

Abstract
Background
Idiopathic dilated cardiomyopathy is one of the most common types of cardiomyopathy. It has been proposed that an increase in oxidative stress in heart failure leads to a decrease in nitric oxide signaling, leading to impaired nitroso–redox signaling. To test this hypothesis, we investigated the occurrence of protein S-nitrosylation (SNO) and oxidation in biopsies from explanted dilated cardiomyopathy and nonfailing donor male and female human hearts.
Methods and Results
Redox-based resin-assisted capture for oxidation and SNO proteomic analysis was used to measure protein oxidation and SNO, respectively. In addition, 2-dimensional difference gel electrophoresis using maleimide sulfhydryl-reactive fluors was used to identify the SNO proteins. Protein oxidation increased in dilated cardiomyopathy biopsies in comparison with those from healthy donors. Interestingly, we did not find a consistent decrease in SNO in failing hearts; we found that some proteins showed an increase in SNO and others showed a decrease, and there were sex-specific differences in the response. We found 10 proteins with a significant decrease in SNO and 4 proteins with an increase in SNO in failing female hearts. Comparing nonfailing and failing male hearts, we found 9 proteins with a significant decrease and 12 proteins with a significant increase. We also found an increase in S-glutathionylation of endothelial nitric oxide synthase in failing female versus male hearts, suggesting an increase in uncoupled nitric oxide synthase in female hearts.
Conclusion
These findings highlight the importance of nitroso–redox signaling in both physiological and pathological conditions, suggesting a potential target to treat heart failure.
Keywords: heart failure, nitroso–redox signaling, oxidation, S-nitrosylation
Dilated cardiomyopathy (DCM) is a myocardial disease leading to congestive heart failure (HF). DCM is clinically characterized by the presence of left ventricular dilatation and systolic dysfunction.1 The most common known causes of DCM are inflammatory heart disease, myocardial toxins, and genetic mutations of cardiac proteins, and many cases are classified as idiopathic.2,3 It is well known that oxidative and nitrosative stress are involved in many cardiovascular diseases and in the development of HF,4–7 but there are still open questions regarding the role of reactive oxygen species (ROS) and reactive nitrogen species in normal and failing myocardium.8–10 Although several experimental and clinical studies reported an increase of ROS in HF, trials with antioxidant treatment failed,11–13 suggesting that we do not fully understand the role of ROS in HF. An excess of oxidative stress may damage lipids, DNA, and proteins, whereas a low amount of ROS can regulate signaling pathways (redox signaling).14 Under physiological conditions, redox signaling can modulate excitation–contraction coupling,15,16 cell differentiation, and the adaptation to hypoxia or ischemia damage.6 ROS, however, is also involved in adverse cardiac remodeling, contractile dysfunction, hypertrophy, and fibrosis, processes that lead to dysfunction in HF.17,18 In addition, it has been suggested that ROS can buffer or quench nitric oxide (NO) levels and thereby change NO signaling.19,20 This has led to the suggestion that an increase in ROS during HF could lead to a decrease in NO signaling and that this may contribute to the pathogenesis of HF.21
NO is a soluble and diffusible gaseous molecule involved in the regulation of signaling pathways. In the myocardium, NO is produced by 3 isoforms of nitric oxide synthase (NOS): neuronal NOS, localized to the sarcoplasmic reticulum, endothelial NOS (eNOS), localized to caveolae, and inducible NOS (iNOS). NO signaling occurs via cyclic guanosine monophosphate–dependent and –independent pathways.22 In the latter case, NO can act directly on protein function by protein S-nitrosylation (SNO), a posttranslational modification occurring on a free thiol of a cysteine residue of a protein.23 SNO can modify protein activity,24 protein stability,25 and protein localization.26 SNO has also been shown to shield free thiols from the increased oxidation that occurs during reperfusion following ischemia.27 A modest increase in SNO has been shown to be associated with cardioprotection induced by ischemic preconditioning.28 Furthermore, Sun et al demonstrated in mice that female subjects had increased expression of eNOS, an increase in protein SNO at baseline, and a greater increase in SNO with brief ischemia and reperfusion.29
With oxidative stress, NOS can become uncoupled due to either oxidation of the cofactor tetrahydrobiopterin or direct oxidation of NOS. Uncoupled NOS leads to generation of superoxide rather than NO, thereby enhancing oxidative stress. During HF, the increase in oxidative stress can decrease NO signaling by uncoupling of NOS, and ROS can also react with NO, leading to its breakdown.
This paper is focused on nitroso–redox balance in human failing hearts. The aim of the study was to investigate (1) whether an increase of oxidative stress during HF leads to a decrease in protein SNO and (2) whether female and male hearts have different nitroso–redox balance and different targets during the development of HF. To test these hypotheses, protein oxidation and SNO were measured in DCM human samples and in nonfailing human donor hearts using proteomics approaches.
Material and Methods
Patient Tissues Collection
Human left ventricular myocardium was obtained from explanted cardiomyopathic hearts from patients undergoing heart transplantation; the discarded tissue samples received an exemption following institution review board guidelines. The nonfailing hearts were excluded from transplantation due to technical problems or donor age. Following gross inspection to avoid areas of scar, samples were snap frozen in liquid nitrogen within 15 minutes of excision in the case of the failing hearts. All tissues remained frozen in liquid nitrogen until total homogenate preparation.
Sample Preparation
Myocardial homogenate was prepared for all analyses in the dark to prevent SNO decomposition. Snap-frozen heart tissue was powdered on liquid nitrogen and homogenized with a tight-fitting glass Dounce homogenizer on ice in 2 mL homogenate buffer (pH 7.8) containing 300 mmol/L sucrose, 250 mmol/L HEPES-NaOH, 1 mmol/L EDTA, 0.1 mmol/L neocuproine, and an EDTA-free protease inhibitor tablet (Roche Diagnostics Corporation). The samples were centrifuged at 1000g for 10 minutes. The supernatant was recovered as total whole-heart homogenate. The Bradford protein assay was used to determine protein concentration. Heart homogenates were aliquoted and stored at −80°C.
