

Abstract
Background
Intracellular Na+ concentration ([Na+]i) regulates Ca2+ cycling, contractility, metabolism, and electrical stability of the heart. [Na+]i is elevated in heart failure, leading to arrhythmias and oxidative stress. We hypothesized that myocyte [Na+]i is also increased in type 2 diabetes (T2D) due to enhanced activity of the Na+–glucose cotransporter.
Methods and Results
To test this hypothesis, we used myocardial tissue from humans with T2D and a rat model of late-onset T2D (HIP rat). Western blot analysis showed increased Na+–glucose cotransporter expression in failing hearts from T2D patients compared with nondiabetic persons (by 73±13%) and in HIP rat hearts versus wild-type (WT) littermates (by 61±8%). [Na+]i was elevated in HIP rat myocytes both at rest (14.7±0.9 versus 11.4±0.7 mmol/L in WT) and during electrical stimulation (17.3±0.8 versus 15.0±0.7 mmol/L); however, the Na+/K+-pump function was similar in HIP and WT cells, suggesting that higher [Na+]i is due to enhanced Na+ entry in diabetic hearts. Indeed, Na+ influx was significantly larger in myocytes from HIP versus WT rats (1.77±0.11 versus 1.29±0.06 mmol/L per minute). Na+–glucose cotransporter inhibition with phlorizin or glucose-free solution greatly reduced Na+ influx in HIP myocytes (to 1.20±0.16 mmol/L per minute), whereas it had no effect in WT cells. Phlorizin also significantly decreased glucose uptake in HIP myocytes (by 33±9%) but not in WT, indicating an increased reliance on the Na+–glucose cotransporter for glucose uptake in T2D hearts.
Conclusions
Myocyte Na+–glucose cotransport is enhanced in T2D, which increases Na+ influx and causes Na+ overload. Higher [Na+]i may contribute to arrhythmogenesis and oxidative stress in diabetic hearts.
Keywords: heart, intracellular Na+ concentration, Na+–glucose cotransporter, type 2 diabetes
Type 2 diabetes (T2D) heightens the risk of developing heart failure (HF), arrhythmias, and sudden cardiac death, even in the absence of vascular complications such as coronary artery disease and hypertension.1–5 Severe heart dysfunction often occurs before full-blown diabetes is diagnosed (prediabetes),6–9 suggesting an early and critical maladaptation of myocardial structure and function. The mechanisms that link T2D to the electrical and structural remodeling of the myocardium are multifactorial but poorly understood.
In the heart, intracellular Na+ concentration ([Na+]i) and Na+ transport are critical modulators of Ca2+ cycling, contractility, energy supply and demand, oxidative state, action potential waveform, and propensity for arrhythmias.10 [Na+]i is elevated in HF,11–15 which mitigates contractile dysfunction by favoring more Ca2+ influx via the Na+/Ca2+ exchanger (NCX), thus augmenting the cellular and sarcoplasmic reticulum Ca2+ load; however, increased sarcoplasmic reticulum Ca2+ load also raises the risk for arrhythmias.16 At the same time, high [Na+]i induces oxidative stress by activating the mitochondrial NCX, leading to lower free Ca2+ in the mitochondria and slower regeneration of NADPH, which is needed to neutralize the reactive oxygen species produced in the mitochondria.17–19 Both decreased Na+ extrusion and increased Na+ entry may contribute to the rise in [Na+]i in HF, depending on the etiology of the disease.
Few studies have so far investigated myocardial Na+ transport in diabetes. Decreased activity of the Na+/K+ pump20,21 (NKA) and NCX22,23 were reported in hearts from animals with type 1 diabetes. The myocardial Na+/H+ exchanger was found to be enhanced in hearts from T2D animals,24,25 which contributed to left ventricular hypertrophy24 and led to higher [Na+]i gain during ischemia–reperfusion.25 Na+/H+ exchanger inhibition was associated with a lower incidence of ventricular tachycardia and fibrillation in T2D hearts during ischemia–reperfusion25; however, it is currently unknown whether myocyte [Na+]i is altered in T2D and how such alterations affect the progression of diabetic heart disease.
We hypothesized that [Na+]i is elevated in diabetic hearts due to enhanced Na+ entry through the Na+–glucose cotransporter (SGLT), which couples the uptake of Na+ ions and glucose. SGLT is present in the heart,26,27 but its physiological and pathophysiological roles are not fully understood. Recent data demonstrated that cardiac-specific overexpression of the SGLT1 isoform causes hypertrophy and left ventricular dysfunction.28 The mRNA level of cardiac SGLT1 is increased in HF and T2D,27 2 pathological conditions that render the heart insulin resistant. SGLT1 seems to be required for activation of NADPH oxidase by hyperglycemia.29 Moreover, SGLT-mediated glucose uptake has been linked to PRKAG2 cardiomyopathy,30 a glycogen storage cardiomyopathy caused by mutations in the gene encoding the γ2 subunit of AMP-activated protein kinase. The activity of cardiac SGLT may be increased in T2D as an adaptation to both chronic hyperglycemia and the reduced ability of myocytes to take up glucose through insulin-dependent pathways. We tested this hypothesis by analyzing [Na+]i and SGLT expression and function in hearts from rats and humans with T2D versus nondiabetic controls.
Methods
Experimental Animals
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996) and was approved by the institutional animal care and use committee at the University of Kentucky. The T2D animal model used in this study is the HIP rat. HIP rats are obese Sprague-Dawley rats that express human amylin, an amyloidogenic hormone cosecreted with insulin, in the pancreatic β cells on the insulin II promoter.31 Increased amylin secretion in the insulin-resistant prediabetic state causes its aggregation and deposition in pancreas in both humans31 and HIP rats,32 which leads to gradual decline of β-cell mass and development of full T2D.31–33 Because rodent amylin is not amyloidogenic, HIP rats are unique within rodent T2D models in showing amylin amyloid deposition. Together with the late-onset development of T2D, this characteristic makes the HIP rat a “humanized” T2D animal model. We recently demonstrated that amylin accumulation in humans with obesity or T2D and in HIP rats is not limited to the pancreas but also occurs in peripheral organs, including the heart.34,35 HIP rats were monitored biweekly for nonfasting blood glucose (with a glucometer) and used in experiments when glucose was >400 mg/dL (22.5 mmol/L) for >8 consecutive weeks (ie, late-stage T2D). Age-matched wild-type (WT) littermates were used as nondiabetic controls. Because the genetic background of HIP rats is the Sprague-Dawley rat, the WT animals are also obese. In fact, the body weight of the WT rats (698±20 g) was larger than that of HIP rats (611±25 g) at the time they were used for experiments, due to weight loss in HIP rats after development of full-blown diabetes. Sample sizes of 20 diabetic male HIP rats and 20 WT male rats were used in this study.
