

Abstract
Background
The aim of this study was to explore the influence of extended-release niacin/laropiprant (ERN/LRP) versus placebo on high-density lipoprotein (HDL) antioxidant function, cholesterol efflux, apolipoprotein B100 (apoB)-containing lipoproteins, and mediators of vascular inflammation associated with 15% increase in high-density lipoprotein cholesterol (HDL-C). Study patients had persistent dyslipidemia despite receiving high-dose statin treatment.
Methods and Results
In a randomized double-blind, placebo-controlled, crossover trial, we compared the effect of ERN/LRP with placebo in 27 statin-treated dyslipidemic patients who had not achieved National Cholesterol Education Program-ATP III targets for low-density lipoprotein cholesterol (LDL-C). We measured fasting lipid profile, apolipoproteins, cholesteryl ester transfer protein (CETP) activity, paraoxonase 1 (PON1) activity, small dense LDL apoB (sdLDL-apoB), oxidized LDL (oxLDL), glycated apoB (glyc-apoB), lipoprotein phospholipase A2 (Lp-PLA2), lysophosphatidyl choline (lyso-PC), macrophage chemoattractant protein (MCP1), serum amyloid A (SAA) and myeloperoxidase (MPO). We also examined the capacity of HDL to protect LDL from in vitro oxidation and the percentage cholesterol efflux mediated by apoB depleted serum. ERN/LRP was associated with an 18% increase in HDL-C levels compared to placebo (1.55 versus 1.31 mmol/L, P<0.0001). There were significant reductions in total cholesterol, triglycerides, LDL cholesterol, total serum apoB, lipoprotein (a), CETP activity, oxLDL, Lp-PLA2, lyso-PC, MCP1, and SAA, but no significant changes in glyc-apoB or sdLDL-apoB concentration. There was a modest increase in cholesterol efflux function of HDL (19.5%, P=0.045), but no change in the antioxidant capacity of HDL in vitro or PON1 activity.
Conclusions
ERN/LRP reduces LDL-associated mediators of vascular inflammation, but has varied effects on HDL functionality and LDL quality, which may counter its HDL-C-raising effect.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT01054508.
Keywords: cholesterol efflux, extended-release niacin, HDL functionality, inflammation, laropiprant, LDL quality, oxidation
In 1975, Miller and Miller1 reported an inverse association between plasma high-density lipoprotein cholesterol (HDL-C) concentration and coronary heart disease. It has been postulated that HDL-C may be an independent predictor of cardiovascular (CV) events in statin-treated patients at all levels of low-density lipoprotein cholesterol (LDL-C).2,3 In 2 recent large clinical trials (AIM-HIGH4 and HPS2 THRIVE),5 raising HDL-C levels with extended-release niacin (ERN) did not reduce CV events. Mean LDL-C in these studies was lower than LDL-C in previous ERN trials where the outcome favored ERN use. HPS2 THRIVE used ERN combined with laropiprant (LRP) (Tredaptive®, Merck Sharpe and Dohme). LRP reduces niacin-induced flushing by blocking prostaglandin D2 receptors. Niacin has a broad spectrum of effects on lipid metabolism, including raising HDL-C.6 Nonetheless, improving HDL functionality, such as its antioxidant capacity and cholesterol efflux capacity, may be more important than raising its cholesterol cargo.7 For example, cholesterol efflux capacity from macrophages has a strong inverse association with both carotid intima-media thickness and the likelihood of angiographic coronary artery disease, independently of the HDL cholesterol level,8 and antioxidative and cholesterol efflux capacities of HDL are reduced in ischemic cardiomyopathy.9 Similarly, in the Dal-OUTCOMES trial where 15 871 patients with a recent acute coronary syndrome were randomized to a cholesteryl ester transfer protein (CETP) inhibitor (dalcetrapib) or placebo, there was no reduction in the primary CV end point events despite a 30% increase in HDL-C.10 There is as yet no explanation for these results.
Atherogenic modifications of LDL such as oxidation and glycation play a key role in promoting atherosclerosis.11,12 Lipoprotein phospholipase A2 (Lp-PLA2) circulates primarily in association with LDL,13 and it has been shown that Lp-PLA2 activity and mass show continuous associations with risk of coronary heart disease, similar in magnitude to that of non-HDL cholesterol or systolic blood pressure.14 Apolipoprotein B100 (ApoB) plays a key role in the association of Lp-PLA2 with LDL.15 LDL oxidation activates Lp-PLA2, which catalyzes the hydrolysis of oxidized phosphatidyl choline to lysophosphatidyl choline (lyso-PC) and oxidized nonesterified fatty acids on the surface of oxidized LDL (oxLDL).16,17 Lyso-PC has been shown to mediate the pro-inflammatory and pro-atherogenic effects of activated Lp-PLA2.18 Furthermore, the association between Lp-PLA2, lyso-PC, and proinflammatory cytokines such as macrophage chemoattractant protein (MCP1) in human plaques suggest that lyso-PC plays a key role in plaque inflammation and vulnerability.19 ApoB, oxLDL, Lp-PLA2, lyso-PC, and MCP1 are important constituents of the LDL-associated pro-atherogenic pathway.4,20–22 We examined the effect of ERN/LRP on these mediators of vascular inflammation as part of a randomized study, as this has not been undertaken previously in either animals or humans.
We report for the first time the effect of ERN/LRP on CETP, and the antioxidant function of HDL and its cholesterol efflux capacity in a randomized, placebo-controlled, crossover study. Although we have not compared ERN with ERN/LRP and so cannot disentangle the effects of LRP, we have explored the effects of ERN/LRP on pro-atherogenic effects of LDL and anti-atherogenic properties of HDL not previously described.
Trial Design and End Points
Patients
Patients were recruited from Central Manchester University Hospitals NHS Foundation Trust and University Hospital of South Manchester. Informed consent was obtained from all patients. The study was approved by the local ethics committee. All study patients were seen in the Cardiovascular Trials Unit at Central Manchester University Hospitals NHS Foundation Trust. Thirty-six patients were recruited and 27 patients completed this randomized placebo-controlled, crossover study (Figure1). Table1 displays basic characteristics of all patients recruited for the study who had at least 1 blood test (n=36). Table2 displays basic characteristics of all patients who completed the study (n=27).
Figure 1.
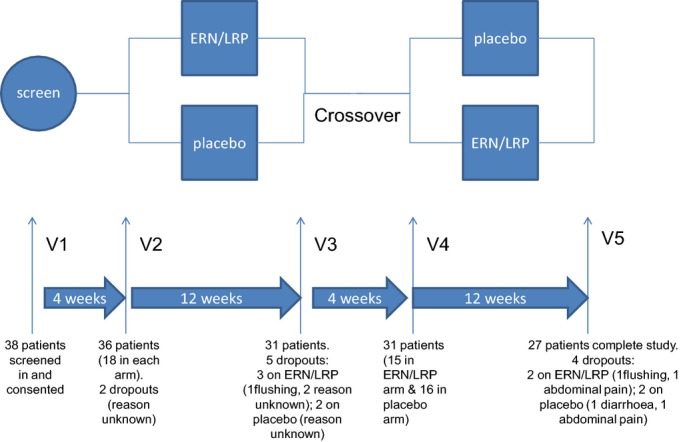
Study overview. Patients randomized to placebo during the first period received extended-release niacin/laropiprant (ERN/LRP) in the second period and vice versa. In each period, the ERN/LRP dose was increased from 1 g/20 mg to 2 g/40 mg after 4 weeks. SC indicates screening visit.
Table 1.
