

Abstract
Background
Although advances in cardiopulmonary resuscitation have improved survival from cardiac arrest (CA), neurologic injury persists and impaired mitochondrial bioenergetics may be critical for targeted neuroresuscitation. The authors sought to determine if excellent cardiopulmonary resuscitation and postresuscitation care and good traditional survival rates result in persistently disordered cerebral mitochondrial bioenergetics in a porcine pediatric model of asphyxia-associated ventricular fibrillation CA.
Methods and Results
After 7 minutes of asphyxia, followed by ventricular fibrillation, 5 female 1-month-old swine (4 sham) received blood pressure–targeted care: titration of compression depth to systolic blood pressure of 90 mm Hg and vasopressor administration to a coronary perfusion pressure >20 mm Hg. All animals received protocol-based vasopressor support after return of spontaneous circulation for 4 hours before they were killed. The primary outcome was integrated mitochondrial electron transport system (ETS) function. CA animals displayed significantly decreased maximal, coupled oxidative phosphorylating respiration (OXPHOSCI+CII) in cortex (P<0.02) and hippocampus (P<0.02), as well as decreased phosphorylation and coupling efficiency (cortex, P<0.05; hippocampus, P<0.05). Complex I– and complex II–driven respiration were both significantly decreased after CA (cortex: OXPHOSCI P<0.01, ETSCII P<0.05; hippocampus: OXPHOSCI P<0.03, ETSCII P<0.01). In the hippocampus, there was a significant decrease in maximal uncoupled, nonphosphorylating respiration (ETSCI+CII), as well as a 30% reduction in citrate synthase activity (P<0.04).
Conclusions
Mitochondria in both the cortex and hippocampus displayed significant alterations in respiratory function after CA despite excellent cardiopulmonary resuscitation and postresuscitation care in asphyxia-associated ventricular fibrillation CA. Analysis of integrated ETS function identifies mitochondrial bioenergetic failure as a target for goal-directed neuroresuscitation after CA. IACUC Protocol: IAC 13-001023.
Keywords: acute brain injury, brain, cardiac arrest, electron transport system, mitochondria, neuroprotection
Pediatric cardiac arrest (CA) is an important public health problem, with thousands of children sustaining one of these potentially devastating events each year in the United States.1 Although survival rates are improving, many survivors have neurologic sequelae, resulting in a major burden for patients, families, and society.2 The brain is particularly susceptible to ischemic injury because of its high metabolic energy demand and limited intrinsic energy supply.
Alterations in mitochondrial bioenergetics may play a pivotal role in secondary neurologic injury cascades, sometimes also referred to as ischemia–reperfusion injury, initiated by CA in mature animals.3,4 Several laboratories report critical alterations in adult brain mitochondrial bioenergetics after ischemia and reperfusion, including diminished mitochondrial mass, population number, decreased function of the electron transport system, reduced antioxidant enzyme activity and content, and damaged and oxidized lipid membrane parameters.3,5,6 Because the immature brain has unique energetics compared with older animals, the effects of CA on cerebral mitochondrial bioenergetics may be different in immature animals.7,8
To address the paucity of data regarding mitochondrial alterations in the immature brain after CA and cardiopulmonary resuscitation (CPR), we performed a detailed evaluation of mitochondrial bioenergetics in a large animal model of hypoxic ventricular fibrillation (VF) pediatric CA that results in good survival rates.9,10 We implemented high-resolution respirometry to ascertain integrated global mitochondrial function, as well as relative contributions of individual electron transport complexes by using specific substrate and inhibitor combinations, together with measures of mitochondrial functional content. We hypothesized that despite good survival parameters after CA and excellent CPR with explicit, protocolized postresuscitative care, pediatric animals would have global persistent alterations in cerebral mitochondrial bioenergetics, mitochondrial content, and specific patterns of individual electron complex dysfunction.
Materials and Methods
All procedures were approved by The Children’s Hospital of Philadelphia’s Institutional Animal Care and Use Committee. Four-week-old female piglets (N=9, 8 to 10 kg), which have neurodevelopment comparable to that of a human toddler, were used for the study.11,12 Piglets were designated into 2 cohorts: (1) the CA group (n=5) killed 4 hours after CPR and return of spontaneous circulation (ROSC), ≈5 hours after the initiation of anesthesia; and (2) anesthetized sham animals (n=5) with no CA and no CPR killed 5 hours after the induction of anesthesia.
Animal Preparation
A porcine model was chosen because of the similarities between swine and humans in regard to anterior–posterior chest depth and compression characteristics (eg, chest stiffness), because the neuroanatomy and neurodevelopment are similar to those of a human toddler, and because of improved survival with the use of blood pressure–targeted CPR in an asphyxia-associated VF CA.10,11,13,14 To keep the population as homogeneous as possible, we did not investigate male animal responses based on prior work.15 All piglets were anesthetized and mechanically ventilated on a mixture of room air and titrated isoflurane (≈1.0% to 2.5%) with a tidal volume of 10 to 12 mL/kg, positive end-expiratory pressure of 6 cm H2O, and titration of rate to maintain end-tidal carbon dioxide at 38 to 42 mm Hg (NICO, Novametrix Medical Systems Inc). High-fidelity, solid-state, micromanometer-tipped catheters (MPC-500; Millar Instruments) were used to measure continuous aortic and right atrial pressures, respectively. A bipolar pacing catheter (Edwards Lifesciences) was used to induce VF. All catheter placements were confirmed with the use of fluoroscopy. Unfractionated heparin 200 U/kg was provided to prevent catheter clotting. All animals received a 20 mL/kg 0.9% normal saline bolus intravenously before we obtained baseline measurements to replace overnight fasting deficits. Animals were prepared in a similar fashion as detailed in our previous publications.16,17
Experimental Protocol: CA Animals
Our group has established that hemodynamically targeted CPR titrated to arterial blood pressure improves short-term survival compared with standard CPR targeted to a fixed depth of compressions and a fixed frequency of vasoactive medications after hypoxic and normoxic VF CA.15,16 This goal-directed approach optimizes coronary perfusion pressure (CPP) and cerebral perfusion pressure and presumably coronary and cerebral blood flow during CPR. In addition, our well-established protocol used in this experiment was designed to mimic resuscitation occurring in an intensive care unit after a respiratory deterioration with an initial documented rhythm of VF (Figure 1).10,16 We selected 7 minutes of asphyxia, plus 10 minutes of CPR, for a total 17 minutes of CA because this insult is clinically relevant and because our prior experience suggested that the majority of these animals would be successfully resuscitated so that we could examine mitochondria 4 hours after the injury.15,16 This model maintains important clinical relevance because most in-hospital pediatric arrests occur in invasively monitored intensive care unit patients.17,18 Ten minutes of CPR was performed before the first defibrillation attempt to mimic the median duration of clinical CPR in the in-hospital CPR setting.10,18,19
Figure 1.