Human Samples
Human left ventricular tissue was from hearts obtained at the time of orthotopic heart transplantation at the Hospital of the University of Pennsylvania (for failing hearts) or organ donation (for nonfailing hearts), in accordance with institutional review board approval and after obtaining informed consent. The hearts were divided into 5 pathological groups, depending on the presence of T2D and HF and body weight. The average age and body mass index, sex distribution, and HF etiology (ischemic versus nonischemic) of the patients in the 5 groups are shown in Table 1. The first group is that of hearts from patients with both T2D and HF (T2D-HF group; 4 hearts). T2D in these patients was controlled with either insulin (for 2 cases) or oral glucose-lowering drugs (n=2). Failing hearts from non-T2D patients were divided into the failing-obese (OB-HF; n=6) and failing-lean (L-HF; n=7) groups based on the body mass index of the patient (≥30 kg/cm2 for the obese group; <30 kg/cm2 for lean). Nonfailing hearts were also divided into OB-NF (n=6) and L-NF (n=6) groups.
Table 1.
Average Age and BMI (±SD), Sex Distribution, and HF Etiology (Ischemic Versus Nonischemic) of the Patients Whose Hearts Were Used in this Study
| Group | Age | BMI (kg/cm2) | Sex | HF Etiology |
|---|---|---|---|---|
| T2D-HF | 56±8 | 32.6±1.5 | All male | 3 ischemic, 1 dilated |
| OB-HF | 51±12 | 33.5±3.7 | 5 male, 1 female | 2 ischemic, 4 dilated |
| L-HF | 53±9 | 21.8±2.0 | 4 male, 3 female | 5 dilated, 2 hypertrophic |
| OB-NF | 55±13 | 33.0±2.3 | 4 male, 2 female | NA |
| L-NF | 61±18 | 25.5±1.8 | 3 male, 3 female | NA |
BMI indicates body mass index; HF, heart failure; L, lean; NA, not available; NF, nonfailing heart; OB, obese; T2D, type 2 diabetes.
Western Blot
Left ventricular tissue was homogenized in homogenization buffer containing 10 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 0.1% sodium dodecyl sulfate, 1% Triton X-100, 1% sodium deoxycholate, 5 mmol/L EDTA, 1 mmol/L NaF, 1 mmol/L sodium orthovanadate, and protease and phosphatase inhibitor cocktail (Calbiochem). After SDS-PAGE electrophoresis, proteins were transferred on nitrocellulose membranes, blocked, and probed with primary antibodies against SGLT1 (Santa Cruz Biotechnology) and the α1 (Millipore) and α2 (Millipore) isoforms of the Na+/K+-ATPase. Equal loading was verified by reprobing with anti-GAPDH (Abcam). Signal intensity analysis was performed in ImageJ (National Institutes of Health). For each gel, we averaged the signal intensity of the corresponding bands for the control samples. We then normalized the signal intensity in all lanes to this average. This procedure was repeated on at least 4 gels, and for each sample, the normalized signal intensity was averaged. In the end, we calculated averages over all groups used.
Immunofluorescence
Freshly isolated myocytes were plated on laminin-coated coverslips, fixed with 4% paraformaldehyde, permeabilized with 50 μg/mL saponin (15 minutes), blocked with 2% goat serum, and labeled with a polyclonal primary antibody against SGLT1 (Santa Cruz Biotechnology). Anti-rabbit Alexa Fluor 488 was used as a secondary antibody, and fluorescence images were collected with a laser scanning confocal microscope.
Cardiac Myocyte Isolation
Rats were anesthetized with 3% to 5% isoflurane (100% O2), then hearts were excised quickly, placed on a gravity-driven Langendorff perfusion apparatus, and perfused with 1 mg/mL collagenase. When the heart became flaccid (≈15 minutes), the left ventricular tissue was cut into small pieces, dispersed, and filtered, and the myocyte suspension was rinsed several times. The standard Tyrode’s solution used in experiments with live myocytes contained (in mmol/L) 140 NaCl, 4 KCl, 1 MgCl2, 10 glucose, 5 HEPES, and 1 CaCl2 (pH 7.4). All experiments were done at room temperature (23°C to 25°C).
[Na+]i Measurements
[Na+]i was measured using the fluorescent indicator SBFI (TefLabs), as described previously.11 The SBFI ratio was calibrated at the end of each experiment using divalent-free solutions with 0, 10, or 20 mmol/L of extracellular Na+ in the presence of 10 μmol/L gramicidin and 100 μmol/L strophanthidin. With our experimental setup, the SBFI ratio increased linearly with [Na+]i up to 50 mmol/L.
To measure NKA function in intact myocytes, cells were Na+ loaded by blocking the pump in K+-free solution. [Na+]i decline was measured in Na+-free solution, differentiated, plotted as a function of [Na+]i, and fitted with the Hill equation.11 Such measurements were done in the absence and presence of 10 mmol/L ouabain in the Na+-free solution. In the first case, Na+ efflux was mediated by both NKA and passive leak, whereas in the second case, the only contribution was that of the passive Na+ leak.
Resting Na+ influx was taken as the initial rate of [Na+]i rise following abrupt NKA inhibition with 10 mmol/L ouabain.
Glucose Uptake Measurements
Myocyte glucose uptake was measured using the fluorescent glucose analog 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose, or 2-NBDG (Life Technologies).36,37 Myocytes were preincubated in glucose-free Tyrode’s solution for 60 minutes, and then they were incubated with 1 mmol/L 2-NBDG for 30 minutes in the absence or presence of 250 μmol/L phlorizin or 50 nmol/L insulin. External 2-NBDG was washed off, and the cells were imaged with a confocal microscope using identical settings for all measurements. For each rat, we calculated the mean fluorescence intensity per group (a minimum of 20 cells per group), and we normalized the fluorescence in the phlorizin and insulin groups to the control. Measurements were repeated in cells from 6 HIP and 6 WT rats.
