

Abstract
Background
CSL112 is a new formulation of human apolipoprotein A-I (apoA-I) being developed to reduce cardiovascular events following acute coronary syndrome. This phase 2a, randomized, double-blind, multicenter, dose-ranging trial represents the first clinical investigation to assess the safety and pharmacokinetics/pharmacodynamics of a CSL112 infusion among patients with stable atherosclerotic disease.
Methods and Results
Patients were randomized to single ascending doses of CSL112 (1.7, 3.4, or 6.8 g) or placebo, administered over a 2-hour period. Primary safety assessments consisted of alanine aminotransferase or aspartate aminotransferase elevations >3× upper limits of normal and study drug–related adverse events. Pharmacokinetic/pharmacodynamic assessments included apoA-I plasma concentration and measures of the ability of serum to promote cholesterol efflux from cells ex vivo. Of 45 patients randomized, 7, 12, and 14 received 1.7-, 3.4-, and 6.8-g CSL112, respectively, and 11 received placebo. There were no clinically significant elevations (>3× upper limit of normal) in alanine aminotransferase or aspartate aminotransferase. Adverse events were nonserious and mild and occurred in 5 (71%), 5 (41%), and 6 (43%) patients in the CSL112 1.7-, 3.4-, and 6.8-g groups, respectively, compared with 3 (27%) placebo patients. The imbalance in adverse events was attributable to vessel puncture/infusion-site bruising. CSL112 resulted in rapid (Tmax≈2 hours) and dose-dependent increases in apoA-I (145% increase in the 6.8-g group) and total cholesterol efflux (up to 3.1-fold higher than placebo) (P<0.001).
Conclusions
CSL112 infusion was well tolerated in patients with stable atherosclerotic disease. CSL112 immediately raised apoA-I levels and caused a rapid and marked increase in the capacity of serum to efflux cholesterol. This potential novel approach for the treatment of atherosclerosis warrants further investigation.
Clinical Trial Registration
URL: http://www.ClinicalTrials.gov. Unique identifier: NCT01499420.
Keywords: apolipoprotein, atherosclerosis, clinical trial, coronary disease, plaque
Atherosclerotic coronary disease is caused by the growth and subsequent instability of cholesterol-rich plaques in the artery wall.1 Current pharmacologic strategies to reduce recurrent events after acute coronary syndromes (ACS) have placed emphasis on antithrombotic agents and reduction of low-density lipoprotein cholesterol (LDL-C) with statins.2 Despite the use of these therapies, patients with ACS continue to experience a substantial rate of recurrent ischemic complications. Moreover, strategies with increased potency of antithrombotic therapies have been limited by risk of severe bleeding.3–5
Abundant evidence documents the association of low levels of high-density lipoprotein cholesterol (HDL-C) with increased risk of atherosclerosis and suggests that elevation of HDL-C may be a novel target.6–8 However, recent large-scale clinical trials have failed to demonstrate a clinical benefit of HDL-C–raising therapies.9–11 Nevertheless, HDL-C level itself may not be an adequate marker of antiatherosclerotic activity and may not reflect HDL function.12,13 Thus, increasing HDL function is now considered to be the goal of HDL-targeting therapies.
It is widely accepted that apolipoprotein A-I (apoA-I), the dominant protein of HDL, selectively promotes cholesterol efflux from arterial wall macrophages via the ABCA1 transporter (ATP-binding cassette transporter A1), and this may account for the antiatherosclerotic effect of HDL.13 Higher cholesterol efflux capacity has been recently shown to be independently correlated with a reduction in risk of cardiovascular events.14
Unfortunately, robust elevations of apoA-I have been difficult to achieve by pharmacotherapy. Fibrates and niacin typically achieve <10% elevation,15 while dalcetrapib and torcetrapib achieved only 10% to 25% elevation.10,11 Additionally, the predominant change caused by these agents is an increase in HDL particle size, and larger HDL particles do not efficiently interact with the ABCA1 transporter.13
An alternative approach to elevate the functional activity of plasma HDL is the direct infusion of lipid-poor apoA-I particles designed to favor interaction with the ABCA1 transporter.16 This approach may be particularly attractive for the prevention of recurrent acute ischemic events in patients with unstable disease.17,18 Infusion of HDL-like particles has been shown in 3 separate studies to modify plaque characterization on intravascular ultrasonography (IVUS).19–21 One of these studies used a prototype formulation termed CSL111, which was discontinued from development due to the occurrence of transient elevations of hepatic enzymes.21
CSL112 is a novel formulation of human apoA-I. The apoA-I is reconstituted with phosphatidylcholine to form disc-shaped HDL particles, each bearing 2 molecules of human apoA-I and ≈110 molecules of phosphatidylcholine.22 CSL112 preparations contain sucrose as a stabilizing agent. In ex vivo studies, CSL112 was an efficient acceptor of cholesterol from J774 macrophages.22 In the presence of plasma, CSL112 preferentially supported ABCA1-dependent cholesterol efflux, an activity attributed to active remodeling in plasma.22 The ability of CSL112 to promote ABCA1-dependent cholesterol efflux has recently been reported in healthy adults.23 CSL112 has also been shown to be safe and well tolerated in healthy adults with predictable and robust pharmacokinetic and pharmacodynamic responses.24
The present phase 2a randomized clinical trial is the first experience with CSL112 in a stable patient population with atherosclerosis. The aim was to assess the safety and pharmacokinetic and pharmacodynamic effects of a single intravenous infusion of CSL112.
Methods
Objectives, Study Rationale, and Design
The main objective of the study was to assess the safety of CSL112 after a single intravenous infusion in patients with stable atherosclerotic disease who were receiving standard-of-care therapy, including aspirin and either clopidogrel or prasugrel. The primary safety evaluations were study drug–related adverse events (AEs) and liver safety. Risk of renal toxicity has been described with intravenous immunoglobulin containing high doses of intravenous sucrose, and we assessed renal function following infusion of low-sucrose–containing preparations of CSL112.25 Finally, the trial further characterized the pharmacokinetics and pharmacodynamics of CSL112.
The current study was a phase 2a, randomized, multicenter, parallel-group, double-blind, placebo-controlled, single-infusion, ascending-dose study (ClinicalTrials.gov identifier NCT01499420) conducted at 11 centers in the United States. It targeted 40 patients for randomization into 3 ascending-dose groups: 1.7 g (n=8), 3.4 g (n=16), and 6.8 g (n=16) (Figure 1). Within each dose group, patients were randomized 3:1 to receive a single infusion of CSL112 or placebo. Randomization was stratified by renal function: normal renal function (creatinine clearance ≥90 mL/min) or mild renal insufficiency (creatinine clearance ≥60 to <90 mL/min), with at least 50% of patients in each group having mild renal insufficiency.
Figure 1.
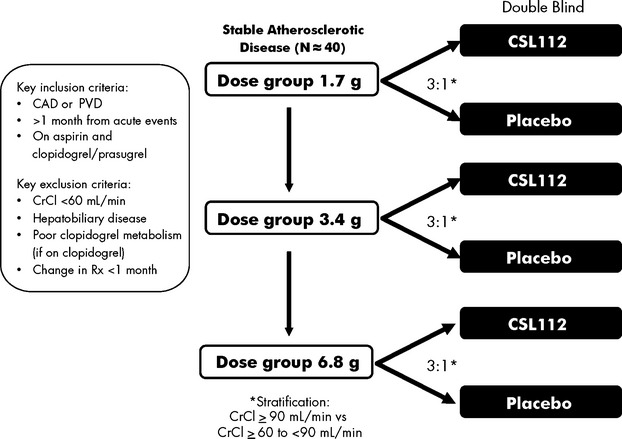
Trial design. CAD indicates coronary artery disease; CrCl, creatinine clearance; PVD, peripheral vascular disease; Rx, prescription.
