

Abstract
Background
Interleukin (IL)-1β, IL-18, and downstream IL-6 are key inflammatory cytokines in the pathogenesis of coronary artery disease. Colchicine is believed to block the NLRP3 inflammasome, a cytosolic complex responsible for the production of IL-1β and IL-18. In vivo effects of colchicine on cardiac cytokine release have not been previously studied. This study aimed to (1) assess the local cardiac production of inflammatory cytokines in patients with acute coronary syndromes (ACS), stable coronary artery disease and in controls; and (2) determine whether acute administration of colchicine inhibits their production.
Methods and Results
Forty ACS patients, 33 with stable coronary artery disease, and 10 controls, were included. ACS and stable coronary artery disease patients were randomized to oral colchicine treatment (1 mg followed by 0.5 mg 1 hour later) or no colchicine, 6 to 24 hours prior to cardiac catheterization. Blood samples from the coronary sinus, aortic root (arterial), and lower right atrium (venous) were collected and tested for IL-1β, IL-18, and IL-6 using ELISA. In ACS patients, coronary sinus levels of IL-1β, IL-18, and IL-6 were significantly higher than arterial and venous levels (P=0.017, <0.001 and <0.001, respectively). Transcoronary (coronary sinus-arterial) gradients for IL-1β, IL-18, and IL-6 were highest in ACS patients and lowest in controls (P=0.077, 0.033, and 0.014, respectively). Colchicine administration significantly reduced transcoronary gradients of all 3 cytokines in ACS patients by 40% to 88% (P=0.028, 0.032, and 0.032, for IL-1β, IL-18, and IL-6, respectively).
Conclusions
ACS patients exhibit increased local cardiac production of inflammatory cytokines. Short-term colchicine administration rapidly and significantly reduces levels of these cytokines.
Keywords: atherosclerosis, coronary sinus sampling, cytokines, inflammation
Multiple pathways are responsible for inflammation in acute coronary syndromes.1,2 Monocytes/macrophages are key effector cells in atherosclerosis-associated inflammation, leading to both plaque progression and instability.3 Moreover, it has recently been shown that the nucleotide-binding oligomerization domain-like receptors, pyrin domain-containing 3 (NLRP3) inflammasome—a cytosolic protein complex that serves as sensor of either pathogen invasion or cellular stress4–6—promotes caspase-1 activation, and therefore the production of active interleukin (IL)-1β and IL-18, key mediators in atherosclerotic plaque development, progression, and destabilization.7,8 Furthermore, this inflammatory milieu leads to increased levels of IL-6 and C-reactive protein (CRP), which significantly correlate with the incidence of future cardiovascular events in patients with atherosclerosis.9,10
Colchicine is a simple, inexpensive, yet potent anti-inflammatory drug that is approved for the management of patients with acute gout, and other inflammatory conditions such as pericarditis.11,12 Purported mechanisms of action in leukocytes include its ability to suppress NLRP3 inflammasome13 through disruption of microtubule formation,14 impairing mitochondrial colocalization of the inflammasome proteins and thus inflammasome activation and cytokine production.
Here, we hypothesize that (1) local coronary production of the inflammasome-specific cytokines IL-1β and IL-18 together with IL-6, a key downstream inflammatory cytokine, will be higher in patients with acute coronary syndromes (ACS) than in patients with stable coronary artery disease (CAD) or subjects without significant CAD; and (2) administration of colchicine to ACS patients will result in lower transcoronary release of these cytokines.
Methods
Consecutive adult patients (>21 years old) with a clinical indication for cardiac catheterization at Royal Prince Alfred Hospital (Sydney, Australia) were invited to participate in the study. According to their clinical presentation they were divided in 3 groups:
ACS—patients with recent-onset chest pain, associated with ST segment and/or T wave ECG changes and/or positive cardiac enzymes (creatine kinase or troponin T). This group included patients with unstable angina as well as non-ST-segment elevation myocardial infarction (as per American College of Cardiology/American Heart Association 2007 guidelines15).
Stable CAD—This group included symptomatic patients with stable angina (chest pain on exertion for >3 months) and asymptomatic patients with positive functional tests and obstructive coronary disease (defined as diameter stenosis >50%).
Controls—patients found to have no significant coronary disease (absence of angiographic evidence of any stenosis >20%).
Patients included in the ACS and stable CAD groups were randomized in a 1:1 fashion to receive either colchicine (1 mg, followed by 0.5 mg 1 hour later) or no colchicine therapy, 6 to 24 hours prior to coronary angiography. This dose regimen was chosen in accordance with current recommendations for the treatment of acute gout.11
Patients with the following characteristics were prospectively excluded from the study:
Patients with >50% stenosis in the left main coronary artery
ST elevation myocardial infarction
Cardiogenic shock or hemodynamic instability
Known hypersensitivity to colchicine
Moderate renal dysfunction (creatinine clearance <45 mL/min) or hepatic dysfunction (alanine aminotransferase 1.5× upper limit of normal range)
Thrombocytopenia or leukopenia
Pregnant or lactating women and women at risk of pregnancy
Patients taking moderate-strong CYP3A4 inhibitors
Patients already taking colchicine
Those with evidence of active infection or inflammatory conditions that might be associated with markedly elevated CRP levels in the blood (eg, active rheumatoid arthritis) and those taking other anti-inflammatory therapies (eg, corticosteroids)
Demographic and clinical data were recorded. The local Ethics Review Committee approved the study protocol and all patients gave written informed consent before participating.
Blood Sampling
A standard coronary angiogram was performed in all patients. Subsequently, right femoral vein access was obtained with a 5F sheath and a 5F Simmons catheter was advanced into the right atrium. Our technique to cannulate the coronary sinus (CS) ostium has been detailed previously.16 This technique allows for a safe and reliable sampling of the CS blood. Twenty milliliters of blood was drawn from the CS, lower right atrium–inferior vena cava junction (venous), and ascending aorta (arterial). For those patients proceeding to clinically indicated percutaneous coronary intervention, blood sampling was performed immediately after the diagnostic angiogram but before the intervention. All patients received 2500 units of heparin prior to CS sampling; if percutaneous coronary intervention was performed, patients received additional heparin aiming for an activated clotting time >250.