Statistical Analysis
The data are presented as mean±SE. For data that passed the D’Agostino and Pearson normality test, statistical discriminations were performed using a 2-tailed unpaired Student t test when comparing 2 groups and 1-way ANOVA with Bonferroni’s post hoc test when comparing multiple groups. When the sample size was too small to perform the normality test, data were analyzed with nonparametric tests (Mann–Whitney when comparing 2 columns, Kruskal–Wallis followed by Dunn’s post test when comparing multiple columns). Differences were considered statistically significant when P<0.05.
Results
Increased SGLT1 Protein Expression in Hearts From Diabetic and Obese Humans and Rats
We started to test the hypothesis that Na+–glucose cotransport is enhanced in T2D hearts by evaluating the effect of T2D on the protein expression of SGLT1 in human hearts, using Western blots (Figure 1A). Because a previous study reported an increased mRNA level of SGLT1 in HF,27 we compared failing hearts from T2D, obese (likely in an insulin-resistant prediabetic state), and lean (metabolically normal) patients. We found that SGLT1 expression was upregulated (by 73±13%) in hearts from patients with T2D versus nondiabetic, lean participants (Figure 1A). Moreover, SGLT1 expression was even increased in hearts from obese patients who were not diagnosed with T2D (by 31±5%) (Figure 1A). We also noted that the presence of HF alone resulted in higher cardiac SGLT1 expression in both lean and obese participants (Figure 1B), in agreement with prior mRNA data.27
Figure 1.
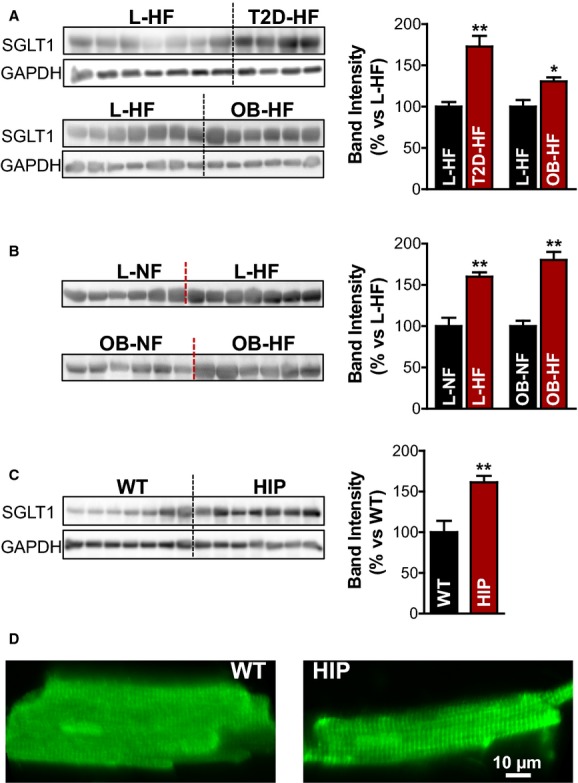
Increased SGLT1 protein expression in hearts from humans and rats with T2D. A, Western blots with an anti-SGLT1 antibody in homogenates of failing hearts from patients with T2D (T2D-HF group; 4 hearts) or obese (OB-HF group; 6 hearts) vs lean participants (L-HF group; 7 hearts). GAPDH was used as loading control, and experiments were repeated 4 times. Bar graph in the right panel shows the relative band intensity. B, SGLT1 expression in homogenates from failing vs nonfailing human hearts from lean (top) and obese (bottom) participants. C, Western blots with an anti-SGLT1 antibody in diabetic HIP vs WT heart homogenates. D, Representative immunofluorescence images of rat (WT and HIP) myocytes labeled with an anti-SGLT1 antibody. In both cases, SGLT1 is localized at the T-tubules. HF indicates heart failure; HIP, model of late-onset T2D; L, lean; NF, nonfailing heart; OB, obese; SGLT, Na+-glucose cotransporter; T2D, type 2 diabetes; WT, wild-type. *P<0.05, **P<0.01.
A comparable level of SGLT1 upregulation occurred in hearts from diabetic HIP rats versus WT littermates (61±8%) (Figure 1C). HIP rats develop late-onset T2D32,33 and, as we previously showed, manifest a cardiac phenotype that provides a good resemblance of the diabetic cardiomyopathy in humans with T2D.34,35
To transport Na+ and glucose, SGLT has to be localized at the membrane. We assessed the distribution of SGLT1 in cardiac myocytes from HIP and WT rats by immunofluorescent labeling with an anti-SGLT1 antibody (Figure 1D). These experiments showed that in both WT and HIP rat myocytes, SGLT1 is positioned in a striated pattern with ≈2 μm between the transverse striations, which strongly suggests localization at the T-tubules. Consequently, SGLT1 is localized at the sarcolemma, and its expression is elevated in hearts from humans and rats with T2D.
Elevated [Na+]i in Myocytes From Diabetic Hearts
To determine whether the increase in SGLT expression in diabetic hearts affects myocyte Na+ homeostasis, we measured [Na+]i in myocytes isolated from diabetic HIP and WT littermate rats using the fluorescent indicator SBFI (Figure 2). After measuring the resting [Na+]i, myocytes were electrically stimulated to contract at 2 Hz (Figure 2A). As in our previous studies,11,38,39 [Na+]i increased when the myocytes started to contract, reaching a new steady state within 5 to 8 minutes. [Na+]i was significantly higher in both resting (14.7±0.9 versus 11.4±0.7 mmol/L in WT) and contracting (17.3±0.8 mmol/L versus 15.0±0.7 mmol/L) HIP rat myocytes compared with WT (Figure 2B). Consequently, [Na+]i is elevated in diabetic hearts.
Figure 2.