Basic Characteristics of All Patients Recruited for the Study Who Had at Least 1 Blood Test (n=36)
| Age, y | 56±11 |
| Gender (M, F) | 22, 14 |
| Smoker, n (%) | 7 (20) |
| Type 2 diabetes, n (%) | 4 (10) |
| Hypertension, n (%) | 18 (50) |
| Known IHD, n (%) | 16 (45) |
| HeFH, n (%) | 16 (45) |
| BMI (kg/m2), mean±SD | 30±4 |
| Systolic BP (mm Hg), mean+SD | 133±15 |
| Diastolic BP (mm Hg), mean+SD | 77±9 |
| Hormone replacement therapy in postmenopausal females, n (%) | 1/9 (10) |
| Patients on niacin prior to entering the trial | 0 |
| Total cholesterol (mmol/L), median (range) | 6.09 (5.15 to 6.71) |
| Triglycerides (mmol/L), median (range) | 1.63 (1.22 to 2.59) |
| HDL-C (mmol/L), median (range) | 1.44 (1.21 to 1.64) |
| LDL-C (mmol/L), median (range) | 3.52 (1.78 to 4.31) |
| ApoAI (g/L), median (range) | 1.42 (1.29 to 1.53) |
| ApoB (g/L), median (range) | 1.32 (1.04 to 1.48) |
| sdLDL-apoB (g/L), median (range) | 0.271 (0.139 to 0.362) |
| PON1 activity (nmol/mL per minute), median (range) | 138 (68 to 176) |
Eighteen patients were on rosuvastatin 40 mg, 16 on atorvastatin 80 mg, and 2 on simvastatin 40 mg daily. Values are mean±SD. ApoAI indicates apolipoprotein AI; ApoB, apolipoprotein B; BMI, body mass index; BP, blood pressure; HDL-C, high-density lipoprotein cholesterol; HeFH, heterozygous familial hypercholesterolemia; IHD, ischemic heart disease; LDL-C, low-density lipoprotein cholesterol; PON1, paraoxonase 1; sdLDL, small dense LDL.
Table 2.
Basic Characteristics of Patients Who Completed the Study (n=27)
| Age, y | 57±10 |
| Gender (M, F) | 18, 9 |
| Smoker, n (%) | 3 (10) |
| Type 2 diabetes, n (%) | 3 (10) |
| Hypertension, n (%) | 13 (48) |
| Known IHD, n (%) | 11 (40) |
| HeFH, n (%) | 11 (40) |
| BMI (kg/m2), mean | 31±4 |
| Systolic BP (mm Hg), mean±SD | 135±14 |
| Diastolic BP (mm Hg), mean+SD | 76±10 |
| Hormone replacement therapy in postmenopausal females, n (%) | 1 (14) |
| Patients on niacin prior to entering the trial | 0 |
| Total cholesterol (mmol/L), median (range) | 6.14 (5.08 to 6.49) |
| Triglycerides (mmol/L), median (range) | 1.55 (1.08 to 2.42) |
| HDL-C (mmol/L), median (range) | 1.39 (1.19 to 1.71) |
| LDL-C (mmol/L), median (range) | 3.43 (2.86 to 4.27) |
| ApoAI (g/L), median (range) | 1.39 (1.29 to 1.51) |
| ApoB (g/L), median (range) | 1.27 (1.04 to 1.47) |
| PON1 activity (nmol/mL per minute), median (range) | 145 (70 to 183) |
| sdLDL-apoB (g/L), median (range) | 0.27 (0.15 to 0.33) |
Fifteen patients were on rosuvastatin 40 mg, 11 on atorvastatin 80 mg, and 1 on simvastatin 40 mg daily. Values are in mean±SD for parametric data and median (interquartile range) for nonparametric data. ApoAI indicates apolipoprotein AI; ApoB, apolipoprotein B; BMI, body mass index; BP, blood pressure; HDL-C, high-density lipoprotein cholesterol; HeFH, heterozygous familial hypercholesterolemia; IHD, ischemic heart disease; LDL-C, low-density lipoprotein cholesterol; PON1, paraoxonase 1; sdLDL, small dense LDL.
Inclusion and Exclusion Criteria
Men and women aged 20 to 75 years with dyslipidemia on lipid-lowering treatment (maximum tolerated statins and/or ezetimibe), but not achieving the NCEP ATP III (National Cholesterol Education Program Adult Treatment Panel III) target of LDL-C <1.8 mmol/L (70 mg/L) were recruited. Patients were excluded if they were pregnant and/or breast-feeding, if they had significant renal impairment (estimated glomerular filtration rate ≤59), alanine aminotransferase >1.5 upper limit of normal, if they were receiving niacin, fibrates, or omega-3 ethyl esters (Omacor, GlaxoSmithKline), if they were allergic to niacin, and if they had active peptic ulcer disease.
Trial Design
The trial medication ERN/LRP and image-matched placebo were supplied by Merck, Sharp & Dohme Ltd for this randomized crossover trial. The Clinical Trials Pharmacist was responsible for the randomization procedure. Block randomization was carried out with equal numbers of participants per group using a computer-generated list. Patients and investigators were blinded to individual assignments and to the randomization procedure.
Crossover study design
Study patients acted as their own controls. The patients were asked to attend for 5 visits. Patients who fulfilled the inclusion and exclusion criteria were invited for a screening visit where informed consent was obtained. On the first visit, patients taking both statin and ezetimibe were asked to stop ezetimibe. Statin dose was unchanged throughout the trial period. All patients received placebo for 4 weeks (placebo run-in period) and were then randomized to either placebo or ERN/LRP for 12 weeks (treatment period). During the treatment period, patients randomized to study medication received ERN/LRP 1 g/20 mg for 4 weeks followed by an increased dose of ERN/LRP 2 g/40 mg for 8 weeks. After the first treatment period, all patients repeated the placebo period (4 weeks) before the second treatment period (12 weeks). Finally, they attended at the end of the placebo or treatment period. During the second treatment period, patients who were randomized to placebo in the first treatment period received ERN/LRP and vice versa. Fasting blood samples were taken at all the study visits except the first visit (screening). Concordance with medication was assessed by a pill count at each visit (Figure1).
End Points
Primary end point
The primary end point was to determine whether treatment with ERN/LRP leads to an increase in HDL-C compared with placebo in statin-treated dyslipidemic patients. A difference of 15% between the groups was defined as clinically significant.
Secondary end points and descriptive analysis
We studied the effect of ERN/LRP on the following:
HDL functionality: apolipoprotein AI (apoAI), paraoxonase 1 (PON1) activity, apolipoprotein M (apoM), CETP, myeloperoxidase (MPO), serum amyloid A (SAA), HDL in vitro anti-oxidant function, and cholesterol efflux.
Lipids and apoB lipoproteins: total cholesterol, triglycerides (TG), LDL-C, non-HDL cholesterol, VLDL cholesterol (VLDL-C), VLDL triglycerides (VLDL-TG), apoB, VLDL apoB, LDL apoB, buoyant LDL apoB, small-dense LDL apoB (sdLDL-apoB) and lipoprotein(a).
Mediators of vascular inflammation associated with LDL: total glycated apoB (glyc-apoB), oxLDL, Lp-PLA2, lyso-PC, and MCP1.
Sample Size and Power Calculation
The sample size calculation was based on the primary outcome (change in HDL-C). Earlier clinical trials vary greatly in the scale of this outcome, particularly against a background of statin therapy. We based the calculation of our sample size using Stata 11 statistical software (College Station, TX) on an anticipated typical increase of 15%. This was recently supported by a study in which this was defined as a good response.23 To achieve a 15% increase in HDL-C with 90% statistical power at P<0.02 in a crossover trial, we concluded we would require 26 participants. We planned to recruit 38 patients to allow for dropouts.
Statistical Analyses
Data distribution was determined using the Kolmogorov–Smirnov test, D’Agastino and Pearson omnibus normality test, and Shapiro–Wilk normality test on GraphPad Prism (GraphPad Software Inc, CA). Stats direct (Stats direct, Altrincham, Cheshire, UK) was used to calculate any significant differences in period effect or treatment–period interaction. Level of significance in the results has been described as treatment or relative effectiveness of drug/placebo. Results are expressed as mean±SD for parametric data and median (interquartile range) for nonparametric data. P<0.05 was regarded as statistically significant.
Materials and Methods
Separation of Serum and Plasma
Blood samples were collected between 9 am and 11 am after participants had fasted from 10 pm the previous day. Serum and EDTA-plasma were isolated by centrifugation at 2000g for 15 minutes at 4°C within 2 hours of collection and were maintained at that temperature until further use.