Identification of Protein Oxidation With Oxidation Resin-Assisted Capture
The oxidation resin-assisted capture (RAC) method was used to identify protein oxidation, as described previously by Kohr et al.27 Briefly, samples (1 mg) were diluted in HEN buffer (pH7.8) containing HEPES-NaOH (250 mmol/L), EDTA (1 mmol/L), and neocuproine (0.1 mmol/L) with 2.5% SDS, and an EDTA-free protease inhibitor tablet (Roche Diagnostics Corporation). To remove SNO, each sample was incubated with 20 mmol/L ascorbate for 45 minutes at room temperature. Free cysteine residues were then blocked with 50 mmol/L N-ethylmaleimide (Sigma-Aldrich) for 20 minutes at 50°C. Ascorbate and N-ethylmaleimide were removed via −20°C acetone precipitation. Homogenates were then resuspended in HEN with 1% SDS, and oxidized thiols were reduced with 10 mmol/L dithiothreitol (DTT) for 10 minutes at room temperature; DTT was removed via acetone precipitation. Samples were resuspended in HEN with 1% SDS and then added to the thiopropyl sepharose–containing columns and rotated for 4 hours in the dark at room temperature to allow the reduced cysteine residues of the protein to bind to the thiol groups of the resin by disulfide cross-bridge formation. Resin-bound proteins were then subjected to trypsin digestion (sequencing grade modified; Promega) overnight at 37°C with rotation in buffer containing NH4HCO3 (50 mmol/L) and EDTA (1 mmol/L). Resin-bound peptides were washed and eluted twice for 30 minutes at room temperature in elution buffer containing DTT (20 mmol/L), NH4CO3 (10 mmol/L), and 50% methanol. The resin was washed with an additional volume of elution buffer, followed by 2 volumes of diethylpyrocarbonate-treated water. All fractions were combined and concentrated via SpeedVac (Thermo Scientific). Samples were then resuspended in 0.1% formic acid and cleaned with a C18 column (ZipTip; Millipore). Liquid chromatography–tandem mass spectrometry (MS) was then performed using an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific). The Mascot database search engine (Matrix Science) was used for protein identification.
Identification of Protein SNO With SNO-RAC
Heart homogenates were treated with a modified biotin switch protocol.27 All buffers were degassed before use with the SNO RAC (SNO-RAC) protocol to prevent oxidation of the resin. Briefly, samples (1 mg) were diluted in HEN buffer (as described for the oxidation RAC method). Free cysteine residues were then blocked with 50 mmol/L N-ethylmaleimide (Sigma-Aldrich) for 20 minutes at 50°C. N-ethylmaleimide was removed via −20°C acetone precipitation. Homogenates were then resuspended in HEN with 1% SDS and were added to the thiopropyl sepharose–containing columns, along with 20 mmol/L ascorbate, and rotated for 4 hours in the dark at room temperature. Resin-bound proteins were subjected to trypsin digestion (sequencing grade modified; Promega) overnight at 37°C with rotation in buffer containing 50 mmol/L NH4HCO3 and 1 mmol/L EDTA. Resin-bound peptides were eluted and processed as described for the oxidation RAC method.
Label-Free Peptide Quantification and Analysis
Relative quantification of protein oxidation was performed using QUOIL (Quantification Without Isotope Labeling), an in-house software program designed as a label-free approach to peptide quantification by liquid chromatography–tandem MS, as described previously by Wang et al.30 Quantitative ratios were obtained by normalizing the peptide peak areas against a chosen reference.
Identification of Protein SNO With 2-Dimensional Difference Gel Electrophoresis
To assess differences in protein fluorescence using 2-dimensional difference gel electrophoresis (2D-DIGE), we used a modified biotin switch protocol31 with CyDye maleimide monoreactive sulfhydryl-reactive fluorescent dyes (GE Healthcare Life Sciences). We used 4 independently prepared heart tissue for each analyzed group (nonfailing male, nonfailing female, DCM male, and DCM female) subjected to the CyDye sample preparations, as described previously.32 In each 2D-DIGE experiment, 1 fluorescent component (in this case, Cy2Dye binding lysine residues) comprised a pooled standard of equal amounts of protein from each heart tissue used (16 in total); the Cy5Dye component of each gel was an individual group, and the Cy3Dye component was used for the compared group. Consequently, each 2D-DIGE analysis consisted of 8 gels from 4 samples for each group: 2 gels compared the nonfailing female and nonfailing male groups, 2 gels compared the nonfailing female and DCM female groups, 2 gels compared the nonfailing male and DCM male groups, and 2 gels compared the DCM male and DCM female groups. After the CyDye-maleimide switch and 2D-DIGE, each gel was scanned at the unique excitation/emission wave length of each dye using a Typhoon 9400 imager (GE Healthcare Life Sciences) at a resolution of 100 μm. The background subtracted spot values obtained by the analysis with Progenesis Discovery software (Nonlinear Dynamics) were normalized to a spot that was representative in the Cy2Dye image for each gel. In this way, we were able to normalize the difference between the different gels and compare all 16 samples. The gel was poststained with Flamingo staining (Bio-Rad). Differences across the 2 groups were considered significant at P<0.05, and these protein spots were picked for protein identification. The Ettan Spot Handling Workstation (GE Healthcare Life 147 Sciences) was used for automated extraction of the selected protein spots followed by in-gel trypsin digestion. After sample extraction from the spot handling workstation, each sample was manually desalted using Millipore C18 ZipTips (following the manufacturer’s procedures).