The study consisted of 3 periods: the screening period, the active study period, and the follow-up period (Figure 2). Patients were screened between 3 and 50 days before randomization and infusion. Eligible patients provided written informed consent and were admitted to the study unit 2 days before drug administration (day −2). Eligibility was reassessed and local laboratory measurements were repeated before randomization. Once eligibility was confirmed, patients began the active treatment period. On day 1, patients received a 2-hour infusion of the allocated study drug via an indwelling catheter through a peripheral vein. Patients remained in the study unit until at least day 3 (ie, ≈48 hours after study drug administration) to perform additional safety and laboratory assessments. After discharge, patients returned for safety and laboratory assessments at days 3, 4, 5, 6, 7, and 9. An additional clinical follow-up for AE assessment was performed at day 14, which completed the active study period. A standard plasma-based product serology follow-up visit included nucleic acid testing for a virus panel and was performed 90 days after administration of study product.
Figure 2.
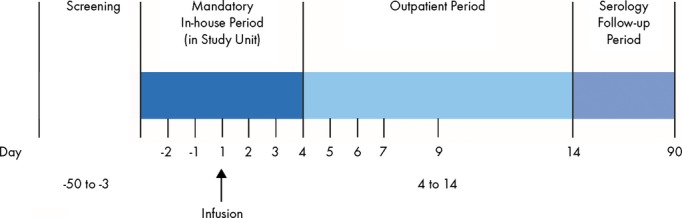
Study design.
Standard medium-fat (≈30% fat) meals (typical of a Western diet; at ≈2020 calories [8457 kJ] per day) were served, according to the clinic’s schedule, to maintain body weight while the subjects were in the study unit. During nonfasting periods, fluids were allowed ad libitum. To reduce variability in pharmacokinetic and pharmacodynamic assessments, patients were required to fast overnight starting 8 hours before study drug administration and ending not before 8 hours after the end of the infusion. There was also an 8-hour fasting period before pharmacokinetic assessments on other days. Alcohol was prohibited from 48 hours before randomization until day 9. Smoking was prohibited from 48 hours before randomization until discharge from the study unit.
The study was approved by institutional review boards governing participating study sites.
CSL112
CSL112 is an investigational, reconstituted HDL product containing apoA-I purified from human plasma and formulated with phosphatidylcholine.22 Lyophilized CSL112 was reconstituted with sterile water for injection and was dosed based on total protein content. The placebo consisted of 0.9% sodium chloride solution for intravenous injection. Both CSL112 and placebo were administered as an intravenous 2-hour infusion.
Patients
A detailed list of inclusion and exclusion criteria is provided in Data S1. Briefly, the study included male and female patients, aged 18 to 80 years, with a history of atherosclerotic coronary artery disease or peripheral vascular disease. All patients were clinically stable, which was defined as a minimum of 1 month without any acute event, including ACS, or hospitalization for chest pain and revascularization procedures. Patients were receiving a stable medical regimen for the past month that was expected to continue during the active study period. Dual antiplatelet therapy was required for at least 1 month before randomization; the regimen had to include aspirin and either clopidogrel or prasugrel. No other antiplatelet medications were permitted, including those with potential antiplatelet effects such as nonsteroidal anti-inflammatory drugs. Concomitant medications required to treat chronic medical conditions were continued. Key exclusion criteria were (1) moderate and severe renal disease (creatinine clearance <60 mL/min), (2) evidence of hepatobiliary disease, (3) any unstable medical condition within 30 days, (4) poor clopidogrel metabolism in patients taking clopidogrel as indicated by rapid genotype testing, and (5) concomitant omeprazole and clopidogrel therapy within 1 month.
Study Procedures
Safety measurements and end points
The primary safety end points were postrandomization frequency of study drug–related AEs and clinically significant elevation (>3× the upper limit of normal [ULN]) of alanine aminotransferase (ALT) or aspartate aminotransferase (AST) based on central laboratory determinations. Additional hepatic function assessments included total bilirubin, direct bilirubin, indirect bilirubin, alkaline phosphatase, and γ-glutamyl transferase. Standard enzymatic assays were used for laboratory determinations and were performed centrally (Eurofins Scientific Laboratories). Several additional laboratory safety assessments were performed, including cystatin C, creatinine, blood urea nitrogen, and kidney injury molecule-1. The immunogenicity of CSL112 was also determined by measurement of serum antibodies to CSL112 and to apoA-I.
Study drug–related AEs were reported by investigators and defined as (1) an AE that began or worsened during study product infusion up to 72 hours after the end of infusion and/or (2) any AE considered possibly, probably, or definitely related to treatment by the local investigator or (3) an AE that occurred during the active study period for which the investigator’s causality assessment was missing or indeterminate.
Pharmacokinetics and pharmacodynamics
The pharmacokinetic profile of CSL112 was determined by measuring apoA-I in plasma samples obtained at study-specified time points before and after the intravenous infusion of study product (Pacific Biomarkers). Pharmacokinetic assessments were performed at the screening visit, on the day of admission to the study unit (day −2), before study product administration (to determine baseline endogenous levels), and at the following times after start of the infusion: 2, 4, 6, 8, 12, 24, 36, 48, 72, 96, 120, and 144 hours. The following pharmacokinetic parameters were calculated as follows: AUC0-last, area under the plasma concentration time curve from time point 0 (start of infusion) to the last quantifiable time point before the analyte first returns to baseline; AUC0-∞, area under the plasma concentration time curve from time 0 to infinity; Cmax, observed maximum concentration in plasma; Tmax, time to reach maximum concentration in plasma; and t1/2, plasma half-life. The geometric mean of screening, day −2, and the pre–study drug administration concentrations were calculated and set as the baseline. For calculation of baseline-corrected parameters, the time 0 measurement was set to the calculated baseline. This baseline was then subtracted from all postdose concentrations, and time 0 became 0 for the baseline-corrected values.
Several exploratory biomarkers were assessed as part of this study to investigate the pharmacodynamics of CSL112. Time points of collections were before dosing and the following postdosing times: 2, 4, 8, 24, 48, 72, 96, and 144 hours. Standard lipid panels were performed. Here, we report total cholesterol efflux capacity measured by incubation of serum from study participants with ABCA1-expressing macrophages in vitro as previously described.23 Key pharmacodynamic parameters assessed were AUEC0-last, area under the effect curve from time point 0 (baseline) to the last time point above baseline; AUEC0-x, area from time point 0 to a meaningful time after infusion (x); Rmax, maximum efflux capacity biomarker response (concentration or activity); and Tmax, time to reach maximum efflux capacity.
Statistical Analysis
Because the study was not designed to test specific hypotheses, the safety data analysis was descriptive. No formal sample-size calculation was conducted. The target sample size of the study was to have at least 40 subjects enrolled, with 10 subjects in the placebo group and 30 in the active treatment groups. We estimated that with 30 active subjects enrolled and observed for 14 days, the chances of observing AEs were as follows: 36% for events with 1% incidence, 59% for events with 2% incidence, and 99% for events with 10% incidence.
The predefined population for safety analyses consisted of all randomized patients who received any amount of study drug. For purposes of the primary safety analysis, a clinically significant elevation of AST/ALT was defined as test results that were >3× ULN in any 2 consecutive blood samples collected during the active study period and ≥24 hours apart.
The pharmacokinetic analysis population consisted of all patients who received an infusion of CSL112 with ≥1 quantifiable concentration of apoA-I during the active study period. All subjects randomized to a CSL112 treatment group contributed to the analysis. Noncompartmental pharmacokinetic analyses were performed using model 202 (constant infusion) in WinNonlin version 5.2 (or higher) from the concentration-time data.
Statistical comparisons for the pharmacokinetic/pharmacodynamic parameters were performed using ANOVA with statistical significance set at P<0.05.
Results
Patient Allocation and Baseline Characteristics
A total of 45 patients were randomized. One patient in the 6.8-g CSL112 group withdrew from the study before receiving study product and, therefore, was not considered in further analyses. Of the 44 remaining patients, 11 were randomly assigned to receive placebo; of the 33 patients randomized to receive CSL112, 7 received 1.7 g, 12 received 3.4 g, and 14 received 6.8 g. Complete treatment administration and follow-up through the active study period (day 14) were achieved in all 44 patients (Figure 3).
Figure 3.
