

Abstract
Background
Human genetically inherited cardiac diseases have been studied mainly in heterologous systems or animal models, independent of patients’ genetic backgrounds. Because sources of human cardiomyocytes (CMs) are extremely limited, the use of urine samples to generate induced pluripotent stem cell–derived CMs would be a noninvasive method to identify cardiac dysfunctions that lead to pathologies within patients’ specific genetic backgrounds. The objective was to validate the use of CMs differentiated from urine-derived human induced pluripotent stem (UhiPS) cells as a new cellular model for studying patients’ specific arrhythmia mechanisms.
Methods and Results
Cells obtained from urine samples of a patient with long QT syndrome who harbored the HERG A561P gene mutation and his asymptomatic noncarrier mother were reprogrammed using the episomal-based method. UhiPS cells were then differentiated into CMs using the matrix sandwich method.
UhiPS-CMs showed proper expression of atrial and ventricular myofilament proteins and ion channels. They were electrically functional, with nodal-, atrial- and ventricular-like action potentials recorded using high-throughput optical and patch-clamp techniques. Comparison of HERG expression from the patient’s UhiPS-CMs to the mother’s UhiPS-CMs showed that the mutation led to a trafficking defect that resulted in reduced delayed rectifier K+ current (IKr). This phenotype gave rise to action potential prolongation and arrhythmias.
Conclusions
UhiPS cells from patients carrying ion channel mutations can be used as novel tools to differentiate functional CMs that recapitulate cardiac arrhythmia phenotypes.
Keywords: arrhythmia, cardiomyocytes, HERG gene, long QT syndrome, urine-derived induced pluripotent stem cells
Despite important achievements in the understanding of cardiovascular diseases over the past decade, heart disease remains the principal cause of death in developed societies. Intense research efforts have been directed at using human induced pluripotent stem (hiPS) cells to invoke cardiac regeneration for heart repair and to model human cardiac development and diseases in vitro. Recent studies have demonstrated that hiPS cell–derived cardiomyocytes (CMs) can be used to model several human genetically inherited cardiac diseases that manifest defects in specific cardiac structural components, signaling pathways, or electrophysiological properties1, validating the technique as a powerful system with which to gain mechanistic insights into human cardiac disease. Currently, a wide variety of molecular, cellular, and physiological assays are optimized to investigate disease phenotypes in hiPS cell–derived CMs. Experimental drugs have also been tested in this setting, inducing alleviation of the disease phenotypes and further paving the way for new therapeutic interventions for cardiac diseases. Importantly, as recently developed by Yamanaka, iPS cell technology can contribute to micromedicine (eg, applied to an individual patient) and personalized medicine, including drug discovery based on cellular and molecular analyses.2 It may also contribute in the future to macromedicine (eg, applied to cohorts of patients), including patient stratification, based on cellular and molecular analyses of participants in clinical trials or cohort studies.2
Nevertheless, the standard approach by which hiPS cells are generated from skin biopsy as raw material limits the extensive use of this technology. Skin biopsy requires an invasive procedure, accompanied by discomfort and risk of bleeding, infection, and permanent scars. Skin biopsy collection also raises ethical issues in the case of young children and patients’ healthy relatives. Furthermore, the obtained cells expand slowly in culture, and the reprogramming efficacy is low. This may require several skin samples; however, the procedure cannot easily be performed several times on the same person. There is a growing consensus that an ideal source for hiPS cells should provide cells easily and noninvasively, with high reprogramming efficacy. Progress has been made regarding this issue with the reprogramming of cells from blood samples, but there is still room for improvement.
Recent reports showed that cells contained in a simple urine sample can be an easy and noninvasive source for generating hiPS cells.3–6 Reprogramming efficacy of urine-derived cells is higher than for human dermal fibroblasts: Efficacy for both viral and nonintegrative methods was consistently higher in urine cells compared with dermal cells, including retroviral reprogramming that was ≈50 times more efficient in urine cells3,7–9 and episomal reprogramming that was ≈10 times more efficient.4,10,11 Furthermore, the cardiac molecular phenotype of a dystrophin-deficient patient was recapitulated in CMs derived from the patient’s urine cells.12 Producing urine-derived CMs from patients with genetic disorders and from their healthy relatives can facilitate study of the impact of individual genetic variation on the risk and progression of the disease of interest.
In the present study, starting from a urine sample, we developed an in vitro model for a patient suffering from a mild form of type 2 long QT (LQT2) syndrome. The long QT syndrome is an inherited cardiac channelopathy that causes potentially fatal cardiac arrhythmia and was one of the first successfully modeled human diseases using hiPS cells.1,13 We investigated the mutation A561P located in the KCNH2 gene encoding the HERG channel. This mutation was the focus of an initial study conducted in the laboratory.14 The patient harboring this mutation presented arrhythmias only when treated with clobutinol, an antitussive drug. Due to the lack of a cardiac cellular model, the analyses were performed in transfected COS-7 cells, and the overall effects on cardiac action potential (AP) were extrapolated with an in silico analysis. In the present study, we used CMs obtained from urine-derived hiPS cells (UhiPS-CMs) to investigate both the molecular and functional phenotypes of the syndrome in a native cellular model. We observed AP changes, characteristic for the long QT syndrome, that were exacerbated by a HERG inhibitor, thus modeling the patient-specific arrhythmic drug sensitivity. We demonstrated that the use of UhiPS-CMs is a convenient and powerful approach to finely model human arrhythmic diseases.
Methods
Patient Characteristics
The study was conducted in compliance with current good clinical practice standards and in accordance with the principles set forth under the Declaration of Helsinki (1989). Institutional review board approvals of the study were obtained before initiation of patient enrollment. Each participant entering the study agreed to and signed an institutional review board–approved statement of informed consent.
Somatic cells from a urine sample were obtained from a man aged 22 years who presented syncope and torsades de pointes arrhythmia at age 13 years during treatment with the antitussive drug clobutinol.14 ECG analysis showed prolonged QT duration (corrected QT interval of 628 ms with Bazett’s formula and 597 ms with Fredericia’s formula). The patient carries a missense mutation in the KCNH2 gene, encoding the HERG K+ channel α-subunit, causing an alanine-to-proline substitution at position 561 (chromosome 7: 150 648 800G>C; NM_000238 A561P). As a control, somatic cells from a urine sample were also obtained from the patient’s mother, aged 46 years, who had no clinical symptoms and a normal ECG and who was negative for the KCNH2 mutation. An additional control, the previously described foreskin fibroblast-derived hiPS (FhiPS) cell clone iPS.C2a, was also used.15
Urine Cell Collection, Isolation, and Culture
Urine cells were isolated and cultured, as described previously.3 Briefly, cell pellets were collected from whole urine samples (130 to 265 mL) via centrifugation (5 minutes at 1200g) and washed with PBS. Pellets were resuspended in RE/MC (renal epithelial/mesenchymal cell) medium composed of 50% RE medium (renal epithelial cell growth medium SingleQuot supplement and growth factors [catalog number CC-3190; Lonza]) and 50% MC medium (DMEM/high glucose supplemented with 10% [vol/vol] FBS, 1% [vol/vol] GlutaMAX, 1% [vol/vol] nonessential amino acid [NEAA], 100 U/mL penicillin, 100 μg/mL streptomycin, 5 ng/mL basic fibroblast growth factor [Miltenyi Biotec], 5 ng/mL PDGF-AB [Cell Guidance] and 5 ng/mL epidermal growth factor [PeproTech]) and cultured on 0.1% gelatin-coated plates. Urine cell clusters were passaged using TripLE (Life Technologies) and characterized.