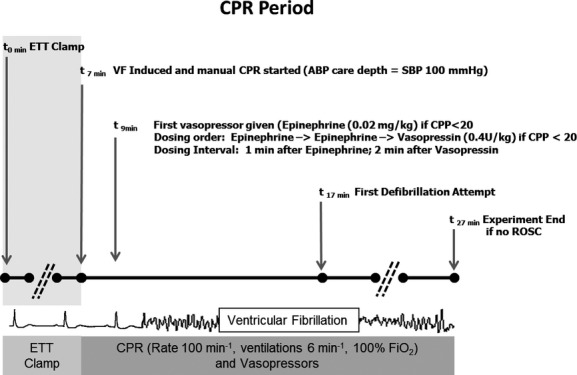
Protocol design. Shaded area denotes period of “acute respiratory insufficiency,” induced by endotracheal tube clamping. During the CPR period, animals received chest compressions with depth titrated to an SBP of 90 mm Hg and vasopressors when the CPP was <20 mm Hg. ABP indicates arterial blood pressure; CPP, coronary perfusion pressure; CPR, cardiopulmonary resuscitation; ETT, endotracheal tube; ROSC, return of spontaneous circulation; SBP, systolic blood pressure; VF, ventricular fibrillation.
Asphyxial and Resuscitation Periods: CA Animals
We selected 7 minutes of asphyxia, plus 10 minutes of CPR for a total 17 minutes of CA because this insult is both clinically relevant and our prior experience suggested that the majority of these animals would be successfully resuscitated so that we could examine mitochondria 4 hours after the injury.17 This model maintains important clinical relevance because most in-hospital pediatric arrests occur in invasively monitored intensive care unit patients.18,19 Endotracheal tube clamping was confirmed by absence of exhaled CO2 (Time 0). After 7 minutes of tracheal tube clamping with severe arterial hypoxemia, VF was induced and maintained by electrical pacing to ensure that the animal would not have ROSC from CPR alone before 10 minutes of CPR.20,21 Once VF was confirmed, the tracheal tube was unclamped and CPR was started. Compressions were provided with a target rate of 100 min−1 and ventilations at 6 min−1 with 100% oxygen. To ensure rigorous adherence to CPR rate goals, a metronome was used. Brief, planned interruptions in CPR of 4 seconds every 2 minutes mimicked pulse checks/rhythm analyses. Animals received arterial blood pressure–targeted care defined as a compression depth titrated to a target systolic blood pressure of >90 mm Hg and, starting at minute 9 of the protocol, vasopressor administration (epinephrine 0.02 mg/kg and vasopressin 0.4 U/kg) if and only if the CPP was <20 mm Hg. After 10 minutes of CPR (minute 17 of the protocol), the initial 200-J biphasic waveform defibrillation attempt was provided. Resuscitation continued until sustained ROSC was attained in the CA animals or until minute 27 of the protocol (an additional 10 minutes of resuscitation post initial defibrillation attempt). If ROSC was attained, the animals then received explicit, protocolized post-CA care (see later).
Post-CA Care: CA Animals
After ROSC, animals received explicit protocolized intensive care, including (1) titration of inspired oxygen concentration to maintain oxygen saturation at 92% to 96%; (2) titration of ventilation to maintain end-tidal carbon dioxide between 38 and 42 mm Hg; and (3) intravenous infusions of dopamine (up to 20 μg/kg per minute) to maintain mean arterial pressure >55 mm Hg. Anesthesia was maintained with inhaled isoflurane (≈1.0%) during this period.
Experimental Protocol: Sham Animals
Sham animals were also instrumented with central venous catheters, and anesthesia was maintained with inhaled isoflurane (≈1.0%). Blood pressure, end-tidal carbon dioxide, oxygen saturation, temperature, and anesthesia were maintained by using the identical explicit post-ROSC protocol as the treatment group.
Sample Acquisition for Mitochondrial Assessments
At 4 hours post ROSC in CA animals (5 hours post induction of anesthesia) and 5 hours post induction of anesthesia in sham animals, a bilateral craniectomy was performed to expose the brain. Tissue was rapidly extracted from 2 regions of interest (a 2-cm2 region of left frontal cortex and both hippocampal regions) while simultaneously receiving a sodium pentobarbital overdose (150 mg/kg). Tissue was removed in <10 seconds and placed in ice-cold isolation buffer (320 mmol/L sucrose, 1.21 g/L tris base, and 2 mmol/L EGTA), where blood, blood vessels, or necrotic tissue was dissected and disposed from the cortex and hippocampus. In addition, subcortical white matter was isolated from the cortex for study. Tissue was then gently homogenized on ice in MiR05 (110 mmol/L sucrose, 0.5 mmol/L EGTA, 3.0 mmol/L MgCl2, 60 mmol/L K-lactobionate, 10 mmol/L KH2PO4, 20 mmol/L taurine, 20 mmol/L HEPES, and 1.0 g/L fatty acid–free BSA) by using a 5-mL Potter-Elvehjem Teflon-glass homogenizer (5 strokes over 30 seconds, 3 sets with a pause of 30 seconds between each set) to a concentration of 1 mg wet weight tissue/10 μL MiR05 buffer. We then compared each measure per milligram of tissue (pmol O2/s×mg) and per citrate synthase activity (pmol O2/s×mg×CS) in sham and CA animals, region by region.
