obtain these skeletal muscle samples from otherwise healthy individuals undergoing limb amputations. Clearly, additional studies of CLI in clinically relevant models are required to validate the therapeutic effects of H2S in this devastating disease.
Sources of Funding
This work was supported by grants from the National Heart, Lung, and Blood Institute (National Institutes of Health; 1R01 HL092141 (Lefer), 1R01 HL093579 (Lefer), 1U24 HL 094373 (Lefer), 1P20 HL113452 (Lefer), and 1KO8HL119592 (Brewster) and supported in part by PHS (Public Health Service) Grant UL1TR000454 from the Clinical and Translational Science Award Program, National Institutes of Health, National Center for Advancing Translational Sciences (Brewster) and the American Heart Association 13IRG14740001 (Brewster).
Disclosures
None.
References
- Adam DJ, Bradbury AW. TASC II document on the management of peripheral arterial disease. Eur J Vasc Endovasc Surg. 2007;33:1–2. doi: 10.1016/j.ejvs.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Henderson PW, Jimenez N, Ruffino J, Sohn AM, Weinstein AL, Krijgh DD, Reiffel AJ, Spector JA. Therapeutic delivery of hydrogen sulfide for salvage of ischemic skeletal muscle after the onset of critical ischemia. J Vasc Surg. 2011;53:785–791. doi: 10.1016/j.jvs.2010.10.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG Group TIW. Inter-society consensus for the management of peripheral arterial disease (TASC II) J Vasc Surg. 2007;45(suppl S):S5–S67. doi: 10.1016/j.jvs.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, Kevil CG, Lefer DJ. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res. 2009;105:365–374. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano M. Epidemiology of critical limb ischaemia: North Italian data. Eur J Med. 1993;2:11–14. [PubMed] [Google Scholar]
- Fosse S, Hartemann-Heurtier A, Jacqueminet S, Ha Van G, Grimaldi A, Fagot-Campagna A. Incidence and characteristics of lower limb amputations in people with diabetes. Diabet Med. 2009;26:391–396. doi: 10.1111/j.1464-5491.2009.02698.x. [DOI] [PubMed] [Google Scholar]
- Jensen SA, Vatten LJ, Myhre HO. The prevalence of chronic critical lower limb ischaemia in a population of 20,000 subjects 40-69 years of age. Eur J Vasc Endovasc Surg. 2006;32:60–65. doi: 10.1016/j.ejvs.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Paraskevas KI, Hamilton G, Mikhailidis DP, Liapis CD. Optimal medical management of peripheral arterial disease. Vasc Endovascular Surg. 2007;41:87. doi: 10.1177/1538574406297268. [DOI] [PubMed] [Google Scholar]
- Gregorakos L, Dimopoulos G, Liberi S, Antipas G. Hydrogen sulfide poisoning: management and complications. Angiology. 1995;46:1123–1131. doi: 10.1177/000331979504601208. [DOI] [PubMed] [Google Scholar]
- Lopez A, Prior MG, Reiffenstein RJ, Goodwin LR. Peracute toxic effects of inhaled hydrogen sulfide and injected sodium hydrosulfide on the lungs of rats. Fundam Appl Toxicol. 1989;12:367–373. doi: 10.1016/0272-0590(89)90053-5. [DOI] [PubMed] [Google Scholar]
- Stine RJ, Slosberg B, Beacham BE. Hydrogen sulfide intoxication: A case report and discussion of treatment. Ann Intern Med. 1976;85:756–758. doi: 10.7326/0003-4819-85-6-756. [DOI] [PubMed] [Google Scholar]
- Dombkowski RA, Doellman MM, Head SK, Olson KR. Hydrogen sulfide mediates hypoxia-induced relaxation of trout urinary bladder smooth muscle. J Exp Biol. 2006;209:3234–3240. doi: 10.1242/jeb.02376. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Distrutti E, Cirino G, Wallace JL. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology. 2006;131:259–271. doi: 10.1053/j.gastro.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Lowicka E, Beltowski J. Hydrogen sulfide (H2S)—the third gas of interest for pharmacologists. Pharmacol Rep. 2007;59:4–24. [PubMed] [Google Scholar]
- Tang C, Li X, Du J. Hydrogen sulfide as a new endogenous gaseous transmitter in the cardiovascular system. Curr Vasc Pharmacol. 2006;4:17–22. doi: 10.2174/157016106775203144. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob C, Anwar A, Burkholz T. Perspective on recent developments on sulfur-containing agents and hydrogen sulfide signaling. Planta Med. 2008;74:1580–1592. doi: 10.1055/s-0028-1088299. [DOI] [PubMed] [Google Scholar]