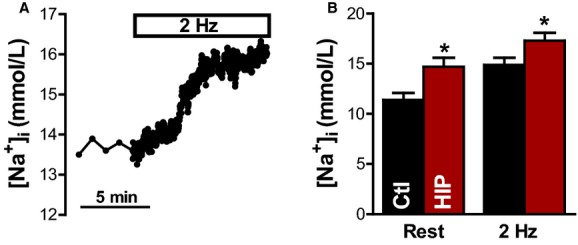
[Na+]i is elevated in myocytes from diabetic HIP rats vs WT. A, Representative example of [Na+]i measurements in a HIP rat myocyte. [Na+]i was first measured at rest, then cells were electrically stimulated at 2 Hz. B, Mean steady-state [Na+]i at rest and during electrical stimulation in myocytes from HIP (9 cells from 5 rats) and WT rats (7 myocytes from 5 animals). Ctl indicates control; HIP, model of late-onset T2D; [Na+]i, intracellular Na+ concentration; WT, wild-type. *P<0.05
Unchanged NKA Function in Diabetic Hearts at Physiological [Na+]i
Higher [Na+]i in diabetic hearts could be caused by reduced Na+ extrusion through NKA or increased Na+ influx (or a combination of both). To uncover the mechanisms responsible for elevated [Na+]i in T2D, we assessed NKA function in myocytes from HIP and WT rats (Figure 3). Intact myocytes were Na+ loaded by blocking NKA in K+-free solution (Figure 3A). NKA was reactivated (by reintroducing external K+), and [Na+]i decline was measured in an Na+-free external solution. Because cell volume does not change in these conditions,11 [Na+]i decline reflects Na+ extrusion. The rate of Na+ extrusion was then calculated as a function of [Na+]i by numerically differentiating the [Na+]i decline. The decrease in [Na+]i was mediated mainly by NKA, but there was also a small component due to passive Na+ leak (because the normal transmembrane Na+ gradient is now inverted). We separately measured this Na+ leak in experiments in which [Na+]i decay occurred in the presence of 10 mmol/L ouabain, which completely blocks NKA (Figure 3A). The rate of NKA-mediated Na+ extrusion was then calculated as the difference between the total rate of [Na+]i decline and the rate of passive Na+ leak. As shown in Figure 3B, there were no significant differences between HIP and WT myocytes at [Na+]i in the physiological range (0 to 20 mmol/L). The curves were fitted with a Hill expression to derive the maximum NKA rate (6.6±0.6 versus 7.5±0.6 mmol/L per minute in HIP versus WT, respectively; P=0.33), the apparent binding constant for internal Na+ (8.9±1.3 versus 12.1±1.2 mmol/L in HIP versus WT, respectively; P=0.11), and the Hill coefficient (2.0±0.6 versus 2.2±0.5 in HIP versus WT, respectively; P=0.82).
Figure 3.
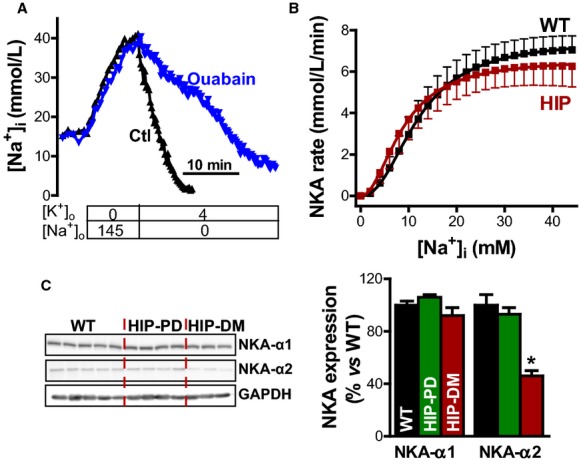
Unchanged NKA function in myocytes from diabetic rats. A, Representative example of Na+ extrusion measurements in 2 myocytes from HIP rats. Intact myocytes were Na+ loaded by blocking NKA in K+ free solution, and then external K+ was reintroduced, and [Na+]i decline was measured in the absence (Ctl) or presence (ouabain) of 10 mmol/L ouabain. [Na+]i decline was then numerically differentiated to calculate the rate of Na+ extrusion. B, Rate of NKA-mediated Na+ extrusion as a function of [Na+]i in intact myocytes from HIP and WT rats (9 cells from 5 HIP rats and 6 cells from 5 WT rats). C, Protein expression of the α1 and α2 isoforms of the NKA in hearts from WT rats and HIP rats in the prediabetic (HIP-PD; nonfasting blood glucose in the 150 to 200 mg/dL range) and fully diabetic (HIP-DM; blood glucose >400 mg/dL for >8 consecutive weeks) groups. NKA indicates Na+/K+ pump; HIP, model of late-onset T2D; [Na+]i, intracellular Na+ concentration; WT, wild-type. *P<0.05
In agreement with largely unaltered NKA function in myocytes from diabetic rats versus control, Western blots showed little change in the expression of NKA-α isoforms (Figure 3C). Indeed, NKA-α1 expression was similar in myocytes from HIP (in the prediabetic and fully diabetic states) and WT rats. The expression of the α2 subunit was reduced significantly (by 54±4%) in the fully diabetic HIP rats (Figure 3C); however, NKA-α2 represents <25% of the total NKA in rat myocytes.40,41 Consequently, a 50% reduction in NKA-α2 means that total NKA expression is reduced by ≈12.5%, which has only a minor effect on total NKA function. Nonetheless, several studies have demonstrated that NKA-α2 has a preferential role in regulating Ca2+ cycling and cardiac hypertrophy.41–44 A reduction in NKA activity (local or global) improves contractility; therefore, the selective downregulation of NKA-α2 may be an adaptation meant to limit the decrease in cardiac function in diabetic hearts, but lower NKA-α2 also promotes arrhythmogenesis and hypertrophy.
In summary, these data indicate that NKA function is not significantly altered in HIP rat myocytes; therefore, elevated [Na+]i must be due to increased Na+ influx.
Enhanced Na+ Entry Through SGLT in Myocytes From Diabetic Rats
We measured Na+ influx in resting myocytes from control and diabetic rats as the slope of the initial increase in [Na+]i on blocking NKA with 10 mmol/L ouabain (Figure 4A and 4B). As expected from the findings that [Na+]i is elevated and NKA function is unchanged, Na+ influx was significantly larger in HIP myocytes versus WT (1.77±0.11 versus 1.29±0.06 mmol/L per minute) (Figure 4B).
Figure 4.