Detection of IL-1β, IL-18, and IL-6 in Plasma
Blood samples were placed into vacuum vials containing EDTA. Samples were immediately centrifuged and plasma was collected and stored at −80°C. Plasma IL-1β, IL-18, and IL-6 concentrations were measured by highly sensitive ELISA using the specific antibodies (IL-1β and IL-6: R&D Systems, Minneapolis, MN; IL-18: Medical and Biological Laboratories, Woburn, MA). The protocols used were those recommended by the antisera suppliers. For IL-1β, IL-18, and IL-6 the minimum detection limit were 1, 12.5, and 0.70 pg/mL, respectively, and inter- and intra-assay coefficient of variances were <10% for each assay.
Statistical Analysis
Continuous variables are reported as mean± SD or SEM and categorical variables as number (percentage). Normal distribution was tested by the Shapiro–Wilk test. Differences in continuous variables (eg, IL-1β concentration) were tested via the use of an independent-samples t test or nonparametric test for non-normally distributed data (Mann–Whitney U test), while proportional differences in categorical variables (eg, patient did or did not have hypertension) were tested via the use of Fisher’s Exact Test. Multiple, nonpaired samples were tested with 1-way ANOVA or Kruskal–Wallis test and multiple paired samples with Friedman test. All tests were 2-tailed with the acceptable type 1 error set at P<0.05. Statistical analysis was performed with SPSS 21 software (IBM, Armonk, NY) and figures were created with GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA).
Results
From September 2013 to August 2014, 83 patients were included in the study. Of these, 40 patients presented with an ACS, 33 patients with stable CAD, and 10 patients had no angiographically significant CAD and served as controls. Within the ACS group, 21 patients were randomized to colchicine treatment and 19 individuals to “no treatment.” In the stable CAD group, 7 nontreated patients were initially studied, while sampling techniques were being optimized, after which 26 patients were randomized to colchicine or no therapy. The initial 7 patients were included in the final analysis, with 13 colchicine-treated patients and 20 untreated patients.
Baseline Characteristics
Baseline characteristics for ACS, stable CAD, and control groups and characteristics by colchicine treatment randomization groups are presented in Tables1 and 2. Groups were comparable for most risk factors, treatments, and blood test results. However, within the ACS group, those allocated to colchicine treatment were more likely to have been diabetic (P=0.04) and therefore more likely to be taking oral hypoglycemics (P=0.02). In the stable CAD group there was a trend to more diabetics in the colchicine-treated group, although this was not statistically significant (P=0.06).
Table 1.
Baseline Characteristics for ACS, Stable CAD, and Control Groups
| ACS | Stable CAD | Controls | P Value | |
|---|---|---|---|---|
| Number | 40 | 33 | 10 | — |
| Age, y—mean (SD) | 64.5 (10.2) | 61.1 (10.3) | 61.3 (6.7) | 0.32 |
| Female (%) | 3 (8) | 3 (9) | 3 (30) | 0.29 |
| Diabetes mellitus (%) | 13 (33) | 13 (39) | 2 (20) | 0.42 |
| Hypertension (%) | 27 (68) | 19 (58) | 3 (30) | 0.06 |
| Dyslipidemia (%) | 27 (68) | 23 (70) | 5 (50) | 0.68 |
| Current smoker (%) | 10 (25) | 6 (18) | 3 (30) | 0.70 |
| Previous MI (%) | 7 (18) | 10 (30) | 1 (10) | 0.28 |
| Previous PCI (%) | 7 (18) | 9 (27) | 0 (0) | 0.17 |
| Previous CABG (%) | 1 (3) | 6 (18) | 0 (0) | 0.05 |
| Medications | ||||
| Aspirin (%) | 40 (100) | 33 (100) | 8 (80) | 0.134 |
| Thienopyridines (%) | 38 (95) | 26 (79) | 6 (60) | 0.017 |
| β-Blockers (%) | 30 (75) | 17 (52) | 3 (30) | 0.023 |
| ACE-I/ARA-2 (%) | 25 (63) | 20 (61) | 4 (40) | 0.331 |
| Statins (%) | 36 (90) | 27 (82) | 7 (70) | 0.115 |
| Hypoglycemics (%) | 12 (30) | 12 (36) | 1 (10) | 0.231 |
| Insulin (%) | 3 (8) | 2 (6) | 0 (0) | 1 |
ACE-I indicates angiotensin-converting enzyme inhibitors; ACS, acute coronary syndrome; ARA-2, angiotensin 2 receptor antagonist; CABG, coronary artery bypass graft surgery; MI, myocardial infarction; PCI, percutaneous coronary intervention; CAD, coronary artery disease.
Table 2.
Baseline Characteristics for ACS and Stable CAD Groups, by Colchicine Administration
| ACS (n=40) | Stable CAD (n=33) | |||||
|---|---|---|---|---|---|---|
| Colchicine | No Colchicine | P Value* | Colchicine | No Colchicine | P Value† | |
| Number | 21 | 19 | — | 13 | 20 | — |
| Age, y—mean (SD) | 65.0 (8.9) | 64.0 (11.7) | 0.76 | 59.9 (11.3) | 61.8 (10.0) | 0.63 |
| Female (%) | 3 (14) | 0 (0) | 0.23 | 1 (8) | 2 (10) | 1 |
| Diabetes mellitus (%) | 10 (48) | 3 (16) | 0.04 | 8 (62) | 5 (25) | 0.06 |
| Hypertension (%) | 13 (62) | 14 (74) | 0.51 | 8 (62) | 11 (55) | 0.71 |
| Dyslipidemia (%) | 13 (62) | 14 (74) | 0.51 | 11 (85) | 12 (60) | 1 |
| Current smoker (%) | 6 (29) | 4 (21) | 0.72 | 1 (8) | 5 (25) | 0.63 |
| Previous MI (%) | 2 (10) | 5 (26) | 0.23 | 6 (46) | 4 (20) | 0.23 |
| Previous PCI (%) | 3 (14) | 4 (21) | 0.69 | 1 (8) | 8 (40) | 0.19 |
| Previous CABG (%) | 0 (0) | 1 (5) | 0.48 | 2 (15) | 4 (20) | 1 |
| Medications | ||||||
| Aspirin (%) | 21 (100) | 19 (100) | — | 13 (100) | 20 (100) | — |
| Thienopyridines (%) | 20 (95) | 18 (95) | 1 | 11 (85) | 15 (75) | 1 |
| β-Blockers (%) | 16 (76) | 14 (74) | 1 | 7 (54) | 10 (50) | 1 |
| ACE-I/ARA-2 (%) | 11 (52) | 14 (74) | 0.20 | 9 (69) | 11 (55) | 0.44 |
| Statins (%) | 19 (91) | 17 (89) | 1 | 11 (85) | 16 (80) | 1 |
| Hypoglycemics (%) | 10 (48) | 2 (11) | 0.02 | 7 (54) | 5 (25) | 0.24 |
| Insulin (%) | 1 (5) | 2 (11) | 0.59 | 1 (8) | 1 (5) | 1 |
ACE-I indicates angiotensin-converting enzyme inhibitors; ACS, acute coronary syndrome; ARA-2, angiotensin 2 receptor antagonist; CABG, coronary artery bypass graft surgery; MI, myocardial infarction; PCI, percutaneous coronary intervention; CAD, coronary artery disease.
