

Abstract
Background
ATP-sensitive potassium (KATP) channel openers provide cardioprotection in multiple models. Ion flux at an unidentified mitochondrial KATP channel has been proposed as the mechanism. The renal outer medullary kidney potassium channel subunit, potassium inward rectifying (Kir)1.1, has been implicated as a mitochondrial channel pore-forming subunit. We hypothesized that subunit Kir1.1 is involved in cardioprotection (maintenance of volume homeostasis and contractility) of the KATP channel opener diazoxide (DZX) during stress (exposure to hyperkalemic cardioplegia [CPG]) at the myocyte and mitochondrial levels.
Methods and Results
Kir subunit inhibitor Tertiapin Q (TPN-Q) was utilized to evaluate response to stress. Mouse ventricular mitochondrial volume was measured in the following groups: isolation buffer; 200 μmol/L of ATP; 100 μmol/L of DZX+200 μmol/L of ATP; or 100 μmol/L of DZX+200 μmol/L of ATP+TPN-Q (500 or 100 nmol/L). Myocytes were exposed to Tyrode’s solution (5 minutes), test solution (Tyrode’s, cardioplegia [CPG], CPG+DZX, CPG+DZX+TPN-Q, Tyrode’s+TPN-Q, or CPG+TPN-Q), N=12 for all (10 minutes); followed by Tyrode’s (5 minutes). Volumes were compared. TPN-Q, with or without DZX, did not alter mitochondrial or myocyte volume. Stress (CPG) resulted in myocyte swelling and reduced contractility that was prevented by DZX. TPN-Q prevented the cardioprotection afforded by DZX (volume homeostasis and maintenance of contractility).
Conclusions
TPN-Q inhibited myocyte cardioprotection provided by DZX during stress; however, it did not alter mitochondrial volume. Because TPN-Q inhibits Kir1.1, Kir3.1, and Kir3.4, these data support that any of these Kir subunits could be involved in the cardioprotection afforded by diazoxide. However, these data suggest that mitochondrial swelling by diazoxide does not involve Kir1.1, 3.1, or 3.4.
Keywords: cardioplegia, ion channels, myocardial stunning, potassium
In an isolated model of myocardial stunning, cardiac myocytes demonstrate significant swelling and reduced contractility during exposure to 3 different stresses: hypothermic hyperkalemic cardioplegia (CPG), hypo-osmotic stress, or metabolic inhibition.1–3 The ATP-sensitive potassium channel (KATP) opener, diazoxide (DZX), ameliorates these stress-induced derangements in mouse, rabbit, and human tissue.1–5 A role of KATP channels in ischemic preconditioning, by coupling membrane conductance of K+ ions to the metabolic state of the cell, has also been well defined in multiple animal models.6–10 However, no precise mechanism of cardioprotection has been identified.
KATP channels are formed as hetero-octamers of subunits that include 4 potassium inward rectifying (Kir) subunits and 4 regulatory subunits from the sulfonylurea receptor family (SUR1/2).11–13 Different combinations of Kir and SUR isoforms exist in different tissues, each with unique properties and function. For example, the predominant combination of subunits found in murine ventricle is Kir6.2 and SUR2A, whereas murine atria mainly consist of Kir6.2 and SUR1.14 Identification of these subunits has allowed for genetic manipulation to more specifically evaluate the mechanism of DZX.