Urine Cell Reprogramming and UhiPS Cell Characterization
Urine cells were reprogrammed into UhiPS cells, as described previously.6 Briefly, 3×105 to 5×105 urine cells were nucleofected (Lonza) with episomal vectors coding for OCT3/4, SOX2, KLF4, MYC, LIN28, NANOG, and SV40LT (catalog numbers 20922, 20923, 20924, 20925, and 20927; Addgene), and a nonepisomal vector coding for miR302/367 (catalog number TDH101PA-GP; System Biosciences) and cultured 14 to 21 days with E7 medium (Stemcell Technologies). UhiPS clones were manually picked on mouse embryonic fibroblasts (MEFs). Control UhiPS and A561P-UhiPS cells were characterized by reverse transcription polymerase chain reaction to verify the expression of endogenous pluripotent factors OCT3/4, NANOG, and SOX2 and the loss of episomal vectors; immunostaining; flow cytometry; and the ability to form teratoma.
Flow Cytometry Analysis
A total of 1×105 cells in suspension for each condition were washed 3 times with PBS/BSA 0.1% and then incubated with phycoerythrin (PE)-labeled antibodies SSEA3-PE, SSEA4-PE, and TRA1-60-PE (eBioscience) for 30 minutes at 4°C in the dark and further rinsed 3 times with fluorescence-activated cell sorting buffer. Data acquisition was performed using FACSDiva software with an LSR II instrument (Becton Dickinson) or the Accury C6 (Becton Dickinson).
Episomal Vector Detection
Total RNA was extracted from hiPS cells at passage 10 using the RNeasy Mini Kit (Qiagen). Following the manufacturer’s instructions, 1 µg of total RNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Reverse transcription polymerase chain reaction studies were conducted in triplicate using Mesa Green 2X PCR Master Mix for SYBR (Eurogentec). DNA concentration was 5 ng per well. Conditions were 2 seconds at 90°C, followed by 40 cycles of 10 seconds at 95°C and 30 seconds at 60°C, and ending by 60 seconds at 60°C. Cycle threshold was calculated using default settings for the real-time sequence detection software (Applied Biosystems). Primer sequences are listed in Table S1.
Teratoma Formation
Undifferentiated UhiPS cells cultured on MEFs were mechanically dissociated with EZ Passage tools (Life Technologies), resuspended in Matrigel (2 mg/mL; Corning), and injected subcutaneously into NOD/SCID IL2R-γ mice (Charles River). Tumor samples were collected after 8 to 10 weeks, fixed in 4% paraformaldehyde, embedded in paraffin, sectioned, and stained with hematoxylin and eosin for analysis. The animal experiments were approved by the regional ethics commitee and the ministry of national education (protocol 02017.01).
Culture of hiPS Cells
Established hiPS cell lines were maintained on mitotically inactivated MEFs in DMEM/F12 medium supplemented with 2 mmol/L l-glutamine, 0.07% β-mercaptoethanol, 20% knockout serum replacement, 5 ng/mL basic fibroblast growth factor, and 1% NEAA in a low-oxygen atmosphere (4% O2). Cells were passaged by manual dissection of cell clusters every 6 to 7 days. Before differentiation, hiPS cells were manually transferred from MEFs to Matrigel-coated plates (0.05 mg/mL; BD Biosciences) and cultured on mTeSR1 medium (Stemcell Technologies). Passages were performed using Gentle Cell Dissociation Buffer (Stemcell Technologies).
Differentiation of hiPS Cells Into CMs and Dissociation
Human iPS cells were differentiated into CMs using the established matrix sandwich method16 with modifications. Briefly, 6 days before initiating differentiation, hiPS cell colonies were passaged on human embryonic stem cell–qualified Matrigel-coated plates (0.05 mg/mL; BD Biosciences) using Gentle Cell Dissociation Buffer (Stemcell Technologies) and cultured as a monolayer in mTeSR1 with 1× Y-27632 ROCK inhibitor (Stemcell Technologies) in a normal oxygen atmosphere. When cells reached 80% confluence, cold mTeSR1 with Growth Factor Reduced Matrigel (0.033 mg/mL) was added to create an overlay of Matrigel. Differentiation was initiated 24 hours later (day 0) by culturing the cells in RPMI-1640 medium (Life Technologies) supplemented with B27 (without insulin; Life Technologies), 100 ng/mL activin A (Miltenyi Biotec), and 10 ng/mL FGF2 (Miltenyi Biotec) for 24 hours. On the next day, the medium was replaced by RPMI-1640 medium supplemented with B27 without insulin, 10 ng/mL BMP4 (Miltenyi Biotec), and 5 ng/mL FGF2 for 4 days. By day 5, cells were cultured in RPMI-1640 medium supplemented with B27 complete (Life Technologies) and 1% NEAA and changed every 2 to 3 days.
For protein and electrophysiological analyses, CMs were dissociated around day 20 of differentiation by 20-minute incubation with collagenase II (200 U/mL; Gibco) and 0.2 U/mL protease XIV (Sigma-Aldrich) at 37°C. Isolated cells were then incubated at room temperature for 30 minutes in Kraft–Bruhe solution containing 85 mmol/L KCl, 30 mmol/L K2HPO4, 5 mmol/L MgSO4, 1 mmol/L EGTA, 2 mmol/L Na2-ATP, 5 mmol/L Na-pyruvate, 5 mmol/L creatinine, 20 mmol/L taurine, and 20 mmol/L glucose (pH 7.2).17
Gene Expression Analysis
Total RNA was extracted from hiPS colonies or during differentiation at days 5, 18, and 28 using the RNeasy Mini Kit (Qiagen). Following the manufacturer’s instructions, 1 μg of total RNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Polymerase chain reaction amplification was performed using FAM-labeled TaqMan probes (Applied Biosystems) (Table S1).
Protein Expression Analysis
For histological analysis, 2 cell types were analyzed: Type 1 comprised undifferentiated UhiPS cells, which were cultured in 12-well plates on MEF feeders, and type 2 comprised dissociated hiPS-derived CMs, which were plated on Ibidi plates (Biovalley) and further cultured for 6 to 10 days. For type 1 cells, UhiPS cells on MEFs were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and blocked with 3% PBS-BSA. Cells were then stained with primary antibodies directed against OCT3/4 (Santa Cruz Biotechnology) and TRA1-60 (eBioscience) diluted in 1% PBS-BSA. For type 2 cells, after methanol fixation, cells were permeabilized with 0.1% Triton X-100, blocked with 5% PBS-BSA and stained with primary antibodies against α-actinin (Abcam), troponin I (Santa Cruz Biotechnology), MLC2a (Abcam), MLC2v (Proteintech Europe), connexin 43 (Chemicon), HERG (Santa Cruz Biotechnology), or Nav1.5 (Alomone Labs). Secondary antibody staining was performed using Alexa 488– and Alexa 568–conjugated antibodies (Molecular Probes). DAPI was used for nuclear staining.