Difference between colchicine vs. no colchicine within ACS group.
Difference between colchicine vs. no colchicine within stable CAD group.
Interleukin-1β
In ACS colchicine-naïve patients, CS IL-1β levels were significantly higher than arterial and venous levels (13.42, 9.68, and 9.49 pg/mL, for CS, arterial and venous, respectively; P=0.017) (Figure 1), whereas in stable CAD patients, IL-1β levels were nonsignificantly higher in the CS versus artery and vein (9.29, 8.42, and 8.55 pg/mL, for CS, arterial and venous, respectively; P=0.058) (Figure 1A). In the control group, there was no difference in IL-1β concentrations between the 3 sampling sites (7.88, 7.93, and 7.75 pg/mL, for CS, arterial and venous, respectively; P=1). There appeared to be a relationship between IL-1β CS—arterial or transcoronary gradient (a measure of intracardiac production) and clinical category, with the highest gradients found in ACS patients, and lowest in control patients; however, this was not statistically significant (P=0.077) (Figure 2A). Notably, the CS-arterial (A) gradient from ACS but not stable CAD patients was significantly higher compared to CS-A gradient from control patients (P=0.028). On the other hand, no difference in the venous concentration of IL-1β was seen between groups (P=0.611).
Figure 1.

Venous, arterial, and coronary sinus (A) IL-1β, (B) IL-18, and (C) IL-6 concentrations in colchicine-naïve ACS and stable CAD patients. Results expressed as mean and SEM. A indicates arterial; ACS, acute coronary syndrome; CAD, coronary artery disease; CS, coronary sinus; IL, interleukin; V, venous.
Figure 2.
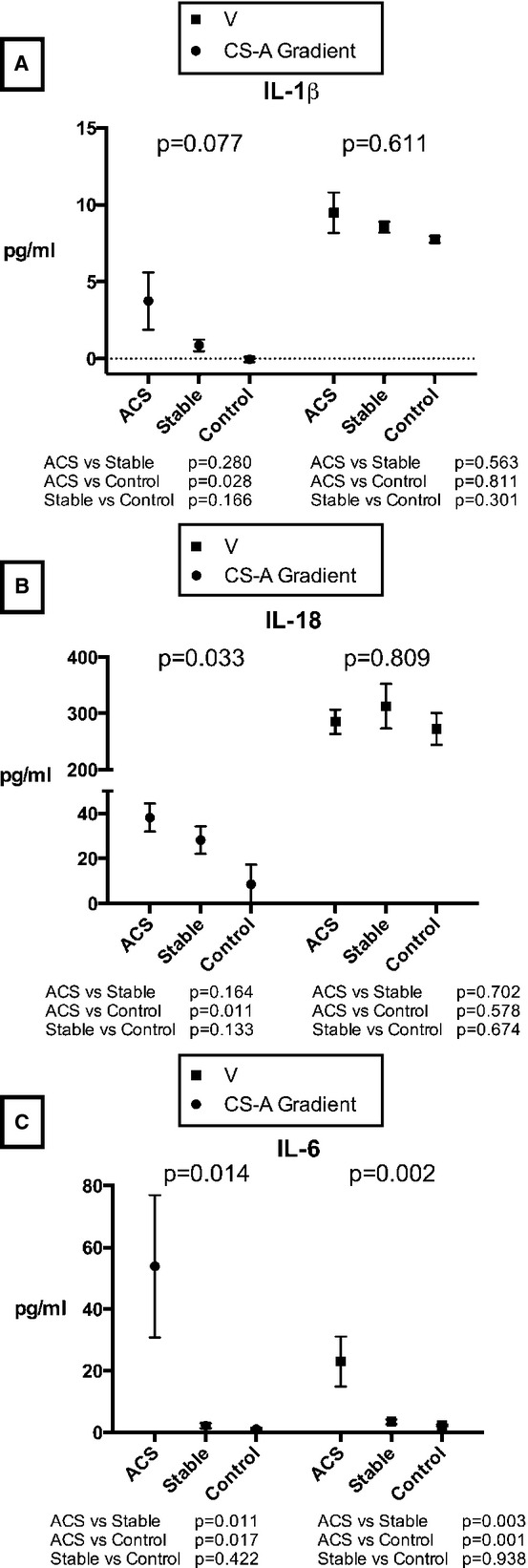
Transcoronary gradients and venous concentrations of (A) IL-1β, (B) IL-18, and (C) IL-6 according to clinical presentation. Results expressed as mean and SEM. A indicates arterial; ACS, acute coronary syndrome; CS, coronary sinus; IL, interleukin; V, venous.