Using KATP subunit-knockout animal models, we have shown that the cardioprotection afforded by DZX requires the regulatory subunit, SUR1, but the pore-forming subunit involved remains elusive.15,16 We have also demonstrated that the sarcolemmal KATP channel (sKATP) is implicated in myocyte swelling secondary to stress, and that knockout of the pore-forming subunit of this channel (Kir6.2) provides resistance to myocyte swelling.2,17 However, there is evidence that DZX cardioprotection does not require the sKATP channel, leading us to propose mechanisms that may include non-KATP channel or mitochondrial KATP channel (mKATP) locations of action.15,16,18–21
Regulation of mitochondrial volume through a purported mKATP channel is one of the proposed mechanisms of action of DZX.22–24 Because no molecular composition of mKATP has been determined, identification of mKATP channel subunits has to date involved indirect methods, such as pharmacological inhibition or observed changes in mitochondrial volume (proposed assay of mKATP activity).24,25 Using such strategies, the inward rectifier potassium channel subunit, Kir1.1 (renal outer medullary kidney channel; ROMK) has been implicated as a pore-forming subunit of the mKATP channel utilizing the nonspecific inhibitor Tertiapin Q (TPN-Q).24
The present study was conducted to assess potential roles of Kir1.1, Kir3.1, and Kir3.4 in the cardioprotection of DZX at the cellular and mitochondrial levels using the Kir subunit inhibitor, TPN-Q.
Methods
All animal procedures were approved by the Animal Studies Committee at Washington University School of Medicine, and all animals received humane care in compliance with the National Institute of Health’s (NIH) Guide to Care and Use of Laboratory Animals.26
Mitochondrial Isolation
Mice (both sexes, 6 weeks to 5 months and 15 to 30 g in weight) were anesthetized with 3% Avertin (0.3 g of 2,2,2-tribromoethanol, 0.186 mL of 2-methyl2-butanol, and 9.814 mL of sterile water) intraperitoneally, and rapid cardiectomy was performed. To isolate mitochondria, atria were excised and discarded whereas ventricular tissue was rapidly minced in cold buffer (in mmol/L: 10 HEPES (N-[2-hydroxyethyl]1,3–5 piperazine-N-[4-butanesulfonic acid]), 1 EDTA-potassium, 250 sucrose, adjusted to a pH of 7.1 with 20% KOH and transferred to a 10-mL glass tube with a Teflon pestle (Glas-Col Homogenizer, Terre Haute, IN), and volume was adjusted to 7 mL with buffer. Tissue was mechanically homogenized with a Teflon pestle driven by a low-speed motor drive shaft set at 120 rpm. The homogenate was transferred to 6 microcentrifuge tubes and centrifuged at 900g for 10 minutes at 4°C. The supernatant was combined into a clean test tube and mixed to get a homogeneous solution that was divided equally between 6 clean microcentrifuge tubes and centrifuged at 5000g for 15 minutes. One pellet was resuspended in 100 μL of buffer, and 10 μL was taken in duplicate for the Bradford protein assay (Thermo Scientific; Rockford, IL) to determine total protein. Each pellet was maintained on ice and was resuspended in test media volume to equal 0.3 μg/μL in order to standardize protein content.
Mitochondrial Volume Measurement
The volume of isolated mitochondria was measured after suspension in test solution: (1) isolation buffer (No ATP; n=13); (2) 200 μmol/L of ATP (n=12); (3) 200 μmol/L of ATP and 100 μmol/L of DZX (Sigma-Aldrich, St. Louis, MO) (n=12); (4) 200 μmol/L of ATP, 100 μmol/L of DZX, and 500 nmol/L of TPN-Q (n=7); or (5) 200 μmol/L of ATP, 100 μmol/L of DZX, and 100 nmol/L of TPN-Q (n=6). Isolation buffer (10 mmol/L of HEPES, 200 mmol/L of mannitol, 50 mmol/L of sucrose, 1 mmol/L of EGTA; pH 7.2) was used as a control solution. ATP has been shown to close mitochondrial KATP channels, so 200 μmol/L of ATP was used to slow the initial rate of mitochondrial swelling (0% Mito KATP activity).22 Conversely, DZX activates “Mito KATP” channels, and 100 μmol/L of DZX was added to achieve maximal activation of mitochondrial KATP channels (100% Mito KATP activity). We were unable to reproduce the TPN-Q dose-response relationships on mitochondrial volume demonstrated by other investigators using 4 TPN-Q concentrations (0.5, 10, 90, or 1000 pmol/L) despite attempts utilizing 3 different vehicles for TPN-Q: 20% acetonitrile, water, and HEPES.24 In the present study, water was used as a vehicle for TPN-Q because of its stability in the medium and because 20% acetonitrile alone resulted in myocyte swelling owing to its cyanide moiety.