Liquid Chromatography–Tandem MS Analysis
The samples were analyzed on an LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) coupled with an Eksigent NanoLC-Ultra 1D Plus system. Peptides were loaded onto a Zorbax 300SB-C18 trap column (Agilent) at a flow rate of 6 μL/min for 6 minutes and then separated on a reversed-phase PicoFrit analytical column (New Objective) using a short 15-minute linear gradient of 5% to 40% acetonitrile in 0.1% formic acid at a flow rate of 250 nL/min for 2D-DIGE protein spots and a 40-minute linear gradient for SNO-RAC samples. Mass analysis was carried out in data-dependent analysis mode, in which MS initially scanned the full MS mass range from m/z 300 to 2000 at 60 000 mass resolution, and then 6 collision-induced dissociation MS scans were sequentially carried out in the Orbitrap and the ion trap, respectively.
Mascot Database Search
The raw file generated from the LTQ Orbitrap Elite was analyzed using Proteome Discoverer version 1.3 software (Thermo Fisher Scientific) with the Mascot server (version 2.4) as the search engine. The liquid chromatography–tandem MS data were searched against the Swiss-Prot database (Sprot_030613, 539616 sequences, http://www.uniprot.org/). Search parameters were set as follows: taxonomy, human; enzyme, trypsin; miscleavages, 2; variable modifications, oxidation (M), deamidation (N,Q), acetylation (protein N-term), N-ethylmaleimide (C); precursor mass (MS) tolerance at 20 ppm; fragment ion (MS/MS) mass tolerance at 0.8 Da. Peptides were accepted positive identifications based on a Mascot ions score >20 and a false discovery rate of ≤1%. Protein identifications from 2D gels were accepted based on the above criteria but also had to match the isoelectric point (pI) and molecular weight from the location at which the spot was picked on the 2D gel.
Human Heart Total Homogenate Immunoprecipitation
Immunoprecipitation of the left ventricular myocardium homogenates (300 μg) was carried out using Dynabeads Protein G (Life Technologies), according to the manufacturer’s instructions. Briefly, the beads were incubated with 5 μg of anti-eNOS antibody (Santa Cruz Biotechnology) for 3 hours at 4°C. The bead–antibody complex was resuspended in PBS with 0.02% Tween. To avoid coelution of the antibody, the crosslinking reagent BS3 was added to the Dynabeads. The supernatant was removed; the samples were added to the bead–antibody complex and incubated overnight with rotation at 4°C. The Dynabead–antibody proteins were washed 3 times using PBS with 0.02% Tween. The proteins were eluted with 20 μL of elution buffer (50 mmol/L glycine, pH 2.8) by gently pipetting and incubating for 10 minutes at room temperature to dissociate the complex. The supernatants containing eluted antibody and proteins were transferred to new tubes, added to Leammli buffer, and loaded to NuPAGE 4% to 12% Bis-Tris gels (Invitrogen) and transferred to nitrocellulose membranes. The resulting blots were probed with anti-glutathione (ViroGen Corporation) and anti-eNOS (Santa Cruz Biotechnology, Inc) antibodies.
Western Blot
Snap-frozen heart tissue was powdered on liquid nitrogen and homogenized with a tight-fitting glass Dounce homogenizer on ice in 1 mL RIPA buffer plus an EDTA-free protease inhibitor tablet (Roche Diagnostics Corporation). The samples were centrifuged at 1000g for 10 minutes. The supernatant was recovered as total whole-heart homogenate. The Bradford protein assay was used to determine protein concentration. Equivalent amounts of protein (20 μg) from each sample were separated on NuPAGE 4% to 12% Bis-Tris gels (Invitrogen) and transferred to nitrocellulose membrane. Gel-transfer efficiency was verified using Ponceau S staining. The resulting blot was probed with anti-eNOS (Santa Cruz Biotechnology), anti-inducible NOS (Santa Cruz Biotechnology), and anti-neuronal NOS (Santa Cruz Biotechnology) antibodies, and equal loading was verified by probing the blot with anti-GAPDH antibody (Santa Cruz Biotechnology).
Data Analysis
Results are expressed as mean±SE. The 2D-gel analysis was done with Progenesis Discovery software (Nonlinear Dynamics). The statistical significance for SNO-RAC and oxidation RAC data was determined using the Mann-Whitney rank sum test. Western blotting analysis was done using 2-way ANOVA, except for eNOS immunoprecipitation analysis, which was done by 1-way ANOVA. For all tests, P<0.05 was considered significant.
Results
As described in the methods, left ventricular samples from failing and nonfailing donor hearts not suitable for transplantation were studied. The age and sex of the patients and donors are shown in Table 1. Table 1 also includes the cause of death of the donors. We examined changes in oxidation using the previously described oxidation RAC protocol, comparing 11 (7 males and 4 females) failing hearts and 6 (2 males and 4 females) nonfailing hearts. For analysis, we included all oxidized peptides that were present in at least 3 of the 11 failing and 2 of the 6 nonfailing samples. This analysis identified 275 unique peptides that were oxidized in either nonfailing or failing human hearts. As shown in Figure 1A, this includes 117 peptides that were found in only the failing hearts and 158 common peptides that were found in both failing and nonfailing hearts. Using label-free analysis, we determined that 6 of the 158 common peptides showed significantly higher oxidation in failing than nonfailing hearts, whereas most showed a small, nonsignificant increase. These 6 peptides were serum albumin, α-2-HS glycoprotein, glyceraldehyde-3-phosphate dehydrogenase, cytochrome c oxidase protein-20, Igγ1 chain C region, and Igγ3 chain C region. All 275 peptides are listed in Table S1. Of interest is a mutation in cytochrome c oxidase protein-20 that has been reported to be associated with a cardiomyopathy.33 These data support the concept that cysteine oxidation is greater in DCM in comparison with nonfailing myocardium.