Interleukin-18
In both ACS and stable CAD colchicine-naïve groups, CS levels of IL-18 were significantly higher than in the aorta or peripheral vein (ACS: 325.82, 287.64, and 285.11; stable CAD: 346.50, 318.43, and 312.82 pg/mL, for CS, arterial and venous, respectively; P<0.001 for both comparisons) (Figure 1B), whereas no difference was found in the control group (280.77, 271.30, and 272.31 pg/mL, for CS, arterial and venous, respectively; P=0.174). In similar fashion to IL-1β, there was a significant relationship between the CS-A gradient and clinical presentation, with highest transcoronary gradients found in ACS patient, and lowest gradients in controls (P=0.033). However, peripheral venous concentrations of this cytokine did not change across groups (Figure 2B).
Interleukin-6
In patients with both ACS and stable CAD, there were significantly higher concentrations of this cytokine in the CS (70.13, 21.47, 22.96 pg/mL, for CS, arterial and venous in ACS patients, respectively; P<0.001; 6.64, 4.44, 3.47 pg/mL, for CS, arterial and venous in stable CAD patients; P<0.001) (Figure 1C). Both transcoronary concentration gradients and venous concentrations were markedly higher in ACS patients versus stable CAD and control groups (P=0.014) (Figure 2C).
Of note, no differences in IL-1β, IL-18, or IL-6 CS or peripheral blood levels were seen between diabetic and nondiabetic patients in both stable and unstable CAD patient groups.
Effect of Colchicine Treatment on IL-1β, IL-18, and IL-6 Levels
The results for CS and CS-A gradient plasma concentrations of IL-1β, IL-18, and IL-6 in ACS and stable CAD patients according to colchicine treatment are summarized in Table 3. Colchicine treatment reduced absolute CS IL-1β concentration as well as the CS-A gradient in patients with ACS (P=0.024 and 0.028, respectively) (Figure 3A). These results represented a 33% reduction in the CS plasma concentration of IL-1β with colchicine treatment in ACS patients, in contrast with only a 6% difference in patients with stable CAD (P<0.001 for the difference in effect between ACS and stable CAD groups). For IL-18, colchicine treatment similarly resulted in a significant reduction in CS-A gradient, again only in the ACS group (Figure 3B). Finally, colchicine therapy in ACS patients resulted in an 88% lower CS concentration of IL-6 (12.79 versus 77.00 pg/mL) and an 83% lower CS-A gradient (6.02 versus 53.90 pg/mL) versus untreated patients (P=0.004 and 0.032, respectively) (Figure 3C). In stable CAD patients, colchicine treatment was associated with a very low IL-6 transcoronary gradient; however, this was not statistically significant (0.29 pg/mL for colchicine-treated versus 2.20 pg/mL for colchicine naïve versus 1.01 pg/mL for controls, P=0.057). In contrast to the inflammasome-specific cytokines, IL-6 venous levels did significantly change with colchicine administration (22.86 versus 5.96 versus 2.32 pg/mL for ACS untreated, ACS treated, and control patients, P=0.011). As there were significantly more diabetic patients in the ACS colchicine group than the ACS noncolchicine group, a sensitivity analysis with adjustment for diabetes and oral hypoglycemic agents was performed. This yielded similar significant differences in cytokine levels between groups. Groups were otherwise well matched with respect to other baseline characteristics.
Table 3.
Coronary Sinus and Transcoronary Gradient Plasma Concentrations of IL-1β and IL-18, by Colchicine Administration
| ACS | Stable CAD | |||||||
|---|---|---|---|---|---|---|---|---|
| Colchicine | No Colchicine | Controls | P Value† | Colchicine | No Colchicine | Controls | P Value* | |
| Number | 21 | 19 | 10 | — | 13 | 20 | 10 | — |
| CS IL-1β | 8.95 (0.26) | 13.42 (2.59) | 7.88 (0.18) | 0.024 | 8.54 (0.38) | 9.29 (0.43) | 7.88 (0.18) | 0.074 |
| Gradient IL-1β | 0.87 (0.32) | 3.75 (1.87) | −0.05 (0.17) | 0.028 | 0.53 (0.41) | 0.87 (0.38) | −0.05 (0.17) | 0.313 |
| CS IL-18 | 301.42 (18.17) | 325.88 (24.49) | 280.66 (30.25) | 0.239 | 349.54 (41.21) | 346.50 (40.46) | 280.66 (30.25) | 0.434 |
| Gradient IL-18 | 22.85 (6.50) | 38.20 (6.25) | 9.33 (9.04) | 0.032 | 32.42 (4.43) | 28.14 (6.10) | 9.33 (9.04) | 0.077 |
| CS IL-6 | 12.79 (2.97) | 77.00 (26.28) | 4.31 (0.42) | 0.004 | 4.75 (0.86) | 6.64 (1.07) | 4.31 (0.42) | 0.617 |
| Gradient IL-16 | 6.02 (2.47) | 53.90 (23.07) | 1.01 (0.36) | 0.032 | 0.29 (0.47) | 2.20 (0.85) | 1.01 (0.36) | 0.057 |
All data: mean (SEM) (pg/mL). ACS indicates acute coronary syndrome; CS, coronary sinus; IL, interleukin; CAD, coronary artery disease.
Difference between ACS colchicine vs. ACS no colchicine vs. controls.
Difference between stable CAD colchicine vs. stable CAD no colchicine vs. controls.
Figure 3.