Mitochondrial matrix volume measurements were obtained using a light-scattering technique,27 where the absorbance, at 520 nm, of a solution of isolated mitochondria was obtained every 14 seconds for the period of 3 minutes using UV Probe 2.33 (Shimadzu Scientific Instruments, Columbia, MD) and a spectrophotometer (UV-1700 Spectrophotometer; Shimadzu Scientific Instruments, Columbia, MD).
Myocyte Isolation
Ventricular myocytes were isolated from C57BL/6J mice of either sex (age 6 weeks to 5 months and 15 to 30 g in weight), as previously described.15 Mice were anesthetized with 2.5% Avertin intraperitoneally. Heparin (0.1 mL) was administered intraperitoneally. Rapid cardiectomy was performed and solution A (described below) was perfused through the aorta for 5 minutes. The heart was then perfused at 37°C for 12 to 20 minutes with solution B (described below). Ventricles were removed, minced, and placed into solution C (described below) and gently dispersed by glass pipette. Cells were allowed to centrifuge by gravity, and serial washings were performed every 10 minutes for 15 to 20 minutes. Cells were used within 5 hours and randomized to treatment group. A typical yield of viable myocytes was 65% to 75%.
Solution A consisted of (in mmol/L, except as noted) 116 NaCl; 5.36 KCl; 0.97 Na2HPO4; 1.47 KH2PO4; 21.10 HEPES (N-[2-hydroxyethyl] piperazine-N′-[4-butanesulfonic acid]); 11.65 glucose; 26.50 μmol/L of phenol red (Sigma-Aldrich); 3.72 MgCl2; 4.40 NaHCO3; essential vitamins (100×, 10 mL; GIBCO, Grand Island, NY); and amino acids (50×, 20 mL; GIBCO). Solution B consisted of solution A plus 10 μmol/L of CaCl2; 1.2 mg/mL of collagenase (Type 2; Worthington Biochemical Corporation, Freehold, NJ). Solution C consisted of solution A plus 5 mg/mL of BSA (Sigma-Aldrich); 1.25 mg/mL of taurine; and 150 μmol/L of CaCl2.
Myocytes were exposed to 37°C control TYR for 5 minutes to obtain baseline volume. Any changes in cell volume secondary to the isolation would be evident during this period. Myocytes were then exposed to test solution (10 minutes) followed by re-exposure to TYR solution (5 minutes). Test solutions included the following groups (n=12 for each): (1) TYR; (2) CPG; (3) CPG+100 μmol/L of DZX; (4) CPG+100 μmol/L of DZX+TPN-Q 200 nm/L; (5) TYR+TPN-Q; and (6) CPG+TPN-Q.
CPG consisted of (in mmol/L): NaCl 110, NaHCO3 10, KCl 16, MgCl2 16, and CaCl2 1.2 and was equilibrated with 95% O2 to 5% CO2 and titrated to the pH of 7.3 with 10% NaHCO3 solution. Diazoxide (7-chloro-3-methyl-1,2,4-benzothiadiazine-1,1-dioxide4; Sigma-Aldrich) dose of 100 μmol/L was utilized because it was effective in ameliorating cell swelling secondary to stress in previous studies.1–4 A stock solution of DZX was made by dissolving DZX in 0.1% dimethyl sulfoxide (DMSO), at which concentration DMSO has no effect on cell volume.28
Myocyte Volume Measurement
Myocytes were used on the day of isolation and were not cultured. Myocytes were visualized on an inverted microscope stage (IX-51; Olympus, Tokyo, Japan), as previously described.15 After 5 minutes, the chamber was perfused at a rate of 3 mL/min with TYR (in mmol/L): NaCl 130, KCl 5, CaCl2 2.5, MgSO4 1.2, NaHCO3 24, Na2HPO4 1.75, and glucose 10 (buffered to a pH of 7.4 using 95% O2 to 5% CO2). After viability was confirmed, myocyte images were captured using video-based edge detection software (IonOptix, Milton, MA) and volume measured every 5 minutes, as previously described.5
Myocyte Contractility
Myocyte contractility was measured using a video-based edge detection system (IonOptix). Cells were paced using a field stimulator (MyoPacer; IonOptix) at a voltage of 105 above threshold at a frequency of 1 Hz with a 5-ms duration to avoid occurrence of fusion beats. After 5 minutes of stimulation, data were obtained from 12 to 30 consecutive beats and averaged. Parameters of contractility included percentage of cell shortening, maximal velocity of shortening, and percentage of cell relengthening, as previously described.5 Contractility was measured at baseline and after 5 minutes of re-exposure to TYR. Cells that showed less than 7% cell shortening at baseline were excluded.