For Western blot analysis, 40 μg of protein lysate from UhiPS-CMs were incubated with antisera against HERG (Santa Cruz Biotechnology) protein. Secondary antibody staining was performed using goat anti-mouse IgG-HRP antibody (Santa Cruz Biotechnology). Stain Free gel technology (Bio-Rad) was used as a loading control for protein normalization.18,19 The amount of total proteins in each lane on the blot was calculated and used for normalization.
Cellular Electrophysiology
AP optical recordings were performed on CMs after 28 days of differentiation, with 5 μmol/L di-8-ANEPPS (Life Technologies) as the voltage-sensitive dye, using the CellOptiq technology (Clyde Biosciences). Cells were incubated at 37°C, 5% CO2, and 20% O2 during the 2-hour incubation of the dye and during the recording.
Dissociated hiPS-derived CMs were resuspended in RPMI plus B27 supplemented with insulin and 1% NEAA and then plated on Matrigel-coated 35-mm plastic Petri dishes. Isolated beating cells were used for patch-clamp recordings using whole-cell configurations between days 10 and 14 after dissociation. The rapid component of the delayed rectifier K+ current (IKr) and APs were recorded in the ruptured and permeabilized patch-clamp configurations, respectively. Cells were continuously superfused with a Tyrode solution containing (in mmol/L): NaCl 140, KCl 4, CaCl2 1, MgCl2 1, glucose 10, HEPES 10; pH 7.4 (NaOH). During recording, a local gravity microperfusion system allowed application of drugs, dissolved in Tyrode solution in which glucose was replaced by mannitol 20 mmol/L.20 For AP recording, the pipette solution contained (in mmol/L): k-gluconate 125, KCl 20, NaCl 5, amphotericin B 0.85, HEPES 10; pH 7.2 (KOH). For IKr recording, the pipette was filled with a solution containing (in mmol/L): k-gluconate 125, KCl 20, K2ATP 5, HEPES 10, EGTA 10; pH 7.2 (KOH).21 All products were purchased from Sigma-Aldrich. Data were recorded through an A/D converter (Digidata 1440A; Molecular Devices) using an Axopatch 200B amplifier (Molecular Devices). Membrane currents and APs were analyzed with an Axon pClamp 10 (Molecular Devices). Borosilicate patch pipettes had a tip resistance of 2 to 2.5 MΩ. All experiments were made at 35±2°C. During voltage-clamp experiments, nifedipine (4 μmol/L) was added to the external solution to block the l-type calcium current (ICa,L). IKr was measured as a 1-μmol/L E-4031-sensitive current by digitally subtracting the current in the presence of E-4031 from that in its absence. IKr densities were calculated by dividing current amplitude, measured at the peak of the tail current at −50 mV following depolarization to 60 mV (prepulse), by cell membrane capacitance. Activation curves, determined from peak tail current normalized to the maximal value and plotted against the prepulse voltage, were fitted with a single Boltzmann function, G/Gmax=1/(1+exp[(V1/2−V)/k]), in which G/Gmax is the conductance normalized with respect to the maximal conductance, V1/2 is half activation potential, V is the prepulse voltage prior to the tail current measurement, and k is the slope factor. Data extracted from this equation are presented as mean±SEM. Statistical significance of differences in current densities or activation properties was calculated using the Student t test.
Statistical Analysis
Data are expressed as mean±SEM. Statistical analysis was performed with Prism 5 (GraphPad Software, Inc). Significant differences between mean values were determined with the Mann–Whitney U test for comparison of 2 groups or paired Student t test if appropriate. For more than 2 groups, 2-way ANOVA was performed. A P value <0.05 was considered to indicate significance.
Results
Generation of Patient-Specific hiPS Cells From a Urine Sample Using Episomal-Based Reprogramming
Cells isolated from urine samples from the patient carrying the HERG A561P mutation and from his healthy mother displayed a mesenchymal stem cell phenotype, including spindle-shaped morphology and expression of cell surface markers CD49a, CD73, CD90, CD105, and CD146. They did not express the hematopoietic stem cell markers CD14, CD45, and CD184 (data not shown). Cells were reprogrammed on transfection of episomal vectors. Control UhiPS clones and A561P-UhiPS clones carrying the A561P mutation were manually picked for further characterization. Endogenous expression of the pluripotent stem cell markers OCT3/4,SOX2, and NANOG, absent in the urine cells, was detected by quantitative reverse transcription polymerase chain reaction in both UhiPS cells types, and the loss of episomal vector expression was verified (Figure S1B). Endogenous expression of OCT3/4 and TRA1-60 proteins was also visualized by immunofluorescence staining in control and A561P-UhiPS cells (Figure S1A). Flow cytometry analysis showed that >90% of control and A561P-UhiPS cells were positive for the expression of pluripotency markers TRA1-60, SSEA4, and SSEA3 (Figure S1C). Finally, the ability of UhiPS cells to form teratoma with all 3 germ layers was observed (Figure S1D). Urine cells were successfully reprogrammed into bona fide hiPS cells, and 2 to 3 clones per line were selected to perform the following experiments.
UhiPS Cells Can Be Differentiated Into Functional CMs
Using the matrix sandwich method, CMs were differentiated (1) from control hiPS cells derived from foreskin fibroblasts (FhiPS-CMs), a reference tissue source that has previously been used successfully for differentiation into functional CMs22,23, and from hiPS cells derived from urine samples; (2) from the control healthy mother’s cells (control UhiPS-CMs); and (3) from the HERG A561P-mutated cells (A561P-UhiPS CMs). All 3 hiPS cells showed comparable cardiac differentiation potential (Figure 1), giving rise to spontaneously contracting cell areas after 6 to 8 days of differentiation. Gene expression profiling of the 3 hiPS cells revealed a similar differentiation pattern into cardiac cells over time, including significant reduction of pluripotent stem cell markers OCT3/4,NANOG, and SOX2, and progressive upregulation of a cardiac transcription factor, NKX2-5; connexins (GJA1 and GJA5); and a regulator of calcium homeostasis, RYR2, by day 18 (Figure 1A). Similar gene expression profiles were also observed for SCN5A, CACNA1C, CACNA1G, KCND3, KCNQ1, KCNH2, and KCNJ2, the genes encoding the major cardiac ion channel α-subunits. We also analyzed the protein expression patterns of several structural and functional proteins that characterize CMs, using immunofluorescence on cells isolated after 30 days of differentiation (Figure 1B). CMs displayed organized cross-striations, resembling sarcomeres with proper alignment of the myofibrils, that were positive for cardiac and muscle-specific cytoskeletal proteins troponin I, α-actinin, ventricular myosin light chain (MLC2V), and atrial myosin light chain (MLC2A). The sodium channel Nav1.5, responsible for the AP upstroke in atrial, Purkinje, and ventricular CMs, was properly addressed at the plasma membrane (data not shown). Finally, dual staining of troponin I and connexin 43, essential for coordinated cell depolarization, showed that the transmembrane proteins were located, as expected, in contact areas between CMs (Figure 1B). These results show that UhiPS cells could be differentiated into CMs using the matrix sandwich method.