Table 1.
Patients Details and Assay Performed
| Heart Number | Diagnosis | Age, y | Sex | Ox-RAC | SNO-RAC | 2D CyDye-DIGE | Cause of Death |
|---|---|---|---|---|---|---|---|
| 2 | DCM | 55 | M | Y | Y | N | |
| 16 | DCM | 65 | M | Y | N | Y | |
| 21 | NF | 63 | M | Y | Y | Y | Head trauma |
| 26 | NF | 33 | F | Y | Y | Y | ICH |
| 28 | DCM | 45 | M | Y | Y | Y | |
| 29 | NF | 66 | F | Y | Y | Y | Stroke |
| 31 | NF | 54 | M | N | Y | Y | MVA |
| 33 | NF | 69 | F | Y | Y | Y | ICB |
| 34 | NF | 69 | M | N | Y | N | Head trauma |
| 39 | DCM | 51 | F | Y | Y | Y | |
| 42 | DCM | 56 | M | Y | Y | N | |
| 43 | DCM | 30 | F | N | Y | N | |
| 44 | DCM | 42 | M | Y | Y | Y | |
| 46 | DCM | 37 | F | Y | Y | N | |
| 48 | DCM | 54 | M | Y | Y | N | |
| 50 | DCM | 41 | M | N | Y | N | |
| 52 | DCM | 29 | F | N | Y | Y | |
| 53 | DCM | 65 | M | Y | Y | Y | |
| 63 | DCM | 41 | F | Y | Y | Y | |
| 64 | DCM | 28 | F | N | Y | N | |
| 71 | DCM | 45 | F | Y | Y | Y | |
| 76 | DCM | 65 | F | N | Y | N | |
| 85 | NF | 59 | F | Y | Y | Y | ICB |
| 93 | NF | 54 | M | Y | Y | Y | CVA |
| 95 | NF | 67 | M | N | Y | Y | ICH |
2D indicates 2-dimensional; CVA, cerebrovascular accident; DCM, dilated cardiomyopathy; DIGE, difference gel electrophoresis; F, female; ICB, intracerebral bleed; ICH, intracerebral hemorrhage; M, male; MVA, motor vehicle accident; N, no; NF, nonfailing; Ox-RAC, oxidation resin-assisted capture; SNO-RAC, S-nitrosylation resin-assisted capture; Y, yes.
Figure 1.
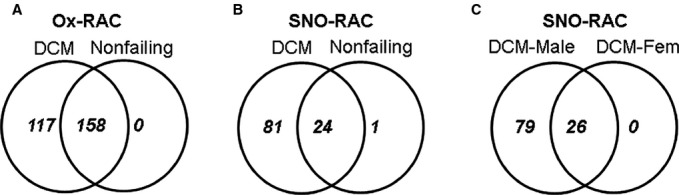
Venn diagrams of Ox-RAC and SNO-RAC analysis of nonfailing and DCM human hearts. A, Ox-RAC analysis from 6 nonfailing hearts and 11 DCM hearts. B, SNO-RAC analysis from 9 nonfailing hearts and 16 DCM hearts. C, SNO-RAC analysis from 7 DCM male and 9 DCM female hearts. DCM indicates dilated cardiomyopathy; Ox-RAC, oxidation resin-assisted capture; SNO-RAC, S-nitrosylation resin-assisted capture.
To test the hypothesis that oxidative stress associated with DCM leads to a decrease in NO signaling, we used the SNO-RAC method to measure levels of the NO-dependent posttranslational modification SNO in nonfailing and failing human hearts. Because we expected SNO levels to be lower than oxidation, we increased the number of hearts in each group: We compared SNO in 16 failing and 9 nonfailing hearts. We identified 106 unique peptides in the nonfailing and failing hearts, using the criterion that a modified peptide must be present in at least 3 of the 9 nonfailing or 3 of the 16 failing samples. As shown in Figure 1B, 81 SNO peptides were found in only the failing hearts, 24 were common between failing and nonfailing hearts, and 1 was found in only the nonfailing hearts. As shown in Table S2, most of the common peptides showed an increase in the failing hearts; however, we noticed that among the common SNO peptides, most of the peptides that showed an increase with failure were from male hearts. Consequently, and because we have previously observed sex differences in SNO,29 we examined whether there were sex-specific differences between the 7 male and 8 female failing hearts. As shown in Figure 1C, 79 unique SNO peptides were found in only the male failing hearts, there were no unique peptides in female hearts, and 26 peptides were common between male and female samples. As shown in Table S2, most of the 26 peptides that were common between male and female failing hearts tended to have higher SNO in male compared with female samples, and 4 were found to be significantly higher in male samples (2 peptides from GAPDH and 2 peptides from cytosolic malate dehydrogenase). Taken together, these data suggest a sex-specific difference in SNO in failing hearts, with a larger increase in SNO in failing male myocardium than in female myocardium.
We next used an alternative, fluorescent method to confirm the sex-specific difference in SNO in the failing hearts. We used a modified biotin switch method based on CyDye-maleimide monoreactive fluorescence dyes and 2D-DIGE proteomics analysis,34 and we compared 4 female nonfailing hearts, 4 male nonfailing hearts, 4 female failing hearts, and 4 male failing hearts. We ran eight 2D gels on a total of 16 samples. An internal standard was run on all gels, allowing us to normalize samples among gels.