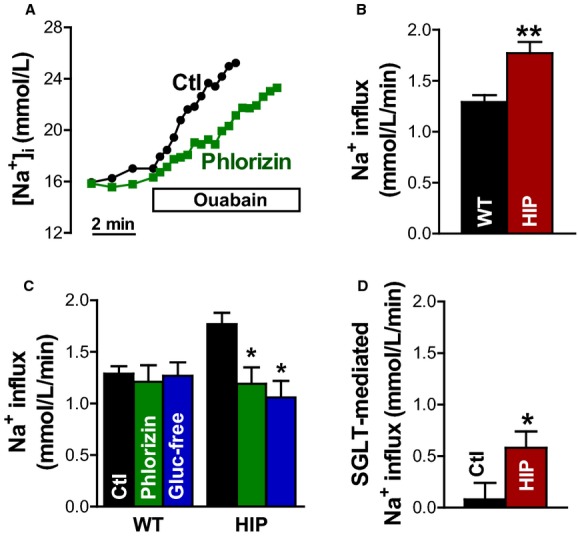
Na+ influx in myocytes from control and diabetic rats. A, Representative example of Na+ influx measurements in resting HIP rat myocytes in the absence (Ctl) and presence of the SGLT inhibitor phlorizin (250 μmol/L). B, Na+ influx calculated as the slope of the initial increase in [Na+]i on blocking the Na+/K+ pump with 10 mmol/L ouabain in myocytes from HIP and WT rats. C, Effect of SGLT inhibition with phlorizin and glucose-free external solution on the rate of Na+ entry in myocytes from WT and diabetic HIP rats. D, SGLT-mediated Na+ influx derived by subtracting the rate of Na+ entry with SGLT blocked from the total Na+ influx. Mean of >7 cells from at least 5 rats in each group. HIP indicates model of late-onset T2D; [Na+]i, intracellular Na+ concentration; SGLT, Na+–glucose cotransporter; WT, wild-type. *P<0.05, **P<0.01
Multiple pathways mediate Na+ entry in cardiac myocytes. To assess the contribution of SGLT to the excess Na+ entry in myocytes from diabetic rats, we measured Na+ influx with SGLT blocked by a specific pharmacological inhibitor (phlorizin, 250 μmol/L) or by omission of glucose from the external solution (Figure 4A and 4C). SGLT inhibition through either method had no effect on WT myocytes but significantly reduced Na+ entry in myocytes from diabetic rats (to 1.20±0.16 and 1.09±0.16 mmol/L per minute for phlorizin and glucose-free solution, respectively) (Figure 4C). In fact, with SGLT blocked, Na+ influx was similar in WT and HIP myocytes. The SGLT-mediated Na+ entry, calculated by subtracting the Na+ influx with SGLT blocked from the total rate of Na+ entry, was greatly increased in HIP versus WT myocytes (Figure 4D). Consequently, SGLT activity is augmented in diabetic hearts, in which it becomes an important factor in regulating [Na+]i.
Increased SGLT-Mediated Glucose Uptake in Myocytes From Diabetic Rats
To further investigate SGLT function in myocytes from diabetic and control rats, we measured glucose uptake using a fluorescent glucose analog, 2-NBDG (Figure 5).36,37 2-NBDG is taken up into cells through both Glut transporters45,46 and SGLT.47,48 Glucose uptake was measured under control conditions, with SGLT blocked by 250 μmol/L phlorizin, and in the presence of 50 nmol/L insulin (Figure 5). As expected, insulin significantly enhanced glucose uptake in myocytes from WT rats (by 43±12%), whereas phlorizin yielded no significant effect (Figure 5A and 5B). In contrast, insulin produced only a small (not significant) effect in myocytes from T2D rats, but SGLT inhibition greatly reduced the glucose uptake (by 33±9%) (Figure 5A and 5C). These results suggest that diabetic hearts rely more heavily on SGLT for taking up glucose, possibly as a maladaptation to insulin resistance.
Figure 5.
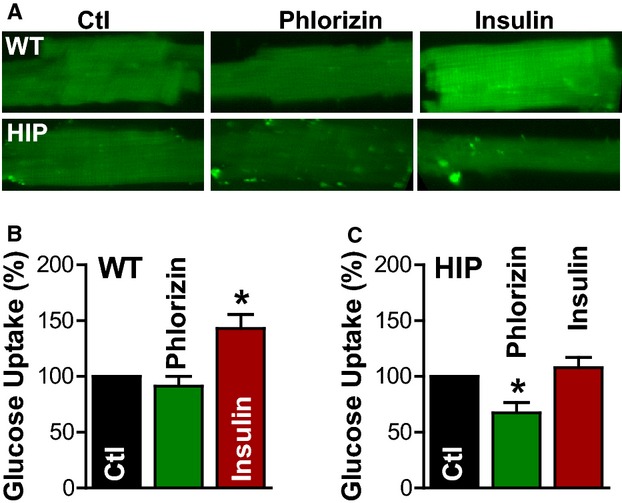
Increased SGLT-mediated glucose uptake in myocytes from diabetic rats. Glucose uptake was measured using the fluorescent glucose analog 2-NBDG. A, Representative fluorescent images of myocytes incubated with 1 mmol/L 2-NBDG for 30 minutes under control conditions (Ctl), with SGLT blocked by 250 μmol/L phlorizin and in the presence of 50 nmol/L insulin. B and C, Mean fluorescence intensity (relative to the control condition) in myocytes from WT rats (B) and diabetic HIP rats (C); n=6 rats, ≥20 cells/rat for both WT and HIP. HIP indicates model of late-onset T2D; SGLT, Na+–glucose cotransporter; WT, wild-type. *P<0.05
Discussion
We found increased SGLT expression in failing hearts from T2D patients compared with those of nondiabetic participants and in diabetic HIP versus WT rat hearts. HIP rat myocytes exhibited enhanced SGLT-mediated Na+ (and glucose) uptake, which resulted in higher [Na+]i.
Altered Myocyte Na+ Homeostasis in Diabetic Hearts
There are few reports of altered cardiac Na+ transport in diabetic animal models (mostly type 1 diabetes)20–25; however, it is currently unclear whether myocyte Na+ homeostasis is perturbed in T2D. We found that [Na+]i was elevated in myocytes from rats with late-onset T2D (HIP rats). In prior studies, we demonstrated that HIP rats manifest cardiac dysfunction that closely resembles, as phenotype and time course, the heart disease in patients with T2D.34,35 Briefly, early diastolic dysfunction and mild hypertrophy, which start in the prediabetic stage, evolve to systolic dysfunction, overt hypertrophy, and left ventricular dilation with the advancement of T2D.
The increase in [Na+]i in HIP rat hearts (≈3 mmol/L) is comparable to the [Na+]i rise reported by us11 and others12–15 in myocytes from failing hearts. In HF, elevated [Na+]i was directly linked to a higher propensity for arrhythmias,16,49 oxidative stress17–19 and hypertrophy.50 HIP rats also display increased arrhythmogenicity,51 oxidative stress, and hypertrophy.34,35 Our current data suggest that high [Na+]i may contribute to the multifactorial mechanisms through which T2D heightens the risk of developing HF and sudden cardiac death.