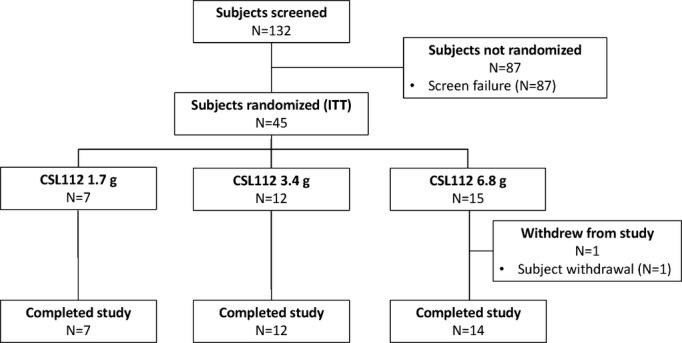
Subject disposition. ITT indicates intention-to-treat.
The median age was 60 years (range 40 to 77 years) in CSL112 patients and 56 years (range 47 to 71) in placebo patients (Table 1). The majority of randomized patients were male and white. The majority of patients in all treatment groups were overweight or obese; the median body mass index in the treatment groups ranged from 28.5 to 31.8 kg/m2. Baseline HDL-C and apoA-I were comparable among all treatment groups. Creatinine clearance was similar between placebo (median 89, range 65 to 118 mL/min) and CSL112 (median 84.0, range 42 to 249 mL/min) groups.
Table 1.
Baseline Characteristics
| Placebo (n=11) | CSL112 1.7 g (n=7) | CSL112 3.4 g (n=12) | CSL112 6.8 g (n=14) | CSL112 Overall (n=33) | P Value | |
|---|---|---|---|---|---|---|
| Demographic | ||||||
| Age, y; median (min, max) | 56 (47, 71) | 65 (54, 77) | 57 (40, 73) | 60 (41, 76) | 60 (40, 77) | 0.839 |
| Male sex | 7 (63.6) | 4 (57.1) | 9 (75.0) | 12 (85.7) | 25 (75.8) | 0.434 |
| Weight, kg; median (min, max) | 91.6 (68.8, 120.4) | 82.7 (53.2, 101.5) | 99.5 (63.0, 145.0) | 83.9 (70.1, 117.5) | 84.0 (53.2, 145.0) | 0.871 |
| BMI, kg/m2; median (min, max) | 28.5 (21.7, 45.5) | 29.3 (20.8, 34.4) | 31.8 (24.8, 54.8) | 28.7 (23.6, 40.0) | 30.5 (20.8, 54.8) | 0.464 |
| Race and ethnicity | ||||||
| White | 8 (72.7) | 6 (85.7) | 9 (75.0) | 12 (85.7) | 27 (81.8) | 0.517 |
| Not Hispanic or Latino | 10 (90.9) | 7 (100.0) | 11 (91.7) | 12 (85.7) | 30 (90.9) | 1.000 |
| Baseline HDL <40 mg/dL | 5 (45.5) | 1 (14.3) | 6 (50.0) | 8 (57.1) | 15 (45.5) | 1.000 |
| Baseline apoA-I | 0.223 | |||||
| <1.32 g/L | 7 (63.6) | 3 (42.9) | 6 (50.0) | 5 (35.7) | 14 (42.4) | |
| ≥1.32 g/L | 4 (36.4) | 4 (57.1) | 6 (50.0) | 9 (64.3) | 19 (57.6) | |
| Baseline CrCl, mL/min; median (min, max) | 89.0 (65.0, 118.0) | 70.0 (42.0, 119.0) | 85.0 (64.0, 249.0) | 92.0 (55.0, 170.0) | 84.0 (42.0, 249.0) | 0.626 |
| Medical history* (ITT population) | ||||||
| Coronary artery disease | 11 (100) | 5 (71.4) | 10 (83.3) | 11 (73.3) | 26 (76.5) | 0.096 |
| Prior myocardial infarction | 6 (54.5) | 4 (57.1) | 4 (33.3) | 5 (33.3) | 13 (38.2) | 0.380 |
| Prior stenting | 6 (54.5) | 3 (42.9) | 3 (25.0) | 5 (33.3) | 11 (32.4) | 0.211 |
| Prior coronary artery bypass graft | 3 (27.3) | 3 (42.9) | 3 (25.0) | 1 (6.7) | 7 (20.6) | 0.678 |
| Peripheral artery disease | 1 (9.1) | 2 (28.6) | 1 (8.3) | 1 (6.7) | 4 (11.8) | 0.784 |
| Hypertension | 7 (63.6) | 6 (85.7) | 11 (91.7) | 11 (73.3) | 28 (82.4) | 0.131 |
| Type 2 diabetes mellitus | 5 (45.5) | 2 (28.6) | 3 (25.0) | 1 (6.7) | 6 (17.6) | 0.070 |
| Hyperlipidemia | 5 (45.4) | 3 (42.9) | 6 (50.0) | 5 (33.3) | 14 (41.2) | 0.861 |
| Concomitant medications | ||||||
| Aspirin | 10 (90.9) | 7 (100) | 12 (100) | 14 (100) | 33 (100) | 0.080 |
| Clopidogrel | 10 (90.9) | 6 (85.7) | 11 (91.7) | 9 (64.3) | 26 (78.8) | 0.367 |
| Prasugrel | 0 (0) | 1 (14.3) | 1 (8.3) | 5 (35.7) | 7 (21.2) | 0.096 |
| Angiotensin-converting enzyme inhibitors | 4 (36.34) | 5 (71.4) | 9 (75.0) | 7 (50.0) | 21 (63.6) | 0.114 |
| Angiotensin receptor blockers | 3 (27.3) | 1 (14.3) | 2 (16.7) | 1 (7.1) | 4 (12.1) | 0.234 |
| β-Blockers | 5 (45.5) | 5 (71.5) | 11 (91.7) | 10 (71.4) | 26 (78.8) | 0.036 |
| Statins | 10 (90.9) | 6 (85.7) | 10 (83.3) | 13 (92.9) | 29 (87.9) | 0.7839 |
| Other lipid-modifying agents | 2 (18.2) | 2 (28.6) | 3 (25.0) | 1 (7.1) | 6 (18.2) | 1.0000 |
Data presented as N (%), unless otherwise noted. ApoA-I indicates apolipoprotein A-I; BMI, body mass index; CrCl, creatinine clearance; HDL, high-density lipoprotein; ITT, intention-to-treat.
n=15 in 6.8-g dose group.
There was no indication of a major imbalance of prevalent diseases or concurrent medications among the treatment groups. The population was composed of patients with coronary artery disease or peripheral artery disease with high prevalence of cardiovascular risk factors. All patients were on dual antiplatelet therapy during the active treatment period. The use of statins was high in the placebo and overall CSL112 groups.
Safety Analyses
Adverse events
During the active treatment period, 3 (27.3%) of 11 patients in the placebo group and 16 (48.5%) of 33 in the combined CSL112 group experienced a study drug–related AE (Table 2). All AEs were mild in intensity except 1 AE of moderate intensity (recurrence of atrial fibrillation) observed with placebo. There was a numerical increase of causally related AEs in the 6.8-g CSL112 group (n=5, 35.7%) compared with the other CSL112 groups (n=1 in each, 8.3% to 14.3%) and placebo (n=1, 9.1%). About half of all AEs observed in the CSL112 groups were infusion-site–related (hematomas reported as bruising or local reactions at the administration site, including coldness, phlebitis, and erythema), accounting for the numerical difference between placebo and CSL112. In addition, mild transient headache, fatigue, nausea, and vessel puncture-site reactions not at the infusion site (including bruising/hematoma and erythema due to venipuncture) were reported more frequently with CSL112. All other AEs, excluding infusion-site–related AEs, reported in the CSL112 groups occurred in a single subject (Table 3).
Table 2.