We analyzed sex differences in nonfailing hearts. Figure 2A and 2B show SNO levels in a nonfailing female heart and a nonfailing male heart, respectively. SNO proteins in female hearts were labeled with Cy3-malemide (green) and male hearts with Cy5-maleimide (red). Figure 1C shows an overlay of SNO in male and female hearts. Proteins that exhibited an increase in SNO in female hearts appear green, whereas proteins with lower SNO in female hearts appear red, and proteins that were similar between male and female hearts appear yellow. The data from all 8 gels were analyzed using Progenesis software, and proteins that showed a significant difference between nonfailing male and female hearts are listed in Table 2. The Progenesis Discovery software identified 14 proteins that showed a significant increase in SNO in female hearts, and 6 proteins showed a significant decrease in SNO compared with the male donor hearts. Interestingly, 8 of the 14 proteins that showed an increase in SNO in female hearts are mitochondrial proteins. In contrast, 5 of the 6 proteins with increased SNO in male hearts are cytosolic proteins, and just 1 is a mitochondrial protein. These data suggest an increase in mitochondrial SNO in female hearts at baseline. We also examined the SNO-RAC data to compare nonfailing male and female hearts and found that the SNO-RAC data were consistent with the 2D-gel data. In the SNO-RAC data, we found that most SNO peptides were found in only female hearts or that SNO was higher in female groups (Table S3).
Figure 2.
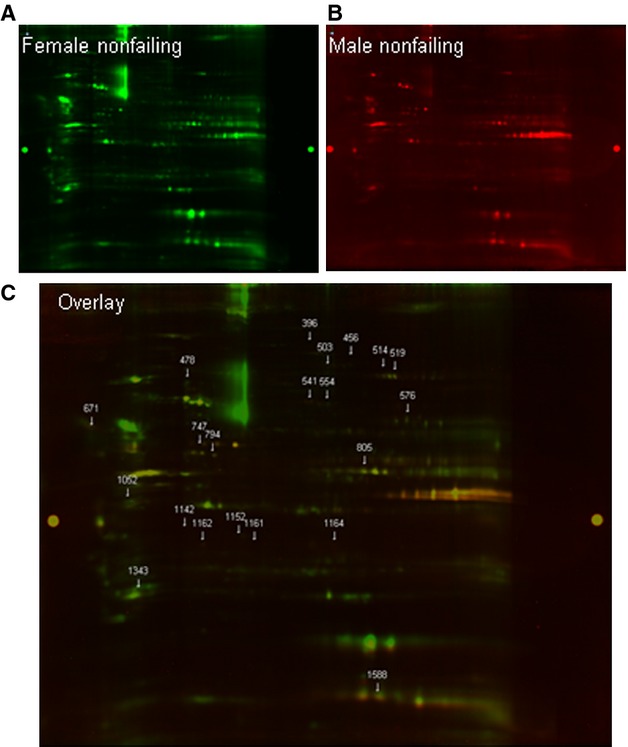
Sex-based differences in protein SNO. Representative 2D CyDye-maleimide DIGE from 4 experiments (4 female nonfailing and 4 male nonfailing hearts). A, Female nonfailing groups were labeled with Cy3-malemide (green). B, Male nonfailing hearts were labeled with Cy5-maleimide (red). C, The overlay of SNO in female and male donor hearts.
Table 2.
Proteins Identified by 2D CyDye-Maleimide DIGE
| Spot No | Protein Name | Accession Number | Mw (kDa) | SNO Level (Fold Change) | ||
|---|---|---|---|---|---|---|
| NF F/NF M | NF F/DCM F | NF M/DCM M | ||||
| 28 | Protein niban | Q9BZQ8 | 103.7 | NS | ↓ in DCM(2.3) | NS |
| 456 | Aconitate hydratase, mitochondrial | Q99798 | 85.4 | F>M (2.2) | NS | ↑ in HF (−1.8) |
| 396 | Mitochondrial inner membrane protein | Q16891 | 83.6 | F>M (2.1) | NS | NS |
| 436 | Mitochondrial inner membrane protein | Q16891 | 83.6 | NS | NS | ↓ in HF (2) |
| 465 | Trifunctional enzyme subunit alpha, mitochondrial | P40939 | 82.9 | NS | NS | ↑ in HF (−2) |
| 478 | NADH-ubiquinone oxidoreductase 75 kDa subunit, mitochondrial | P28331 | 79.4 | F>M (2.1) | ↑ in HF (−2.7) | ↑ in HF (−2.7) |
| 503 | Long-chain fatty-acid CoA ligase 1 | P33121 | 77.9 | F>M (2.4) | NS | NS |
| 519 | Carnitine O-palmitoyltransferase 2, mitochondrial | P23786 | 73.7 | F>M (2.2) | NS | ↑ in HF (−2.8) |
| 514 | Tyrosine-protein phosphatase nonreceptor type 11 | Q06124 | 68.4 | F>M (2.3) | NS | NS |
| 541 | Moesin | P26038 | 67.8 | F>M (2) | NS | NS |
| 554 | Mitochondrial import receptor sub TOM70 | O94826 | 67.4 | F>M (1.9) | NS | NS |
| 604 | Dihydropyrimidinase-related protein 2 | Q16555 | 62.3 | NS | NS | ↓ in HF (2.3) |
| 597 | Phosphoglucomutase-1 | P36871 | 61.4 | NS | ↓ in HF (1.7) | NS |
| 744 | 60 kDa heat shock protein, mitochondrial | P10809 | 61.0 | NS | NS | ↑ in HF (−2.3) |
| 576 | Amine oxidase (flavin-containing) B | P27338 | 58.7 | F>M (2.6) | NS | NS |
| 692 | Annexin A11 | P50995 | 54.4 | NS | NS | ↑ in HF (−2.2) |
| Dihydrolipoyl dehydrogenase, mitochondrial | P09622 | 54.1 | ||||
| 671 | Target of Myb protein 1 | O60784 | 53.8 | M>F (−1.9) | ↓ in HF (1.6) | ↓ in HF (2.3) |
| 729 | Desmin | P17661 | 53.5 | NS | NS | ↑ in HF (−2.6) |
| 757 | Cytochrome b-c1 complex subunit 1, mitochondrial | P31930 | 52.6 | NS | ↑ in HF (−1.9) | NS |
| 745 | Glutathione synthetase | P48637 | 52.3 | NS | ↑ in HF (−1.7) | ↑ in HF (−2.3) |
| 722 | Peptidyl-prolyl cis-trans isomerase FKBP4 | Q02790 | 51.7 | NS | ↑ in HF (−2.3) | NS |
| 747 | Tubulin beta-4B chain | P68371 | 49.8 | F>M (1.7) | NS | ↑ in HF (−2.1) |
| 794 | Secernin-3 | Q0VDG4 | 48.5 | F>M (2.1) | NS | ↑ in HF (−5) |
| 805 | Cytocrome b-c1 complex sub 2, mitochondrial | P22695 | 48.4 | F>M (1.9) | NS | NS |
| 845 | Aspartate aminotransferase, mitochondrial | P00505 | 47.5 | NS | NS | ↓ in HF (2.3) |
| Acetyl-CoA acetyltransferase, mitochondrial | P24752 | 45.2 | ||||
| 961 | Succinyl-CoA ligase (ADP/GDP-forming) subunit alpha, mitochondrial | P53597 | 36.2 | NS | ↓ in HF (7.4) | NS |