Figure 1.

Human UhiPS cells differentiated into functional cardiomyocytes. A, Transcriptional profile of control hiPS cells derived from skin fibroblasts (control FhiPS-CMs) and from urine cells (control UhiPS-CMs and A561P-UhiPS CMs) at days 5, 18, and 28 of cardiac differentiation. Quantitative reverse transcription polymerase chain reaction analysis was performed on pluripotent stem cell markers (OCT3/4,NANOG,SOX2), on cardiomyocyte markers (NKX2-5,GJA1,GJA5,RYR2), and on key genes encoding cardiac ion channels (SCN5A,CACNA1C,CACNA1G,KCND3,KCNQ1,KCNH2 and KCNJ2) to show enrichment for the cardiomyocyte population. The expression for each gene and sample (n=5 per condition) was normalized to the ß-actin gene ACTB and was calculated relative to the median expression level. Raw minimum and maximum values were taken as a reference for heat map representation. B, (top) In control UhiPS-CMs, representative immunofluorescence images of the cardiac sarcomeric protein α-actinin (green, left) and costaining of myosin light chain 2a (MLC2a; green, middle) and myosin light chain 2v (MLC2v; red, middle) and troponin I and connexin 43 (green and red, respectively, right); (bottom) in A561P-UhiPS CMs, representative immunofluorescence images of α-actinin (red, left), costaining of MLC2a (red; middle) and MLC2v (green, middle) and troponin I and connexin 43 (green and red, respectively, right). Scale=5 μm. CM indicates cardiomyocytes; FhiPS, foreskin fibroblast–derived human induced pluripotent stem cells; max, maximum; min, minimum; UhiPS, urine-derived human induced pluripotent stem cells.
CMs Differentiated From UhiPS Cells Are Electrically Functional
APs of the differentiated CMs were assessed using both patch-clamp and optical recording with the voltage-sensitive dye di-8-ANEPPS. Isolated CMs derived from both FhiPS and UhiPS cells started beating by day 6 after dissociation. Using the patch-clamp technique, spontaneous APs from control and A561P-UhiPS CMs were recorded and classified as nodal-, atrial-, and ventricular-like APs based on their duration, maximum upstroke velocity (dV/dtmax), peak-to-peak duration, and maximum diastolic potential (Figure 2A, Table S2). The 3 types of APs were also obtained during CellOptiq AP optical recordings (Figure 2B), and their quantification showed that, regardless of hiPS cell origin and genotype, ventricular-like APs were the most frequently recorded (Figure 2C). Inversely, nodal-like APs were less frequently recorded, with none observed in A561P-UhiPS CMs using patch-clamp technique (Figure 2A).
Figure 2.
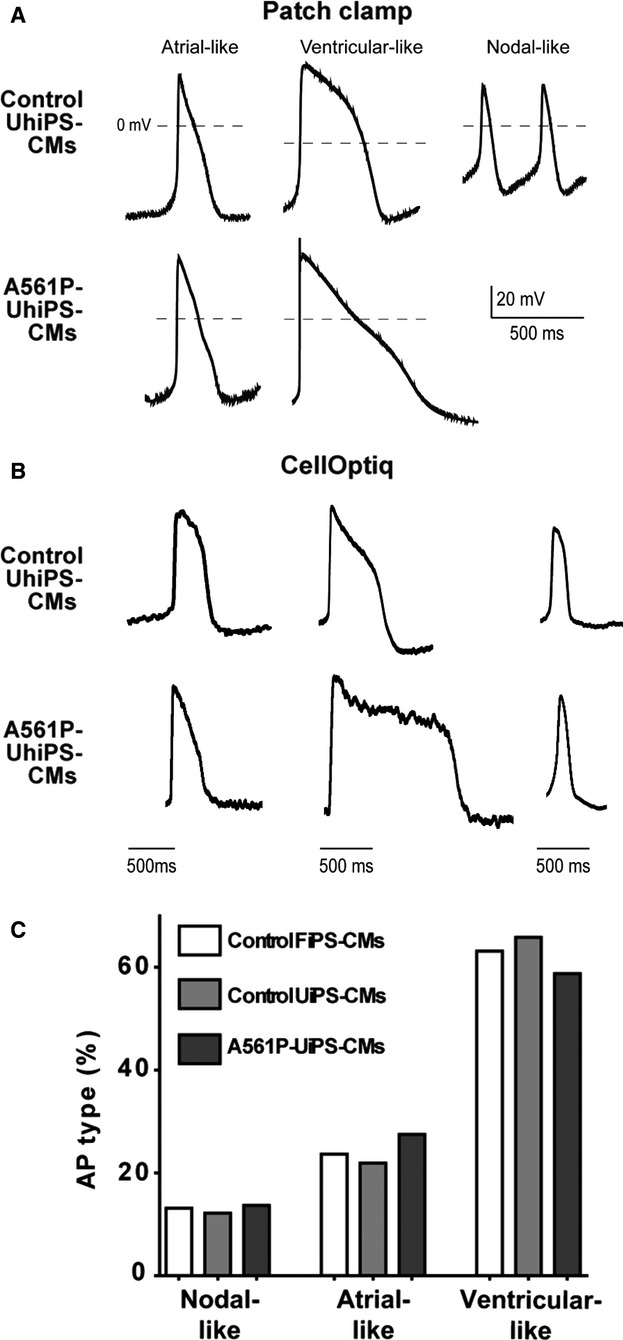
Human UhiPS cells differentiated into electrically functional cardiomyocytes. Representative traces of spontaneous nodal-, atrial- and ventricular-like action potential recordings using (A) patch-clamp or (B) optical dye (CellOptiq) in control UhiPS-CMs and A561P-UhiPS CMs (no nodal-like AP was obtained in A561P-UhiPS-CMs using patch-clamp technique). C, Distribution of the 3 types of action potentials obtained by optical measurement. Regardless of the hiPS cell type, the majority of the action potentials were ventricular-like. Control FhiPS-CMs, n=38; control UhiPS-CMs, n=41; A561P-UhiPS CMs, n=51. AP indicates action potential; CM, cardiomyocytes; FhiPS, foreskin fibroblast–derived human induced pluripotent stem cells; UhiPS, urine-derived human induced pluripotent stem cells.