Summary of AEs Reported
| Placebo (n=11) | CSL112 1.7 g (n=7) | CSL112 3.4 g (n=12) | CSL112 6.8 g (n=14) | CSL112 Overall (n=33) | |
|---|---|---|---|---|---|
| No. (%) Patients With Event | |||||
| Primary end point: study product–related AE* | 3 (27.3) | 5 (71.4) | 5 (41.7) | 6 (42.9) | 16 (48.5) |
| Nonserious AE | 3 (27.3) | 5 (71.4) | 5 (41.7) | 6 (42.9) | 16 (48.5) |
| Serious AE | 1 (9.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Any AE | 3 (27.3) | 5 (71.4) | 6 (50.0) | 8 (57.1) | 19 (57.6) |
| Most frequent AEs | |||||
| Infusion- site–related AE† | 1 (9.1) | 0 (0) | 2 (16.7) | 4 (28.6) | 6 (18.2) |
| Vessel puncture-site hematoma | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 2 (6.1) |
| Fatigue | 0 (0) | 1 (14.3) | 1 (8.3) | 0 (0) | 2 (6.1) |
| Headache | 0 (0) | 0 (0) | 1 (8.3) | 1 (7.1) | 2 (6.1) |
| Nausea | 0 (0) | 1 (14.3) | 1 (8.3) | 0 (0) | 2 (6.1) |
| Types of AE | |||||
| Causally related AE | 1 (9.1) | 1 (14.3) | 1 (8.3) | 5 (35.7) | 7 (21.2) |
| Maximum intensity‡ | |||||
| Mild | 2 (18.2) | 5 (71.4) | 6 (50) | 8 (57.1) | 19 (57.6) |
| Moderate | 1 (9.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
AE indicates adverse event.
Occurring within 72 hours of infusion or considered related by investigator (independent of time).
Includes intravenous infusion-site ecchymosis/hematoma, erythema, coldness, and phlebitis.
Common Terminology Criteria for Adverse Events v.4 grade at any time point.
Table 3.
Summary of Study Product-Related SAEs/AEs
| Preferred Term | Placebo (n=11) | CSL112 1.7 g (n=7) | CSL112 3.4 g (n=12) | CSL112 6.8 g (n=14) | CSL112 Overall (n=33) |
|---|---|---|---|---|---|
| No. (%) | |||||
| Subjects with ≥1 SAE | 1 (9.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Atrial fibrillation | 1 (9.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Subjects with ≥1 AE | 3 (27.3) | 5 (71.4) | 5 (41.7) | 6 (42.9) | 16 (48.5) |
| Fatigue | 0 (0) | 1 (14.3) | 1 (8.3) | 0 (0) | 2 (6.1) |
| Headache | 0 (0) | 0 (0) | 1 (8.3) | 1 (7.1) | 2 (6.1) |
| Infusion-site hematoma | 0 (0) | 0 (0) | 1 (8.3) | 1 (7.1) | 2 (6.1) |
| Nausea | 0 (0) | 1 (14.3) | 1 (8.3) | 0 (0) | 2 (6.1) |
| Vessel puncture site hematoma | 0 (0) | 1 (14.3) | 0 (0) | 1 (7.1) | 2 (6.1) |
| Alanine aminotransferase increased | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 1 (3.0) |
| Aspartate aminotransferase increased | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 1 (3.0) |
| Blood creatinine increased | 1 (9.1) | 1 (14.3) | 0 (0) | 0 (0) | 1 (3.0) |
| Blood amylase increased | 0 (0) | 1 (14.3) | 0 (0) | 0 (0) | 1 (3.0) |
| Blood glucose increased | 0 (0) | 1 (14.3) | 0 (0) | 0 (0) | 1 (3.0) |
| Blood pressure increased | 1 (9.1) | 0 | 0 (0) | 1 (7.1) | 1 (3.0) |
| Blood urea increased | 1 (9.1) | 1 (14.3) | 0 (0) | 0 (0) | 1 (3.0) |
| Constipation | 0 (0) | 0 | 0 (0) | 1 (7.1) | 1 (3.0) |
| Creatinine renal clearance decreased | 0 (0) | 1 (14.3) | 0 (0) | 0 (0) | 1 (3.0) |
| Dermatitis contact | 0 (0) | 0 (0) | 1 (8.3) | 0 (0) | 1 (3.0) |
| Diarrhea | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 1 (3.0) |
| Dizziness | 1 (9.1) | 0 (0) | 0 (0) | 1 (7.1) | 1 (3.0) |
| Dysgeusia | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 1 (3.0) |
| Glucose urine | 0 (0) | 0 (0) | 1 (8.3) | 0 (0) | 1 (3.0) |
| Infusion-site coldness | 0 (0) | 0 (0) | 1 (8.3) | 0 (0) | 1 (3.0) |
| Injection-site hematoma | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 1 (3.0) |
| Injection-site phlebitis | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 1 (3.0) |
| Rash | 0 (0) | 0 (0) | 1 (8.3) | 0 (0) | 1 (3.0) |
| Urine output decreased | 0 (0) | 1 (14.3) | 0 (0) | 0 (0) | 1 (3.0) |
| Vessel–puncture-site reaction | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 1 (3.0) |
| Catheter-site erythema | 1 (9.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Flatulence | 1 (9.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Muscular weakness | 1 (9.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
AE indicates adverse event; SAE, serious adverse event.
During the serology follow-up period, a second serious AE was reported for a patient in the 6.8-g CSL112 group. This AE was an episode of unstable angina occurring ≈11 weeks after the infusion and was considered to not be related to the study product by the investigator. There was no pattern of higher frequency of study drug–related AEs in patients with mild or moderate renal insufficiency compared with those with normal renal function (data not shown).
Laboratory abnormalities
No patient had an increase from baseline in ALT or AST >3× ULN (Table 4). There were no increases >2× ULN in bilirubin observed. Similar increases in the mean serum creatinine from baseline were seen in all groups, including placebo, with peaks observed between 12 and 36 hours after study product infusion and without sustained changes (Figure 4). A transient mild (1 to 1.5× baseline) elevation (peak 12 to 36 hours after infusion) in serum creatinine was common and occurred in both placebo (n=11, 100%) and CSL112 (n=31, 93.9%) groups (Table 4). A clinically significant increase in serum creatinine (defined as serum creatinine >2× baseline or shift in Common Terminology Criteria for Adverse Events [CTCAE] v.4 creatinine grade) was observed in 7 (63.6%) placebo patients and 25 (75.8%) CSL112 patients. No consistent increases in other renal biomarkers were observed, including blood urea nitrogen, cystatin-C, and kidney injury molecule-1 (Table 5). Because blood samples for clinical laboratory tests were obtained in the fasting state, the observed increases in serum creatinine observed in both placebo and active treatment groups are most likely due to mild volume depletion as a result of fasting.
Table 4.
Summary of Clinical Laboratory Results for Hepatic and Renal Toxicity
| Placebo (n=11) | CSL112 1.7 g (n=7) | CSL112 3.4 g (n=12) | CSL112 6.8 g (n=14) | CSL112 Overall (n=33) | |
|---|---|---|---|---|---|
| Liver safety laboratory | |||||
| ALT | |||||
| No elevation | 10 (90.9) | 7 (100) | 11 (91.7) | 12 (85.7) | 30 (90.9) |
| >1× ULN | 1 (9.1) | 0 (0) | 1 (8.3) | 2 (14.3) | 3 (9.1) |
| >3× ULN | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| AST | |||||
| No elevation | 10 (90.9) | 6 (85.7) | 12 (100) | 14 (100) | 32 (97.0) |
| >1× ULN | 1 (9.1) | 1 (14.3) | 0 (0) | 0 (0) | 1 (3.0) |
| >3× ULN | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Bilirubin >2× ULN | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Renal safety laboratory | |||||
| Overall | |||||
| Clinically significant deterioration of S-creatinine | 7 (63.6) | 6 (85.7) | 7 (58.3) | 12 (85.7) | 25 (75.8) |
| S-Creatinine increase | |||||
| No increase | 0 (0) | 0 (0) | 1 (8.3) | 0 (0) | 1 (3.0) |
| >1 to 1.5× baseline | 11 (100) | 6 (85.7) | 11 (91.7) | 14 (100) | 31 (93.9) |
| >1.5 to 3.0× baseline | 0 (0) | 1 (14.3) | 0 (0) | 0 (0) | 1 (3.0) |
| >3.0× baseline | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| CrCl ≥90 mL/min | |||||
| Clinically significant deterioration of S-creatinine | 5 (100.0) | 3 (100.0) | 4 (80.0) | 7 (100.0) | 14 (93.3) |
| S-Creatinine increase | |||||
| No increase | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| >1 to 1.5× baseline | 5 (100) | 2 (66.7) | 5 (100) | 7 (100) | 14 (93.3) |
| >1.5 to 3.0× baseline | 0 (0) | 1 (33.3) | 0 (0) | 0 (0) | 1 (6.7) |
| >3.0× baseline | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| CrCl ≥60 to <90 mL/min | |||||
| Clinically significant deterioration of S-creatinine | 2 (33.3) | 2 (100.0) | 3 (42.9) | 5 (83.3) | 10 (66.7) |
| S-Creatinine increase | |||||
| No increase | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 1 (6.7) |
| >1 to 1.5× baseline | 6 (100) | 2 (100) | 6 (85.7) | 6 (100) | 14 (93.3) |
| >1.5 to 3.0× baseline | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| >3.0× baseline | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| CrCl ≥30 to <60 mL/min | |||||
| Clinically significant deterioration of S-creatinine | 0 (0) | 1 (50.0) | 0 (0) | 0 (0) | 1 (33.3) |
| S-Creatinine increase | |||||
| No increase | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| >1 to 1.5× baseline | 0 (0) | 2 (100) | 0 (0) | 1 (100) | 3 (100) |
| >1.5 to 3.0× baseline | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| >3.0× baseline | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Data presented as n (%). ALT indicates alanine transaminase; AST, aspartate transaminase; CrCl, creatinine clearance; ULN, upper limit of normal.