Na+–Glucose Cotransport Is Upregulated in Diabetic Hearts
We found that the increased [Na+]i in diabetic myocytes is due to a larger Na+ influx (by ≈40% versus control) as opposed to reduced Na+ extrusion via the NKA. There are multiple routes for Na+ entry in myocytes. Quantitatively, NCX brings in the largest amount (≈40% of the total Na+ influx in resting rat myocytes38). It is unlikely, however, that NCX contributes to the enhanced Na+ entry in T2D myocytes because our previous studies revealed reduced NCX expression (by about 20%34) and function (measured as the rate of decline of the caffeine induced Ca2+ transients35) in HIP rat hearts. Consequently, Na+ uptake through NCX may actually be reduced in diabetic hearts.
A pathway rarely mentioned when discussing cardiac Na+ influx is SGLT, which couples ion transport to energy substrate metabolism by taking up 2 Na+ ions together with 1 glucose molecule. SGLT expression was initially thought to be limited to the epithelial cells from the intestine and kidneys, but more recent data have demonstrated that at least the SGLT1 isoform is also present in the heart.26,27 The physiological and pathophysiological roles of SGLT in the heart are poorly understood. We found that SGLT-mediated Na+ and glucose entry are negligible in myocytes from control rats, and that implies a minor role for SGLT in myocyte Na+ and glucose homeostasis under physiological conditions. Recent studies revealed that the expression of SGLT1 mRNA in the heart is increased in T2D.27 We hypothesized that SGLT is upregulated in diabetic hearts to compensate for the reduced ability of myocytes to take up glucose through insulin-dependent pathways and/or as an adaptation to chronic hyperglycemia. Indeed, external glucose modulates SGLT expression through a glucose-sensing mechanism.26 In support of this hypothesis, we found that the protein expression of SGLT1 is increased in hearts from humans and rats with T2D. Obesity, in the absence of T2D, was also associated with elevated levels of cardiac SGLT1 in human hearts. SGLT inhibition (both pharmacological and through omission of glucose from external solution) greatly reduced the rate of Na+ influx in myocytes from diabetic rats to a level comparable to that measured in control myocytes. This indicates that SGLT is the main pathway responsible for the excess Na+ entry and thus [Na+]i elevation in T2D myocytes. The SGLT-mediated Na+ flux in our measurements is normalized to cell volume. Because many cardiomyopathies are associated with detubulation and thus lower membrane surface/volume ratio, our measurement might slightly underestimate the increase in SGLT-mediated Na+ flux density (ie, normalized to membrane area). We also found that a significantly larger fraction of glucose uptake is mediated by SGLT in diabetic versus control myocytes. Together, these data indicate that T2D triggers an increase in the activity of cardiac SGLT, which leads to larger myocyte Na+ entry and Na+ overload.
Recent studies point to a pathological role of SGLT upregulation in the heart. Mice with cardiac-specific overexpression of the SGLT1 isoform develop hypertrophy and left ventricular dysfunction.28 SGLT1 is upregulated in transgenic mice with a mutation in the gene encoding the γ2 subunit of AMP-activated protein kinase that is known to cause a glycogen storage cardiomyopathy (PRKAG2 cardiomyopathy)30 and mediates, at least in part, the increased cardiac glucose uptake. Moreover, SGLT1 inhibition attenuated the disease phenotype. SGLT activity is also needed for NADPH activation by hyperglycemia.29 However, the mechanisms that link SGLT function to such alterations in the structure and function of the heart are unclear. We propose that an increase in myocyte [Na+]i due to enhanced Na+ entry through SGLT is a key contributor to these pathologies.
In several HF models, the increase in myocyte [Na+]i is due to larger Na+ influx mediated by Na+ channels or the Na+/H+ exchanger.11,13 The expression of the NCX, the main route for Na+ entry, is increased in HF, which may also cooperate to raise Na+ influx. In contrast, our data show that in diabetic hearts, the excess Na+ entry occurs mostly through SGLT. Thus, distinct pathways are responsible for myocyte Na+ overload in T2D and HF. A previous study demonstrated that SGLT expression is upregulated in HF,27 a result confirmed by our results in human hearts; however, no data indicated a possible contribution of SGLT to Na+ entry in HF. This suggests that in addition to upregulating SGLT expression, T2D also creates a milieu that promotes activation of the cotransporter. Our finding that SGLT-mediated Na+ influx is ≈7 times higher in HIP versus WT myocytes, whereas SGLT expression increases of <2-fold support this hypothesis. Although SGLT regulation is incompletely elucidated, glucose (via n-glycosylation),52 insulin,53 and protein kinases A and C26,53 raise its activity by modifying either the affinity for glucose or the maximal turnover rate.
In summary, we have demonstrated that myocyte Na+–glucose cotransport is enhanced in T2D, probably as a maladaptation to reduced insulin-mediated glucose uptake and chronic hyperglycemia. Elevated SGLT activity increases Na+ influx into myocytes and causes Na+ overload, which may contribute to arrhythmogenesis and oxidative stress in diabetic hearts. Although elevated [Na+]i and Na+ influx are also common in other pathological conditions (eg, HF), the major underlying mechanism in diabetic heart disease is specific to T2D.
Sources of Funding
This work was supported by NIH (R01-HL109501 to S. Despa, R01-HL118474 to F. Despa and R01-HL105993 and R01-HL089847 to Margulies).
Disclosures
None.