Mitochondrial Assessments: High-Resolution Respirometry
A final concentration of tissue homogenate suspended in MiR05 solution was analyzed at a 1 mg/mL concentration, at a constant 37°C, and the rate of oxygen consumption was measured by using a high-resolution oxygraph and expressed in pmol/[s×mg of tissue homogenate] (OROBOROS, Oxygraph-2K, and DatLab software; all from OROBOROS Instruments). All experiments were performed at 75 to 220 μmol/L oxygen, and reoxygenation was performed routinely before addition of the complex IV electron donor as described later. The oxygraph was calibrated daily: oxygen concentration was automatically calculated from barometric pressure and MiR05 oxygen solubility factor was set at 0.92 relative to pure water.22 A substrate, uncoupler, inhibitor titration protocol (Figure 2) previously used for rodent brain tissue22 was further developed and optimized in previous studies in porcine brain tissue.23 Respiratory capacities with electron flow through both complex I and complex II, as well as the convergent electron input through the Q-junction (complexes I and II), were evaluated sequentially by using succinate and NADH-linked substrates.24 Plasma membranes were permeabilized with the detergent digitonin to allow non–membrane-permeable substrates and ADP access to the subpopulations of mitochondria trapped within synaptosomes. Further, to achieve similar results in brain tissue homogenates and isolated brain mitochondria, with a combination of both subpopulations, Pecinova and colleagues demonstrated that digitonin is necessary in brain homogenate preparations.25,26 Without the addition of digitonin, oxidative phosphorylation capacity would likely be greatly underestimated. Thus, in preliminary experiments, a careful digitonin dose titration was completed, in the presence of exogenously administered cytochrome c, which did not induce any significant effect on respiration with the digitonin dose used in the present study, indicating intact integrity of the outer mitochondrial membrane (data not shown). The same concentration of digitonin was used for both sham and injured tissue. Routine mitochondrial respiration was established by the concomitant addition of malate (5 mmol/L) and pyruvate (5 mmol/L), followed by ADP (1 mmol/L) and glutamate (5 mmol/L), to measure the oxidative phosphorylation capacity of complex I (OXPHOSCI [where CI is complex I]), driven by the NADH-related substrates. Sequential additions followed: Succinate (10 mmol/L) was added to stimulate maximal phosphorylating respiration capacity via convergent input through complexes I and II (OXPHOSCI+CII [where CII is complex II]). Oligomycin, an inhibitor of ATP-synthase, induced mitochondrial respiration independent of ATP production across the inner mitochondrial membrane, commonly referred to as LEAK respiration (LEAKCI+CII) or state 4o. Maximal convergent nonphosphorylating respiration of the ETS (ETSCI+CII) was evaluated by titrating the protonophore, carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone until no further increase in respiration was detected. Rotenone was added to inhibited complex I–driven respiration and remaining measured complex II–driven respiration; the ETS capacity through complex II alone (ETSCII) was measured. The complex III inhibitor antimycin-A (1 μg/mL) was added to measure the residual oxygen consumption that is independent of the ETS. This residual value was subtracted from each of the measured respiratory states to report ETS function. Finally, complex IV activity was determined by the addition of ascorbate (0.8 mmol/L) and N,N,N′,N′-tetramethyl-p-phenylenediamine (0.5 mmol/L), an electron donor to complex IV. Due to the high level of auto-oxidation of N,N,N′,N′-tetramethyl-p-phenylenediamine, sodium azide (10 mmol/L), an inhibitor of complex IV, was added, and the remaining chemical background was subtracted to assess complex IV activity. The substrate, uncoupler, inhibitor titration protocol was identical for shams and injured tissue. Tissue harvest to completion of respirometry measurements were completed in all animals in <1.5 hours. Varying the order of the substrate, uncoupler, inhibitor titration protocol additions did not affect individual respiratory state values. Integrated ETS analysis with internal normalization was generated using flux control ratios (FCRs) calculated by dividing each respiratory state by the maximal uncoupled mitochondrial respiration (ETSCI+CII).
Figure 2.
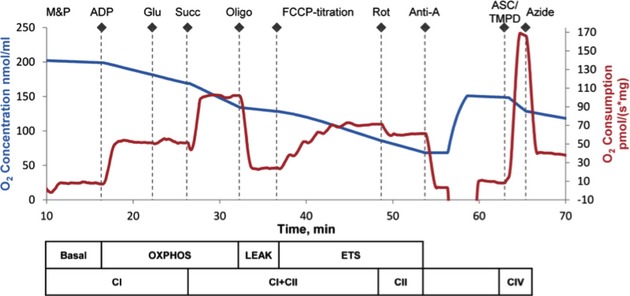
Oxygen concentration and consumption example trace from the SUIT protocol. Mitochondrial respiratory states and respiratory complexes are outlined below the x-axis. The chamber was opened to increase oxygen concentration to 150 nmol/mL before the addition of TMPD. Anti-A indicates antimycin-A; ASC/TMPD, ascorbate and N,N,N′,N′-tetramethyl-p-phenylenediamine; ETS, electron transport system; FCCP, carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone; Glu, glutamate; M&P, malate+pyruvate; Oligo, oligomycin; OXPHOS, oxidative phosphorylation; Rot, rotenone; Succ, succinate; SUIT, substrate, uncoupler, inhibitor titration.
Citrate Synthase Determination
On completion of the high-resolution respirometry measurements, chamber contents were frozen at −80°C for subsequent citrate synthase (CS) activity quantification (Citrate Synthase Assay Kit, CS0720; Sigma). CS activity (given in micromoles per milliliter per minute) was used as a marker of brain metabolism and surrogate for mitochondrial content or mass secondary to its location within the mitochondrial matrix and its ability to catalyzes the condensation of oxaloacetate and the acetyl coenzyme-A, yielding citrate and coenzyme-A in the first step of the tricarboxylic acid cycle.27 Currently, to our knowledge, there is limited research on biomarkers of mitochondrial content (CS activity, cardiolipin content, mitochondrial DNA content, complexes I to IV protein, and complexes I to IV) that validates these surrogates against morphologic measures after acute brain injury, such as transmission electron microscopy.28,29 To differentiate whether changes in absolute rates of respiration per amount of tissue were due to changes in global mitochondrial content and function or specific to each respiratory state, we used 2 strategies to normalize the values. First, we used the common approach of measuring CS activity as a mitochondrial marker of content. However, the enzyme itself may be altered after injury devoid of any alteration in mitochondrial content.28 In addition, flux control ratios were assessed as an internal normalization of the respiratory states to the maximal uncoupler-induced rate of respiration within the ETS, a method that may be independent of tissue heterogeneity.30 All of these methods have strengths and weaknesses; thus, we decided to present data for each respiratory state in response to injury in multiple ways: tissue mass, FCR, CS activity, and ratios for evaluating coupling of respiration to ATP production and the relative contribution of complex I– and II–driven respiration to maximal flux. This approach revealed distinct patterns of mitochondrial respiration after CA in the immature brain.
Data Analysis/Outcomes
The primary outcomes of this study were ETS respiration and mitochondrial content. Secondary outcomes consisted of other recorded variables, including physiologic and hemodynamic measures, CPR quality variables, and 4-hour survival. The oxygen flux traces were analyzed using a customized Matlab (Mathworks) program designed to extract the oxygen flux plateau at each phase of the substrate, uncoupler, inhibitor titration protocol, with the exception of N,N,N′,N′-tetramethyl-p-phenylenediamine/ascorbate, when the peak oxygen flux was extracted.
Individual assessments of mitochondrial function between groups (unpaired analysis) were described as median (25th to 75th percentile range) and were compared by using nonparametric Wilcoxon rank sums. Comparison of continuous variables between regions within animals was similarly evaluated with use of a sign rank test. Statistical analysis was completed by using the Stata-IC statistical package (Version 12.0; StataCorp).
Results
Physiologic and Hemodynamic Variables
In the CA animals, average Pao2 after 6.5 minutes of endotracheal tube–clamped asphyxia (Time 6.5) was 11±3 mm Hg, and after 4.5 minutes of CPR (Time 11.5), it was 110±14 mm Hg. Hemodynamic measurements (ie, arterial systolic blood pressure, diastolic blood pressure and CPP) during CPR in the CA animals demonstrated adherence to experimental protocol goals (Figure 3). All 5 CA animals studied survived to 4 hours post ROSC. Median number of vasopressor doses given during the CPR period was 5.5 (range 4 to 6). All animals achieved sustained ROSC on first defibrillation attempt. No additional vasopressor support was needed post ROSC. In addition, after ROSC post-CA care, vital signs were arterial systolic blood pressure 99±0.6 mm Hg, arterial diastolic blood pressure 64±0.5 mm Hg, CPP 39±0.8 mm Hg, end-tidal carbon dioxide 40±0.1 mm Hg, and pulse oximetry 94±0.4%. There was no statistical difference in mean vital signs between CA and sham animals in the post-CA period.