Laboratory Analyses
Total serum cholesterol was measured using the cholesterol oxidase phenol 4-aminoantipyrine peroxidase method, triglycerides by glycerol phosphate oxidase phenol 4-aminoantipyrine peroxidase method, and apoAI and apoB (ABX Diagnostics, Shefford, UK) were assayed using immunoturbidimetric assays. Serum HDL-C was assayed using a second-generation homogenous direct method (Roche Diagnostics, Burgess Hill, UK). A Cobas Mira analyzer was deployed for all these analysis (Horiba ABX Diagnostics, Nottingham, UK). The laboratory participated in the RIQAS (Randox Laboratories, Dublin, Ireland) scheme, which is calibration and repair centre calibrated. LDL-C was estimated using the Friedewald formula.
Different density lipoproteins were isolated using ultracentrifugation of 1-mL volumes of plasma adjusted to the desired density (D) in polycarbonate tubes with the Beckman Optima TLX ultracentrifuge.24 VLDL-TG and VLDL-C (D<1.006 g/mL), LDL cholesterol at D>1.006 g/mL, and small-dense LDL apo B (D>1.044 g/mL) were isolated with ultracentrifugation for 4 hours in a fixed-angle TLA120.2 rotor (Beckman Coulter, High Wycombe, UK). Cholesterol, TG, and apoB were determined in lipoprotein fractions using the methods described above. Serum PON1 activity was determined by a semiautomated microtiter plate method using paraoxon (O,O-diethyl O-(4-nitrophenyl) phosphate) as a substrate and read by spectrophotometer at 405 nm.25 Lipoprotein(a) was determined using ELISA kits from Mercodia (Upsala, Sweden).
Biomarkers
Total glyc-apoB was assayed in plasma using Glyacor ELISA kits from bioGnosis Ltd, Hailsham, UK. Ox-LDL was measured in plasma by a direct ELISA sandwich technique with a kit from Mercodia, Upsala, Sweden. Lp-PLA2 mass was determined in plasma using USCN Lifescience ELISA kits from Hölzel Diagnostika Handels GmbH, Köln, Germany. Lyso-PC was measured in serum using Azwell lyso-PC enzymatic assay kits from Cosmo Bio, Japan. MCP-1 was measured in serum using DuoSet® ELISA development kits from R&D Systems (Abingdon, UK).
ApoM was assayed in serum using Bluegene E01A0522 kits from Hölzel Diagnostika Handels GmbH, Köln, Germany. CETP activity was measured using a fluorometric method using assay kits (ab65383) from Abcam, UK. MPO mass was determined in plasma by ELISA using kits from R&D Systems Ltd (Abingdon, UK). SAA was assayed by ELISA using kits from Invitrogen Ltd, Paisley, UK. High-sensitivity C-reactive protein was measured in serum by an in-house, antibody sandwich ELISA technique using anti-human C-reactive protein antibodies, calibrators, and controls from Abcam (Cambridge, UK).
Table3 displays the intra-assay and inter-assay coefficient of variance (CV) for various biomarkers measured.
Table 3.
Intra-assay and Inter-assay CV for Various Biomarkers Measured
| Biomarker | Intra-assay CV (%) | Inter-assay CV (%) |
|---|---|---|
| Apolipoprotein M | <10 | <10 |
| PON1 activity | 3.5 | 3 |
| Serum amyloid A | 6 | 7 |
| Myeloperoxidase | <8 | <8 |
| Cholesterol efflux* | 4 | 5 |
| Adiponectin | 3 | 9 |
| Insulin | <5 | <6 |
| Lipoprotein(a) | <4 | <6 |
| Small dense LDL apoB | 2 | 2 |
| Glycated apoB | 3.5 | 15 |
| Oxidized LDL | 6 | 5 |
| Lipoprotein phospholipase A2 | <10 | <10 |
| Lysophosphatidyl choline | <5 | <5 |
| Macrophage chemoattractant protein 1 | 7 | 7 |
| Lipid peroxides | <10 | <10 |
All samples were run in singlicates except serum PON1 activity where samples were run in duplicates. ApoB indicates apolipoprotein B; CV, coefficient of variance; LDL, low-density lipoprotein; PON1, paraoxonase 1.
Results were not normalized to a pool, all assays were performed on the same day using the same cells and settings.
In Vitro Oxidation Studies
LDL (D=1.019 to 1.063 g/mL) and HDL (D=1.063 to 1.21 g/mL) were isolated from serum by sequential density gradient ultracentrifugation without the addition of EDTA in a Beckman L7 ultracentrifuge with a 50.4 Ti fixed-angle rotor using 13×64 mm polyallomer tubes at speeds of 34 000 r/min (144 361g) for 22 hours 17 minutes.26 The isolated fractions were dialyzed against PBS overnight at 4°C followed by sterile filtration (0.2 μm) and stored at 4°C under nitrogen for not more than 16 hours before in vitro oxidation studies. LDL and HDL protein concentration were determined using bicinchoninic acid.27
Susceptibility of lipoprotein fractions to oxidation by CuSO4 in vitro was assessed immediately after isolation by incubating 0.25 mg protein/mL of lipoprotein fractions with 5 μL CuSO4 for 3 hours at 37°C in a Gallenkamp Economy Size 1 Incubator (Gallenkamp, Leicester, UK). Lipid peroxides (LPO) production was measured by spectrophotometer at 365 nmol at baseline and 3 hours.28 LDL was incubated separately and together with HDL in order to evaluate the effect of HDL on in vitro LDL oxidation.
Cholesterol Efflux
The cholesterol efflux capacity of HDL was determined in an assay that has been validated previously.29 In summary, J774A.1 cells were incubated with radiolabeled cholesterol. These cells were then incubated with apoB-depleted serum for 4 hours. After incubation, the cell media were collected and cells were washed with PBS and dissolved in 0.5 mL 0.2 N NaOH to determine radioactivity. Cellular cholesterol efflux was expressed as the percentage of radioactivity in the medium from the radioactivity in the cells+medium. Cholesterol efflux was linear over 4 hours and with concentration. Cholesterol efflux was calculated using the following formula:
To calculate cholesterol efflux on placebo and on ERN/LRP, we subtracted efflux to serum-free media (control) from efflux to apoB depleted serum.
Results
In this study, patients acted as their own controls and 27 patients completed all 5 visits. Two thirds of these patients were male. Ten percent were current smokers and 10% had diabetes (Table2). Nearly half of the patients had hypertension, history of ischemic heart disease, and/or heterozygous familial hypercholesterolemia. The mean body mass index was 31 kg/m2. Of the 27 participants, 15 received rosuvastatin 40 mg, 11 atorvastatin 80 mg, and 1 simvastatin 40 mg daily.
End Points
Primary end point
Treatment with ERN/LRP was associated with an increase in HDL-C of 18% compared to placebo (1.55 versus 1.31 mmol/L, P<0.0001) (Table4).
Table 4.
Primary End Point and HDL Functionality on Treatment With ERN/LRP and Placebo
| Variable | Placebo (n=27) | ERN/LRP (n=27) | P Value |
|---|---|---|---|
| HDL-C, mmol/L | 1.31 (1.12 to 1.59) | 1.55 (1.22 to 1.73) | <0.0001* |
| CETP activity, nmol/mL per hour | 26.1 (19.6 to 32.3) | 19.8 (16.4 to 25.7) | 0.02* |
| ApoAI, g/L | 1.33 (1.21 to 1.48) | 1.34 (1.22 to 1.51) | 0.75 |
| PON1 activity, nmol/mL per minute | 144 (86 to 179) | 147 (56 to 187) | 0.42 |
| ApoM, mg/L | 30.1 (27.2 to 36.4) | 30.9 (23.4 to 37.7) | 0.83 |
| MPO, ng/mL | 92.1 (34.4 to 490) | 90.2 (36.8 to 1670) | 0.14 |
| SAA, mg/L | 17.6 (11.1 to 36.4) | 16.2 (9.9 to 56.6) | 0.03* |
| Cholesterol efflux, % (SD, CI) | 10.15 (2.6, 0.98) | 12.10 (3.5, 1.32) | 0.045* |
Values are in mean (SD) for parametric data and median (interquartile range) for nonparametric data. Cholesterol efflux: J774A.1 cells were incubated initially with radiolabeled cholesterol and later with apoB depleted serum. Cellular cholesterol efflux was expressed as the percentage of radioactivity in the medium from the radioactivity in the cells+medium. The P value for the significance of the difference between placebo and ERN/LRP is shown. ApoAI indicates apolipoprotein AI; ApoM, apolipoprotein M; CETP, cholesteryl ester transfer protein; ERN/LRP, extended release niacin/laropiprant; HDL-C, high-density lipoprotein cholesterol; MPO, myeloperoxidase; PON1, paraoxonase 1; SAA, serum amyloid A.