Statistical Analysis
Data were analyzed using SYSTAT 13 (Systat Software, Inc, Point Richmond, CA). All data are presented as mean±SEM relative to baseline. A repeated-measures ANOVA was used for sequential time-based measurements for each test solution against its own baseline and control values. Using Fisher’s least significant different test, post-hoc multiple comparisons were done between different test groups at different time points during test solution and re-exposure periods. Probability values <0.05 were considered significant. A Shapiro-Wilk test was used to test for normality. If the data failed the normality test, a nonparametric (Friedman’s nonparametric repeated-measures comparison) was used. Statistical analysis was performed using SyStat 10.2 (Systat Software).
Results
Mitochondrial Matrix Volume
Isolated mitochondrial volume is represented in Figure 1. There were no statistically significant differences in mitochondrial volume between any of the groups.
Figure 1.
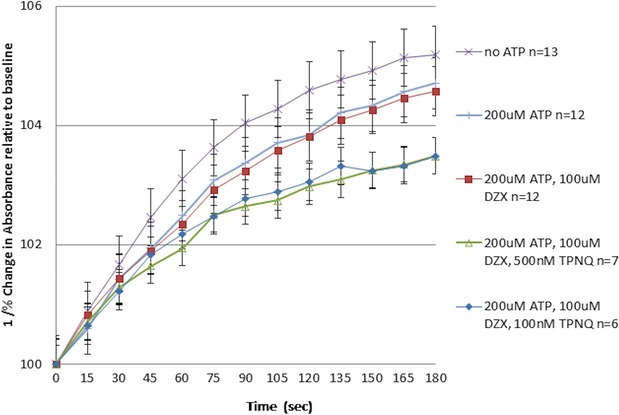
Mitochondrial matrix volume. Isolated mouse mitochondria were exposed to: (1) isolation buffer (no ATP); (2) 200 μmol/L of ATP; (3) 200 μmol/L of ATP and 100 μmol/L of DZX; (4) 200 μmol/L of ATP, 100 μmol/L of DZX, and 500 nmol/L of TPN-Q; or (5) 200 μmol/L of ATP, 100 μmol/L of DZX, and 100 nmol/L of TPN-Q and volume measured using light scattering (at 520 nm) for 2 minutes. Mitochondrial matrix volume is represented as 1/percent change in absorbance (mitochondrial matrix swelling is inversely related to absorbance measured at 520 nm) over time. Data are represented as mean±SEM. DZX indicates diazoxide; TPN-Q, tertiapin Q.
Myocyte Volume
Myocyte volume over time is represented in Figure 2. Myocytes demonstrated no significant volume change from baseline during exposure to TYR with or without TPN-Q. Myocytes did demonstrate significant swelling when exposed to CPG (P<0.05 vs. TYR). This swelling was prevented by addition of DZX (P<0.05 vs. CPG). The further addition of TPN-Q prevented this benefit (volume homeostasis) of DZX (CPG+DZX+TPN-Q P<0.05 vs. CPG+DZX). As expected, the interaction between experimental groups and time was significant (P<0.001).