Figure 3.
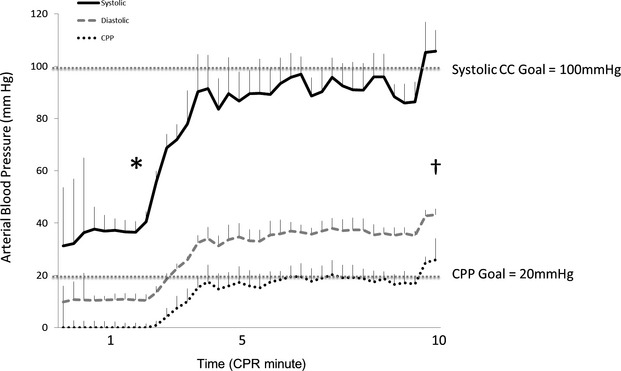
Hemodynamic resuscitation showing systolic, diastolic, and coronary perfusion pressures during CPR period. *First vasopressor dose. †First shock. Error bars represent SEM. CC indicates chest compression; CPP, coronary perfusion pressure; CPR, cardiopulmonary resuscitation.
Citrate Synthase
There were no differences in CS activity between regions in sham animals (Figure 4). Further analysis displayed no differences in CS activity post CA in the cortex (16.8 [14.3 to 17.9] μmol/mL per minute) compared with sham (14.5 [12.8 to 15.6] μmol/mL per minute; P=0.15). However, post CA, the mitochondrial content as measured by CS activity in the hippocampus decreased significantly, by nearly 30%, compared with sham (13.9 [10.7 to 15.2] versus 16.9 [15.0 to 18.3] μmol/mL per minute; P<0.04). The results of all measures of respiratory capacity within the mitochondrial ETS were also normalized to CS activity for each animal (Table 1).
Figure 4.
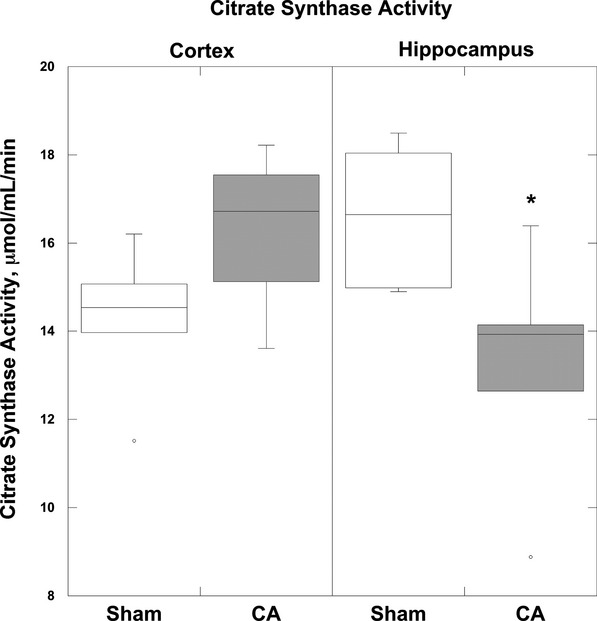
Citrate synthase activity. Four hours after CA there was a significant decrease in CS activity in the hippocampus compared with sham animals (*P<0.01). The cortex did not exhibit a significant reduction in CS activity post CA. Boxplots: Horizontal lines represent the median CS activity, with the boxes representing the 25th and 75th percentiles, the whiskers representing the 5th and 95th percentiles, and the dots representing the outlierss. CA indicates cardiac arrest; CS, citrate synthase.
Table 1.
Respiration of Brain Tissue Homogenates From Sham and Cardiac Arrest Cohorts
| Per mg of tissue (pmols O2/s×mg) | ||||
|---|---|---|---|---|
| Sham (n=5) | Cardiac Arrest (n=6) | |||
| Respiratory Parameters | Cortex | Hippo | Cortex | Hippo |
| OXPHOSCI | 42.7 (37 to 49) | 50 (30 to 54) | 25.0 (22 to 31)* (−39%) | 22.8 (12 to 26)* (−45%) |
| ETSCII | 50.4 (42 to 52) | 65.5 (57 to 74) | 24.6 (21 to 48)* (−50%) | 30.8 (21 to 40)* (−53%) |
| OXPHOSCI+CII | 85.8 (79 to 89) | 105.3 (85 to 115) | 47.3 (46 to 81)* (−49%) | 53.1 (35 to 64)* (−50%) |
| ETSCI+CII | 59.2 (51 to 66) | 79.1 (65 to 91) | 39.4 (37 to 62) (−33%) | 45.5 (31 to 54)* (44%) |
| Complex IV | 116.0 (110 to 127) | 151.4 (135 to 166) | 66.8 (51 to 138) (−40%) | 71.6 (68 to 94)* (−50%) |
| LEAKCI+CII | 12.9 (12.5 to 14.6) | 15.8 (15.0 to 17.2) | 13.6 (12.6 to 16.0) (+5%) | 12.6 (8.7 to 15.6)* (−20%) |
| Per mg of tissue (pmols O2/s×mg) | ||||
|---|---|---|---|---|
| Sham (n=5) | Cardiac Arrest (n=6) | |||
| Respiratory Parameters | Cortex | Hippo | Cortex | Hippo |
| OXPHOSCI | 2.9 (2.5 to 3.8) | 2.9 (2.2 to 3.4) | 1.6 (1.3 to 1.8)* (−39%) | 1.5 (1.1 to 1.7)* (−45%) |
| ETSCII | 3.4 (2.8 to 3.8) | 4.1 (3.9 to 4.2) | 1.6 (1.4 to 2.7)* (−44%) | 2.1 (1.6 to 3.1)* (−50%) |
| OXPHOSCI+CII | 6.0 (5.2 to 6.6) | 6.4 (5.9 to 6.7) | 3.3 (2.9 to 4.6)* (−45%) | 3.7 (2.8 to 4.9)* (−50%) |
| ETSCI+CII | 4.0 (3.4 to 4.6) | 5.0 (4.6 to 5.1) | 2.3 (1.9 to 3.5)* (−41%) | 3.2 (2.3 to 3.8)* (36%) |
| Complex IV | 8.0 (7.5 to 9.4) | 9.3 (9.1 to 9.6) | 4.1 (3.4 to 7.9) (−50%) | 5.4 (4.7 to 7.9)* (−42%) |
| LEAKCI+CII | 0.93 (0.84 to 1.16) | 1.02 (0.92 to 1.18) | 0.90 (0.77 to 0.96) (+4%) | 0.98 (0.53 to 1.16) (−4%) |
Mitochondrial respiration of brain tissue homogenates from the cortex and hippocampus (Hippo) side in 2 cohorts: sham and 4 hours post CA. Respiration is expressed per mg of tissue (pmols O2/s×mg), and per citrate synthase activity (pmols O2/s×CS). Presented as median (25th to 75th percentile range). CA indicates cardiac arrest; CS, citrate synthase; ETS, electron transport system; OXPHOSCI, oxidative phosphorylation capacity of complex I.