Figure 4.
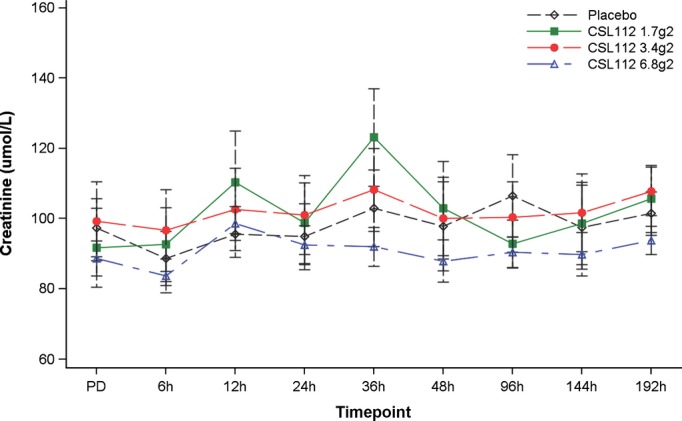
Variation in serum creatinine before and after study drug administration.
Table 5.
Change From Baseline at 24 Hours in Lipid Profile and Biomarkers
| Placebo (n=11) | CSL112 1.7 g (n=7) | CSL112 3.4 g (n=12) | CSL112 6.8 g (n=14) | CSL112 Overall (n=33) | |
|---|---|---|---|---|---|
| Lipids | |||||
| LDL cholesterol, mmol/L | −0.142 (0.180) | −0.143 (0.140) | −0.008 (0.263) | −0.230 (0.295) | −0.131 (0.270) |
| HDL cholesterol, mmol/L | −0.085 (0.095) | 0.056 (0.186) | 0.153 (0.134) | 0.439 (0.150) | 0.254 (0.222) |
| Total cholesterol, mmol/L | −0.240 (0.271) | −0.216 (0.319) | 0.047 (0.307) | 0.089 (0.441) | 0.009 (0.381) |
| Triglycerides, mmol/L | −0.120 (0.353) | −0.406 (0.456) | −0.070 (0.656) | −0.111 (0.538) | −0.159 (0.567) |
| Other renal biomarkers | |||||
| BUN, mmol/L | −0.01 (0.829) | 0.67 (2.191) | 0.30 (0.842) | 0.13 (1.496) | 0.31 (1.448) |
| Cystatin-C, mg/L | 0.005 (0.058) | 0.031 (0.148) | 0.049 (0.057) | 0.064 (0.093) | 0.052 (0.094) |
| KIM-1, pg/mL | 101.1 (649.3) | 95.7 (394.3) | 144.9 (615.7) | 283.1 (1169.0) | 196.2 (859.7) |
Data presented as mean (SD). BUN indicates blood urea nitrogen; HDL, high-density lipoprotein; KIM-1, kidney injury molecule-1; LDL, low-density lipoprotein.
No seroconversion to any virus was detected after infusion. No patient developed anti-CSL112/apoA-I antibodies, and no trends were observed in other biochemistry, coagulation, or hematology parameters assessed.
Pharmacokinetic parameters of apoA-I
CSL112 infusion resulted in a rapid increase in plasma apoA-I concentration (Table 6 and Figure 5). At peak, the apoA-I level was 25% of the baseline level in the 1.7-g dose group and 145% of the baseline level in the 6.8-g dose group. The Cmax occurred at the end of the infusion at ≈2 hours in all CSL112 dose groups. Both Cmax and AUC increased in a dose-proportional manner.
Table 6.
Summary of Baseline-Corrected Pharmacokinetic Parameters of Apolipoprotein A-I
| Pharmacokinetic Parameter | CSL112 1.7 g (n=7) | CSL112 3.4 g (n=12) | CSL112 6.8 g (n=14) | P Value (ANOVA) |
|---|---|---|---|---|
| Cmax, g/L | <0.001 | |||
| Mean (CV %) | 0.34 (26.9) | 0.77 (16.9) | 1.84 (19.1) | |
| Median (range) | 0.33 (0.23 to 0.49) | 0.79 (0.51 to 0.92) | 1.82 (1.26 to 2.38) | |
| Tmax, h | 0.12 | |||
| Mean (CV %) | 2.70 (57.8) | 2.03 (4.4) | 1.93 (13.5) | |
| Median (range) | 2.00 (2.0 to 6.2) | 2.00 (1.9 to 2.3) | 2.00 (1.0 to 2.0) | |
| AUC0-last, g·h/L | 0.005 | |||
| Mean (CV %) | 8.69 (85.3) | 20.44 (41.3) | 53.31 (33.9) | |
| Median (range) | 5.72 (1.22 to 19.93) | 20.83 (7.92 to 37.59) | 53.21 (17.78 to 87.41) | |
| AUC0-∞, g·h/L | 0.45 | |||
| Mean (CV %) | 4.02 (56.9) | 23.49 (49.5) | 63.78 (43.0) | |
| Median (range) | 4.47 (1.54 to 6.05) | 23.69 (8.57 to 48.16) | 58.85 (22.79 to 120.66) | |
| t1/2, h* | 0.48 | |||
| Mean (CV %) | 13.6 (81.6) | 29.6 (55.7) | 49.1 (62.1) | |
| Median (range) | 12.5 (3.2 to 25.3) | 23.2 (8.4 to 59.1) | 45.4 (15.9 to 123) |
ANOVA indicates analysis of variance; AUC, area under the curve; Cmax, observed maximum concentration in plasma; CV, coefficient of variance; t1/2, plasma half-life; Tmax, time to reach maximum concentration in plasma.
n=3, 9, and 13 in CSL112 1.7-, 3.4-, and 6.8-g groups, respectively.
Figure 5.
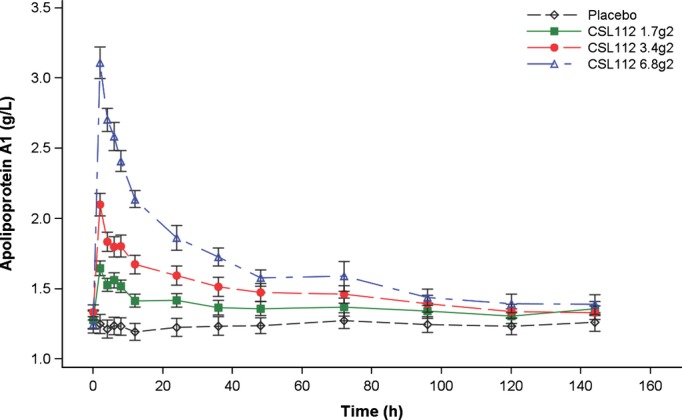
Change in mean apolipoprotein A-I concentration after infusion of ascending doses of CSL112. Error bars represent the 95% CI for the mean.