A561P KCNH2 Mutation Causes a Trafficking Defect of the HERG Channel in Human CMs
We next investigated whether we could model and characterize the LQT2 syndrome molecular phenotype using UhiPS-CMs. The A561P mutation studied in COS-7 cells induced a decrease in plasma membrane expression of the channel that potentially causes the disease phenotype.14 We initially examined cellular localization of HERG by immunofluorescence in UhiPS-CMs. As compared with control FhiPS- and UhiPS-CMs, HERG plasma membrane expression was reduced in A561P-UhiPS CMs (Figure 3A). Higher magnification analyses confirmed that in both FhiPS-CMs and UhiPS-CMs, HERG staining was prominently observed at the cell plasma membrane (Figure 3B, arrows and plot profiles). In contrast, HERG A561P protein was detected at the plasma membrane at the same intensity as in intracellular compartments (Figure 3B, plot profile). Further analysis of HERG protein cellular localization was performed, combining plot profile analysis of multiple CMs differentiated from each hiPS cell line. We found statistically significant enrichment of HERG protein at the membrane, compared with the intracellular compartment, in the 2 control cell lines. In opposition, the A561P-UhiPS CMs did not show membrane enrichment of HERG protein (Figure 3C). These findings suggest a trafficking defect of the HERG ion channel in UhiPS-CMs carrying the HERG A561P mutation. To further confirm this result, we investigated whether this mutation had an effect on KCNH2 gene expression. Interestingly, the A561P mutation did not alter transcriptional expression of the KCNH2 gene (Figure S2). Using Western blot analysis, the differential expression of the immature cytosolic protein was quantified compared with the mature plasma membrane protein (Figure S3A).24 The difference in mature over immature forms of HERG was abolished in A561P-CMs (Figure S3B), confirming a trafficking defect of mutated HERG proteins.
Figure 3.
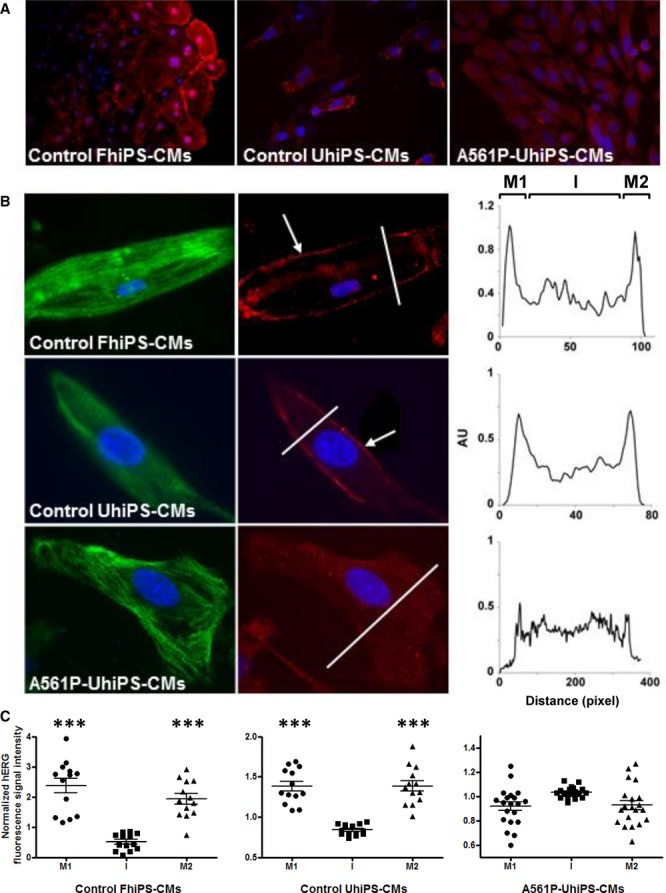
HERG trafficking defect in A561P-UhiPS CMs. A, HERG protein localization (red) after cardiac differentiation of hiPS cells, highlighting the low membrane expression of HERG in A561P-UhiPS CMs relative to intracellular signal (magnification ×20) compared with both controls (control FhiPS-CMs and UhiPS-CMs). B, (left) Specific cellular localization of HERG (red) in cardiomyocytes with counterstaining for troponin I (green, magnification ×65); (right) surface plots of HERG staining in each cell at the level of the line. C, Statistical analysis of surface plots of HERG fluorescence intensity signal. Control FhiPS-CMs, n=12; control UhiPS-CMs, n=12; A561P-UhiPS CMs, n=20. The surface plot was arbitrarily segmented in 3 different areas: the first and last 15% of the surface plot, corresponding to plasma membrane (M1 and M2, as described in Figure 3B), and the remaining intermediate 70% of the surface plot, corresponding to the intracellular compartments. For each of these areas in each cell, the HERG fluorescence intensity values were averaged and normalized to the average cell HERG fluorescence intensity signal. Paired t tests were performed between membrane and intracellular signals. ***P<0.001, for membrane vs intracellular fluorescence intensity signal. CM indicates cardiomyocytes; FhiPS, foreskin fibroblast–derived human induced pluripotent stem cells; I, intracellular compartments; UhiPS, urine-derived human induced pluripotent stem cells.
IKr Density Is Altered in Patient-Derived UhiPS-CMs
Using the patch-clamp technique, we investigated whether this trafficking defect had an effect on HERG-associated ionic current (IKr). As shown in Figure 4A and 4B, the IKr was significantly reduced in A561P-UhiPS CMs (P=0.007) in comparison to control UhiPS-CMs. The tail K+ current density measured at −50 mV was markedly reduced to ≈50% to 60% of the control values (Table 1); however, the activation kinetics were not modified by the A561P mutation (Figure 4C and Table 1). The current density reduction observed in A561P-UhiPS CMs was consistent with the membrane expression reduction described above (Figure 3 and Figure S3).
Figure 4.

Reduction of delayed rectifier K+ current (IKr) density in A561P-UhiPS CMs. A, Representative raw data of currents recorded from control UhiPS-CMs (left) and A561P-UhiPS-CMs (right); protocol is shown as inset (holding potential: −50 mV; stimulation frequency: 1/12.5 Hz; traces with test pulse from −40 to 0 mV are shown). Currents are shown before and after 1 μmol/L E-4031 application. E-4031-sensitive currents are considered as IKr. B, Tail current density measurement obtained on repolarization after test pulse at +60 mV in control UhiPS-CMs (n=13) and A561P-UhiPS CMs (n=11), highlighting a significant reduction in A561P-UhiPS CMs. Protocol is shown in inset. **P<0.01 vs control UhiPS-CMs. C, Voltage-dependent activation curves obtained by plotting the relative E-4031-sensitive current on repolarization at −50 mV (arrows in A) in control UhiPS-CMs (n=13) and A561P-UhiPS CMs (n=8) (arrows in A). CM indicates cardiomyocytes; Ctrl, control; UhiPS, urine-derived human induced pluripotent stem cells.
Table 1.
Effects of A561P Mutation on Delayed Rectifier K+ Current in UhiPS-CMs
| Control UhiPS-CMs | A561P-UhiPS CMs | |
|---|---|---|
| Tail current density(n) | 1.79±0.2 pA/pF(13) | 0.89±0.1 pA/pF**(11) |
| Activation | ||
| V1/2 | −26.49±1.7 mV | −23.81±1.5 mV |
| k(n) | 7.75±0.7 mV(13) | 7.1±0.7 mV(8) |
The tail current density was measured at −50 mV following a depolarization to +60 mV. CMs indicates cardiomyocytes; k, slope factor (see protocol in Figure 4A); pA, current amplitude; pF, cell membrane capacitance; UhiPS, urine-derived human induced pluripotent stem cells; V1/2, prepulse voltage for which the tail current is half of its maximal value.