P<0.05 significantly different from corresponding sham region. Percent change from sham indicated in parentheses. For definitions of respiratory states and substrates used, please see Figure 2.
Cerebral Mitochondrial Respiration After Hypoxic VF Pediatric CA
Complex I–driven (OXPHOSCI) respiration and complex II–driven respiration (ETSCII) were both significantly decreased after CA, in the cortex (OXPHOSCI: P<0.01, ETSCII: P<0.05) and hippocampus (OXPHOSCI: P<0.03, ETSCII: P<0.01) when compared per milligram of tissue (Table 1). Reductions in both complex I– and complex II–driven respiration compared with sham animals remained significant when the data were normalized for CS (OXPHOSCI: cortex P<0.04, hippocampus P<0.01; ETSCII: cortex P<0.05, hippocampus P<0.01) (Table 1) and FCR (OXPHOSCI: cortex P<0.01, hippocampus P<0.05; ETSCII: cortex P<0.05, hippocampus P<0.01) (Figure 5A). Consequently, maximal coupled, oxidative phosphorylating respiration (OXPHOSCI+CII), stimulated by both complex I and complex II substrates, was significantly decreased in the cortex (P<0.02) and hippocampus (P<0.02) per milligram of tissue and remained significant when normalized to CS activity (cortex P<0.01, hippocampus P<0.01) and FCR (cortex P<0.05, hippocampus P<0.02) (Figure 5B).
Figure 5.

Mitochondrial FCR normalized by ETS capacity (ETSCI+CII) measured 4 hours post CA. A, CA resulted in a significant decrease in complex I–driven respiration (OXPHOSCI) in both regions: cortex and hippocampus. Complex II–driven respiration (ETSCII) in CA tissue is significantly decreased bilaterally, compared with corresponding sham tissue. B, Maximal coupled, phosphorylating respiration (OXPHOSCI+CII), stimulated by both complex I and complex II substrates, was significantly decreased post CA in both regions compared with shams. State 4o (LEAKCI+CII) displayed a significant increase 4 hours post CA compared with shams in the cortex and the hippocampus. For definitions of respiratory states and substrates, please see Figure 1. *P<0.05. Boxplots: Horizontal lines represent the median FCR, with the boxes representing the 25th and 75th percentiles, the whiskers representing the 5th and 95th percentiles, and the dots representing the outliers. CA indicates cardiac arrest; ETS, electron transport system; FCR, flux control ratios; OXPHOSCI+CII, oxidative phosphorylation capacity of complexes I and II.
Maximal uncoupled, nonphosphorylating respiration (ETSCI+CII), initiated by the protonophore carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone after ATP synthase inhibition with oligomycin, was significantly reduced in the hippocampus (P<0.01) per all measures: milligram of tissue and normalized to CS, while cortical ETSCI+CII post CA was significantly reduced only per CS (P<0.03) (Table 1). Complex IV respiration was significantly decreased in the hippocampus per milligram of tissue and normalized to CS activity (P<0.02) but did not display a significant difference in either the cortex or the hippocampus post CA via FCR. Finally, LEAK (LEAKCI+CII), or state 4o respiration, did not display a significant alteration post CA compared with sham in either region, except when normalized to FCR in the cortex and hippocampus, where there was a significant increase (P<0.03) (Figure 5B).
Respiratory ratios evaluating phosphorylation coupling efficiency were calculated for sham and CA animals (Table 2). The respiratory control ratio for OXPHOSCI+CII (OXPHOSCI+CII/LEAK) was calculated to determine the efficiency of coupled phosphorylating respiration, a global measure of overall mitochondrial function, and was significantly decreased in the cortex (P<0.05) and the hippocampus (P<0.05) in CA compared with sham.
Table 2.
Respiratory Ratio of Brain Tissue Homogenates From Sham and Cardiac Arrest Cohorts
| Respiratory Ratio | Sham (n=5) | Cardiac Arrest (n=6) | ||
|---|---|---|---|---|
| Cortex | Hippo | Cortex | Hippo | |
| OXPHOSCI+CII/LEAKCI+CII | 6.5 (5.6 to 6.8) | 6.4 (5.6 to 6.7) | 3.8 (3.4 to 5.1)*(−42%) | 3.9 (3.5 to 4.7)*(−40%) |
The respiratory control ratio OXPHOSCI+CII/LEAKCI+CII is calculated and presented as median (25th to 75th percentile range). OXPHOSCI+CII indicates the oxidative phosphorylation capacity of complexes I and II.
P<0.05 significantly different from corresponding sham region. Percent change from sham is indicated in parentheses.
Discussion
These data establish that pediatric animals with ROSC after severe hypoxia, followed by VF and excellent CPR for 10 minutes, survive; however, they have diffuse cerebral mitochondrial bioenergetics dysfunction 4 hours after ROSC compared with sham controls. The pattern of mitochondrial bioenergetics dysfunction differs from previously described findings in mature animals, as we observe significant decreases in both complex I– and complex II–driven respiration, suggesting an important limitation in the immature brain’s ability to maintain or even accelerate oxidative phosphorylation in the first few hours after CA and challenges the notion that these animals should expect good neurologic recovery. This persistent cerebral mitochondrial bioenergetic dysfunction could play an important role in the development of secondary neurologic injury and reduce the likelihood of long-term recovery in the at-risk immature brain after survival from prolonged CA.
Our biochemical analysis of complex interdependent pathways of electron flow through the ETS revealed an overall injury pattern with a significant decrease in both complex I– and complex II–driven respiration in the brain 4 hours after CA despite maintenance of adequate CPP and cerebral perfusion pressure for successful clinical ROSC. Our methodology extends conventional bioenergetic protocols that use only complex I NADH–linked substrates (pyruvate+malate+glutamate) with the addition of the complex II substrate, succinate (pyruvate+malate+glutamate+succinate), to maximize oxidative phosphorylation capacity (ATP production) by simulating the in situ tricarboxylic acid cycle and maintaining key substrates for convergent flow of electrons through complexes I and II.22,31 Consequently, the reductions in convergent pathways of complexes I and II translated into significant reductions in oxidative phosphorylation post CA, likely placing the immature brain at significant risk due to a reduced ability to increase its ATP production and maintain bioenergetic output. Rosenthal and colleagues reported, in another large animal model of CA with dogs, that mitochondrial respiratory inhibition recovered to sham levels by 2 hours after a 10-minute VF CA.32 Our data suggest a more prolonged period of mitochondrial respiratory inhibition; however, this may be due to model differences where animals underwent a prolonged period of hypoxia before VF CA to simulate pediatric CA. Other confounders complicating the comparison with Rosenthal and colleagues’ work is the developmental age of the animal, as well as the techniques used to obtain mitochondrial respiration.