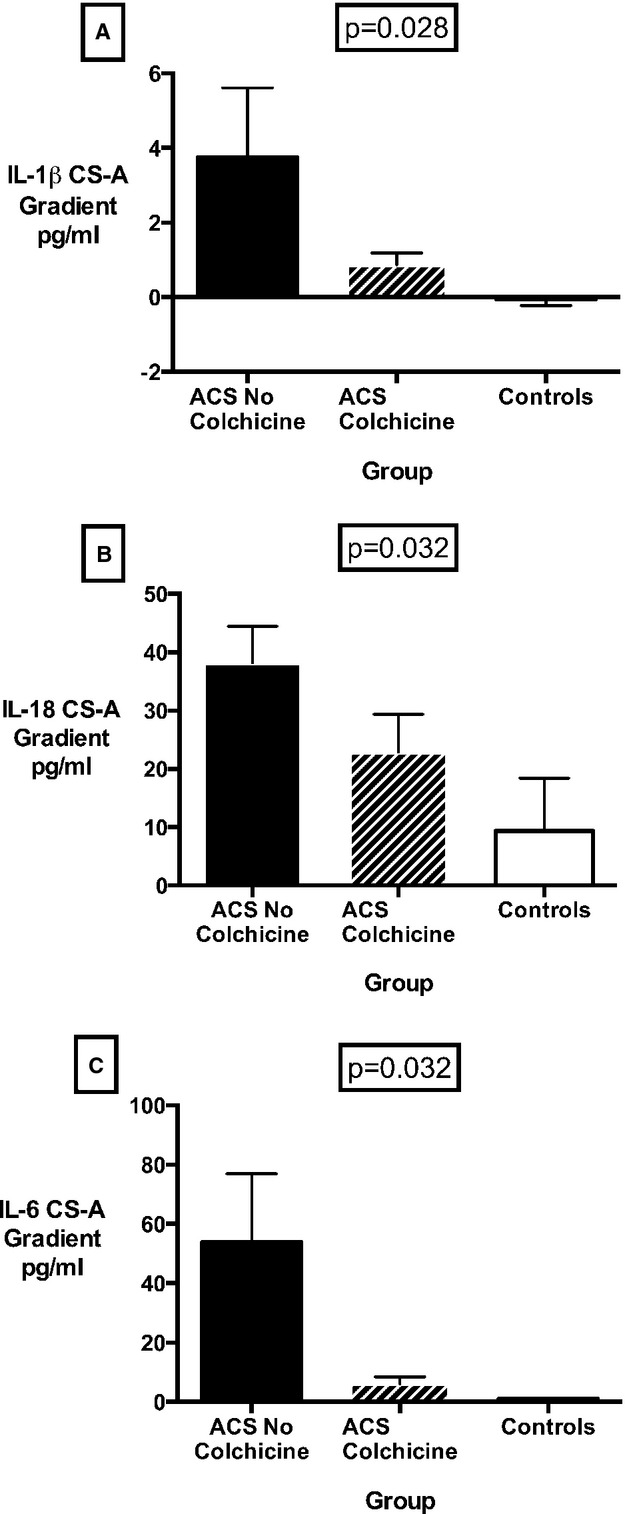
(A) IL-1β, (B) IL-18, and (C) IL-6 transcoronary gradients in ACS patients and controls, according to colchicine treatment. Results expressed as mean and SEM. ACS indicates acute coronary syndrome; CS-A gradient, coronary sinus–arterial gradient; IL, interleukin.
Discussion
There are 3 main findings in this study. First, the transcoronary gradients of all 3 inflammatory cytokines studied strongly correlated with disease activity, with the highest gradients found in ACS patients, and lowest in patients without any obstructive CAD. Second, venous plasma concentrations of the inflammasome-specific cytokines, IL-1β and IL-18, do not reliably identify increased transcoronary production. Finally, short-term colchicine treatment of ACS patients was associated with significantly lower transcoronary release of IL-1β and IL-18, as well as IL-6, a key downstream pro-inflammatory cytokine strongly associated with atherosclerosis-associated inflammation (Figure 4).
Figure 4.
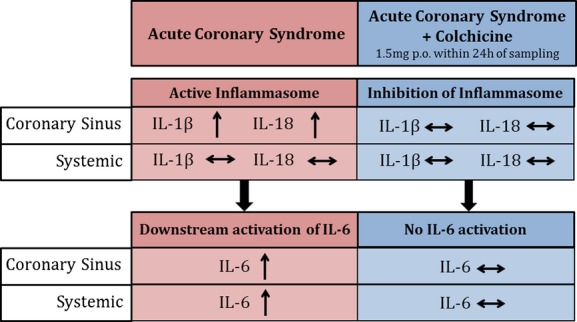
Effect of short-term colchicine on local cardiac and systemic inflammation in acute coronary syndrome patients. IL indicates interleukin.
Increased Transcoronary Production of IL-1β and IL-18 Is Found in ACS Patients
IL-1β is a key inflammatory cytokine and has been identified in multiple disease states.17 It exerts pro-inflammatory (neutrophil recruitment, increase in IL-6 and tumor necrosis factor-α, stimulation of phagocytosis, increase of intracellular adhesion molecule-1 and vascular permeability, CD4 and B-cell activation) and prothrombogenic effects (increase in plasminogen activator inhibitor 1 and fibrinogen), by binding to the IL-1 type 1 receptor.18 IL-1β might also be directly involved in plaque destabilization by the stimulation of matrix metalloproteinases.19 Abundant evidence supports the participation of IL-1β in atherothrombosis, although most of this comes from studies in animal models or ex vivo human samples. IL-1β or IL-1 type 1 receptor-deficient mice are characterized by reduced atherosclerotic lesions,20,21 and IL-1β exposure in Apo E−/− mice resulted in enhanced aortic plaque development.7 In humans, a study in explanted hearts demonstrated increased IL-1β levels in arteries with coronary disease.22 Our results show that there is higher local cardiac production of IL-1β in patients with acute coronary events. It remains unclear, however, whether IL-1β production is derived from macrophages in acute plaques, from macrophages in the infarcted myocardium, or both. However, given that there was no statistically significant difference in IL-1β levels in patients with and without troponin elevations (P=0.308), this strongly suggests a vascular rather than a myocardial origin of this cytokine. Also, it has been suggested that statin type may influence inflammasome activation, with rosuvastatin having a greater inhibitory effect than atorvastatin.23 In our study, however, no differences were seen, irrespective of statin type, with ≈40% of statin-treated patients on rosuvastatin and the other 60% on atorvastatin. As the majority of these patients were on dual antiplatelet inhibition and statins, these findings also suggest ongoing inflammation in ACS patients despite best available therapy. Accordingly, further inhibition of the IL-1β pathway seems attractive as a potential treatment for the acute period of unstable coronary disease. Indeed, the CANTOS trial is currently testing the hypothesis that canakinumab, a specific IL-1β human monoclonal antibody, would reduce events in stable but persistently inflamed post–myocardial infarction patients.24 In contrast to canakinumab, colchicine also has inhibitory effects on neutrophils, which also contribute to plaque progression and plaque destabilization;25 however, the clinical benefits of colchicine versus canakinumab are still to be determined.