| 958 | l-lactate dehydrogenase A chain | P00338 | 36.0 | NS | ↓ in HF (3.7) | NS |
| 928 | Glyceraldehyde-3-phosphate dehydrogenase | P04406 | 35.5 | NS | ↓ in HF (2.8) | NS |
| 929 | Glyceraldehyde-3-phosphate dehydrogenase | P04406 | 35.5 | NS | ↓ in HF (2.7) | NS |
| 968 | Malate dehydrogenase, mitochondrial | P40926 | 35.2 | NS | ↓ in HF (6.3) | NS |
| 1052 | Elongation factor 1-delta | P29692 | 31.1 | F>M (2) | NS | NS |
| 1152 | Glutathione S-transferase omega-1 | P78417 | 27.5 | M>F (−3.4) | NS | ↓ in HF (3.4) |
| 1161 | Glutathione S-transferase omega-1 | P78417 | 27.5 | M>F (−1.8) | NS | NS |
| 1142 | Proteasome activator complex subunit 2 | Q9UL46 | 27.4 | M>F (−2.9) | NS | NS |
| 1108 | Voltage-dependent anion-selective channel protein 3 | Q9Y277 | 30.6 | NS | NS | ↑ in HF (−2.7) |
| 1164 | Phosphoglycerate mutase 1 | P18669 | 28.8 | M>F (−3.5) | NS | ↓ in HF (4.5) |
| 1162 | Chloride intracellular channel protein 4 | Q9Y696 | NS | NS | ↓ in HF (2.1) | |
| 1220 | Coiled-coil-helix-coiled-coil-helix domain-containing protein 3, mitochondrial | Q9NX63 | 26.1 | NS | NS | ↑ in HF (−2.2) |
| 1250 | Flavin reductase (NADPH) | P30043 | 22.1 | NS | ↓ in HF (1.5) | NS |
| 1273 | Adenylate kinase isoenzyme 1 | P00568 | 21.6 | NS | ↓ in HF (1.6) | NS |
| 1343 | Heat shock protein beta-2 | Q16082 | 20.2 | F>M (2.5) | NS | NS |
| 1612 | Fatty acid-binding protein, heart | P05413 | 14.8 | NS | NS | ↓ in HF (2.8) |
| 1611 | Fatty acid-binding protein, heart | P05413 | 14.8 | NS | NS | ↓ in HF (1.8) |
| 1588 | Cytochrome c oxidase subunit 5B, mitochondria | P10606 | 13.7 | M>F (−3.6) | NS | NS |
Protein identifications were accepted based on ≥2 unique peptides with a false discovery rate of ≥99% and a correct molecular mass identification (n=4 in each group). In all, P<0.05. 2D indicates 2-dimensional; DCM, dilated cardiomyopathy hearts; DIGE, difference in gele electrophoresis; F, protein was found just in the female group; HF, heart failure; M, protein was found just in the male group; NF, nonfailing hearts; NS, nonsignificantly different; SNO, protein S-nitrosylation.
We compared sex-specific differences between failing and nonfailing hearts. Figure 3A shows a representative 2D gel comparing nonfailing and failing female hearts, and Table 2 shows proteins with a significant difference between the 4 nonfailing female and the 4 failing female hearts. We found 10 proteins with a significant decrease in SNO in failing hearts and 4 proteins with an increase in SNO. These results are also consistent with the SNO-RAC data, in which we found most of the SNO peptides to be present in only or at higher levels in nonfailing female compared with failing female hearts (Table S3). We next examined SNO differences between nonfailing and failing male hearts. As illustrated in Figure 3B and Table 2, in failing male hearts, we found 9 proteins with a significant decrease and 12 proteins with a significant increase. This is consistent with the SNO-RAC data, which showed that the majority of the peptides (81 of 106) were found in only the failing male hearts (Table S3). Interestingly, in both male and female samples with HF, some proteins showed an increase in SNO, whereas others showed a decrease. Only 1 protein, glutathione synthetase, showed a similar significant increase in both male and female samples with HF. These data raise the question of why SNO decreases more in failing female hearts than in failing male hearts. We considered the possibility that perhaps there was increased glutathionylation of eNOS in female hearts that might lead to an increase in uncoupled NOS in female samples and a resultant decrease in NO production. To test this hypothesis, we immunoprecipitated eNOS and then probed with an antibody against glutathione. As shown in Figure 4A, failing female hearts had a significant increase in glutathionylation of eNOS compared with failing male hearts. No differences were found in eNOS protein level between female and male failing hearts (Figure 4B). We also checked the protein levels for inducible and neuronal NOS in the myocardial homogenates from nonfailing (4 female and 5 male) and failing (7 female and 8 male) hearts. Consistent with previously published data,35 we found a significant increase in inducible NOS expression during HF (Figure 4C) but no sex difference. Immunoblotting with anti-neuronal NOS antibody showed no difference between the failure and nonfailure groups (Figure 4D) and no sex difference.