P<0.05.
Secondary end points and descriptive analysis
HDL functionality
Treatment with ERN/LRP did not affect apoAI, PON1 activity, apoM levels, or MPO. CETP activity and SAA were significantly reduced on ERN/LRP treatment (19.8 versus 26.1 nmol/mL per hour, P=0.02 and 16.2 versus 17.6 mg/L, P=0.03, respectively) (Table4).
In vitro oxidation studies
There was the previously described substantial decrease in LPO over 3 hours when LDL was incubated with CuSO4 in the presence of HDL as opposed to LDL alone.30 There was a significant reduction in LPO generated in 3 hours when LDL was incubated with HDL as opposed to LDL alone in all patients post ERN/LRP (44.2 [25.1 to 54.0] versus 144 [105 to 164] nmol/mL, respectively, P<0.0001) (Figure2). Similarly, there was a significant reduction in LPO generated in 3 hours when LDL was incubated with HDL as opposed to LDL alone in all patients on placebo (41.1 [15.1 to 69.9] versus 125 [76.0 to 158] nmol/mL, respectively, P<0.0001) (Figure2). However, there was no significant difference in LPO generated in 3 hours when LDL was incubated alone, on treatment with ERN/LRP versus placebo (144 [105 to 164] versus 125 [76.0 to 158] nmol/mL, respectively, P=0.2); or when LDL was incubated with CuSO4 in the presence of HDL on treatment with ERN/LRP versus placebo (44.2 [25.1 to 54.0] versus 41.1 [15.1 to 69.9] nmol/mL, P =0.4) (Figure2).
Figure 2.
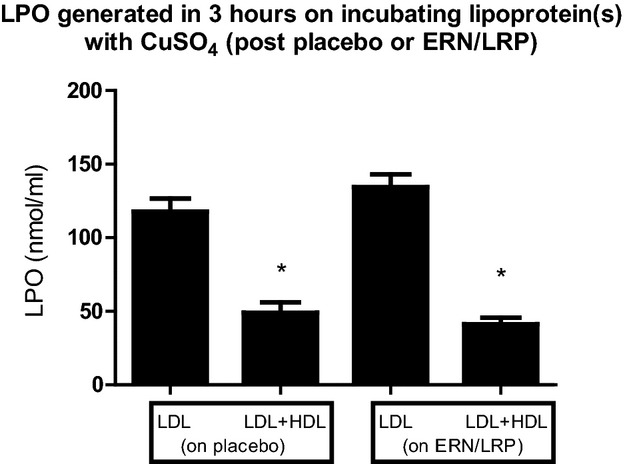
LPO generated over 3 hours on incubating LDL or LDL+HDL with CuSO4 on treatment with ERN/LRP and placebo. HDL on both placebo and ERN/LRP markedly decreased generation of lipid peroxides, *P<0.0001. However, this antioxidant property of HDL was not enhanced by ERN/LRP therapy. Values are in median (interquartile range). ERN/LRP indicates extended release niacin/laropiprant; HDL, high-density lipoprotein; LDL, low-density lipoprotein; LPO, lipid peroxides.
Cholesterol efflux studies
Treatment with ERN/LRP led to a modest increase in cholesterol efflux compared to placebo (+19%, P=0.045) (Table4, Figure3).
Figure 3.
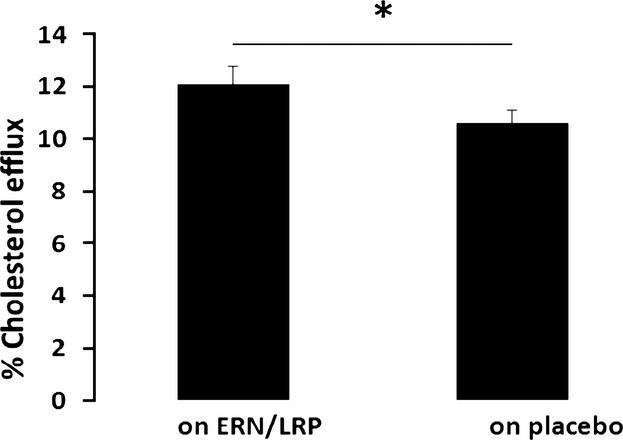
Effect of extended-release niacin/laropiprant (ERN/LRP) (treatment) and placebo on % cholesterol efflux from J774A.1 cells over 4 hours (when control is subtracted), *P=0.045. Control is percent efflux to serum-free media.
Cholesterol efflux showed a positive correlation with HDL-C on treatment with ERN/LRP (Spearman’s Rho=0.4, P=0.04) and also on placebo (ρ=0.4, P=0.04), suggesting that cholesterol efflux capacity influences HDL cholesterol cargo. Furthermore, treatment-induced changes in percentage cholesterol efflux correlated to changes in HDL-C concentration (P<0.01, R2=0.24) with 24% of the treatment-induced increase in cholesterol efflux accounted for by changes in HDL-C concentration.
Lipids and apoB-containing lipoproteins
ERN/LRP therapy compared to placebo resulted in significant reductions in total cholesterol (5.06 versus 5.74 mmol/L, P value=0.005), TG (1.05 versus 1.48 mmol/L, P value=0.01), LDL-C (2.65 versus 3.32 mmol/L, P=0.01), non-HDL cholesterol (3.41 versus 4.24 mmol/L, P=0.0002), VLDL-TG (0.60 versus 0.74 mmol/L, P=0.007), total apoB (0.99 versus 1.28 g/L, P<0.0001), and lipoprotein(a) (24.4 versus 35.0 nmol/L, P<0.0001). The reduction in total apoB was mainly due to a significant reduction in buoyant LDL apoB (0.68 versus 0.89 g/L, P=0.0006). There was a small reduction in sdLDL-apoB with ERN/LRP treatment versus placebo (0.16 versus 0.18 g/L, P=0.23), but this did not reach statistical significance (Table5).
Table 5.