P<0.01 vs control UhiPS-CMs.
A561P HERG Mutation Impairs IKr Contribution to AP in UhiPS-CMs
As expected, A561P-UhiPS CMs had statistically significant longer ventricular-like AP durations compared with control UhiPS-CMs, mainly APD75 and APD90 (Figure 5A). To exclude that the AP frequency difference was responsible for these changes, we compared AP duration in the 2 cell groups at comparable frequencies. To do so, we used the CellOptiq method to record a high amount of APs at spontaneous automatic rates. APs were then classified and compared according to the beating frequency. The data show that in A561P-UhiPS CMs, APD90 was significantly longer than in control cells regardless of the frequency and that, as expected, the AP prolongation in A561P-UhiPS CMs was potentiated at slower rates25 (Figure 5B). In addition, statistical analyses with 2-way ANOVA show that the effect of the frequency on APDs was minor compared with the effect of the HERG mutation. Consequently, the APD prolongation observed in our LQT2 model likely was mainly due to the mutation. These data show that UhiPS-CMs model the typical AP prolongation associated with LQT2 syndrome.
Figure 5.

Action potentials recorded from A561P-UhiPS CMs model type 2 long QT syndrome. A, Ventricular-like APDs using patch-clamp (left, n=7 in each group) and optical measurement (right, n=27 and 30 for control and A561P cells, respectively). B, APD90 at 2 different AP frequencies, showing a larger prolongation in A561P-UhiPS CMs at slow rate (white circles) compared with control UhiPS-CMs (black circles; n=13 and 6 for control and A561P cells, respectively). Statistical significance: *P<0.05, **P<0.01, ***P<0.001, for A561P-UhiPS CMs vs control UhiPS-CMs. C, Increased arrhythmia susceptibility in A561P-UhiPS CMs in the presence of E-4031: representative action potential patch-clamp recordings before and after 1 μmol/L E-4031 treatment. D, Quantification of the prolongation of the APD after E-4031 treatment of control UhiPS-CMs (gray bars) and A561P-UhiPS CMs (white bars). Statistical significance of APD prolongation after E-4031 treatment in each cell type: *P<0.05. AP indicates action potential; APD, action potential duration; CM, cardiomyocytes; UhiPS, urine-derived human induced pluripotent stem cells.
UhiPS-CMs Carrying the HERG A561P Mutation Have Increased Susceptibility to Arrhythmia
The HERG A561P mutation carrier experienced arrhythmic episodes (ie, torsades de pointes) when treated with the HERG channel blocker clobutinol. Because this drug is no longer available, we used the HERG blocker E-4031 to evaluate susceptibility to arrhythmia in control and A561P-UhiPS CMs. E-4031 did not trigger any arrhythmic event in control UhiPS-CMs recorded with the CellOptiq method (n=10), but an arrhythmic event was recorded in 1 A561P-UhiPS CM of 6 cells upon E-4031 treatment. Ventricular-like APs recorded with the patch-clamp technique exhibited spontaneous Early after-depolarizations (EADs) only in A561P-UiPS CMs (in 2 of 7 cells). As shown in Figure 5C, EADs were exacerbated in the presence of E-4031, a reflection of the patient’s phenotype, which was characterized by arrhythmias when treated with the antitussive drug clobutinol. Quantitative analysis of the prolongation of APD50, APD75, and APD90 showed statically significant prolongation of APD75 and APD90 in both control and A561P-UhiPS CMs, with, as expected, greater prolongation in the mutated cells (Figure 5D).
Discussion
In the present study, we demonstrated that the use of patient-specific iPS cells obtained from urine samples is a powerful approach to modeling the subtlety of human cardiac arrhythmic disease in vitro. The cells were differentiated into functional CMs that reproduced both phenotypic characteristics of this LQT2 mutation, namely, (1) prolonged repolarization and (2) increased drug-induced arrhythmogenicity.
The opportunity of producing unique patient- and disease-specific hiPS cell lines7,26,27 provides genetically defined cellular human models for studying diseases, taking in consideration the patient’s specific genetic background. This approach has been applied successfully to cardiac arrhythmic diseases28, including LQT2 syndrome29–32, in which hiPS cell lines were derived from dermal fibroblasts; however, owing to the complications associated with skin biopsy collection, in all these studies, control hiPS cells were derived from genetically unrelated persons30–32 or even from human embryonic stem cell–derived fibroblasts and thus had a different genetic context.29 Nevertheless, direct reprogramming of somatic cells into pluripotent cells should address the need to study patient-specific cells as well as cells derived from control subjects, of any age and, ideally, from family relatives. This would allow testing of the specific role of a mutation in a pathological phenotype by comparing phenotypes in similar genetic backgrounds.
To avoid skin biopsy—an invasive procedure that raises ethical issues when collected from young persons or healthy relatives—less invasive methods are required. Recently, Zhou et al.3 were able to reprogram urine-derived cells, collected using a completely noninvasive method, into iPS cells. Our study shows that these cells can be differentiated into CMs and used to model an arrhythmic cardiac disease. There are, however, some limitations. First, in our laboratory, 79 urine samples were collected from the same number of persons for several ongoing studies. Using both penicillin and streptomycin in culture medium at usual concentrations, 8 samples were contaminated (data not shown). Cells from urine samples were isolated and expanded from only 45 (57%) donors. These results are comparable to those obtained by one of the first studies using urine cells to generate iPS cells.3 Nevertheless, the fact that cells cannot be obtained from all urine samples is an important limitation in comparison to peripheral blood mononuclear cells, which can usually be obtained from any participant. Consequently, if no urine cells can be isolated from urine samples of donors, either more urine samples must be harvested and cultivated or the use of peripheral blood mononuclear cells or skin biopsy may be necessary. Second, no “quarantine” time was necessary to eliminate contaminated samples, but usually 2 to 3 weeks were needed to obtain a sufficient number of cells for reprogramming. This culture duration is usual with fibroblasts from skin biopsies but is a limitation in comparison to peripheral blood mononuclear cells, which can be used immediately after purification; however, urine samples are easy to obtain from any participant, including intrafamilial controls and pediatric patients. Consequently, the use of urine samples may be a good starting point to obtain iPS cells, followed by blood harvesting in case of lack of urine cells. Finally, the study was performed with only 1 patient and 2 controls. Similar investigations with other patients with other HERG mutations would be useful to confirm the strength of this new model.