Complex I–driven respiratory dysfunction is a persistent characteristic of early mitochondrial dysfunction within 6 to 8 hours after traumatic and ischemic injury in the brain and in other organ systems such as the heart.33–37 Interestingly, our data show that both complex I– and complex II–driven respiration decreased in the immature brain after CA and resuscitation. This differs from measurements in mature rat brains exposed to forebrain ischemia and 6 hours of reperfusion, which did not show significant inhibition of complex II-–driven respiration with succinate.38 It should be noted that this model may be distinctly different from the ischemia–reperfusion injury that occurs after CA. Despite this difference, several studies suggest that complex II–driven respiration is critical to mitochondrial respiration and salvaging ATP generation in the first several hours of reperfusion after cerebral ischemia.39,40 Future experiments will focus on measurements of substrate quantification and enzyme activity for succinate-dependent respiration. Thus, we provide preliminary support for the hypothesis that complex II–targeted substrate resuscitation may present a plausible candidate target for therapeutic intervention given its central role in cerebral energy production after hypoxic–ischemic injury.
Concomitant reduction in both complex I and II respiration in this study reveals a pattern of injury that suggests specific opportunities for mitochondrial-targeted interventions for brain injury after CA, beyond substrate supplementation. Mitochondria are a major source of reactive oxygen species generation in mature animals after reperfusion of the brain after hypoxia,41 and complex I is an important source of cerebral reactive oxygen species in ischemia and reperfusion.42,43 Niatsetskaya et al reported that inhibition of complex I with pyridaben was beneficial and led to a significant reduction in oxidative injury and cerebral infarct volume after hypoxic ischemia in neonatal mice.33 It is possible that acute attenuation of complex I respiration after CA in immature animals may be beneficial despite reductions in ATP production because of the potential to limit reactive oxygen species generation by attenuating reverse electron transport during reperfusion.33 However, chronic reductions in complex I function have been implicated in exacerbating chronic inflammation and cellular destruction in disease states such as Parkinson’s disease, and may play a prominent role in persistent bioenergetic dysfunction in other chronic neurodegenerative processes, such as Alzheimer’s disease, and, potentially, chronic encephalopathy triggered by acute brain injury, such as CA.44,45 Future research should focus on temporal variations in bioenergetic demands and mechanistic alterations that will inform manipulation of the mitochondrial respiration in survivors after CA to limit ongoing neurologic injury.
In contrast to a previous study using mature rodent models of VF and CPR,46 we did not find significant global alterations in LEAKCI+CII, or state 4o, respiration early after ROSC (with the exception of results normalized to FCR in the hippocampus). In the context of respiratory control ratios, OXPHOSCI+CII (state 3) respiration/LEAKCI+CII (state 4o), the reductions in coupled respiration seem to be driven by significant reductions in OXPHOSCI+CII post CA rather than significant alterations in LEAKCI+CII, or state 4o. Therefore, we suspect that the global reductions in mitochondrial respiration, in both the cortex and the hippocampus, were not due to significant uncoupling and increased permeability of the inner membrane at this time point. Our data suggest that mitochondrial permeability transition pores do not seem to be open at 4 hours after CA and add to an important understanding of the cerebral bioenergetic time-course after CA. Mitochondrial permeability transition activation is likely an early and potentially reversible event in ischemia–reperfusion injury, leading to mitochondrial swelling and release of cytochrome c that may contribute to global reductions in mitochondrial respiration seen in our study at 4 hours post CA.47,48
Finally, decreased CS activity in the hippocampus may parallel significant oxidative injury in the at-risk selectively vulnerable hippocampus after CA.28 Importantly, our data reveal a substantial reduction of CS activity, by nearly 30%, within the hippocampus 4 hours after CA. This is presumably relevant because CS is generally assumed to be the rate-limiting enzyme of the tricarboxylic acid cycle and, possibly, a biomarker of mitochondrial content. Future studies will be performed to design and assess other markers of mitochondrial content, such as mitochondrial DNA, which may add to the assessment of surrogates for evolving mitochondrial content alterations after injury.
Limitations
Time-course likely plays a significant role in alterations of mitochondrial function after CA. Evaluation of mitochondrial function over a more comprehensive time interval post injury will further improve our understanding of bioenergetic alterations post CA. In an attempt to limit heterogeneity, we also limited our investigations only to female animals at this time; however, future investigations should involve male animals to determine the applicability of these findings across sexes in the immature brain. Other limitations are that this is a descriptive study that has yet to be correlated with neurologic outcome measures. Additionally, we have reported that physiologic directed resuscitation improves neurologic outcomes; however, the Swine Cerebral Performance Category scales at 24 hours are a gross measure of both short- and long-term neurologic dysfunction. In fact, good neurologic outcome was defined by whether an animal could stand, but could still possess an unsteady gait, be able to eat, and exhibit a slow response to environmental stimuli.10,49 Based on this gross scale, there could still be significant neurologic injury. In addition, bioenergetic dysfunction has been shown to correlate with neurologic injury in other models; including human studies.50–52 Our current study generates several hypothesis-driven questions that future research will address, including the development of and correlation to short- and long-term neurologic outcomes. Finally, the use of an inhaled volatile anesthetic, isoflurane, may affect mitochondrial respiration. However, both the sham and experimental groups received the same concentrations and duration of isoflurane53; therefore, the differences are likely to remain meaningful despite the anesthetic.
Conclusions
We have evaluated the mitochondrial bioenergetic response in the pediatric brain after CA and conclude that there are significant alterations in cerebral mitochondrial respiration and mitochondrial content in the pediatric brain 4 hours post ROSC in a large animal model despite excellent CPR, protocolized postresuscitative care, and improved traditional survival rates.10 Significant alterations in complex I– and complex II–driven mitochondrial respiration may offer opportunities for targeted therapeutic interventions in the immature brain to support post-CA bioenergetic demand by using substrate-directed resuscitation.
Sources of Funding
This study was funded by the National Institute of Child Health and Human Development (RMS K23), NIH UO1 NS069545, the Laerdal Foundation for Acute Care Medicine, and The Children’s Hospital of Philadelphia Critical Care Medicine Endowed Chair Funds.
Disclosures
Dr. Hansson owns shares in and has received salary support from NeuroVive Pharmaceutical AB, a public company active in the field of mitochondrial medicine. Dr. Karlsson has received salary support from NeuroVive Pharmaceutical AB.