Lipids and apoB Lipoproteins on Treatment With ERN/LRP and Placebo
| Variable | Placebo (n=27) | ERN/LRP (n=27) | P Value |
|---|---|---|---|
| TC, mmol/L | 5.74 (4.73 to 6.61) | 5.06 (4.12 to 6.64) | 0.005* |
| TG, mmol/L | 1.48 (1.12 to 2.24) | 1.05 (0.79 to 1.68) | 0.01* |
| LDL-C, mmol/L | 3.32 (2.63 to 4.38) | 2.65 (2.11 to 4.23) | 0.01* |
| Non-HDL cholesterol, mmol/L | 4.24 (3.29 to 5.21) | 3.41 (2.68 to 4.62) | 0.0002* |
| VLDL-C, mmol/L | 0.53 (0.29 to 0.72) | 0.34 (0.21 to 0.67) | 0.08 |
| VLDL-TG, mmol/L | 0.74 (0.62 to 1.47) | 0.60 (0.41 to 1.14) | 0.007* |
| VLDL-TG/VLDL-C ratio | 1.84 (1.21 to 2.52) | 1.87 (1.36 to 2.43) | 0.94 |
| Total apoB, g/L | 1.28 (0.99 to 1.58) | 0.99 (0.83 to 1.21) | <0.0001* |
| VLDL apoB, g/L | 0.11 (0.07 to 0.17) | 0.09 (0.06 to 0.12) | 0.08 |
| LDL apoB, g/L | 1.10 (0.91 to 1.40) | 0.96 (0.75 to 1.13) | <0.0001* |
| Buoyant LDL apoB, g/L | 0.89 (0.71 to 1.19) | 0.68 (0.62 to 0.98) | 0.0006* |
| sdLDL-apoB, g/L | 0.18 (0.11 to 0.22) | 0.16 (0.10 to 0.26) | 0.23 |
| Lp(a), nmol/L | 35.0 (15.4 to 113) | 24.4 (8.07 to 89.1) | <0.0001* |
Values are in median (interquartile range) as data are nonparametric. The P value for the significance of the difference between placebo and ERN/LRP is shown. ApoB indicates apolipoprotein B; ERN/LRP, extended release niacin/laropiprant; HDL, high-density lipoprotein; LDL-C, low-density lipoprotein cholesterol; Lp(a), lipoprotein(a); sdLDL, small dense LDL; TC, total cholesterol; VLDL-C, very low-density lipoprotein cholesterol; VLDL-TG, very low-density lipoprotein triglycerides.
P<0.05.
Mediators of vascular inflammation associated with LDL
OxLDL (16.7 versus 18.4 mg/L, P=0.01), Lp-PLA2 (509 versus 730 ng/mL, P<0.0001), lyso-PC (257 versus 285 μmol/L, P=0.007) and MCP1 (188 versus 205 pg/mL, P=0.01) were significantly reduced with ERN/LRP treatment compared to placebo. There was an apparent reduction in glyc-apoB with ERN/LRP treatment versus placebo (15.5 versus 16.7 mg/dL, P=0.43, respectively), but this did not reach statistical significance (Table6).
Table 6.
Mediators of Vascular Inflammation Associated With LDL on Treatment With ERN/LRP and Placebo
| Variable | Placebo (n=27) | ERN/LRP (n=27) | P Value |
|---|---|---|---|
| oxLDL, mg/L | 18.4 (11.9 to 22.6) | 16.7 (9.30 to 20.6) | 0.01* |
| Lp-PLA2, ng/mL | 730 (589 to 813) | 509 (398 to 657) | <0.0001* |
| Lyso-PC, μmol/L | 285 (262 to 354) | 257 (230 to 335) | 0.007* |
| MCP1, pg/mL | 205 (155 to 268) | 188 (136 to 246) | 0.01* |
| Total glyc-apoB, mg/L | 16.7 (12.7 to 31.1) | 15.5 (11.4 to 27.4) | 0.43 |
| Hs-CRP, mg/L | 0.95 (0.48 to 1.45) | 0.85 (0.43 to 1.88) | 0.22 |
Values are in median (interquartile range) as data are nonparametric. The P value for the significance of the difference between placebo and ERN/LRP is shown. ERN/LRP indicates extended-release niacin/laropiprant; glyc-apoB, glycated apolipoprotein B; hs-CRP, high-sensitivity C-reactive protein; LDL, low-density lipoprotein; Lp-PLA2, lipoprotein-associated phospholipase A2; Lyso-PC, lysophosphatidyl choline; MCP1, macrophage chemoattractant protein 1; oxLDL, oxidized LDL.
P<0.05.
Adverse Events
ERN/LRP
Twelve patients reported flushing, 1 had diarrhea, and 2 developed a skin rash. Two of the 12 patients who reported flushing withdrew from the study. Another developed impaired fasting glycemia. One patient was noted to have ALT >4× upper limit of normal. The patient was asymptomatic and had a normal abdominal ultrasound scan result. The high ALT resolved after stopping the ERN/LRP.
Placebo
Two patients reported flushing, 2 had diarrhea, and 1 patient developed a skin rash. ALP was elevated in 1 case, and it resolved spontaneously.
Serious adverse events
There were 2 serious adverse events in patients on ERN/LRP needing hospitalization. Neither had any long-term or permanent consequences (1 angina and 1 abdominal pain). Two serious adverse events were reported in patients on placebo needing hospitalization. Neither had any long-term or permanent consequences (1 allergic reaction to another drug and 1 chest pain).
Discussion
Effect on HDL-C and HDL Functionality
In this investigation, treatment with ERN/LRP led to a significant increase in HDL-C compared to placebo. There is evidence from previous studies that treatment with niacin promotes HDL-mediated reverse cholesterol transport31,32 and lowers CETP activity.33 In one of these studies, niacin was added in vitro31 and in the other one apoB-depleted pooled serum and human THP1 cells were used for the cholesterol efflux experiments.32 In our study, we report individual cholesterol efflux experiments using the individual’s own apoB-depleted serum as acceptor.
The reverse cholesterol transport and CETP-lowering actions of niacin should result in an increase in cholesterol content of HDL rather than the number of particles. Our results also show that ERN/LRP significantly reduces CETP activity and increases HDL cholesterol efflux capacity, explaining the increase in HDL-C. The increase in cholesterol efflux with ERN/LRP treatment is modest, but significant. Franceschini et al have, using methods for cholesterol efflux similar to this study, demonstrated that niacin promotes reverse cholesterol efflux by passive diffusion rather than via the ABCA1 receptor.34 Khera et al, who employed a cholesterol efflux method similar to our study in order to assess the effect of niacin, did not show any significant improvement as compared to placebo.35 However, that study was a parallel cohort trial where baseline equivalent rosuvastatin doses were much lower and mean age of participants was much higher than those in our study. The randomized crossover design adopted in our study provides more power to detect small changes in HDL functionality indices, such as efflux. In fact, 24% of the variation in cholesterol efflux is accounted for by changes in HDL-C concentration.
A recent study demonstrated a consistent inverse association between CV end points and nuclear magnetic resonance–derived HDL particle concentration compared with HDL-C.36 However, there was no significant change in apoAI levels on either ERN/LRP or placebo in our study. These findings may suggest that treatment with ERN/LRP leads to an increase in HDL-C cargo rather than number of particles.
apoAI, PON1, apoM, and MPO play a role in antioxidant function of HDL.7 In this study, there was no difference with treatment in apoAI, apoM levels, PON1 activity, and MPO. This may explain lack of improvement in the capacity of HDL to protect LDL from in vitro oxidation on treatment with ERN/LRP compared to placebo. These findings may provide an explanation for failure of niacin in recent trials in reducing CV events, despite increasing HDL-C significantly.
A study with similar size and population to our study did not show a significant improvement in apoAI level,37 although previously larger studies have demonstrated an apoAI-raising effect of niacin.38–40 In our study, all except one patient were on maximum licensed doses of atorvastatin or rosuvastatin. The potent statins may have eroded the capacity of niacin to raise HDL, but it is against such a background of statin therapy that ERN/LRP is of potential value in high-risk patients whose LDL or HDL levels remain suboptimally controlled.
Effect on apoB Lipoproteins
Our results show that treatment with ERN/LRP was associated with a reduction in VLDL-TG. There was also a trend toward reductions in VLDL-C and VLDL apoB on treatment with ERN/LRP, although these did not reach significance. This might indicate that there was a greater reduction in VLDL particle size compared to particle number with ERN/LRP, likely because of reduced nonesterified fatty acids delivery to liver and inhibition of diacylglycerol acyl transferase 2.
ERN/LRP significantly lowered LDL apoB and buoyant LDL apoB in this study. There was reduction in sdLDL-apoB, but it was not statistically significant. Although larger decreases in sd-LDL have been reported with niacin,41 statins in the doses we employed are remarkably effective in decreasing sd-LDL,42 and thus there may have been little scope for niacin in our study. VLDL is a precursor of LDL. Thus, the reduction in VLDL apoB on treatment with ERN/LRP explains at least in part the reduction in LDL apoB.