References
- Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Lee ET, Welty TK, Fabsitz RR, Robbins D, Rhoades ER, Howard BV. Impact of diabetes on cardiac structure and function: the Strong Heart Study. Circulation. 2000;101:2271–2276. doi: 10.1161/01.cir.101.19.2271. [DOI] [PubMed] [Google Scholar]
- Taegtmeyer H, McNulty P, Young ME. Adaptation and maladaptation of the heart in diabetes: part I: general concepts. Circulation. 2002;105:1727–1733. doi: 10.1161/01.cir.0000012466.50373.e8. [DOI] [PubMed] [Google Scholar]
- Guha A, Harmancey R, Taegtmeyer H. Nonischemic heart failure in diabetes mellitus. Curr Opin Cardiol. 2008;23:241–248. doi: 10.1097/HCO.0b013e3282fcc2fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LH, Wackers FJ, Chyun DA, Davey JA, Barrett EJ, Taillefer R, Heller GV, Iskandrian AE, Wittlin SD, Filipchuk N, Ratner RE, Inzucchi SE DIAD Investigators. Cardiac outcomes after screening for asymptomatic coronary artery disease in patients with type 2 diabetes: the DIAD study: a randomized controlled trial. JAMA. 2009;301:1547–1555. doi: 10.1001/jama.2009.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11:31–39. doi: 10.1007/s11154-010-9131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelsson E, Sundstrom J, Arnlov J, Zethelius B, Lind L. Insulin resistance and risk of congestive heart failure. JAMA. 2005;294:334–341. doi: 10.1001/jama.294.3.334. [DOI] [PubMed] [Google Scholar]
- Kostis JB, Sanders M. The association of heart failure with insulin resistance and the development of type 2 diabetes. Am J Hypertens. 2005;18:731–737. doi: 10.1016/j.amjhyper.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Masoudi FA, Inzucchi SE. Diabetes mellitus and heart failure: epidemiology, mechanisms, and pharmacotherapy. Am J Cardiol. 2007;99:113B–132B. doi: 10.1016/j.amjcard.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Anselmino M, Gohlke H, Mellbin L, Ryden L. Cardiovascular prevention in patients with diabetes and prediabetes. Herz. 2008;33:170–177. doi: 10.1007/s00059-008-3105-5. [DOI] [PubMed] [Google Scholar]
- Despa S, Bers DM. Na+ transport in the normal and failing heart—remember the balance. J Mol Cell Cardiol. 2013;61:2–10. doi: 10.1016/j.yjmcc.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure, but Na/K-pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- Pieske B, Maier LS, Piacentino V, III, Weisser J, Hasenfuss G, Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–453. doi: 10.1161/01.cir.0000023042.50192.f4. [DOI] [PubMed] [Google Scholar]
- Baartscheer A, Schumacher CA, van Borren MM, Belterman CN, Coronel R, Fiolet JW. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res. 2003;57:1015–1024. doi: 10.1016/s0008-6363(02)00809-x. [DOI] [PubMed] [Google Scholar]
- Louch WE, Hougen K, Mørk HK, Swift F, Aronsen JM, Sjaastad I, Reims HM, Roald B, Andersson KB, Christensen G, Sejersted OM. Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J Physiol. 2010;588:465–478. doi: 10.1113/jphysiol.2009.183517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schillinger W, Teucher N, Christians C, Kohlhaas M, Sossalla S, Van Nguyen P, Schmidt AG, Schunck O, Nebendahl K, Maier LS, Zeitz O, Hasenfuss G. High intracellular Na+ preserves myocardial function at low heart rates in isolated myocardium from failing hearts. Eur J Heart Fail. 2006;8:673–680. doi: 10.1016/j.ejheart.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2nd ed. Dordrecht/Boston/London: Kluwer Academic Publishers; 2001. p. 427. [Google Scholar]
- Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–182. doi: 10.1161/01.RES.0000232546.92777.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–288. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Böhm M, O’Rourke B, Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeldsen K, Braendgaard H, Sidenius P, Larsen JS, Nørgaard A. Diabetes decreases Na+-K+ pump concentration in skeletal muscles, heart ventricular muscle, and peripheral nerves of rat. Diabetes. 1987;36:842–848. doi: 10.2337/diab.36.7.842. [DOI] [PubMed] [Google Scholar]
- Hansen PS, Clarke RJ, Buhagiar KA, Hamilton E, Garcia A, White C, Rasmussen HH. Alloxan-induced diabetes reduces sarcolemmal Na+-K+ pump function in rabbit ventricular myocytes. Am J Physiol Cell Physiol. 2007;292:C1070–C1077. doi: 10.1152/ajpcell.00288.2006. [DOI] [PubMed] [Google Scholar]
- Chattou S, Diacono J, Feuvray D. Decrease in sodium-calcium exchange and calcium currents in diabetic rat ventricular myocytes. Acta Physiol Scand. 1999;166:137–144. doi: 10.1046/j.1365-201x.1999.00547.x. [DOI] [PubMed] [Google Scholar]
- Hattori Y, Matsuda N, Kimura J, Ishitani T, Tamada A, Gando S, Kemmotsu O, Kanno M. Diminished function and expression of the cardiac Na+-Ca2+ exchanger in diabetic rats: implication in Ca2+ overload. J Physiol. 2000;527:85–94. doi: 10.1111/j.1469-7793.2000.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmellah A, Baetz D, Prunier F, Tamareille S, Rücker-Martin C, Feuvray D. Enhanced activity of the myocardial Na+/H+ exchanger contributes to left ventricular hypertrophy in the Goto-Kakizaki rat model of type 2 diabetes: critical role of Akt. Diabetologia. 2007;50:1335–1344. doi: 10.1007/s00125-007-0628-x. [DOI] [PubMed] [Google Scholar]
- Anzawa R, Bernard M, Tamareille S, Baetz D, Confort-Gouny S, Gascard JP, Cozzone P, Feuvray D. Intracellular sodium increase and susceptibility to ischaemia in hearts from type 2 diabetic db/db mice. Diabetologia. 2006;49:598–606. doi: 10.1007/s00125-005-0091-5. [DOI] [PubMed] [Google Scholar]
- Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91:733–794. doi: 10.1152/physrev.00055.2009. [DOI] [PubMed] [Google Scholar]
- Banerjee SK, McGaffin KR, Pastor-Soler NM, Ahmad F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc Res. 2009;84:111–118. doi: 10.1093/cvr/cvp190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramratnam M, Sharma RK, D’Auria S, Lee SJ, Wang D, Huang XY, Ahmad F. Transgenic knockdown of cardiac sodium/glucose cotransporter 1 (SGLT1) attenuates PRKAG2 cardiomyopathy, whereas transgenic overexpression of cardiac SGLT1 causes pathologic hypertrophy and dysfunction in mice. J Am Heart Assoc. 2014;3:e000899. doi: 10.1161/JAHA.114.000899. doi: 10.1161/JAHA.114.000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balteau M, Tajeddine N, de Meester C, Ginion A, Des Rosiers C, Brady NR, Sommereyns C, Horman S, Vanoverschelde JL, Gailly P, Hue L, Bertrand L, Beauloye C. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc Res. 2011;92:237–246. doi: 10.1093/cvr/cvr230. [DOI] [PubMed] [Google Scholar]