IL-18 has also been implicated in CAD pathogenesis.8,26,27 Systemic concentrations of IL-18 have been shown to predict future cardiovascular events in patients with both stable and unstable symptoms, and its prognostic value is additive to other recognized inflammatory markers, such as C-reactive protein and IL-6.28 Moreover, variations of the IL18 gene influence outcomes in patients with CAD, suggesting a causal role for IL18 in the complications of atherosclerosis.29 Our data suggest increased local production of IL-18 in patients with CAD, in a similar fashion to IL-1β, in agreement with a common inflammasome-mediated pathway underlying their production. The results are also consistent with the significant association between caspase-1 levels and cardiovascular events in CAD patients.30 Accordingly, inhibition of this cytokine could also be attractive; however, to our knowledge, no specific drug is currently under testing for this purpose.
IL-6 Correlates With Levels of Inflammasome-Specific Cytokines
Increased cardiac production of IL-6 in patients with acute myocardial infarction has previously been demonstrated.31 Moreover, IL-6 levels in venous blood are strongly associated with both disease activity in ACS,32 as well as being predictive of future coronary events in patients with stable disease.33,34 Similarly, in our study, both transcoronary and venous levels of IL-6 correlated significantly with disease activity, in parallel with the transcoronary levels of the inflammasome-specific cytokines, IL-1β and IL-18. As IL-1β is known to play a pivotal role in IL-6 production, these data suggest a plausible link between increased local production of inflammasome-specific inflammatory cytokines, increased plaque vulnerability, and unstable coronary events.
Acute Colchicine Treatment Suppresses the Intracardiac Synthesis of IL-1β, IL-18, and IL-6
Colchicine administration in patients with unstable CAD within the 24 hours prior to cardiac catheterization significantly reduced local synthesis of IL-1β, IL-18, and IL-6, as well as venous concentrations of IL-6 (Figure 4). The acute suppression of these pro-inflammatory cytokines in ACS patients potentially has important therapeutic implications. ACS are characterized by widespread vascular inflammation,35 affecting not only the culprit lesion but also multiple coronary segments, thus potentially promoting plaque rupture and erosion and subsequent events.36 Acute inhibition of a number of key effectors in this inflammatory cascade with colchicine—an oral and inexpensive drug—could therefore reduce the incidence of new coronary events, which are common post-ACS,37 by stabilizing both culprit and nonculprit lesions. Indeed, values as high as 15% of new cardiovascular events on 3-year follow-up have been described in recent trials.38 Moreover, IL-6 has been strongly linked with increased risk of cardiovascular events in large epidemiological studies.34 Our study demonstrated a marked reduction in local IL-6 production as well as venous levels with colchicine, suggesting a potential role for colchicine in reducing this risk.
A recently published study showed that a subcutaneous IL-1 receptor antagonist (Anakinra) administered daily for 14 days was associated with a significant reduction in CRP and IL-6 compared to placebo.39 Although both CRP and IL-6 have been shown to predict events in patients with ACS,10,40 Anakinra was associated with an increase in clinical events at 1-year follow-up. A possible explanation is the rebound in the inflammatory response (as revealed by a significant increase of high-sensitivity CRP levels at day 30) after a too-short treatment (14 days) with anakinra. Thus, a more prolonged inhibition of the IL-1 pathway or a broader inhibition of the inflammatory response using an anti-inflammasome agent, such as colchicine, could have clinical potential. Encouragingly, in this regard, the LoDoCo trial showed significant benefit of long-term colchicine treatment in patients with stable CAD, mainly due to a reduced number of acute events during follow-up (that is, predominantly by preventing plaque destabilization).41 The ongoing Cardiovascular Inflammation Reduction Trial (CIRT), testing the effect of methotrexate on cardiovascular outcomes, may also support the use of anti-inflammatories to prevent secondary cardiovascular events.42 It is possible based on the results of the current study that suppression of inflammasome-related cytokines may play a key role in effects of colchicine on acute plaque instability and the reduction of clinical events. This clearly requires further assessment; our current study was powered to examine a reduction in transcoronary cytokine release but not to assess clinical event rates.
The mechanisms by which colchicine inhibits the production of these cytokines are not completely understood. It has been suggested that colchicine might block crystal endocytosis and posterior stimulation of the inflammasome complex.43 Colchicine might have a transcriptional effect by blocking the MEFV gene, thereby inhibiting the production of the inflammasome complex proteins.44 Also, colchicine has been shown to prevent mobilization of a key protein in inflammasome complex assembly, known as apoptosis-associated speck-like protein containing caspase and activation recruitment domain (CARD) (ASC) from the mitochondria to the endoplasmic reticulum and thus prevents its colocalization with the rest of the inflammasome complex.14 It is plausible that colchicine might exert its effects upon the inflammasome complex in more than 1 way. For example, it might rapidly inhibit the inflammasome complex assembly, hence exerting an acute effect, as demonstrated in the present study. It may also have a transcriptional effect, inhibiting the production of IL-1β and IL-18, a mechanism that might take longer to act and be related to more long-term exposure to the drug.
Limitations
Study limitations include a relatively small sample size. Also, although our study demonstrates increased cardiac production of inflammasome-associated cytokines in ACS, we cannot determine whether changes in cytokines contribute to ongoing coronary inflammation or are simply released as a consequence of plaque rupture and erosion. Moreover, mechanisms of inflammasome activation in this setting and the subacute/chronic time course of cytokine release require further exploration. Our study was designed to test the effects of short-term colchicine administration on the local cardiac production of cytokines. However, long-term use of this drug and its possible side effects were not evaluated. Finally, patients were not followed with respect to clinical outcomes.
Conclusions
In the present study, we have demonstrated increased intracardiac production of the inflammasome specific cytokines IL-1β, IL-18 and downstream IL-6 in patients presenting with an ACS.
Furthermore, acute colchicine administration is associated with a significant reduction in the transcoronary production of these cytokines in patients with ACS. These data suggest a possible therapeutic role for colchicine in acutely suppressing atherosclerosis-associated inflammation.
Acknowledgments
The authors would like to acknowledge the cardiac catheterization laboratory staff at the Royal Prince Alfred Hospital, for their assistance in performing the studies.
Sources of Funding
This study was supported by Sydney Medical School Foundation Grant (Patel).