There was a small, but insignificant reduction in glyc-apoB in our study in the treatment arm, as treatment with ERN/LRP led to a similar effect on sdLDL-apoB. SdLDL-apoB is preferentially glycated.12,43 Absence of a significant change in sdLDL-apoB may explain the lack of a significant reduction in glyc-apoB.44
Effect on LDL-Associated Mediators of Vascular Inflammation
In previous studies, anti-inflammatory effects of niacin were thought to be independent of its lipid-lowering functions.45,46 In this study, niacin reduced all constituents of a distinct pro-atherogenic LDL-associated mechanism involving oxLDL, Lp-PLA2, lyso-PC, and MCP1. The significant reduction in oxLDL, Lp-PLA2, lyso-PC, and MCP1 with ERN/LRP treatment could be explained by its ability to reduce apoB levels or number of LDL particles significantly. Also, we have shown that ERN/LRP treatment leads to a reduction in SAA, which is located on HDL and is increased in proinflammatory states. Hence, niacin may mediate anti-inflammatory effects through multiple pathways.
Role of Niacin in the Future
LDL-C lowering with statins remains first-line treatment in CVD, but there is mounting evidence that the residual CV risk of 60% to 70% is also modifiable. This risk may be reduced by optimizing treatment targets based on apo-B measurement,47 but HDL-C also represents an attractive therapeutic target. Statins have a limited effect on HDL-C, whereas niacin reduces TG and all atherogenic apo-B-containing particles as well as increasing HDL-C. Therefore, over the past decade focus has shifted to HDL-C as an additional treatment target, and there has been a steady increase in research elucidating protective effects of HDL-C and identifying drugs that are able to improve the anti-atherogenic properties of HDL.
Niacin was shown to prevent CV events and reduce mortality in initial pre–statin era small-scale studies, but the results of 2 large trials, AIM HIGH and HPS2-THRIVE, were disappointing.5 The addition of ERN to patients with an average LDL-C of 1.80 mmol/L, in order to raise HDL-C, was found to have no additional positive effect on CV outcomes. In contrast to these large studies, we recruited patients who had not reached the NCEP-ATP III targets for LDL-C with current statin therapy. Also, we have shown that despite elevating HDL-C, ERN/LRP did not improve the antioxidant function of HDL in vitro nor potential markers of its antioxidant functionality like PON1 activity, MPO, apoM, and apoAI in these patients. Nonetheless, there was some beneficial effect on cholesterol efflux and CETP activity. However, a large CV outcome trial with the CETP inhibitor dalcetrapib elevated HDL-C levels, but did not reduce the risk of recurrent CV events.10 Capacity of HDL to promote cholesterol efflux remains an interesting in vitro marker of HDL functionality and a potential therapeutic target. It has been shown to correlate with CVD independent of HDL-C.8
Niacin makes a significant impact on the pro-atherogenic lipoproteins and mediators of vascular inflammation. Therefore, niacin may be initiated in patients with high LDL-C despite the use of potent statins. It is worth mentioning here that the only niacin study that recruited patients with high LDL-C and assessed CV events was HATS,48 which did show a significant benefit of niacin added to simvastatin compared to statin alone (24% first CV events in placebo versus 3% in niacin+simvastatin group [P=0.03]). There was a significant reduction in LDL-C in this study, which could have contributed to the positive CV outcome. However, there was no statin-only arm in HATS.
Safety of LRP in Combination With Niacin
LRP competitively and selectively blocks prostaglandin D2 receptor (DP1) in order to reduce flushing. Although LRP also has a weak affinity to the thromboxane A2 receptor, therapeutic doses of LRP have no clinically relevant effect on lipoprotein metabolism or the lipid-modifying actions of niacin.6,49,50 Studies of LRP in hyperlipidemic patients have not demonstrated any clinically significant side effects.50 Concerns about the possible effects of LRP on platelet aggregation have not been substantiated.51–54 Co-administration of LRP with clopidogrel or aspirin is generally well tolerated in healthy subjects.55 Co-administration of multiple doses of LRP 40 mg and clopidogrel 75 mg or aspirin 81 mg have no clinically important effect on bleeding time or platelet aggregation.55 Another study demonstrated that LRP does not enhance platelet reactivity when given alone or with ERN in healthy subjects.52
However, based on murine studies, more recently Song et al have suggested that DP1 antagonism may restrain the atheroprotective effects of niacin56 by enhancing the effect of thromboxane A2. This effect is overridden by aspirin inhibition of both thromboxane A2 and prostaglandin D2 production. They suggest that LRP may be harmful in patients intolerant of aspirin, particularly in the presence of coronary artery disease as DP1 receptors may be protective in atheroma lesions. It is possible that LRP may have had a role to play in the negative outcome of HPS2-THRIVE, but not that of the AIM HIGH, which used only ERN. It would be interesting to establish complications arising in the ERN/LRP arm in the HPS2 THRIVE study, especially around CV risk factors, CV events, and aneurysms and also whether lack of aspirin had any effect.
Limitations
Our study has some limitations. It was powered for a 15% increase in HDL-C. Therefore, the results of other parameters have been categorized as descriptive in the analysis rather than additional end points. Although we introduced washout periods during the study and analyzed the data through formulae recommended for crossover trials, there may yet be a “carryover effect” from the first phase to the second phase. However, this is unlikely, taking into consideration the half-life of ERN, LRP, HDL, and LDL. There remains the possibility that LRP somehow ameliorates some potential benefits of niacin. We have earlier reviewed this possibility, but we concede that a treatment arm with ERN alone may have answered this issue. However, adopting such an approach would not necessarily have done so because it would undoubtedly have led to further dropouts. Some of the more optimistic outcomes of trials before the advent of LRP could have been because of the lack of even single blinding due to flushing in almost all patients receiving active treatment and a select few persisting on active treatment to the end of the trial. Also, our intention was to provide more information about the ENR/LRP combination, which when our study was initiated appeared to be so much better tolerated that the relevance of ERN alone in clinical practice seemed dubious. It is only now that ERN/LRP may have failed to provide the hoped-for decrease in CVD incidence that the role of LRP, along with that of background statin therapy, in ameliorating potentially favorable effects and perhaps amplifying those less beneficial effects, is being questioned. We are the first to report on a wide panoply of lipoprotein and inflammatory mediator effects of the combination of ERN and LRP, which may assist in the interpretation of large randomized, placebo-controlled, clinical events trials such as HPS2 THRIVE and to improve drug design.
Conclusions
In this study, treatment with ERN/LRP resulted in a significant improvement in HDL-C and reduction in pro-atherogenic lipoproteins/apolipoproteins in patients with persistent dyslipidemia despite high doses of potent statins. For the first time we have shown that ERN/LRP reduces mediators of vascular inflammation in addition to improving the cholesterol efflux capacity of HDL in humans. However, ERN/LRP does not influence antioxidant capacity of HDL or any other markers of HDL functionality such as PON1 activity, apoAI, MPO, or apoM. Because niacin has not been shown to reduce mortality or CV morbidity in recent large trials and had varied effects on HDL functionality in our study, we suggest niacin should not be employed solely for its HDL-C-raising effect. However, niacin may be used as a second- or third-line agent for its LDL-C-lowering effect in dyslipidemic patients on maximum tolerated dose of statins where LDL-C or apoB remain considerably off target.
Acknowledgments
We acknowledge support from Manchester Comprehensive Local Research Network and The National Institute for Health Research/Wellcome Trust Clinical Research Facility.
Sources of Funding
This study was supported by a research grant from MSD.
Disclosures
None.