Figure 3.
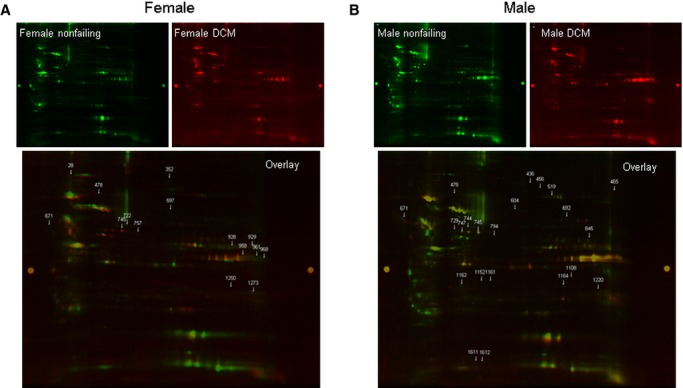
Myocardial protein S-nitrosylation in nonfailing and DCM hearts. A, Representative 2D CyDye-maleimide DIGE from 4 experiments (4 female DCM and 4 female nonfailing hearts). Female nonfailing groups were labeled with Cy3-malemide (green), and female DCM hearts were labeled with Cy5-maleimide (red). B, Representative 2D CyDye-maleimide DIGE from 4 experiments (4 male DCM and 4 male nonfailing hearts). Male nonfailing groups were labeled with Cy3-malemide (green), and male DCM hearts were labeled with Cy5-maleimide (red). 2D indicates 2-dimensional; DCM, dilated cardiomyopathy; DIGE, difference gel electrophoresis.
Figure 4.

Detection of S-glutathionylation of eNOS in DCM female and male hearts. A, Total homogenates were extracted from left ventricular myocardium of the DCM female and male hearts (4 for each group) and subjected to immunoprecipitation with an anti-eNOS antibody. Immunoblot was stained with anti-GSH and with eNOS antibodies to confirm and normalize the immunoprecipitation. Densitometry analysis shows a ratio between the densitometric values of the S-glutathionylated eNOS bands and those bands detected with the eNOS antibody. B, Total homogenates from NF (4 female and 5 male) and DCM (7 female and 8 male) hearts were analyzed by immunoblot probed with anti-eNOS antibody. Equal loading was checked by probing the membrane with anti-GAPDH antibody. Densitometry analysis show the ratio between the densitometric values of the eNOS bands and those bands detected with the anti-GAPDH antibody. C, Total homogenates from NF (4 female and 4 male) and DCM (7 female and 7 male) hearts were analyzed by immunoblot probed with anti-iNOS antibody. Equal loading was checked by probing the membrane with anti-GAPDH antibody. Densitometry analysis show the ratio between the densitometric values of the iNOS bands and those bands detected with the anti-GAPDH antibody. D: Immunoblot of total homogenates from NF (4 female and 4 male) and DCM (7 female and 7 male) hearts probed with anti-nNOS antibody. Equal loading was checked by probing the membrane with anti-GAPDH antibody. Densitometry analysis show the ratio between the densitometric values of the nNOS bands and those bands detected with the anti-GAPDH antibody. *P<0.05. DCM indicates dilated cardiomyopathy; eNOS, endothelial nitric oxide synthase; F, female; GSH, glutathione; iNOS, inducible nitric oxide synthase; IP, immunoprecipitation; M, male; NF, nonfailing; nNOS, neuronal nitric oxide synthase; WB, western blot.
Discussion
HF is known to be associated with increased ROS formation4,14,17 and NOX2 and NOX4 have been proposed as major contributors to ROS in HF.36,37 This increase in oxidative stress during pressure overload and hypertrophy has been suggested to result in NOS uncoupling, a mode in which NOS generates superoxide rather than NO.38 An increase in ROS and NOS uncoupling also leads to a decrease in NO bioavailability, which has been suggested to play a prominent role in the development of HF.5,21,39–41 Consistent with this hypothesis, we reasoned that the increase in oxidative stress during hypertrophy and HF would lead to a decrease in the level of the NO-dependent posttranslational modification protein SNO. We tested this hypothesis in explanted failing human hearts. Consistent with previous studies, we found an increase in cysteine oxidation in failing human hearts; however, we did not find a general decrease in SNO during HF. Contrary to our initial hypothesis, DCM did not lead to a general decrease in SNO; rather, SNO was decreased in some proteins and increased in others.