Intense research efforts have been directed at developing methods to differentiate CMs from hiPS cells by mimicking the cell-signaling environment during the early stages of cardiogenesis. We chose to use the matrix sandwich method, one of the most recently designed protocols, which has been reported to have among the highest efficacy for differentiation16; however, differentiation efficacy can be specific to iPS cell line. This method, for example, has been unsuccessful at differentiating cord blood–derived iPS cells as opposed to skin fibroblast–derived iPS cells due to their parental source memory.33 We tested whether this method would be efficient at differentiating urine-derived iPS cells. In this study, we showed that urine cells reprogrammed with nonintegrating episomal vectors can be differentiated into CMs from a monolayer cell culture using the matrix sandwich method.
To validate the use of urine-derived iPS cells for modeling cardiac arrhythmic disease, we selected a mild form of LQT2 syndrome, related to HERG, because the LQT2 syndrome is the reference model in studies using conventional skin fibroblast–derived iPS cells. Before the use of iPS cells, studies of HERG channel mutations, as for other channels, were conducted in heterologous expression systems, providing insights into the probable mechanism of LQT2. Those studies showed that defects in expression, trafficking, and/or function of the mutated channels can be responsible for the diseased phenotype; however, these systems hampered the complete understanding of this disease mechanism. Indeed, they are noncardiac cell types that do not provide a fully integrated vision, and some of the disease phenotypes cannot be investigated using these systems, forcing investigators to resort to noncellular methods such as mathematical modeling of AP.
The studied patient presented QT prolongation (corrected QT interval 597 ms) and was asymptomatic until he experienced syncope and torsades de pointes arrhythmias during treatment with clobutinol, a common antitussive drug.14 It has been proposed that the population prevalence of milder long QT syndrome mutations might be high,34 and it is now recognized that such form of long QT syndrome could manifest itself as a predisposition to drug-induced torsades de pointes, as in the case of the patient with the A561P mutation presented in this study. In addition, it has been demonstrated by Moss et al.35 that mutations in different locations of the HERG potassium-channel gene are associated with different levels of risk for arrhythmic cardiac events in LQT2, which makes it important to focus on mild phenotypes and to discern them from severe phenotypes. Interestingly, this study demonstrates that the use of urine-derived iPS cell models allows replication of the subtlety of a patient-specific phenotype, a result that widens the usefulness of iPS cells to model arrhythmogenic syndromes with complex phenotypes.
The A561P mutation has been the subject of a previous study conducted in the laboratory using transfected COS-7 cells.14 This study showed that the A561P mutation induced defects in HERG trafficking and biophysical properties. Because of the lack of a cardiac cellular model, its impact on AP duration had been tested using computer simulations.14 In the present study, we investigated the mutation-associated disease mechanism in human CMs differentiated from UhiPS cells carrying the HERG A561P mutation in comparison to 2 controls, 1 of which was UhiPS-CMs generated from a healthy family member (mother). The second control was derived from foreskin fibroblasts of a healthy person.15 Almost all studies that investigated the mutations associated with arrhythmic diseases in hiPS-CMs recognized that those cells are immature, with electrical properties similar to fetal or neonatal CMs; however, our results showed that these cells were capable of capturing specific traits of LQT2 syndrome, namely, reduced IKr amplitude leading to prolongation of AP. A discrepancy between the present study and our former study is that we did not confirm that the HERG A561P mutation results in modification of the voltage dependence of activation, as described in COS-7 cells.14 In COS-7 cells, this phenotype was probably due to the specific protein environment of the exogenous channel in COS-7 that may exacerbate subtle differences that do not exist in the native environment.
Our data further support the use of CMs differentiated from iPS cells rather than noncardiac heterologous expression systems for providing genuine knowledge of the mechanism for cardiac channelopathies. To date, they are the closest model to human CMs sourced directly from cardiac tissue, providing accurate insights into the molecular basis of disease phenotypes. Moreover, we demonstrated that iPS cells derived from a urine sample are an alternative strategy for cardiac arrhythmic disease modeling, with the advantage of facilitating the study of familial controls compared with more invasive fibroblast collection procedures, broadening the potential for iPS cell–guided personalized medicine.
Sources of Funding
This work was funded by grants from the Lefoulon Delalande Foundation, the Fédération Française de Cardiologie, Génavie and the Marie Curie European Actions (PIIF-GA-2012-331436) to Dr Gaborit, by grants from the FP7 Marie Curie IRG 277188/IPSMILD, the Leducq Foundation and the Génavie Foundation to Dr Si-Tayeb, by a grant from the VaCarME project funded by the Région Pays de la Loire to Dr Si-Tayeb, B. Champon and Dr Caillaud, and by a grant from the Lebanese University to Dr Zibara. M. Jouni was awarded a scholarship from Association of Scientific Orientation and Specialization (ASOS) and X. Latypova from Pasteur Mutualité Foundation. Dr Z. Es-Salah-Lamoureux was supported by grants from the Lefoulon Delalande Foundation, the French Foundation for Medical Research (FRM) and Génavie Foundation. Finally, this work was also supported by the Region Pays de Loire (Projet Devips).
Acknowledgments
We thank the cytometry core facility (Cytocell), the imaging core facility (MicroPICell), the animal housing facility (UTE), Therassay and the iPS cells core facility of SFR Santé F. Bonamy (Nantes) for their technical support. We also thank the clinical research assistants from Nantes Clinical Investigation Center for collecting patient’s urine samples. We are grateful to Simon Lecointe and the genomics core facility for their support with DNA sequencing. We also thank Séverine Abramatic and Hervé Le Marec for careful proofreading of the paper.
Disclosures
None.
Supporting Information
Figure S1.Characterization of human induced pluripotent stem cells from urine samples (UhiPS).Figure S2. Analysis of hERG gene expression.
Figure S3. Analysis hERG protein expression.
Table S1. TaqMan probes and SYBR Green primers.
Table S2. Quantitative parameters used for classification of action potentials obtained from patch-clam experiments on control cells.