Figure 2.
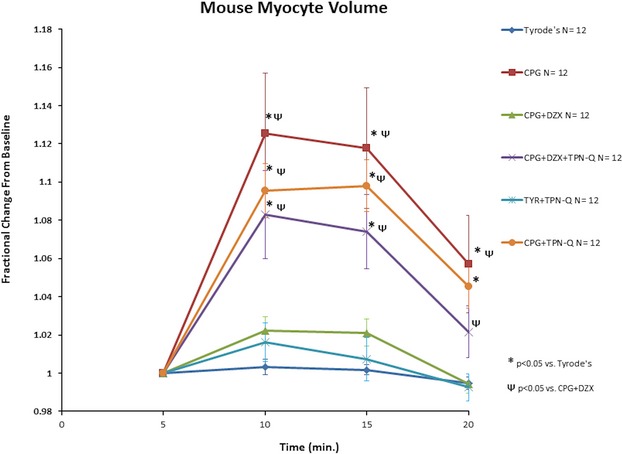
TPN-Q prevents beneficial maintenance of myocyte volume during stress by DZX. Isolated myocytes were exposed to control Tyrode’s physiologic solution (TYR) for 5 minutes (time 0 to 5), stress (CPG, CPG+DZX, CPG+DZX+TPN-Q, CPG+TPN-Q, and TYR+TPN-Q) for 10 minutes (time 5 to 15), followed by TYR for 5 minutes (time 15 to 20). Myocyte volume was compared. Data are represented as mean±SEM. *P<0.05 vs. TYR; ΨP<0.05 vs. CPG+DZX. CPG indicates hypothermic hyperkalemic cardioplegia; DZX, diazoxide; TPN-Q, tertiapin Q.
Myocyte Contractility
Myocyte contractility is presented in Figure 3. Contractility remained unaltered in the TYR group. Myocytes demonstrated a significant decline in all 3 parameters of contractility after exposure to CPG (P<0.05 vs. TYR) that was worsened by the addition of TPN-Q. The addition of DZX to CPG prevented the significant reduction in contractility observed in the CPG group. TPN-Q inhibited the protection provided by DZX.
Figure 3.
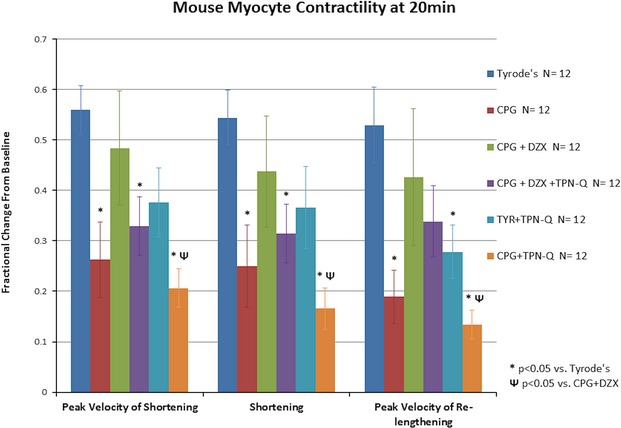
TPN-Q prevents beneficial maintenance of myocyte contractility after stress by DZX. Isolated mouse myocytes were exposed to control Tyrode’s physiologic solution (TYR) for 5 minutes (time 0 to 5), stress (CPG, CPG+DZX, CPG+DZX+TPN-Q, CPG+TPN-Q, and TYR+TPN-Q) for 10 minutes (time 5 to 15), followed by TYR for 5 minutes (time 15 to 20). Myocyte contractility was compared. Data are represented as mean±SEM. *P<0.05 vs. TYR; ΨP<0.05 vs. CPG+DZX. CPG indicates hypothermic hyperkalemic cardioplegia; DZX, diazoxide; TPN-Q, tertiapin Q
Discussion
In previous studies, we have shown that myocyte swelling and reduced contractility in response to stress (hyperkalemic CPG, hypoosmotic stress, and metabolic inhibition) are ameliorated by addition of the KATP channel opener, DZX.1–5 Our efforts to localize the precise mechanism of cardioprotection afforded by DZX have implicated an involvement of the KATP channel, SUR1,16,19 but not the Kir6.2 subunit.2 Other investigators have provided evidence in support of the idea that Kir1.1 (the ROMK) is a pore-forming subunit of the mKATP channel.24 In the current study, changes in mitochondrial and myocyte volume were observed in the presence of ROMK blocker TPN-Q in the presence of DZX. A dose effect on mitochondrial volume was not detectable using TPN-Q in the presence of DZX and multiple doses of TPN-Q. The stability of TPN-Q in the vehicle medium was thus evaluated in the media, in 20% acetonitrile, HEPES, and in water. However, no significant change in mitochondrial volume was observed after the addition of TPN-Q, even at high concentrations, in contrast to other investigators.24 Thus, we conclude that TPN-Q had no effect on mitochondrial volume in the presence of DZX, suggesting a location of action of DZX that was distinct from the channels targeted by TPN-Q.