- Kimura H. Hydrogen sulfide: its production, release and functions. Amino Acids. 2011;41:113–121. doi: 10.1007/s00726-010-0510-x. [DOI] [PubMed] [Google Scholar]
- Kimura H, Shibuya N, Kimura Y. Hydrogen sulfide is a signaling molecule and a cytoprotectant. Antioxid Redox Signal. 2012;17:45–57. doi: 10.1089/ars.2011.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler CW, Parfenova H, Jaggar JH, Wang R. Carbon monoxide and hydrogen sulfide: gaseous messengers in cerebrovascular circulation. J Appl Physiol. 2006;100:1065–1076. doi: 10.1152/japplphysiol.00793.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson KR. A practical look at the chemistry and biology of hydrogen sulfide. Antioxid Redox Signal. 2012;17:32–44. doi: 10.1089/ars.2011.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu K, Lee SW, Bian JS, Low CM, Wong PT. Hydrogen sulfide: neurochemistry and neurobiology. Neurochem Int. 2008;52:155–165. doi: 10.1016/j.neuint.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Stipanuk MH, Ueki I. Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J Inherit Metab Dis. 2011;34:17–32. doi: 10.1007/s10545-009-9006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman M, Moore PK. Hydrogen sulfide and the vasculature: a novel vasculoprotective entity and regulator of nitric oxide bioavailability? J Cell Mol Med. 2009;13:488–507. doi: 10.1111/j.1582-4934.2009.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Predmore BL, Lefer DJ, Gojon G. Hydrogen sulfide in biochemistry and medicine. Antioxid Redox Signal. 2012;17:119–140. doi: 10.1089/ars.2012.4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doeller JE, Isbell TS, Benavides G, Koenitzer J, Patel H, Patel RP, Lancaster JR, Jr, Darley-Usmar VM, Kraus DW. Polarographic measurement of hydrogen sulfide production and consumption by mammalian tissues. Anal Biochem. 2005;341:40–51. doi: 10.1016/j.ab.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discovery. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. 2008;295:H801–H806. doi: 10.1152/ajpheart.00377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan TT, Feng ZN, Lee SW, Moore PK, Bian JS. Endogenous hydrogen sulfide contributes to the cardioprotection by metabolic inhibition preconditioning in the rat ventricular myocytes. J Mol Cell Cardiol. 2006;40:119–130. doi: 10.1016/j.yjmcc.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Fukuto JM, Carrington SJ, Tantillo DJ, Harrison JG, Ignarro LJ, Freeman BA, Chen A, Wink DA. Small molecule signaling agents: the integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem Res Toxicol. 2012;25:769–793. doi: 10.1021/tx2005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furne J, Saeed A, Levitt MD. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1479–R1485. doi: 10.1152/ajpregu.90566.2008. [DOI] [PubMed] [Google Scholar]
- Kabil O, Banerjee R. Redox biochemistry of hydrogen sulfide. J Biol Chem. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavu M, Bhushan S, Lefer DJ. Hydrogen sulfide-mediated cardioprotection: mechanisms and therapeutic potential. Clin Sci. 2011;120:219–229. doi: 10.1042/CS20100462. [DOI] [PubMed] [Google Scholar]
- Li L, Rose P, Moore PK. Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol. 2011;51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- Lynn EG, Austin RC. Hydrogen sulfide in the pathogenesis of atherosclerosis and its therapeutic potential. Expert Rev Clin Pharmacol. 2011;4:97–108. doi: 10.1586/ecp.10.130. [DOI] [PubMed] [Google Scholar]
- Olson KR. Is hydrogen sulfide a circulating “gasotransmitter” in vertebrate blood? Biochim Biophys Acta. 2009;1787:856–863. doi: 10.1016/j.bbabio.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Olson KR. The therapeutic potential of hydrogen sulfide: separating hype from hope. Am J Physiol Regul Integr Comp Physiol. 2011;301:R297–R312. doi: 10.1152/ajpregu.00045.2011. [DOI] [PubMed] [Google Scholar]