References
- Miller GJ, Miller NE. Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet. 1975;1:16–19. doi: 10.1016/s0140-6736(75)92376-4. [DOI] [PubMed] [Google Scholar]
- Jafri H, Alsheikh-Ali AA, Karas RH. Meta-analysis: statin therapy does not alter the association between low levels of high-density lipoprotein cholesterol and increased cardiovascular risk. Ann Intern Med. 2010;153:800–808. doi: 10.7326/0003-4819-153-12-201012210-00006. [DOI] [PubMed] [Google Scholar]
- Barter P, Gotto AM, LaRosa JC, Maroni J, Szarek M, Grundy SM, Kastelein JJ, Bittner V, Fruchart JC. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med. 2007;357:1301–1310. doi: 10.1056/NEJMoa064278. [DOI] [PubMed] [Google Scholar]
- Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- HPS2 THRIVE Group. Landray MJ, Haynes R. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–212. doi: 10.1056/NEJMoa1300955. [DOI] [PubMed] [Google Scholar]
- Yadav R, France M, Younis N, Hama S, Ammori BJ, Kwok S, Soran H. Extended-release niacin with laropiprant: a review on efficacy, clinical effectiveness and safety. Expert Opin Pharmacother. 2012;13:1345–1362. doi: 10.1517/14656566.2012.690395. [DOI] [PubMed] [Google Scholar]
- Soran H, Hama S, Yadav R, Durrington PN. HDL functionality. Curr Opin Lipidol. 2012;23:353–366. doi: 10.1097/MOL.0b013e328355ca25. [DOI] [PubMed] [Google Scholar]
- Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PJ, Khera AV, Wilensky RL, Rader DJ. Anti-oxidative and cholesterol efflux capacities of high-density lipoprotein are reduced in ischaemic cardiomyopathy. Eur J Heart Fail. 2013;15:1215–1219. doi: 10.1093/eurjhf/hft084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- Soran H, Durrington PN. Susceptibility of LDL and its subfractions to glycation. Curr Opin Lipidol. 2011;22:254–261. doi: 10.1097/MOL.0b013e328348a43f. [DOI] [PubMed] [Google Scholar]
- Rizos E, Tambaki AP, Gazi I, Tselepis AD, Elisaf M. Lipoprotein-associated PAF-acetylhydrolase activity in subjects with the metabolic syndrome. Prostaglandins Leukot Essent Fatty Acids. 2005;72:203–209. doi: 10.1016/j.plefa.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Thompson A, Gao P, Orfei L, Watson S, Di Angelantonio E, Kaptoge S, Ballantyne C, Cannon CP, Criqui M, Cushman M, Hofman A, Packard C, Thompson SG, Collins R, Danesh J. Lipoprotein-associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: collaborative analysis of 32 prospective studies. Lancet. 2012;375:1536–1544. doi: 10.1016/S0140-6736(10)60319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafforini DM, Tjoelker LW, McCormick SP, Vaitkus D, McIntyre TM, Gray PW, Young SG, Prescott SM. Molecular basis of the interaction between plasma platelet-activating factor acetylhydrolase and low density lipoprotein. J Biol Chem. 1999;274:7018–7024. doi: 10.1074/jbc.274.11.7018. [DOI] [PubMed] [Google Scholar]
- Steinbrecher UP, Pritchard PH. Hydrolysis of phosphatidylcholine during LDL oxidation is mediated by platelet-activating factor acetylhydrolase. J Lipid Res. 1989;30:305–315. [PubMed] [Google Scholar]
- Benitez S, Sanchez-Quesada JL, Ribas V, Jorba O, Blanco-Vaca F, Gonzalez-Sastre F, Ordonez-Llanos J. Platelet-activating factor acetylhydrolase is mainly associated with electronegative low-density lipoprotein subfraction. Circulation. 2003;108:92–96. doi: 10.1161/01.CIR.0000072791.40232.8F. [DOI] [PubMed] [Google Scholar]
- Vickers KC, Castro-Chavez F, Morrisett JD. Lyso-phosphatidylcholine induces osteogenic gene expression and phenotype in vascular smooth muscle cells. Atherosclerosis. 2010;211:122–129. doi: 10.1016/j.atherosclerosis.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves I, Edsfeldt A. Evidence supporting a key role of Lp-PLA2 generated lysophosphatidylcholine in human atherosclerotic plaque inflammation. Arterioscler Thromb Vasc Biol. 2012;32:1505–1512. doi: 10.1161/ATVBAHA.112.249854. [DOI] [PubMed] [Google Scholar]
- Niu N, Yu YH, Wang Y, Wang LJ, Li Q, Guo LM. Combined effects of niacin and chromium treatment on vascular endothelial dysfunction in hyperlipidemic rats. Mol Biol Rep. 2009;36:1275–1281. doi: 10.1007/s11033-008-9309-1. [DOI] [PubMed] [Google Scholar]
- Kuvin JT, Dave DM, Sliney KA, Mooney P, Patel AR, Kimmelstiel CD, Karas RH. Effects of extended-release niacin on lipoprotein particle size, distribution, and inflammatory markers in patients with coronary artery disease. Am J Cardiol. 2006;98:743–745. doi: 10.1016/j.amjcard.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Wu BJ, Yan L, Charlton F, Witting P, Barter PJ, Rye KA. Evidence that niacin inhibits acute vascular inflammation and improves endothelial dysfunction independent of changes in plasma lipids. Arterioscler Thromb Vasc Biol. 2010;30:968–975. doi: 10.1161/ATVBAHA.109.201129. [DOI] [PubMed] [Google Scholar]
- Christian JB, Olson EJ, Allen JK, Lowe KA. HDL-C response variability to niacin ER in US adults. Cholesterol. 2013 doi: 10.1155/2013/681475. http://dx.doi.org/10.1155/2013/681475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton-Menys V, Chobotova J, Durrington PN. Use of the TLX ultracentrifuge for the isolation of different density lipoproteins and effects of freeze/thawing of human plasma before ultracentrifugation. Clin Chem Lab Med. 2008;46:1285–1288. doi: 10.1515/CCLM.2008.254. [DOI] [PubMed] [Google Scholar]
- Charlton-Menys V, Liu Y, Durrington PN. Semiautomated method for determination of serum paraoxonase activity using paraoxon as substrate. Clin Chem. 2006;52:453–457. doi: 10.1373/clinchem.2005.063412. [DOI] [PubMed] [Google Scholar]
- Havel RJ, Eder HA, Bragdon JH. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest. 1955;34:1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Jarvis KL, Hyland KJ. Protein measurement using bicinchoninic acid: elimination of interfering substances. Anal Biochem. 1989;180:136–139. doi: 10.1016/0003-2697(89)90101-2. [DOI] [PubMed] [Google Scholar]
- el-Saadani M, Esterbauer H, el-Sayed M, Goher M, Nassar AY, Jurgens G. A spectrophotometric assay for lipid peroxides in serum lipoproteins using a commercially available reagent. J Lipid Res. 1989;30:627–630. [PubMed] [Google Scholar]
- de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol. 2010;30:796–801. doi: 10.1161/ATVBAHA.109.199158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackness MI, Abbott C, Arrol S, Durrington PN. The role of high-density lipoprotein and lipid-soluble antioxidant vitamins in inhibiting low-density lipoprotein oxidation. Biochem J. 1993;294:829–834. doi: 10.1042/bj2940829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubic T, Trottmann M, Lorenz RL. Stimulation of CD36 and the key effector of reverse cholesterol transport ATP-binding cassette A1 in monocytoid cells by niacin. Biochem Pharmacol. 2004;67:411–419. doi: 10.1016/j.bcp.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Yvan-Charvet L, Kling J, Pagler T. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler Thromb Vasc Biol. 2010;30:1430–1438. doi: 10.1161/ATVBAHA.110.207142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoorn JW, de Haan W, Berbee JF, Havekes LM, Jukema JW, Rensen PC, Princen HM. Niacin increases HDL by reducing hepatic expression and plasma levels of cholesteryl ester transfer protein in APOE*3Leiden.CETP mice. Arterioscler Thromb Vasc Biol. 2008;28:2016–2022. doi: 10.1161/ATVBAHA.108.171363. [DOI] [PubMed] [Google Scholar]