Changes in the lipid profile
After infusion of CSL112, there was a dose-dependent increase in plasma HDL-C that correlated with the CSL112 dose. Peak plasma concentrations of baseline-corrected total HDL-C were observed at 8 hours after infusion of CSL112 (Figure 6A and Table 5).
Figure 6.
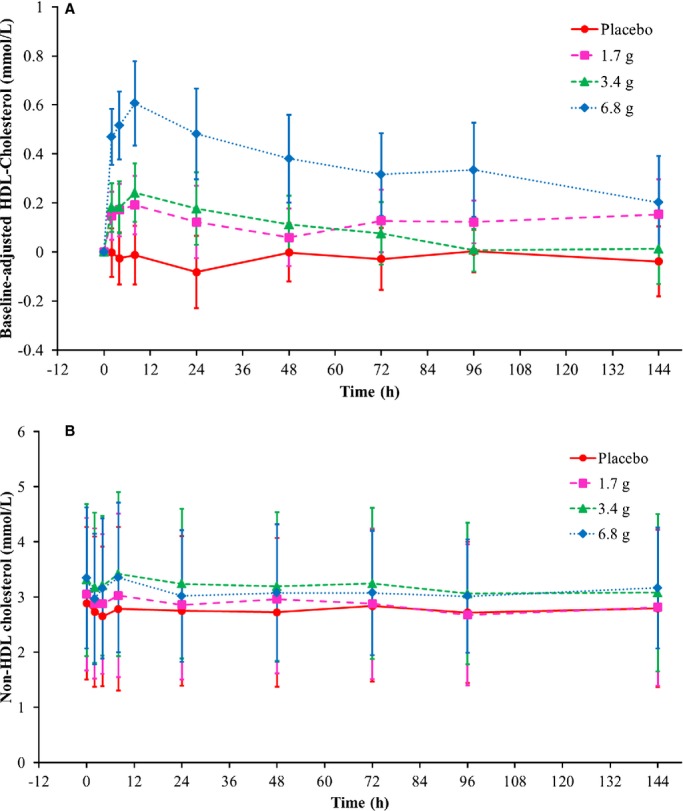
Cholesterol concentrations in lipoprotein fractions following infusion of CSL112 by time and dose group. Shown are means and SD. (A) Baseline-corrected high-density lipoprotein (HDL)-cholesterol, (B) non-HDL cholesterol.
Infusion of CSL112 also caused a time-dependent and dose-related elevation in total cholesterol, with a peak in plasma concentration also observed at 8 hours (Table 5). The change was attributable to HDL-C, because CSL112 did not cause changes in non–HDL-C concentration (Figure 6B), nor were changes in LDL-C and triglyceride levels observed (Table 5).
Serum capacity to promote cholesterol efflux
CSL112 caused a rapid, intense, and dose-dependent increase of the total capacity of serum to cause cholesterol efflux from macrophages that was up to 3.1-fold higher at the peak than at baseline (ie, reflecting effect of native apoA-I only) (Table 7 and Figure 7).
Table 7.
Summary of Pharmacodynamic Parameters of Total Serum Cholesterol Efflux Capacity
| Pharmacodynamic Parameter | Placebo (n=11) | CSL112 1.7 g (n=7) | CSL112 3.4 g (n=12) | CSL112 6.8 g (n=14) | P Value (ANOVA) |
|---|---|---|---|---|---|
| Rmax, % Efflux/4 h | 12.43 (3.22) | 20.21 (4.45) | 23.75 (4.00) | 23.93 (5.51) | NA |
| Tmax, h | 35.53 (32.31) | 2.91 (2.25) | 2.51 (1.68) | 2.22 (0.79) | <0.001 |
| AUEC0-last, % Efflux/4 h | 68.21 (74.34)* | 285.02 (265.24) | 464.93 (364.05) | 721.29 (229.46)† | <0.001 |
| AUEC0-24, % Efflux/4 h | 221.37 (47.6) | 298.56 (66.18) | 349.52 (75.17) | 362.45 (119.12) | <0.001 |
Data are presented as uncorrected mean (SD). ANOVA indicates analysis of variance; AUEC, area under the effect curve; Rmax, maximum efflux capacity biomarker response; Tmax, time to reach maximum efflux capacity.
N=7.
N=13.
Figure 7.
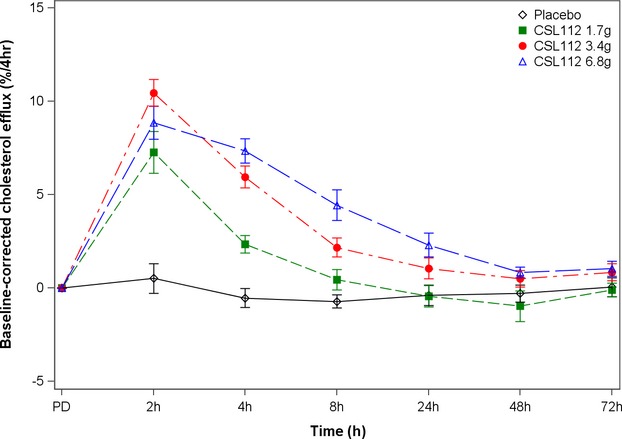
Change in serum total cholesterol efflux capacity after infusion of ascending doses of CSL112 or placebo. Error bars represent the 95% CI for the mean.
Discussion
In this study of patients with stable atherosclerotic vascular disease, CSL112 had a favorable safety profile and predictable dose-proportional pharmacokinetics. CSL112 infusion caused a rapid and marked increase in cholesterol efflux capacity, which was >3-fold higher than in preexposure native serum in the high-dose group.
There was no trend suggesting increased AEs with CSL112, except for an excess in mild infusion-site reactions, mostly bruising, and an excess of mild transient headache, fatigue, and nausea. These results are consistent with prior studies of CSL112 in healthy adults.24 There was no signal of liver toxicity. Renal function and other biochemical, hematologic, or immunologic markers did not show evidence of drug-related changes. In addition, there was no evidence that CSL112 or infused apoA-I was immunogenic.
Notably, this study showed no evidence of elevation in ALT or AST within the dose range of CSL112 studied. Development of the predecessor compound CSL111 was halted after transient asymptomatic elevations in serum transaminase levels were observed.21 Animal studies suggest that these increases were due to cholate and phosphatidylcholine rather than to the active apoA-I component (Samuel Wright, PhD, Personal communication, 2015). CSL112 has been reformulated with reduced amounts of phosphatidylcholine and cholate for improved safety while maintaining its ability to promote reverse cholesterol transport.22,23 The lack of effect of the reformulated product (CSL112) on transaminases in this study and prior phase 1 studies24 indicates that the reduction in excipient levels has provided enhanced safety.
In this study, we did not observe any signal of drug-related renal toxicity with the dose range of CSL112 studied, including in patients with mild renal insufficiency.
The pharmacokinetic analysis of this study indicates that a single intravenous infusion of CSL112 produces a rapid, dose-proportional increase in plasma apoA-I with a maximum concentration reached in ≈2 hours in all dose groups. Notably, the apoA-I levels after infusion with CSL112 were increased up to 244%, which is much higher than that with other apoA-I–enhancing treatments.26 This suggests a higher potential of CSL112 to rapidly act on reverse cholesterol transport.
The recent failure of HDL-C–raising agents to reduce cardiovascular events9–11 has reset thinking on the development of new agents directed at atherosclerotic plaque with a new focus on the role of HDL function.12,13 In particular, recent work has shown that measurement of plasma cholesterol efflux capacity offers a very strong and independent risk marker for atherosclerotic cardiovascular disease.14,27,28 Importantly, the failed HDL-C–raising agents (torcetrapib, dalcetrapib, niacin) have shown negligible effects on cholesterol efflux.29,30 Here, we show that the infusion of CSL112 rapidly and strongly increases cholesterol efflux capacity of plasma sampled from patients with stable atherosclerotic disease. As such, CSL112 may be the first agent available to test the “HDL function hypothesis.”