References
- Merchant RM, Yang L, Becker LB, Berg RA, Nadkarni V, Nichol G, Carr BG, Mitra N, Bradley SM, Abella BS, Groeneveld PW American Heart Association Get With The Guidelines-Resuscitation Investigators. Incidence of treated cardiac arrest in hospitalized patients in the United States. Crit Care Med. 2011;39:2401–2406. doi: 10.1097/CCM.0b013e3182257459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotra S, Chan PS. Trends in survival after in-hospital cardiac arrest. N Engl J Med. 2013;368:680–681. doi: 10.1056/NEJMc1215155. [DOI] [PubMed] [Google Scholar]
- Ayoub IM, Radhakrishnan J, Gazmuri RJ. Targeting mitochondria for resuscitation from cardiac arrest. Crit Care Med. 2008;36:S440–S446. doi: 10.1097/ccm.0b013e31818a89f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazmuri RJ, Radhakrishnan J. Protecting mitochondrial bioenergetic function during resuscitation from cardiac arrest. Crit Care Clin. 2012;28:245–270. doi: 10.1016/j.ccc.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han F, Da T, Riobo NA, Becker LB. Early mitochondrial dysfunction in electron transfer activity and reactive oxygen species generation after cardiac arrest. Crit Care Med. 2008;36:S447–S453. doi: 10.1097/ccm.0b013e31818a8a51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S-D, Yang D-I, Lin T-K, Shaw F-Z, Liou C-W, Chuang Y-C. Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ischemia. Int J Mol Sci. 2011;12:7199–7215. doi: 10.3390/ijms12107199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Brooks KJ, Sammut I, Keelan J, Davey GP, Clark JB, Bates TE. Postnatal development of the complexes of the electron transport chain in synaptic mitochondria from rat brain. Dev Neurosci. 1995;17:212–218. doi: 10.1159/000111289. [DOI] [PubMed] [Google Scholar]
- Del Maestro R, McDonald W. Distribution of superoxide dismutase, glutathione peroxidase and catalase in developing rat brain. Mech Ageing Dev. 1987;41:29–38. doi: 10.1016/0047-6374(87)90051-0. [DOI] [PubMed] [Google Scholar]
- Sutton RM, Friess SH, Maltese MR, Naim MY, Bratinov G, Weiland TR, Garuccio M, Bhalala U, Nadkarni VM, Becker LB, Berg RA. Hemodynamic-directed cardiopulmonary resuscitation during in-hospital cardiac arrest. Resuscitation. 2014;85:983–986. doi: 10.1016/j.resuscitation.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RM, Friess SH, Naim MY, Lampe JW, Bratinov G, Weiland TR, Garuccio M, Nadkarni VM, Becker LB, Berg RA. Patient-centric blood pressure-targeted cardiopulmonary resuscitation improves survival from cardiac arrest. Am J Respir Crit Care Med. 2014;190:1255–1262. doi: 10.1164/rccm.201407-1343OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstead WM. Age and cerebral circulation. Pathophysiology. 2005;12:5–15. doi: 10.1016/j.pathophys.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Durham SR, Duhaime A-C. Basic science; maturation-dependent response of the immature brain to experimental subdural hematoma. J Neurotrauma. 2007;24:5–14. doi: 10.1089/neu.2006.0054. [DOI] [PubMed] [Google Scholar]
- Neurauter A, Nysaether J, Kramer-Johansen J, Eilevstjønn J, Paal P, Myklebust H, Wenzel V, Lindner KH, Schmölz W, Pytte M, Steen PA, Strohmenger H-U. Comparison of mechanical characteristics of the human and porcine chest during cardiopulmonary resuscitation. Resuscitation. 2009;80:463–469. doi: 10.1016/j.resuscitation.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Duhaime A-C. Large animal models of traumatic injury to the immature brain. Dev Neurosci. 2006;28:380–387. doi: 10.1159/000094164. [DOI] [PubMed] [Google Scholar]
- Missios S, Harris BT, Dodge CP, Simoni MK, Costine BA, Lee Y-L, Quebada PB, Hillier SC, Adams LB, Duhaime A-C. Scaled cortical impact in immature swine: effect of age and gender on lesion volume. J Neurotrauma. 2009;26:1943–1951. doi: 10.1089/neu.2009.0956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friess SH, Sutton RM, French B, Bhalala U, Maltese MR, Naim MY, Bratinov G, Arciniegas Rodriguez S, Weiland TR, Garuccio M, Nadkarni VM, Becker LB, Berg RA. Hemodynamic directed CPR improves cerebral perfusion pressure and brain tissue oxygenation. Resuscitation. 2014;85:1298–1303. doi: 10.1016/j.resuscitation.2014.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RM, Friess SH, Bhalala U, Maltese MR, Naim MY, Bratinov G, Niles D, Nadkarni VM, Becker LB, Berg RA. Hemodynamic directed CPR improves short-term survival from asphyxia-associated cardiac arrest. Resuscitation. 2013;84:696–701. doi: 10.1016/j.resuscitation.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg RA, Sutton RM, Holubkov R, Nicholson CE, Dean JM, Harrison R, Heidemann S, Meert K, Newth C, Moler F, Pollack M, Dalton H, Doctor A, Wessel D, Berger J, Shanley T, Carcillo J, Nadkarni VM Eunice Kennedy Shriver National Institute of Child Health and Human Development Collaborative Pediatric Critical Care Research Network and for the American Heart Association’s Get With the Guidelines-Resuscitation (formerly the National Registry of Cardiopulmonary Resuscitation) Investigators. Ratio of PICU versus ward cardiopulmonary resuscitation events is increasing. Crit Care Med. 2013;41:2292–2297. doi: 10.1097/CCM.0b013e31828cf0c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney PA, Nadkarni VM, Kern KB, Indik JH, Halperin HR, Berg RA. Rhythms and outcomes of adult in-hospital cardiac arrest. Crit Care Med. 2010;38:101–108. doi: 10.1097/CCM.0b013e3181b43282. [DOI] [PubMed] [Google Scholar]
- Berg RA, Hilwig RW, Kern KB, Ewy GA. “Bystander” chest compressions and assisted ventilation independently improve outcome from piglet asphyxial pulseless “cardiac arrest”. Circulation. 2000;101:1743–1748. doi: 10.1161/01.cir.101.14.1743. [DOI] [PubMed] [Google Scholar]
- Berg RA, Hilwig RW, Kern KB, Babar I, Ewy GA. Simulated mouth-to-mouth ventilation and chest compressions (bystander cardiopulmonary resuscitation) improves outcome in a swine model of prehospital pediatric asphyxial cardiac arrest. Crit Care Med. 1999;27:1893–1899. doi: 10.1097/00003246-199909000-00030. [DOI] [PubMed] [Google Scholar]
- Karlsson M, Hempel C, Sjövall F, Hansson MJ, Kurtzhals JAL, Elmér E. Brain mitochondrial function in a murine model of cerebral malaria and the therapeutic effects of rhEPO. Int J Biochem Cell Biol. 2013;45:151–155. doi: 10.1016/j.biocel.2012.08.008. [DOI] [PubMed] [Google Scholar]
- Kilbaugh TJ, Karlsson M, Byro M, Bebee A, Ralston J, Sullivan S, Duhaime A-C, Hansson MJ, Elmér E, Margulies SS. Mitochondrial bioenergetic alterations after focal traumatic brain injury in the immature brain. Exp Neurol. 2015;271:136–144. doi: 10.1016/j.expneurol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol. 2009;41:1837–1845. doi: 10.1016/j.biocel.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Pecinová A, Drahota Z, Nůsková H, Pecina P, Houštěk J. Evaluation of basic mitochondrial functions using rat tissue homogenates. Mitochondrion. 2011;11:722–728. doi: 10.1016/j.mito.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Sims NR, Blass JP. Expression of classical mitochondrial respiratory responses in homogenates of rat forebrain. J Neurochem. 1986;47:496–505. doi: 10.1111/j.1471-4159.1986.tb04529.x. [DOI] [PubMed] [Google Scholar]