- Franceschini G, Favari E, Calabresi L, Simonelli S, Bondioli A, Adorni MP, Zimetti F, Gomaraschi M, Coutant K, Rossomanno S, Niesor EJ, Bernini F, Benghozi R. Differential effects of fenofibrate and extended-release niacin on high-density lipoprotein particle size distribution and cholesterol efflux capacity in dyslipidemic patients. J Clin Lipidol. 2013;7:414–422. doi: 10.1016/j.jacl.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Khera AV, Patel PJ, Reilly MP. The addition of niacin to statin therapy improves high-density lipoprotein cholesterol levels but not metrics of functionality. J Am Coll Cardiol. 2013;62:1909–1910. doi: 10.1016/j.jacc.2013.07.025. [DOI] [PubMed] [Google Scholar]
- Mackey RH, Greenland P, Goff DCJ. High density lipoprotein cholesterol and particle concentrations, carotid atherosclerosis, and coronary events: MESA (Multi-Ethnic Study of Atherosclerosis) J Am Coll Cardiol. 2012;60:508–516. doi: 10.1016/j.jacc.2012.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Robson MD, Yu LM, Shirodaria CC, Cunnington C, Kylintireas I, Digby JE, Neubauer S, Choudhury RP. Effects of high-dose modified-release nicotinic acid on atherosclerosis and vascular function: a randomized, placebo-controlled, magnetic resonance imaging study. J Am Coll Cardiol. 2009;54:1787–1794. doi: 10.1016/j.jacc.2009.06.036. [DOI] [PubMed] [Google Scholar]
- Hamoud S, Kaplan M, Meilin E. Niacin administration significantly reduces oxidative stress in patients with hypercholesterolemia and low levels of high-density lipoprotein cholesterol. Am J Med Sci. 2013;345:195–199. doi: 10.1097/MAJ.0b013e3182548c28. [DOI] [PubMed] [Google Scholar]
- Albers JJ, Slee A, Kevin DO. Relationships of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH Trial. J Am Coll Cardiol. 2013;62:1575–1579. doi: 10.1016/j.jacc.2013.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamon-Fava S, Diffenderfer MR, Barrett PH, Buchsbaum A, Nyaku M, Lichtenstein AH, Dolnikowski GG, Schaefer EJ. Extended-release niacin alters the metabolism of plasma apolipoprotein (Apo) A-I and ApoB-containing lipoproteins. Arterioscler Thromb Vasc Biol. 2008;28:1672–1678. doi: 10.1161/ATVBAHA.108.164541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JM, Capuzzi DM, Baksh RI, Intenzo C, Carey CM, Reese D, Walker K. Effects of extended-release niacin on lipoprotein subclass distribution. Am J Cardiol. 2003;91:1432–1436. doi: 10.1016/s0002-9149(03)00394-1. [DOI] [PubMed] [Google Scholar]
- Caslake MJ, Stewart G, Day SP, Daly E, McTaggart F, Chapman MJ, Durrington P, Laggner P, Mackness M, Pears J, Packard CJ. Phenotype-dependent and -independent actions of rosuvastatin on atherogenic lipoprotein subfractions in hyperlipidaemia. Atherosclerosis. 2003;171:245–253. doi: 10.1016/j.atherosclerosis.2003.08.025. [DOI] [PubMed] [Google Scholar]
- Younis N, Charlton-Menys V, Sharma R, Soran H, Durrington PN. Glycation of LDL in non-diabetic people: small dense LDL is preferentially glycated both in vivo and in vitro. Atherosclerosis. 2009;202:162–168. doi: 10.1016/j.atherosclerosis.2008.04.036. [DOI] [PubMed] [Google Scholar]
- Younis NN, Soran H, Sharma R, Charlton-Menys V, Greenstein A, Elseweidy MM, Durrington PN. Small-dense LDL and LDL glycation in metabolic syndrome and in statin-treated and non-statin-treated type 2 diabetes. Diab Vasc Dis Res. 2010;7:289–295. doi: 10.1177/1479164110383063. [DOI] [PubMed] [Google Scholar]
- Digby JE, Martinez F, Jefferson A. Anti-Inflammatory effects of nicotinic acid in human monocytes are mediated by GPR109a dependent mechanisms. Arterioscler Thromb Vasc Biol. 2012;32:669–676. doi: 10.1161/ATVBAHA.111.241836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PH, Lin CP, Wang CH. Niacin improves ischemia-induced neovascularization in diabetic mice by enhancement of endothelial progenitor cell functions independent of changes in plasma lipids. Angiogenesis. 2012;15:377–389. doi: 10.1007/s10456-012-9267-z. [DOI] [PubMed] [Google Scholar]
- Soran H, France MW, Kwok S, Dissanayake S, Charlton-Menys V, Younis NN, Durrington PN. Apolipoprotein B100 is a better treatment target than calculated LDL and non-HDL cholesterol in statin-treated patients. Ann Clin Biochem. 2011;48:566–571. doi: 10.1258/acb.2011.010277. [DOI] [PubMed] [Google Scholar]
- Brown BG, Zhao XQ, Chait A, Fisher LD, Bolson EL, Alaupovic P, Frohlich J, Albers JJ. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345:1583–1592. doi: 10.1056/NEJMoa011090. [DOI] [PubMed] [Google Scholar]
- Maccubbin D, Bays HE, Olsson AG, Elinoff V, Pasternak RC, Paolini JF. Lipid-modifying efficacy and tolerability of extended-release niacin/laropiprant in patients with primary hypercholesterolaemia or mixed dyslipidaemia. Int J Clin Pract. 2008;62:1959–1970. doi: 10.1111/j.1742-1241.2008.01938.x. [DOI] [PubMed] [Google Scholar]
- Paolini JF, Mitchel YB, Reyes R, Kher U, Bays HE, Davidson M, Ballantyne CM. Effects of laropiprant on nicotinic acid-induced flushing in patients with dyslipidemia. Am J Cardiol. 2008;101:625–630. doi: 10.1016/j.amjcard.2007.10.023. [DOI] [PubMed] [Google Scholar]
- Lai E, Wenning LA, Crumley TM, De Lepeleire I, Liu F, Gottesdiener KG, Wagner JA. Pharmacokinetics, pharmacodynamics, and safety of a prostaglandin D2 receptor antagonist. Clin Pharmacol Ther. 2008;83:840–847. doi: 10.1038/sj.clpt.6100345. [DOI] [PubMed] [Google Scholar]
- Lai E, Schwartz JI, Dallob A, Jumes P, Liu F, McCrary Sisk C, Radziszewski W, Wagner JA. Effects of extended release niacin/laropiprant, laropiprant, extended release niacin and placebo on platelet aggregation and bleeding time in healthy subjects. Platelets. 2010;21:191–198. doi: 10.3109/09537100903521611. [DOI] [PubMed] [Google Scholar]
- McKenney J, Bays H, Koren M, Ballantyne CM, Paolini JF, Mitchel Y, Sisk CM, Maccubbin D. Safety of extended-release niacin/laropiprant in patients with dyslipidemia. J Clin Lipidol. 2010;4:105–112.e101. doi: 10.1016/j.jacl.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Lauring B, Dishy V, Luo WL, Laterza O, Patterson J, Cote J, Chao A, Larson P, Gutierrez M, Wagner JA, Lai E. Laropiprant in combination with extended-release niacin does not alter urine 11-dehydrothromboxane B2, a marker of in vivo platelet function, in healthy, hypercholesterolemic, and diabetic subjects. J Clin Pharmacol. 2009;49:1426–1435. doi: 10.1177/0091270009339593. [DOI] [PubMed] [Google Scholar]
- Dallob A, Luo WL, Luk JM, Ratcliffe L, Johnson-Levonas AO, Lepeleire ID, Radziszewski W, Wagner JA, Lai E. The effects of laropiprant, a selective prostaglandin D(2) receptor 1 antagonist, on the antiplatelet activity of clopidogrel or aspirin. Platelets. 2011;22:495–503. doi: 10.3109/09537104.2011.565433. [DOI] [PubMed] [Google Scholar]
- Song WL, Stubbe J, Ricciotti E. Niacin and biosynthesis of PGD2 by platelet COX-1 in mice and humans. J Clin Invest. 2012;122:1459–1468. doi: 10.1172/JCI59262. [DOI] [PMC free article] [PubMed] [Google Scholar]