- Tangerman A. Measurement and biological significance of the volatile sulfur compounds hydrogen sulfide, methanethiol and dimethyl sulfide in various biological matrices. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3366–3377. doi: 10.1016/j.jchromb.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR. Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1930–R1937. doi: 10.1152/ajpregu.00025.2008. [DOI] [PubMed] [Google Scholar]
- Bliksoen M, Kaljusto ML, Vaage J, Stenslokken KO. Effects of hydrogen sulphide on ischaemia-reperfusion injury and ischaemic preconditioning in the isolated, perfused rat heart. Eur J Cardiothorac Surg. 2008;34:344–349. doi: 10.1016/j.ejcts.2008.03.017. [DOI] [PubMed] [Google Scholar]
- Henderson PW, Singh SP, Belkin D, Nagineni V, Weinstein AL, Weissich J, Spector JA. Hydrogen sulfide protects against ischemia-reperfusion injury in an in vitro model of cutaneous tissue transplantation. J Surg Res. 2010;159:451–455. doi: 10.1016/j.jss.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Johansen D, Ytrehus K, Baxter GF. Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury—evidence for a role of K ATP channels. Basic Res Cardiol. 2006;101:53–60. doi: 10.1007/s00395-005-0569-9. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Dargusch R, Schubert D, Kimura H. Hydrogen sulfide protects HT22 neuronal cells from oxidative stress. Antioxid Redox Signal. 2006;8:661–670. doi: 10.1089/ars.2006.8.661. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Goto Y, Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal. 2010;12:1–13. doi: 10.1089/ars.2008.2282. [DOI] [PubMed] [Google Scholar]
- Rossoni G, Sparatore A, Tazzari V, Manfredi B, Del Soldato P, Berti F. The hydrogen sulphide-releasing derivative of diclofenac protects against ischaemia-reperfusion injury in the isolated rabbit heart. Br J Pharmacol. 2008;153:100–109. doi: 10.1038/sj.bjp.0707540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivarajah A, Collino M, Yasin M, Benetti E, Gallicchio M, Mazzon E, Cuzzocrea S, Fantozzi R, Thiemermann C. Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock. 2009;31:267–274. doi: 10.1097/SHK.0b013e318180ff89. [DOI] [PubMed] [Google Scholar]
- Sodha NR, Clements RT, Feng J, Liu Y, Bianchi C, Horvath EM, Szabo C, Stahl GL, Sellke FW. Hydrogen sulfide therapy attenuates the inflammatory response in a porcine model of myocardial ischemia/reperfusion injury. J Thorac Cardiovasc Surg. 2009;138:977–984. doi: 10.1016/j.jtcvs.2008.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res. 2007;76:29–40. doi: 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- Shi YX, Chen Y, Zhu YZ, Huang GY, Moore PK, Huang SH, Yao T, Zhu YC. Chronic sodium hydrosulfide treatment decreases medial thickening of intramyocardial coronary arterioles, interstitial fibrosis, and ROS production in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;293:H2093–H2100. doi: 10.1152/ajpheart.00088.2007. [DOI] [PubMed] [Google Scholar]
- Zhao X, Zhang LK, Zhang CY, Zeng XJ, Yan H, Jin HF, Tang CS, Du JB. Regulatory effect of hydrogen sulfide on vascular collagen content in spontaneously hypertensive rats. Hypertens Res. 2008;31:1619–1630. doi: 10.1291/hypres.31.1619. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhao X, Jin H, Wei H, Li W, Bu D, Tang X, Ren Y, Tang C, Du J. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2009;29:173–179. doi: 10.1161/ATVBAHA.108.179333. [DOI] [PubMed] [Google Scholar]
- Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- Liu H, Bai XB, Shi S, Cao YX. Hydrogen sulfide protects from intestinal ischaemia-reperfusion injury in rats. J Pharm Pharmacol. 2009;61:207–212. doi: 10.1211/jpp/61.02.0010. [DOI] [PubMed] [Google Scholar]
- Chang L, Geng B, Yu F, Zhao J, Jiang H, Du J, Tang C. Hydrogen sulfide inhibits myocardial injury induced by homocysteine in rats. Amino Acids. 2008;34:573–585. doi: 10.1007/s00726-007-0011-8. [DOI] [PubMed] [Google Scholar]