Myocytes demonstrated significant swelling in response to CPG that was prevented by the KATP channel opener, DZX, which is consistent with previous results.2,3 Similarly, a correlation between myocyte volume changes and contractility changes was consistent with previous work, supporting this myocyte model of stunning.4,5 TPN-Q prevented this beneficial effect of DZX, thus implicating TPN-Q targets—Kir1.1, Kir3.1, or Kir3.4—in the cardioprotection of DZX at the cellular level. Other non–Kir TPN-Q targets or undefined channel subunits sensitive to TPN-Q may play a role (GIRK1/4, KACH, and voltage-dependent Ca2+-activated K+ channels) in cardioprotection and will be the subject of future investigations.29–31
The present study further characterizes the mechanism of cardioprotection provided by DZX. TPN-Q did not alter DZX-induced mitochondrial swelling, but it did inhibit myocyte cardioprotection provided by DZX during stress. Given that TPN-Q inhibits Kir1.1-, Kir3.1-, and Kir3.4-dependent K-channel activities, these data support that any of these subunits (as well as undefined subunits sensitive to TPN-Q) could be involved in the cardioprotection afforded by DZX. However, these data also suggest that mitochondrial swelling by DZX does not involve Kir1.1, Kir3.1, or Kir3.4 and likely results from a yet to be identified mechanism.
Future work to identify the site and mechanism of action of KATP channel openers will involve indirect methods until genetic identification is accomplished.
Sources of Funding
This study was supported by NIH RO1 HL098182-01A1 (Lawton), NIH 5T32HL007776 (Henn, Janjua), and the Barnes Jewish Hospital Foundation (Lawton).
Disclosures
None.