References
- Zanella F, Lyon RC, Sheikh F. Modeling heart disease in a dish: from somatic cells to disease-relevant cardiomyocytes. Trends Cardiovasc Med. 2014;24:32–44. doi: 10.1016/j.tcm.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Nagata N, Kurokawa H, Yamanaka S. Ips cells: a game changer for future medicine. EMBO J. 2014;33:409–417. doi: 10.1002/embj.201387098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J, Wang Y, Zhang Y, Zhuang Q, Li Y, Bao X, Tse HF, Grillari J, Grillari-Voglauer R, Pei D, Esteban MA. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc. 2012;7:2080–2089. doi: 10.1038/nprot.2012.115. [DOI] [PubMed] [Google Scholar]
- Xue Y, Cai X, Wang L, Liao B, Zhang H, Shan Y, Chen Q, Zhou T, Li X, Hou J, Chen S, Luo R, Qin D, Pei D, Pan G. Generating a non-integrating human induced pluripotent stem cell bank from urine-derived cells. PLoS One. 2013;8:e70573. doi: 10.1371/journal.pone.0070573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KI, Kim HT, Hwang DY. Footprint- and xeno-free human ipscs derived from urine cells using extracellular matrix-based culture conditions. Biomaterials. 2014;35:8330–8338. doi: 10.1016/j.biomaterials.2014.05.059. [DOI] [PubMed] [Google Scholar]
- Gerbal-Chaloin S, Funakoshi N, Caillaud A, Gondeau C, Champon B, Si-Tayeb K. Human induced pluripotent stem cells in hepatology: beyond the proof of concept. Am J Pathol. 2014;184:332–347. doi: 10.1016/j.ajpath.2013.09.026. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Gao Y, Gordon A, Wang ZZ, Qian Z, Wu WS. Efficient generation of fully reprogrammed human ips cells via polycistronic retroviral vector and a new cocktail of chemical compounds. PLoS ONE. 2011;6:e26592. doi: 10.1371/journal.pone.0026592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Chau KF, Vodyanik MA, Jiang J, Jiang Y. Efficient feeder-free episomal reprogramming with small molecules. PLoS ONE. 2011;6:e17557. doi: 10.1371/journal.pone.0017557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Yamanaka S. Induced pluripotent stem cells: opportunities and challenges. Philos Trans R Soc Lond B Biol Sci. 2011;366:2198–2207. doi: 10.1098/rstb.2011.0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X, Mack DL, Moreno CM, Strande JL, Mathieu J, Shi Y, Markert CD, Wang Z, Liu G, Lawlor MW, Moorefield EC, Jones TN, Fugate JA, Furth ME, Murry CE, Ruohola-Baker H, Zhang Y, Santana LF, Childers MK. Dystrophin-deficient cardiomyocytes derived from human urine: new biologic reagents for drug discovery. Stem Cell Res. 2014;12:467–480. doi: 10.1016/j.scr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Seufferlein T, Illing A, Homann J. Modelling human channelopathies using induced pluripotent stem cells: a comprehensive review. Stem Cells Int. 2013;2013:496501. doi: 10.1155/2013/496501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellocq C, Wilders R, Schott JJ, Louerat-Oriou B, Boisseau P, Le Marec H, Escande D, Baro I. A common antitussive drug, clobutinol, precipitates the long qt syndrome 2. Mol Pharmacol. 2004;66:1093–1102. doi: 10.1124/mol.104.001065. [DOI] [PubMed] [Google Scholar]
- Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Klos M, Wilson GF, Herman AM, Lian X, Raval KK, Barron MR, Hou L, Soerens AG, Yu J, Palecek SP, Lyons GE, Thomson JA, Herron TJ, Jalife J, Kamp TJ. Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: the matrix sandwich method. Circ Res. 2012;111:1125–1136. doi: 10.1161/CIRCRESAHA.112.273144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrenoire C, Wang K, Tung KW, Chung WK, Pass RH, Lu JT, Jean JC, Omari A, Sampson KJ, Kotton DN, Keller G, Kass RS. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol. 2013;141:61–72. doi: 10.1085/jgp.201210899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero-Gutierrez B, Anzola A, Martinez-Augustin O, de Medina FS. Stain-free detection as loading control alternative to ponceau and housekeeping protein immunodetection in western blotting. Anal Biochem. 2014;467:1–3. doi: 10.1016/j.ab.2014.08.027. [DOI] [PubMed] [Google Scholar]
- Gurtler A, Kunz N, Gomolka M, Hornhardt S, Friedl AA, McDonald K, Kohn JE, Posch A. Stain-free technology as a normalization tool in western blot analysis. Anal Biochem. 2013;433:105–111. doi: 10.1016/j.ab.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Loussouarn G, Baro I, Escande D. KCNQ1K+ channel-mediated cardiac channelopathies. Methods Mol Biol. 2006;337:167–183. doi: 10.1385/1-59745-095-2:167. [DOI] [PubMed] [Google Scholar]
- Bellin M, Casini S, Davis RP, D’Aniello C, Haas J, Ward-van Oostwaard D, Tertoolen LG, Jung CB, Elliott DA, Welling A, Laugwitz KL, Moretti A, Mummery CL. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. 2013;32:3161–3175. doi: 10.1038/emboj.2013.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP, Thomson JA, Kamp TJ. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30–e41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepac A, Si-Tayeb K, Sedlic F, Barrett S, Canfield S, Duncan SA, Bosnjak ZJ, Lough JW. Comparison of cardiomyogenic potential among human ESC and IPSC lines. Cell Transplant. 2012;21:2523–2530. doi: 10.3727/096368912X653165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, January CT. Properties of herg channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J. 1998;74:230–241. doi: 10.1016/S0006-3495(98)77782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy CE, Rudy Y. Cellular consequences of herg mutations in the long QT syndrome: precursors to sudden cardiac death. Cardiovasc Res. 2001;50:301–313. doi: 10.1016/s0008-6363(00)00293-5. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Priori SG, Napolitano C, Di Pasquale E, Condorelli G. Induced pluripotent stem cell-derived cardiomyocytes in studies of inherited arrhythmias. J Clin Invest. 2013;123:84–91. doi: 10.1172/JCI62838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A, Denning C. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J. 2011;32:952–962. doi: 10.1093/eurheartj/ehr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M, Gepstein L. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- Lahti AL, Kujala VJ, Chapman H, Koivisto AP, Pekkanen-Mattila M, Kerkela E, Hyttinen J, Kontula K, Swan H, Conklin BR, Yamanaka S, Silvennoinen O, Aalto-Setala K. Model for long QT syndrome type 2 using human IPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech. 2012;5:220–230. doi: 10.1242/dmm.008409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Sequiera GL, Ramachandra CJ, Sudibyo Y, Chung Y, Sheng J, Wong KY, Tan TH, Wong P, Liew R, Shim W. Re-trafficking of herg reverses long QT syndrome 2 phenotype in human IPS-derived cardiomyocytes. Cardiovasc Res. 2014;102:497–506. doi: 10.1093/cvr/cvu060. [DOI] [PubMed] [Google Scholar]
- Ye L, Zhang S, Greder L, Dutton J, Keirstead SA, Lepley M, Zhang L, Kaufman D, Zhang J. Effective cardiac myocyte differentiation of human induced pluripotent stem cells requires vegf. PLoS ONE. 2013;8:e53764. doi: 10.1371/journal.pone.0053764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjamaa A, Salomaa V, Newton-Cheh C, Porthan K, Reunanen A, Karanko H, Jula A, Lahermo P, Vaananen H, Toivonen L, Swan H, Viitasalo M, Nieminen MS, Peltonen L, Oikarinen L, Palotie A, Kontula K. High prevalence of four long QT syndrome founder mutations in the finnish population. Ann Med. 2009;41:234–240. doi: 10.1080/07853890802668530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, Towbin JA, Keating MT, Priori SG, Schwartz PJ, Vincent GM, Robinson JL, Andrews ML, Feng C, Hall WJ, Medina A, Zhang L, Wang Z. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 2002;105:794–799. doi: 10.1161/hc0702.105124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.Characterization of human induced pluripotent stem cells from urine samples (UhiPS).Figure S2. Analysis of hERG gene expression.
Figure S3. Analysis hERG protein expression.
Table S1. TaqMan probes and SYBR Green primers.
Table S2. Quantitative parameters used for classification of action potentials obtained from patch-clam experiments on control cells.