- Sen U, Basu P, Abe OA, Givvimani S, Tyagi N, Metreveli N, Shah KS, Passmore JC, Tyagi SC. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. Am J Physiol Renal Physiol. 2009;297:F410–F419. doi: 10.1152/ajprenal.00145.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi N, Moshal KS, Sen U, Vacek TP, Kumar M, Hughes WM, Jr, Kundu S, Tyagi SC. H2S protects against methionine-induced oxidative stress in brain endothelial cells. Antioxid Redox Signal. 2009;11:25–33. doi: 10.1089/ars.2008.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SK, Chang T, Wang H, Wu L, Wang R, Meng QH. Effects of hydrogen sulfide on homocysteine-induced oxidative stress in vascular smooth muscle cells. Biochem Biophys Res Commun. 2006;351:485–491. doi: 10.1016/j.bbrc.2006.10.058. [DOI] [PubMed] [Google Scholar]
- Jeney V, Komodi E, Nagy E, Zarjou A, Vercellotti GM, Eaton JW, Balla G, Balla J. Suppression of hemin-mediated oxidation of low-density lipoprotein and subsequent endothelial reactions by hydrogen sulfide (H(2)S) Free Radic Biol Med. 2009;46:616–623. doi: 10.1016/j.freeradbiomed.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–1231. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, Cheung NS, Halliwell B, Moore PK. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J Neurochem. 2004;90:765–768. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- Geng B, Yang J, Qi Y, Zhao J, Pang Y, Du J, Tang C. H2S generated by heart in rat and its effects on cardiac function. Biochem Biophys Res Commun. 2004;313:362–368. doi: 10.1016/j.bbrc.2003.11.130. [DOI] [PubMed] [Google Scholar]
- Elrod JW, Greer JJ, Bryan NS, Langston W, Szot JF, Gebregzlabher H, Janssens S, Feelisch M, Lefer DJ. Cardiomyocyte-specific overexpression of NO synthase-3 protects against myocardial ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2006;26:1517–1523. doi: 10.1161/01.ATV.0000224324.52466.e6. [DOI] [PubMed] [Google Scholar]
- Fisher CD, Augustine LM, Maher JM, Nelson DM, Slitt AL, Klaassen CD, Lehman-McKeeman LD, Cherrington NJ. Induction of drug-metabolizing enzymes by garlic and allyl sulfide compounds via activation of constitutive androstane receptor and nuclear factor E2-related factor 2. Drug Metab Dispos. 2007;35:995–1000. doi: 10.1124/dmd.106.014340. [DOI] [PubMed] [Google Scholar]
- Islam KN, Koch WJ. Involvement of nuclear factor kappaB (NF-kappaB) signaling pathway in regulation of cardiac G protein-coupled receptor kinase 5 (GRK5) expression. J Biol Chem. 2012;287:12771–12778. doi: 10.1074/jbc.M111.324566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam KN, Kayanoki Y, Kaneto H, Suzuki K, Asahi M, Fujii J, Taniguchi N. TGF-beta1 triggers oxidative modifications and enhances apoptosis in HIT cells through accumulation of reactive oxygen species by suppression of catalase and glutathione peroxidase. Free Radic Biol Med. 1997;22:1007–1017. doi: 10.1016/s0891-5849(96)00493-5. [DOI] [PubMed] [Google Scholar]
- King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R, Lefer DJ. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc Natl Acad Sci USA. 2014;111:3182–3187. doi: 10.1073/pnas.1321871111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Chen X, Pan TT, Neo KL, Lee SW, Khin ES, Moore PK, Bian JS. Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and Pi3K/Akt pathways. Pflugers Arch. 2008;455:607–616. doi: 10.1007/s00424-007-0321-4. [DOI] [PubMed] [Google Scholar]
- Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G, Sr, Gojon G, Jr, Wang R, Karusula N, Nicholson CK, Calvert JW, Lefer DJ. H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polhemus DJ, Kondo K, Bhushan S, Bir SC, Kevil CG, Murohara T, Lefer DJ, Calvert JW. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ Heart Fail. 2013;6:1077–1086. doi: 10.1161/CIRCHEARTFAILURE.113.000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Guo Y, Ou Q, Cui C, Wu WJ, Tan W, Zhu X, Lanceta LB, Sanganalmath SK, Dawn B, Shinmura K, Rokosh GD, Wang S, Bolli R. Gene transfer of inducible nitric oxide synthase affords cardioprotection by upregulating heme oxygenase-1 via a nuclear factor-{kappa}B-dependent pathway. Circulation. 2009;120:1222–1230. doi: 10.1161/CIRCULATIONAHA.108.778688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um HC, Jang JH, Kim DH, Lee C, Surh YJ. Nitric oxide activates Nrf2 through S-nitrosylation of Keap1 in PC12 cells. Nitric Oxide. 2011;25:161–168. doi: 10.1016/j.niox.2011.06.001. [DOI] [PubMed] [Google Scholar]