References
- Mizutani S, Prasad SM, Sellitto AD, Schuessler RB, Damiano RJ, Jr, Lawton JS. Myocyte volume and function in response to osmotic stress: observations in the presence of an adenosine triphosphate-sensitive potassium channel opener. Circulation. 2005;112:I219–I223. doi: 10.1161/CIRCULATIONAHA.104.523746. [DOI] [PubMed] [Google Scholar]
- Prasad SM, Al-Dadah AS, Byrd GD, Flagg TP, Gomes J, Damiano RJ, Jr, Nichols CG, Lawton JS. Role of the sarcolemmal adenosine triphosphate-sensitive potassium channel in hyperkalemic cardioplegia-induced myocyte swelling and reduced contractility. Ann Thorac Surg. 2006;81:148–153. doi: 10.1016/j.athoracsur.2005.06.055. [DOI] [PubMed] [Google Scholar]
- Mizutani S, Al-Dadah AS, Bloch JB, Prasad SM, Diodato MD, Schuessler RB, Damiano RJ, Jr, Lawton JS. Hyperkalemic cardioplegia-induced myocyte swelling and contractile dysfunction: prevention by diazoxide. Ann Thorac Surg. 2006;81:154–159. doi: 10.1016/j.athoracsur.2005.06.057. [DOI] [PubMed] [Google Scholar]
- Maffit SK, Sellitto AD, Al-Dadah AS, Schuessler RB, Damiano RJ, Jr, Lawton JS. Diazoxide maintains human myocyte volume homeostasis during stress. J Am Heart Assoc. 2012;1:jah3-e000778. doi: 10.1161/JAHA.112.000778. doi: 10.1161/JAHA.112.000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Dadah AS, Voeller RK, Schuessler RB, Damiano RJ, Jr, Lawton JS. Maintenance of myocyte volume homeostasis during stress by diazoxide is cardioprotective. Ann Thorac Surg. 2007;84:857–862. doi: 10.1016/j.athoracsur.2007.04.103. [DOI] [PubMed] [Google Scholar]
- Auchampach JA, Maruyama M, Cavero I, Gross GJ. Pharmacological evidence for a role of ATP-dependent potassium channels in myocardial stunning. Circulation. 1992;86:311–319. doi: 10.1161/01.cir.86.1.311. [DOI] [PubMed] [Google Scholar]
- Galinanes M, Shattock MJ, Hearse DJ. Effects of potassium channel modulation during global ischaemia in isolated rat heart with and without cardioplegia. Cardiovasc Res. 1992;26:1063–1068. doi: 10.1093/cvr/26.11.1063. [DOI] [PubMed] [Google Scholar]
- Grover GJ, Sleph PG, Parham CS. Nicorandil improves postischemic contractile function independently of direct myocardial effects. J Cardiovasc Pharmacol. 1990;15:698–705. doi: 10.1097/00005344-199005000-00003. [DOI] [PubMed] [Google Scholar]
- Pignac J, Bourgouin J, Dumont L. Cold cardioplegia and the K+ channel modulator aprikalim (RP 52891): improved cardioprotection in isolated ischemic rabbit hearts. Can J Physiol Pharmacol. 1994;72:126–132. doi: 10.1139/y94-020. [DOI] [PubMed] [Google Scholar]
- Shigematsu S, Sato T, Abe T, Saikawa T, Sakata T, Arita M. Pharmacological evidence for the persistent activation of ATP-sensitive K+ channels in early phase of reperfusion and its protective role against myocardial stunning. Circulation. 1995;92:2266–2275. doi: 10.1161/01.cir.92.8.2266. [DOI] [PubMed] [Google Scholar]
- Flagg TP, Enkvetchakul D, Koster JC, Nichols CG. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev. 2010;90:799–829. doi: 10.1152/physrev.00027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S, Nichols CG. Octameric stoichiometry of the KATP channel complex. J Gen Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CG, Singh GK, Grange DK. KATP channels and cardiovascular disease: suddenly a syndrome. Circ Res. 2013;112:1059–1072. doi: 10.1161/CIRCRESAHA.112.300514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagg TP, Kurata HT, Masia R, Caputa G, Magnuson MA, Lefer DJ, Coetzee WA, Nichols CG. Differential structure of atrial and ventricular KATP: atrial KATP channels require SUR1. Circ Res. 2008;103:1458–1465. doi: 10.1161/CIRCRESAHA.108.178186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastacio MM, Kanter EM, Keith AD, Schuessler RB, Nichols CG, Lawton JS. Inhibition of succinate dehydrogenase by diazoxide is independent of the ATP-sensitive potassium channel subunit sulfonylurea type 1 receptor. J Am Coll Surg. 2013;216:1144–1149. doi: 10.1016/j.jamcollsurg.2013.