- Banerjee SK, Wang DW, Alzamora R, Huang XN, Pastor-Soler NM, Hallows KR, McGaffin KR, Ahmad F. SGLT1, a novel cardiac glucose transporter, mediates increased glucose uptake in PRKAG2 cardiomyopathy. J Mol Cell Cardiol. 2010;49:683–692. doi: 10.1016/j.yjmcc.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev. 2011;91:795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- Butler AE, Jang J, Gurlo T, Carty MD, Soeller WC, Butler PC. Diabetes due to a progressive defect in beta-cell mass in rats transgenic for human islet amyloid polypeptide (HIP rat): a new model for type 2 diabetes. Diabetes. 2004;53:1509–1516. doi: 10.2337/diabetes.53.6.1509. [DOI] [PubMed] [Google Scholar]
- Matveyenko AV, Butler PC. β-cell deficit due to increased apoptosis in the human islet amyloid polypeptide transgenic (HIP) rat recapitulates the metabolic defects present in type-2 diabetes. Diabetes. 2006;55:2106–2114. doi: 10.2337/db05-1672. [DOI] [PubMed] [Google Scholar]
- Despa S, Margulies KB, Chen L, Knowlton AA, Havel PJ, Taegtmeyer H, Bers DM, Despa F. Hyperamylinemia contributes to heart dysfunction in obesity and diabetes, a study in humans and rats. Circ Res. 2012;110:598–608. doi: 10.1161/CIRCRESAHA.111.258285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Sharma S, Harris TR, Dong H, Li N, Chiamvimonvat N, Taegtmeyer H, Margulies KB, Hammock BD, Despa F. Cardioprotection by controlling hyperamylinemia in a “humanized” diabetic rat model. J Am Heart Assoc. 2014;3:e001015. doi: 10.1161/JAHA.114.001015. doi: 10.1161/JAHA.114.001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreault N, Scriven DR, Laher I, Moore ED. Subcellular characterization of glucose uptake in coronary endothelial cells. Microvasc Res. 2008;75:73–82. doi: 10.1016/j.mvr.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Kim WH, Lee J, Jung DW, Williams DR. Visualizing sweetness: increasingly diverse applications for fluorescent-tagged glucose bioprobes and their recent structural modifications. Sensors (Basel) 2012;12:5005–5027. doi: 10.3390/s120405005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Islam MA, Pogwizd SM, Bers DM. Intracellular [Na+]i and Na+-pump rate in rat and rabbit ventricular myocytes. J Physiol. 2002;539:133–143. doi: 10.1113/jphysiol.2001.012940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camors E, Mohler PJ, Bers DM, Despa S. Ankyrin-B reduction alters Na and Ca transport promoting cardiac myocyte arrhythmic activity. J Mol Cell Cardiol. 2012;52:1240–1248. doi: 10.1016/j.yjmcc.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Bers DM. Functional analysis of Na/K-ATPase isoform distribution in rat ventricular myocytes. Am J Physiol Cell Physiol. 2007;293:C321–C327. doi: 10.1152/ajpcell.00597.2006. [DOI] [PubMed] [Google Scholar]
- Swift F, Tovsrud N, Enger UH, Sjaastad I, Sejersted OM. The Na+/K+-ATPase α2-isoform regulates cardiac contractility in rat cardiomyocytes. Cardiovasc Res. 2007;75:109–117. doi: 10.1016/j.cardiores.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Despa S, Lingrel JB, Bers DM. Na/K-ATPase α2-subunit preferentially affects sarcoplasmic reticulum Ca release in mouse cardiac myocytes. Cardiovasc Res. 2012;95:480–486. doi: 10.1093/cvr/cvs213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift F, Tovsrud N, Sjaastad I, Sejersted OM, Niggli E, Egger M. Functional coupling of alpha(2)-isoform Na+/K+-ATPase and Ca2+ extrusion through the Na+/Ca2+-exchanger in cardiomyocytes. Cell Calcium. 2010;48:54–60. doi: 10.1016/j.ceca.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Correll RN, Eder-Negrin P, Burr AR, Despa S, Davis JM, Bers DM, Molkentin JD. Overexpression of the Na+/K+ ATPase α2 but not α1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ Res. 2014;114:249–256. doi: 10.1161/CIRCRESAHA.114.302293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman Y, Alfonso A, Louzao MC, Vieytes MR, Botana LM. Confocal microscopy study of the different patterns of 2-NBDG uptake in rabbit enterocytes in the apical and basal zone. Pflugers Arch. 2001;443:234–239. doi: 10.1007/s004240100677. [DOI] [PubMed] [Google Scholar]
- Wilson C, Contreras-Ferrat A, Venegas N, Osorio-Fuentealba C, Pávez M, Montoya K, Durán J, Maass R, Lavandero S, Estrada M. Testosterone increases GLUT4-dependent glucose uptake in cardiomyocytes. J Cell Physiol. 2013;228:2399–2407. doi: 10.1002/jcp.24413. [DOI] [PubMed] [Google Scholar]
- Kanwal A, Singh SP, Grover P, Banerjee SK. Development of a cell-based nonradioactive glucose uptake assay system for SGLT1 and SGLT2. Anal Biochem. 2012;429:70–75. doi: 10.1016/j.ab.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Jiang M, Wang Q, Karasawa T, Koo J-W, Li H, Steyger PS. Sodium-glucose transporter-2 (SGLT2; SLC5A2) enhances cellular uptake of aminoglycosides. PLoS One. 2014;9:e108941. doi: 10.1371/journal.pone.0108941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogwizd SM, Sipido KR, Verdonck F, Bers DM. Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc Res. 2003;57:887–896. doi: 10.1016/s0008-6363(02)00735-6. [DOI] [PubMed] [Google Scholar]
- Nakamura TY, Iwata Y, Arai Y, Komamura K, Wakabayashi S. Activation of Na+/H+ exchanger 1 is sufficient to generate Ca2+ signals that induce cardiac hypertrophy and heart failure. Circ Res. 2008;103:891–899. doi: 10.1161/CIRCRESAHA.108.175141. [DOI] [PubMed] [Google Scholar]
- Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–376. doi: 10.1038/nature12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur S, Coon S, Kekuda R, Sundaram U. Regulation of sodium glucose co-transporter SGLT1 through altered glycosylation in the intestinal epithelial cells. Biochim Biophys Acta. 2014;1838:1208–1214. doi: 10.1016/j.bbamem.2014.01.002. [DOI] [PubMed] [Google Scholar]
- Ghezzi C, Wright EM. Regulation of the human Na+-dependent glucose cotransporter hSGLT2. Am J Physiol Cell Physiol. 2012;303:C348–C354. doi: 10.1152/ajpcell.00115.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]