Prior work suggests that infusion of apoA-I–based products, which elevate cholesterol efflux, may withdraw cholesterol from atherosclerotic plaque. Patients with claudication undergoing percutaneous superficial femoral artery revascularization with plaque excision received 1 intravenous infusion of a reconstituted HDL or placebo 5 to 7 days before the procedure.31 A significant reduction in the cholesterol content of the excised plaque was observed in patients treated with the HDL infusion.31 Prior IVUS studies have also shown that infused apoA-I–based products may promote a remodeling of coronary atherosclerotic plaques within a few weeks after an ACS event.19–21 One IVUS study with 4-times weekly infusion of the predecessor compound CSL111 indicated a significant reduction in atheroma volume from baseline, although this was not significantly different from placebo.21 Nonetheless, significant differences between CSL111 and placebo (with background statin treatment) were observed in the plaque characteristic indices, suggesting a relatively rapid effect on atherosclerotic plaque composition. An apparent exception to this pattern was seen in the CHI-SQUARE trial in which infusion of a recombinant apoA-I product failed to affect IVUS parameters.32 Notably, the doses tested in CHI-SQUARE were substantially lower than the doses found to be effective in reducing femoral artery cholesterol31 and lower than the doses tested in the present study. In the present study, we show that CSL112 doses yield increments in levels of both apoA-I and efflux that are dose proportional through the entire dose range. An ongoing phase 2b study (AEGIS I) will explore the safety and efficacy of doses of CSL112 in this range (ClinicalTrials.gov, NCT02108262). A preliminary assessment of efficacy events in AEGIS I will be used instead of a surrogate endpoint, such as the IVUS used in the CHI-SQUARE trial.33
This study has limitations. In particular, the sample size is small, and therefore findings do not establish final evidence but rather serve to inform the design of larger randomized clinical trials of CSL112. While the study was randomized, because of a relatively small number of patients in each group, some unbalance in baseline characteristics (eg, use of β-blocker, prevalence of diabetes mellitus) was observed, which we do not think affects the overall conclusion of the study.
Taken together, overall evidence—including data from the current study—supports infusion of CSL112 as a means to increase reverse cholesterol transport, modify plaque lipid composition, and potentially reduce plaque vulnerability. These premises support the development of CSL112 as potential therapy to reduce recurrent atherosclerotic events. Larger studies will need to define the clinical safety profile and explore clinical benefits of CSL112, in particular in patients with ACS who have a high risk of ischemic outcomes, have a higher risk of adverse events, and receive aggressive concomitant medical treatment and invasive procedures.
Conclusion
CSL112 is a novel apoA-I formulation under development for the treatment of patients with high-risk cardiovascular disease, including ACS. In this phase 2a trial among patients with stable atherosclerotic vascular disease, CSL112 had an overall favorable safety profile and there was no evidence of hepatic or renal toxicity. CSL112 markedly increased apoA-I and total cholesterol efflux capacity, supporting the biological mechanisms for potential clinical efficacy. Our results support the continued assessment of the safety and efficacy of CSL112 as a new therapy for patients with high-risk coronary artery disease.
Acknowledgments
We would like to thank former CSL Behring employee Rachael Easton, MD, PhD, for her contribution in the design and conduct of the CSL112 2a study. Editorial support was provided by Meridian HealthComms, Plumley, UK, funded by CSL Behring.
Sources of Funding
This trial was supported by CSL Behring.
Disclosures
Dr Tricoci: Consultant agreement and research grant: Merck. Dr D’Andrea: Employee of CSL Behring. Dr Gurbel: Consultation: Daiichi Sankyo, Lilly, Pozen, Novartis, Bayer, AstraZeneca, Accumetrics, Nanosphere, Sanofi-Aventis, Boehringer Ingelheim, Merck, Medtronic, Genetics, CSL, and Haemonetics; grants/pending grants: National Institutes of Health, Daiichi Sankyo, Lilly, Pozen, CSL, AstraZeneca, Sanofi-Aventis, Haemoscope, Medtronic, Harvard Clinical Research Institute, and Duke Clinical Research Institute; lectures/speakers’ bureaus: Lilly, Daiichi Sankyo, Nanosphere, Sanofi-Aventis, Merck, and lverson Genetics; patents: personalized antiplatelet therapy and interventional cardiology. Dr Yao: Former employee of CSL Behring. Dr Cuchel: Research support: CSL Behring. Dr Winston: None. Dr Schott: None. Dr Weiss: Research grants: Sanofi, Amgen, and Pfizer. Dr Blazing: Consultant: Merck, AstraZeneca, and Novartis; advisory board: Merck. Dr Cannon: Modest grant: Abbott; consultant and advisory boards: Abbot, Covidien, Medtronic, and Boston Scientific. Dr Bailey: Research support: AstraZeneca. Dr Angiolillo: Institutional payments for grants: Bristol-Myers Squibb, Sanofi-Aventis, GlaxoSmithKline, Eli Lilly, Daiichi Sankyo, The Medicines Company, AstraZeneca, and Evolva; consulting fee or honorarium: Bayer, Bristol-Myers Squibb, Sanofi-Aventis, Eli Lilly, Daiichi Sankyo, The Medicines Company, AstraZeneca, Merck, Evolva, Abbott Vascular, Regado, and PLx Pharma; participation in review activities: CeloNova, Johnson & Johnson, St. Jude, and Sunovion. Dr Gille: Employee of CSL Behring. Dr Shear: Employee of CSL Behring. Dr Wright: Employee of CSL Behring. Dr Alexander: Research support: Bristol-Myers Squibb, CSL Behring, Duke Health System, NIH, and Regado Biosciences; consulting/honoraria: Moerae Matrix, VA Cooperative Studies Program, and Duke Private Diagnostic Clinic.
Supporting Information
Data S1. Inclusion and exclusion criteria.
References
- Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med. 2013;368:2004–2013. doi: 10.1056/NEJMra1216063. [DOI] [PubMed] [Google Scholar]
- 2012 Writing Committee Members; Jneid H, Anderson JL, Wright RS, Adams CD, Bridges CR, Casey DE, Jr, Ettinger SM, Fesmire FM, Ganiats TG, Lincoff AM, Peterson ED, Philippides GJ, Theroux P, Wenger NK, Zidar JP, Anderson JL American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2012;126:875–910. doi: 10.1161/CIR.0b013e318256f1e0. [DOI] [PubMed] [Google Scholar]
- Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van de Werf F, White HD, Aylward PE, Wallentin L, Chen E, Lokhnygina Y, Pei J, Leonardi S, Rorick TL, Kilian AM, Jennings LH, Ambrosio G, Bode C, Cequier A, Cornel JH, Diaz R, Erkan A, Huber K, Hudson MP, Jiang L, Jukema JW, Lewis BS, Lincoff AM, Montalescot G, Nicolau JC, Ogawa H, Pfisterer M, Prieto JC, Ruzyllo W, Sinnaeve PR, Storey RF, Valgimigli M, Whellan DJ, Widimsky P, Strony J, Harrington RA, Mahaffey KW TRACER Investigators. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366:20–33. doi: 10.1056/NEJMoa1109719. [DOI] [PubMed] [Google Scholar]
- Alexander JH, Lopes RD, James S, Kilaru R, He Y, Mohan P, Bhatt DL, Goodman S, Verheugt FW, Flather M, Huber K, Liaw D, Husted SE, Lopez-Sendon J, De Caterina R, Jansky P, Darius H, Vinereanu D, Cornel JH, Cools F, Atar D, Leiva-Pons JL, Keltai M, Ogawa H, Pais P, Parkhomenko A, Ruzyllo W, Diaz R, White H, Ruda M, Geraldes M, Lawrence J, Harrington RA, Wallentin L APPRAISE-2 Investigators. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med. 2011;365:699–708. doi: 10.1056/NEJMoa1105819. [DOI] [PubMed] [Google Scholar]