- He X, Kan H, Cai L, Ma Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J Mol Cell Cardiol. 2009;46:47–58. doi: 10.1016/j.yjmcc.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Yoh K, Hirayama A, Ishizaki K, Yamada A, Takeuchi M, Yamagishi S, Morito N, Nakano T, Ojima M, Shimohata H, Itoh K, Takahashi S, Yamamoto M. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells. 2008;13:1159–1170. doi: 10.1111/j.1365-2443.2008.01234.x. [DOI] [PubMed] [Google Scholar]
- Zhou S, Sun W, Zhang Z, Zheng Y. The role of Nrf2-mediated pathway in cardiac remodeling and heart failure. Oxid Med Cell Longev. 2014;2014:260429. doi: 10.1155/2014/260429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antithrombotic Trialists C. Baigent C, Blackwell L, Collins R, Emberson J, Godwin J, Peto R, Buring J, Hennekens C, Kearney P, Meade T, Patrono C, Roncaglioni MC, Zanchetti A. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849–1860. doi: 10.1016/S0140-6736(09)60503-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DL, Fox KA, Hacke W, Berger PB, Black HR, Boden WE, Cacoub P, Cohen EA, Creager MA, Easton JD, Flather MD, Haffner SM, Hamm CW, Hankey GJ, Johnston SC, Mak KH, Mas JL, Montalescot G, Pearson TA, Steg PG, Steinhubl SR, Weber MA, Brennan DM, Fabry-Ribaudo L, Booth J, Topol EJ Investigators C. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med. 2006;354:1706–1717. doi: 10.1056/NEJMoa060989. [DOI] [PubMed] [Google Scholar]
- Cacoub PP, Bhatt DL, Steg PG, Topol EJ, Creager MA Investigators C. Patients with peripheral arterial disease in the CHARISMA trial. Eur Heart J. 2009;30:192–201. doi: 10.1093/eurheartj/ehn534. [DOI] [PubMed] [Google Scholar]
- Investigators O. Yusuf S, Teo KK, Pogue J, Dyal L, Copland I, Schumacher H, Dagenais G, Sleight P, Anderson C. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547–1559. doi: 10.1056/NEJMoa0801317. [DOI] [PubMed] [Google Scholar]
- Mancia G, De Backer G, Dominiczak A, Cifkova R, Fagard R, Germano G, Grassi G, Heagerty AM, Kjeldsen SE, Laurent S, Narkiewicz K, Ruilope L, Rynkiewicz A, Schmieder RE, Struijker Boudier HA, Zanchetti A, Vahanian A, Camm J, De Caterina R, Dean V, Dickstein K, Filippatos G, Funck-Brentano C, Hellemans I, Kristensen SD, McGregor K, Sechtem U, Silber S, Tendera M, Widimsky P, Zamorano JL, Kjeldsen SE, Erdine S, Narkiewicz K, Kiowski W, Agabiti-Rosei E, Ambrosioni E, Cifkova R, Dominiczak A, Fagard R, Heagerty AM, Laurent S, Lindholm LH, Mancia G, Manolis A, Nilsson PM, Redon J, Schmieder RE, Struijker-Boudier HA, Viigimaa M, Filippatos G, Adamopoulos S, Agabiti-Rosei E, Ambrosioni E, Bertomeu V, Clement D, Erdine S, Farsang C, Gaita D, Kiowski W, Lip G, Mallion JM, Manolis AJ, Nilsson PM, O’Brien E, Ponikowski P, Redon J, Ruschitzka F, Tamargo J, van Zwieten P, Viigimaa M, Waeber B, Williams B, Zamorano JL The task force for the management of arterial hypertension of the European Society of H, The task force for the management of arterial hypertension of the European Society of C. 2007 Guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC) Eur Heart J. 2007;28:1462–1536. doi: 10.1093/eurheartj/ehm236. [DOI] [PubMed] [Google Scholar]
- Radack K, Deck C. Beta-adrenergic blocker therapy does not worsen intermittent claudication in subjects with peripheral arterial disease: a meta-analysis of randomized controlled trials. Arch Intern Med. 1991;151:1769–1776. [PubMed] [Google Scholar]
- Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients: The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–153. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- Jude EB, Jacob K. Managing painful neuropathy in diabetes. Pol Arch Med Wewn. 2008;118:260–261. [PubMed] [Google Scholar]