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellitto AD, Maffit SK, Al-Dadah AS, Zhang H, Schuessler RB, Nichols CG, Lawton JS. Diazoxide maintenance of myocyte volume and contractility during stress: evidence for a non-sarcolemmal K(ATP) channel location. J Thorac Cardiovasc Surg. 2010;140:1153–1159. doi: 10.1016/j.jtcvs.2010.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellitto AD, Al-Dadah AS, Schuessler RB, Nichols CG, Lawton JS. An open sarcolemmal adenosine triphosphate-sensitive potassium channel is necessary for detrimental myocyte swelling secondary to stress. Circulation. 2011;124:S70–S74. doi: 10.1161/CIRCULATIONAHA.110.012039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastacio MM, Kanter EM, Makepeace C, Keith AD, Zhang H, Schuessler RB, Nichols CG, Lawton JS. Cardioprotective mechanism of diazoxide involves the inhibition of succinate dehydrogenase. Ann Thorac Surg. 2013;95:2042–2050. doi: 10.1016/j.athoracsur.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastacio MM, Kanter EM, Makepeace CM, Keith AD, Zhang H, Schuessler RB, Nichols CG, Lawton JS. Relationship between mitochondrial matrix volume and cellular volume in response to stress and the role of ATP-sensitive potassium channel. Circulation. 2013;128:S130–S135. doi: 10.1161/CIRCULATIONAHA.112.000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley PJ, Mickel M, Loffler M, Brandt U, Daut J. K(ATP) channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J Physiol. 2002;542:735–741. doi: 10.1113/jphysiol.2002.023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Sasaki N, Seharaseyon J, O’Rourke B, Marban E. Selective pharmacological agents implicate mitochondrial but not sarcolemmal K(ATP) channels in ischemic cardioprotection. Circulation. 2000;101:2418–2423. doi: 10.1161/01.cir.101.20.2418. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ, Lodge NJ, Smith MA, Grover GJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Dos Santos P, Xie ZJ, Costa AD, Paucek P. Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K(+) channel in cardiac function and cardioprotection. Biochim Biophys Acta. 2003;1606:1–21. doi: 10.1016/s0005-2728(03)00109-9. [DOI] [PubMed] [Google Scholar]
- Foster DB, Ho AS, Rucker J, Garlid AO, Chen L, Sidor A, Garlid KD, O’Rourke B. Mitochondrial ROMK channel is a molecular component of mitoK(ATP) Circ Res. 2012;111:446–454. doi: 10.1161/CIRCRESAHA.112.266445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta. 2007;1767:1007–1031. doi: 10.1016/j.bbabio.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council (US) Committee for the Update of. Guide to Care and Use of Laboratory Animals. 8th ed. Washington, DC: The National Academy Press; 2011. [Google Scholar]
- Beavis AD, Brannan RD, Garlid KD. Swelling and contraction of the mitochondrial matrix. I. A structural interpretation of the relationship between light scattering and matrix volume. J Biol Chem. 1985;260:13424–13433. [PubMed] [Google Scholar]
- Drewnowska K, Clemo HF, Baumgarten CM. Prevention of myocardial intracellular edema induced by St. Thomas’ Hospital cardioplegic solution. J Mol Cell Cardiol. 1991;23:1215–1221. doi: 10.1016/0022-2828(91)90079-2. [DOI] [PubMed] [Google Scholar]
- Kitamura H, Yokoyama M, Akita H, Matsushita K, Kurachi Y, Yamada M. Tertiapin potently and selectively blocks muscarinic K(+) channels in rabbit cardiac myocytes. J Pharmacol Exp Ther. 2000;293:196–205. [PubMed] [Google Scholar]
- Kanjhan R, Coulson EJ, Adams DJ, Bellingham MC. Tertiapin-Q blocks recombinant and native large conductance K+ channels in a use-dependent manner. J Pharmacol Exp Ther. 2005;314:1353–1361. doi: 10.1124/jpet.105.085928. [DOI] [PubMed] [Google Scholar]
- Jin W, Klem AM, Lewis JH, Lu Z. Mechanisms of inward-rectifier K+ channel inhibition by tertiapin-Q. Biochemistry. 1999;38:14294–14301. doi: 10.1021/bi991206j. [DOI] [PubMed] [Google Scholar]