- Mega JL, Braunwald E, Wiviott SD, Bassand JP, Bhatt DL, Bode C, Burton P, Cohen M, Cook-Bruns N, Fox KA, Goto S, Murphy SA, Plotnikov AN, Schneider D, Sun X, Verheugt FW, Gibson CM ATLAS ACS 2–TIMI 51 Investigators. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med. 2012;366:9–19. doi: 10.1056/NEJMoa1112277. [DOI] [PubMed] [Google Scholar]
- Baseline serum cholesterol and treatment effect in the Scandinavian Simvastatin Survival Study (4S) Lancet. 1995;345:1274–1275. [PubMed] [Google Scholar]
- Prospective Studies Collaboration. Lewington S, Whitlock G, Clarke R, Sherliker P, Emberson J, Halsey J, Qizilbash N, Peto R, Collins R. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55 000 vascular deaths. Lancet. 2007;370:1829–1839. doi: 10.1016/S0140-6736(07)61778-4. [DOI] [PubMed] [Google Scholar]
- Emerging Risk Factors Collaboration. Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. 2013;34:1279–1291. doi: 10.1093/eurheartj/eht055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B ILLUMINATE Investigators. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS dal-OUTCOMES Investigators. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- Degoma EM, Rader DJ. Novel HDL-directed pharmacotherapeutic strategies. Nat Rev Cardiol. 2011;8:266–277. doi: 10.1038/nrcardio.2010.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenson RS, Brewer HB, Jr, Ansell B, Barter P, Chapman MJ, Heinecke JW, Kontush A, Tall AR, Webb NR. Translation of high-density lipoprotein function into clinical practice: current prospects and future challenges. Circulation. 2013;128:1256–1267. doi: 10.1161/CIRCULATIONAHA.113.000962. [DOI] [PubMed] [Google Scholar]
- Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, Shaul PW. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays HE, Shah A, Lin J, Sisk CM, Dong Q, Maccubbin D. Consistency of extended-release niacin/laropiprant effects on Lp(a), ApoB, non-HDL-C, Apo A1, and ApoB/ApoA1 ratio across patient subgroups. Am J Cardiovasc Drugs. 2012;12:197–206. doi: 10.2165/11631530-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Kingwell BA, Chapman MJ, Kontush A, Miller NE. HDL-targeted therapies: progress, failures and future. Nat Rev Drug Discov. 2014;13:445–464. doi: 10.1038/nrd4279. [DOI] [PubMed] [Google Scholar]
- Olsson AG, Schwartz GG, Szarek M, Sasiela WJ, Ezekowitz MD, Ganz P, Oliver MF, Waters D, Zeiher A. High-density lipoprotein, but not low-density lipoprotein cholesterol levels influence short-term prognosis after acute coronary syndrome: results from the MIRACL trial. Eur Heart J. 2005;26:890–896. doi: 10.1093/eurheartj/ehi186. [DOI] [PubMed] [Google Scholar]
- Wolfram RM, Brewer HB, Xue Z, Satler LF, Pichard AD, Kent KM, Waksman R. Impact of low high-density lipoproteins on in-hospital events and one-year clinical outcomes in patients with non-ST-elevation myocardial infarction acute coronary syndrome treated with drug-eluting stent implantation. Am J Cardiol. 2006;98:711–717. doi: 10.1016/j.amjcard.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Waksman R, Torguson R, Kent KM, Pichard AD, Suddath WO, Satler LF, Martin BD, Perlman TJ, Maltais JA, Weissman NJ, Fitzgerald PJ, Brewer HB., Jr A first-in-man, randomized, placebo-controlled study to evaluate the safety and feasibility of autologous delipidated high-density lipoprotein plasma infusions in patients with acute coronary syndrome. J Am Coll Cardiol. 2010;55:2727–2735. doi: 10.1016/j.jacc.2009.12.067. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- Tardif JC, Grégoire J, L’Allier PL, Ibrahim R, Lespérance J, Heinonen TM, Kouz S, Berry C, Basser R, Lavoie MA, Guertin MC, Rodés-Cabau J Effect of rHDL on Atherosclerosis-Safety and Efficacy (ERASE) Investigators. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297:1675–1682. doi: 10.1001/jama.297.15.jpc70004. [DOI] [PubMed] [Google Scholar]
- Diditchenko S, Gille A, Pragst I, Stadler D, Waelchli M, Hamilton R, Leis A, Wright SD. Novel formulation of a reconstituted high-density lipoprotein (CSL112) dramatically enhances ABCA1-dependent cholesterol efflux. Arterioscler Thromb Vasc Biol. 2013;33:2202–2211. doi: 10.1161/ATVBAHA.113.301981. [DOI] [PubMed] [Google Scholar]
- Gille A, Easton R, D’Andrea D, Wright SD, Shear CL. CSL112 enhances biomarkers of reverse cholesterol transport after single and multiple infusions in healthy subjects. Arterioscler Thromb Vasc Biol. 2014;34:2106–2114. doi: 10.1161/ATVBAHA.114.303720. [DOI] [PubMed] [Google Scholar]
- Easton R, Gille A, D’Andrea D, Davis R, Wright SD, Shear C. A multiple ascending dose study of CSL112, an infused formulation of ApoA-I. J Clin Pharmacol. 2014;54:301–310. doi: 10.1002/jcph.194. [DOI] [PubMed] [Google Scholar]
- Cantu TG, Hoehn-Saric EW, Burgess KM, Racusen L, Scheel PJ. Acute renal failure associated with immunoglobulin therapy. Am J Kidney Dis. 1995;25:228–234. doi: 10.1016/0272-6386(95)90003-9. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Gordon A, Johansson J, Wolski K, Ballantyne CM, Kastelein JJ, Taylor A, Borgman M, Nissen SE. Efficacy and safety of a novel oral inducer of apolipoprotein a-I synthesis in statin-treated patients with stable coronary artery disease a randomized controlled trial. J Am Coll Cardiol. 2011;57:1111–1119. doi: 10.1016/j.jacc.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleheen D, Scott R, Javad S, Zhao W, Rodrigues A, Picataggi A, Lukmanova D, Mucksavage M, Luben R, Khaw K, Billheimer J, Wareham N, Rader DJ. HDL cholesterol efflux capacity is inversely associated with incident CHD events independent of HDL-C and ApoA-I concentrations. Circulation. 2014;130:A19753. Presented at: American Heart Association Scientific Sessions 2014. [Google Scholar]
- Khera AV, Patel PJ, Reilly MP, Rader DJ. The addition of niacin to statin therapy improves high-density lipoprotein cholesterol levels but not metrics of functionality. J Am Coll Cardiol. 2013;62:1909–1910. doi: 10.1016/j.jacc.2013.07.025. [DOI] [PubMed] [Google Scholar]
- Ray KK, Ditmarsch M, Kallend D, Niesor EJ, Suchankova G, Upmanyu R, Anzures-Cabrera J, Lehnert V, Pauly-Evers M, Holme I, Štásek J, van Hessen MW, Jones P dal-ACUTE Investigators. The effect of cholesteryl ester transfer protein inhibition on lipids, lipoproteins, and markers of HDL function after an acute coronary syndrome: the dal-ACUTE randomized trial. Eur Heart J. 2014;35:1792–1800. doi: 10.1093/eurheartj/ehu105. [DOI] [PubMed] [Google Scholar]
- Shaw JA, Bobik A, Murphy A, Kanellakis P, Blombery P, Mukhamedova N, Woollard K, Lyon S, Sviridov D, Dart AM. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res. 2008;103:1084–1091. doi: 10.1161/CIRCRESAHA.108.182063. [DOI] [PubMed] [Google Scholar]
- Tardif JC, Ballantyne CM, Barter P, Dasseux JL, Fayad ZA, Guertin MC, Kastelein JJ, Keyserling C, Klepp H, Koenig W, L’Allier PL, Lespérance J, Lüscher TF, Paolini JF, Tawakol A, Waters DD Can HDL Infusions Significantly QUicken Atherosclerosis REgression (CHI-SQUARE) Investigators. Effects of the high-density lipoprotein mimetic agent CER-001 on coronary atherosclerosis in patients with acute coronary syndromes: a randomized trial. Eur Heart J. 2014;35:3277–3286. doi: 10.1093/eurheartj/ehu171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BG, Zhao XQ. Is intravascular ultrasound the gold standard surrogate for clinically relevant atherosclerosis progression? J Am Coll Cardiol. 2007;49:933–938. doi: 10.1016/j.jacc.2006.12.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Inclusion and exclusion criteria.