- Bowling AC, Mutisya EM, Walker LC, Price DL, Cork LC, Beal MF. Age-dependent impairment of mitochondrial function in primate brain. J Neurochem. 1993;60:1964–1967. doi: 10.1111/j.1471-4159.1993.tb13430.x. [DOI] [PubMed] [Google Scholar]
- Chepelev NL, Bennitz JD, Wright JS, Smith JC, Willmore WG. Oxidative modification of citrate synthase by peroxyl radicals and protection with novel antioxidants. J Enzyme Inhib Med Chem. 2009;24:1319–1331. doi: 10.3109/14756360902852586. [DOI] [PubMed] [Google Scholar]
- Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, Schroder HD, Boushel R, Helge JW, Dela F, Hey-Mogensen M. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol (Lond) 2012;590:3349–3360. doi: 10.1113/jphysiol.2012.230185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aidt FH, Nielsen SMB, Kanters J, Pesta D, Nielsen TT, Nørremølle A, Hasholt L, Christiansen M, Hagen CM. Dysfunctional mitochondrial respiration in the striatum of the Huntington’s disease transgenic R6/2 mouse model. PLoS Curr. 2013;2:5. doi: 10.1371/currents.hd.d8917b4862929772c5a2f2a34ef1c201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesta D, Gnaiger E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol Biol. 2012;810:25–58. doi: 10.1007/978-1-61779-382-0_3. [DOI] [PubMed] [Google Scholar]
- Rosenthal RE, Bogaert YE, Fiskum G. Delayed therapy of experimental global cerebral ischemia with acetyl-l-carnitine in dogs. Neurosci Lett. 2005;378:82–87. doi: 10.1016/j.neulet.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Niatsetskaya ZV, Sosunov SA, Matsiukevich D, Utkina-Sosunova IV, Ratner VI, Starkov AA, Ten VS. The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia-ischemia in neonatal mice. J Neurosci. 2012;32:3235–3244. doi: 10.1523/JNEUROSCI.6303-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchadolskiene O, Pranskunas A, Baliutyte G, Veikutis V, Dambrauskas Z, Vaitkaitis D, Borutaite V. Microcirculatory, mitochondrial, and histological changes following cerebral ischemia in swine. BMC Neurosci. 2014;15:2. doi: 10.1186/1471-2202-15-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CL, Scafidi S, McKenna MC, Fiskum G. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp Neurol. 2009;218:371–380. doi: 10.1016/j.expneurol.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective. J Cereb Blood Flow Metab. 2006;26:1407–1418. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- Gilmer LK, Roberts KN, Joy K, Sullivan PG, Scheff SW. Early mitochondrial dysfunction after cortical contusion injury. J Neurotrauma. 2009;26:1271–1280. doi: 10.1089/neu.2008.0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilland E, Puka-Sundvall M, Hillered L, Hagberg H. Mitochondrial function and energy metabolism after hypoxia-ischemia in the immature rat brain: involvement of NMDA-receptors. J Cereb Blood Flow Metab. 1998;18:297–304. doi: 10.1097/00004647-199803000-00008. [DOI] [PubMed] [Google Scholar]
- Sims NR. Selective impairment of respiration in mitochondria isolated from brain subregions following transient forebrain ischemia in the rat. J Neurochem. 1991;56:1836–1844. doi: 10.1111/j.1471-4159.1991.tb03438.x. [DOI] [PubMed] [Google Scholar]
- Allen KL, Almeida A, Bates TE, Clark JB. Changes of respiratory chain activity in mitochondrial and synaptosomal fractions isolated from the gerbil brain after graded ischaemia. J Neurochem. 1995;64:2222–2229. doi: 10.1046/j.1471-4159.1995.64052222.x. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke. 1996;27:327–331. doi: 10.1161/01.str.27.2.327. discussion 332. [DOI] [PubMed] [Google Scholar]
- Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci. 2008;1147:37–52. doi: 10.1196/annals.1427.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara EB, Navarete FDT, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med. 2009;46:1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Walker KR, Tesco G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front Aging Neurosci. 2013;5:29. doi: 10.3389/fnagi.2013.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Boveris A. Brain mitochondrial dysfunction and oxidative damage in Parkinson’s disease. J Bioenerg Biomembr. 2009;41:517–521. doi: 10.1007/s10863-009-9250-6. [DOI] [PubMed] [Google Scholar]
- Jiang J, Fang X, Fu Y, Xu W, Jiang L, Huang Z. Impaired cerebral mitochondrial oxidative phosphorylation function in a rat model of ventricular fibrillation and cardiopulmonary resuscitation. Biomed Res Int. 2014;2014:1–9. doi: 10.1155/2014/192769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson MJ, Månsson R, Mattiasson G, Ohlsson J, Karlsson J, Keep MF, Elmér E. Brain-derived respiring mitochondria exhibit homogeneous, complete and cyclosporin-sensitive permeability transition. J Neurochem. 2004;89:715–729. doi: 10.1111/j.1471-4159.2004.02400.x. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci. 1999;19:RC39. doi: 10.1523/JNEUROSCI.19-22-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg RA, Chapman FW, Berg MD, Hilwig RW, Banville I, Walker RG, Nova RC, Sherrill D, Kern KB. Attenuated adult biphasic shocks compared with weight-based monophasic shocks in a swine model of prolonged pediatric ventricular fibrillation. Resuscitation. 2004;61:189–197. doi: 10.1016/j.resuscitation.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Rosenthal G, Hemphill JC, III, Sorani M, Martin C, Morabito D, Obrist WD, Manley GT. Brain tissue oxygen tension is more indicative of oxygen diffusion than oxygen delivery and metabolism in patients with traumatic brain injury. Crit Care Med. 2008;36:1917–1924. doi: 10.1097/CCM.0b013e3181743d77. [DOI] [PubMed] [Google Scholar]
- Kilbaugh TJ, Bhandare S, Lorom DH, Saraswati M, Robertson CL, Margulies SS. Cyclosporin a preserves mitochondrial function after traumatic brain injury in the immature rat and piglet. J Neurotrauma. 2011;28:763–774. doi: 10.1089/neu.2010.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MJ, Mcarthur DL, Alger JR, Van Horn J, Irimia A, Filippou M, Glenn TC, Hovda DA, Vespa PM. Early metabolic crisis-related brain atrophy and cognition in traumatic brain injury. Brain Imaging Behav. 2013;7:307–315. doi: 10.1007/s11682-013-9231-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Dong Y, Wu X, Lu Y, Xu Z, Knapp A, Yue Y, Xu T, Xie Z. The mitochondrial pathway of anesthetic isoflurane-induced apoptosis. J Biol Chem. 2010;285:4025–4037. doi: 10.1074/jbc.M109.065664. [DOI] [PMC free article] [PubMed] [Google Scholar]