Disclosures
None.
References
- Flego D, Severino A, Trotta F, Previtero M, Ucci S, Zara C, Massaro G, Pedicino D, Biasucci LM, Liuzzo G, Crea F. Increased PTPN22 expression and defective CREB activation impair regulatory T-cell differentiation in non-ST-segment elevation acute coronary syndromes. J Am Coll Cardiol. 2015;65:1175–1186. doi: 10.1016/j.jacc.2015.01.027. [DOI] [PubMed] [Google Scholar]
- Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Hansson GK Leducq Transatlantic Network on A. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT, Eklund KK. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765. doi: 10.1371/journal.pone.0011765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ, Moore KJ. IL-1 signaling in atherosclerosis: sibling rivalry. Nat Immunol. 2013;14:1030–1032. doi: 10.1038/ni.2711. [DOI] [PubMed] [Google Scholar]
- Merhi-Soussi F, Kwak BR, Magne D, Chadjichristos C, Berti M, Pelli G, James RW, Mach F, Gabay C. Interleukin-1 plays a major role in vascular inflammation and atherosclerosis in male apolipoprotein E-knockout mice. Cardiovasc Res. 2005;66:583–593. doi: 10.1016/j.cardiores.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Mallat Z, Corbaz A, Scoazec A, Besnard S, Leseche G, Chvatchko Y, Tedgui A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 2001;104:1598–1603. doi: 10.1161/hc3901.096721. [DOI] [PubMed] [Google Scholar]
- Luc G, Bard JM, Juhan-Vague I, Ferrieres J, Evans A, Amouyel P, Arveiler D, Fruchart JC, Ducimetiere P Group PS. C-reactive protein, interleukin-6, and fibrinogen as predictors of coronary heart disease: the PRIME Study. Arterioscler Thromb Vasc Biol. 2003;23:1255–1261. doi: 10.1161/01.ATV.0000079512.66448.1D. [DOI] [PubMed] [Google Scholar]
- Liuzzo G, Biasucci LM, Gallimore JR, Grillo RL, Rebuzzi AG, Pepys MB, Maseri A. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N Engl J Med. 1994;331:417–424. doi: 10.1056/NEJM199408183310701. [DOI] [PubMed] [Google Scholar]
- Terkeltaub RA, Furst DE, Bennett K, Kook KA, Crockett RS, Davis MW. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62:1060–1068. doi: 10.1002/art.27327. [DOI] [PubMed] [Google Scholar]
- Imazio M, Brucato A, Cemin R, Ferrua S, Maggiolini S, Beqaraj F, Demarie D, Forno D, Ferro S, Maestroni S, Belli R, Trinchero R, Spodick DH, Adler Y Investigators I. A randomized trial of colchicine for acute pericarditis. N Engl J Med. 2013;369:1522–1528. doi: 10.1056/NEJMoa1208536. [DOI] [PubMed] [Google Scholar]
- Pope RM, Tschopp J. The role of interleukin-1 and the inflammasome in gout: implications for therapy. Arthritis Rheum. 2007;56:3183–3188. doi: 10.1002/art.22938. [DOI] [PubMed] [Google Scholar]
- Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14:454–460. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- King SB, III, Smith SC, Jr, Hirshfeld JW, Jr, Jacobs AK, Morrison DA, Williams DO, Feldman TE, Kern MJ, O’Neill WW, Schaff HV, Whitlow PL ACC/AHA/SCAI. Adams CD, Anderson JL, Buller CE, Creager MA, Ettinger SM, Halperin JL, Hunt SA, Krumholz HM, Kushner FG, Lytle BW, Nishimura R, Page RL, Riegel B, Tarkington LG, Yancy CW. 2007 Focused update of the ACC/AHA/SCAI 2005 guideline update for percutaneous coronary intervention: a report of the American College of Cardiology/American Heart Association Task Force on Practice guidelines. J Am Coll Cardiol. 2008;51:172–209. doi: 10.1016/j.jacc.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Martínez GJ, Bailey BP, Celermajer DS, Patel S. A safe and easy technique to sample the coronary sinus—facilitating a closer look at cardiac disease. Int J Cardiol. 2014;176:1321–1322. doi: 10.1016/j.ijcard.2014.07.151. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol. 2011;41:1203–1217. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. The role of the interleukin-1-receptor antagonist in blocking inflammation mediated by interleukin-1. N Engl J Med. 2000;343:732–734. doi: 10.1056/NEJM200009073431011. [DOI] [PubMed] [Google Scholar]
- Lee E, Grodzinsky AJ, Libby P, Clinton SK, Lark MW, Lee RT. Human vascular smooth muscle cell-monocyte interactions and metalloproteinase secretion in culture. Arterioscler Thromb Vasc Biol. 1995;15:2284–2289. doi: 10.1161/01.atv.15.12.2284. [DOI] [PubMed] [Google Scholar]
- Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:656–660. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- Chamberlain J, Evans D, King A, Dewberry R, Dower S, Crossman D, Francis S. Interleukin-1beta and signaling of interleukin-1 in vascular wall and circulating cells modulates the extent of neointima formation in mice. Am J Pathol. 2006;168:1396–1403. doi: 10.2353/ajpath.2006.051054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol. 1996;16:1000–1006. doi: 10.1161/01.atv.16.8.1000. [DOI] [PubMed] [Google Scholar]
- Satoh M, Tabuchi T, Itoh T, Nakamura M. NLRP3 inflammasome activation in coronary artery disease: results from prospective and randomized study of treatment with atorvastatin or rosuvastatin. Clin Sci (Lond) 2014;126:233–241. doi: 10.1042/CS20130043. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. 2012;110:875–888. doi: 10.1161/CIRCRESAHA.111.257535. [DOI] [PubMed] [Google Scholar]
- Whitman SC, Ravisankar P, Daugherty A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(−/−) mice through release of interferon-gamma. Circ Res. 2002;90:E34–E38. doi: 10.1161/hh0202.105292. [DOI] [PubMed] [Google Scholar]
- Mallat Z, Corbaz A, Scoazec A, Graber P, Alouani S, Esposito B, Humbert Y, Chvatchko Y, Tedgui A. Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ Res. 2001;89:E41–E45. doi: 10.1161/hh1901.098735. [DOI] [PubMed] [Google Scholar]
- Blankenberg S, Tiret L, Bickel C, Peetz D, Cambien F, Meyer J, Rupprecht HJ, AtheroGene I. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation. 2002;106:24–30. doi: 10.1161/01.cir.0000020546.30940.92. [DOI] [PubMed] [Google Scholar]
- Tiret L, Godefroy T, Lubos E, Nicaud V, Tregouet DA, Barbaux S, Schnabel R, Bickel C, Espinola-Klein C, Poirier O, Perret C, Munzel T, Rupprecht HJ, Lackner K, Cambien F, Blankenberg S, AtheroGene I. Genetic analysis of the interleukin-18 system highlights the role of the interleukin-18 gene in cardiovascular disease. Circulation. 2005;112:643–650. doi: 10.1161/CIRCULATIONAHA.104.519702. [DOI] [PubMed] [Google Scholar]
- Blankenberg S, Godefroy T, Poirier O, Rupprecht HJ, Barbaux S, Bickel C, Nicaud V, Schnabel R, Kee F, Morrison C, Evans A, Lackner KJ, Cambien F, Munzel T, Tiret L, AtheroGene I. Haplotypes of the caspase-1 gene, plasma caspase-1 levels, and cardiovascular risk. Circ Res. 2006;99:102–108. doi: 10.1161/01.RES.0000232324.87983.4b. [DOI] [PubMed] [Google Scholar]
- Maier W, Altwegg LA, Corti R, Gay S, Hersberger M, Maly FE, Sutsch G, Roffi M, Neidhart M, Eberli FR, Tanner FC, Gobbi S, von Eckardstein A, Luscher TF. Inflammatory markers at the site of ruptured plaque in acute myocardial infarction: locally increased interleukin-6 and serum amyloid A but decreased C-reactive protein. Circulation. 2005;111:1355–1361. doi: 10.1161/01.CIR.0000158479.58589.0A. [DOI] [PubMed] [Google Scholar]
- Ikeda U, Ito T, Shimada K. Interleukin-6 and acute coronary syndrome. Clin Cardiol. 2001;24:701–704. doi: 10.1002/clc.4960241103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Howard CP, Walter V, Everett B, Libby P, Hensen J, Thuren T Group CPI. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. 2012;126:2739–2748. doi: 10.1161/CIRCULATIONAHA.112.122556. [DOI] [PubMed] [Google Scholar]
- Kaptoge S, Seshasai SR, Gao P, Freitag DF, Butterworth AS, Borglykke A, Di Angelantonio E, Gudnason V, Rumley A, Lowe GD, Jorgensen T, Danesh J. Inflammatory cytokines and risk of coronary heart disease: new prospective study and updated meta-analysis. Eur Heart J. 2014;35:578–589. doi: 10.1093/eurheartj/eht367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffon A, Biasucci LM, Liuzzo G, D’Onofrio G, Crea F, Maseri A. Widespread coronary inflammation in unstable angina. N Engl J Med. 2002;347:5–12. doi: 10.1056/NEJMoa012295. [DOI] [PubMed] [Google Scholar]
- Kato K, Yonetsu T, Kim SJ, Xing L, Lee H, McNulty I, Yeh RW, Sakhuja R, Zhang S, Uemura S, Yu B, Mizuno K, Jang IK. Nonculprit plaques in patients with acute coronary syndromes have more vulnerable features compared with those with non-acute coronary syndromes: a 3-vessel optical coherence tomography study. Circ Cardiovasc Imaging. 2012;5:433–440. doi: 10.1161/CIRCIMAGING.112.973701. [DOI] [PubMed] [Google Scholar]
- Fox KA, Carruthers KF, Dunbar DR, Graham C, Manning JR, De Raedt H, Buysschaert I, Lambrechts D, Van de Werf F. Underestimated and under-recognized: the late consequences of acute coronary syndrome (GRACE UK-Belgian Study) Eur Heart J. 2010;31:2755–2764. doi: 10.1093/eurheartj/ehq326. [DOI] [PubMed] [Google Scholar]
- O’Donoghue ML, Braunwald E, White HD, Lukas MA, Tarka E, Steg PG, Hochman JS, Bode C, Maggioni AP, Im K, Shannon JB, Davies RY, Murphy SA, Crugnale SE, Wiviott SD, Bonaca MP, Watson DF, Weaver WD, Serruys PW, Cannon CP Investigators S-T. Steen DL. Effect of darapladib on major coronary events after an acute coronary syndrome: the SOLID-TIMI 52 randomized clinical trial. JAMA. 2014;312:1006–1015. doi: 10.1001/jama.2014.11061. [DOI] [PubMed] [Google Scholar]
- Morton AC, Rothman AM, Greenwood JP, Gunn J, Chase A, Clarke B, Hall AS, Fox K, Foley C, Banya W, Wang D, Flather MD, Crossman DC. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA Heart Study. Eur Heart J. 2015;36:377–384. doi: 10.1093/eurheartj/ehu272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisman EZ, Benderly M, Esper RJ, Behar S, Boyko V, Adler Y, Tanne D, Matas Z, Tenenbaum A. Interleukin-6 and the risk of future cardiovascular events in patients with angina pectoris and/or healed myocardial infarction. Am J Cardiol. 2006;98:14–18. doi: 10.1016/j.amjcard.2006.01.045. [DOI] [PubMed] [Google Scholar]
- Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol. 2013;61:404–410. doi: 10.1016/j.jacc.2012.10.027. [DOI] [PubMed] [Google Scholar]
- Everett BM, Pradhan AD, Solomon DH, Paynter N, Macfadyen J, Zaharris E, Gupta M, Clearfield M, Libby P, Hasan AA, Glynn RJ, Ridker PM. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166:199–207.e15. doi: 10.1016/j.ahj.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Nidorf SM, Eikelboom JW, Thompson PL. Targeting cholesterol crystal-induced inflammation for the secondary prevention of cardiovascular disease. J Cardiovasc Pharmacol Ther. 2014;19:45–52. doi: 10.1177/1074248413499972. [DOI] [PubMed] [Google Scholar]