Recently, we and others have reported a role for NO-dependent cardiac protection during myocardial ischemic pre- and postconditioning. We showed that preconditioning increases protein SNO and shields thiols from further irreversible oxidation. Many of the proteins that showed an increase in SNO in failing human hearts (Table 2) have also been reported to show an increase in SNO following preconditioning.24,27,42,43 These proteins include aconitase, NADH-ubiquinone oxidoreductase 75 kDa, trifunctional enzyme subunit α, fumarate hydratase, long-chain fatty-acid CoA ligase 1, 60-kDa heat shock protein, tubulin β-4B chain, aspartate aminotransferase, l-lactate dehydrogenase A chain, GAPDH, malate dehydrogenase, and cytochrome c oxidase subunit 5B. We also found mitochondrial complex I (NADH-ubiquinone oxidoreductase 75 kDa) to have higher SNO in female compared with male nonfailing hearts. SNO of several of these proteins has been shown to contribute to cardioprotection. Recent findings by Chouchani et al, for example, demonstrated that SNO of complex I protects the mitochondria during the first minutes of reperfusion after ischemia injury. SNO of the ND3 subunit of complex I inhibits its activity, shields the thiols from other modifications that can occur during the first minutes of the reperfusion, and decreases ROS production.44
Interestingly, we observed sex differences in SNO in failing human hearts. In male hearts, some protein cysteines showed an increase in SNO and some showed a decrease, whereas the majority of the SNO protein in female hearts tended to decrease. In failing male hearts, 43% of the proteins that showed a significant difference in SNO exhibited a decrease; however, in failing female hearts, 71% of the SNO proteins showed a decrease. Part of the mechanism for the decrease in SNO in failing female hearts can be attributed to an increase in glutathionylation of eNOS in female compared with male samples (Figure 4).
We also observed sex differences in protein SNO in nonfailing hearts. We found that nonfailing female hearts had higher levels of SNO than male nonfailing hearts. These data are consistent with our previous animal studies showing that female hearts have increased SNO at baseline.29 In addition, we found that the majority of the proteins with SNO in female nonfailing hearts were mitochondrial proteins. In contrast, male nonfailing hearts showed higher levels of cytosolic protein SNO.
Study Limitations
For SNO measurement, we used 2 complementary methods: SNO-RAC and a 2D fluorescent gel method. As previously reported and discussed, these complementary methods identify SNO of different proteins.45 SNO-RAC measures SNO of peptides, whereas 2D-DIGE measures SNO of proteins. SNO-RAC allows identification of the sites of SNO. In addition, the 2D gel can identify multiple spots for the same protein due to differences in posttranslational modifications, and different spots of the same protein can respond differently. Three spots, for example, were identified as mitofilin (spots 396, 436, and 352 on the gel); these different spots are likely related to differences in posttranslational modifications such as phosphorylation. Spot 352 showed an insignificant increase in SNO in failing female hearts, whereas spot 436 showed a decrease in failing male hearts. As observed by others, these different methods select for proteins with different chemistry.46 A limitation of the 2D-gel method is that high-molecular-weight and membrane proteins poorly enter the 2D gel and thus are difficult to detect.28,47 The lack of overlap in proteins found with the 2 methods makes it difficult to compare proteins across the 2 methods; however, the use of 2 methods increases the coverage of proteins that are altered in HF. Oxidation of proteins could occur during sample preparation. We performed a small study comparing samples prepared in room air versus nitrogen and found only 4 to 5 proteins that were oxidized in room air.
In summary, the present findings demonstrate an increase in oxidation in failing human hearts. There was no apparent sex difference in protein oxidation in the failing hearts. This is not surprising, given the large number of reactions that have been shown to lead to an increase in ROS in failing hearts. The data in this study are consistent with previous studies showing that increases in NOX2 and NOX4 activity are the main contributors to oxidative stress in failing hearts.36,37 Furthermore, we did not find a correlation between increased oxidation and a decrease in SNO; therefore, it appears that the relationship between oxidation and SNO in HF is complex and that an increase in oxidation per se in HF does not lead to an overall decrease in protein SNO. Interestingly, the data suggest some compartmentalization of SNO signaling, with the decrease in SNO occurring primarily in the cytosolic compartment; however, further studies are needed to better investigate SNO and oxidation signaling compartmentalization. Taken together, the data suggest that altered nitroso–redox signaling is a signature of failing hearts.
Sources of Funding
This work was supported by the NHLBI-NIH Intramural Program (ZO1HL006059 and ZO1HL002066, Menazza, and Murphy; HL005903-08, Aponte and Gucek) and NIH grant 5R01HL039752 (Steenbergen).
Disclosures
None.
Supporting Information
Table S1. Peptides Showing Cysteine Oxidation as Identified by Oxidation Resin-Assisted Capture Proteomic Analysis
Table S2. Peptides Showing Cysteine S-Nitrosylation (SNO) as Identified by SNO Resin-Assisted Capture Proteomic Analysis
Table S3. SNO peptides identified by SNO-RAC proteomic analysis in nonfailing female, nonfailing male, DCM female and DCM male.
References
- Sanbe A. Dilated cardiomyopathy: a disease of the myocardium. Biol Pharm Bull. 2013;36:18–22. doi: 10.1248/bpb.b212023. [DOI] [PubMed] [Google Scholar]
- Morales A, Hershberger RE. Genetic evaluation of dilated cardiomyopathy. Curr Cardiol Rep. 2013;15:375. doi: 10.1007/s11886-013-0375-1. [DOI] [PubMed] [Google Scholar]
- Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, Canter C, Wilkinson JD, Lipshultz SE. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–1876. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva RM, Hare JM. Nitric oxide signaling in the cardiovascular system: implications for heart failure. Curr Opin Cardiol. 2006;21:221–228. doi: 10.1097/01.hco.0000221584.56372.dc. [DOI] [PubMed] [Google Scholar]
- Hafstad AD, Nabeebaccus AA, Shah AM. Novel aspects of ros signalling in heart failure. Basic Res Cardiol. 2013;108:359. doi: 10.1007/s00395-013-0359-8. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmet JM, Hare JM. Nitroso-redox interactions in the cardiovascular system. Circulation. 2006;114:1531–1544. doi: 10.1161/CIRCULATIONAHA.105.605519. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Tocchetti CG, Krieg T, Moens AL. Oxidative and nitrosative stress in the maintenance of myocardial function. Free Radic Biol Med. 2012;53:1531–1540. doi: 10.1016/j.freeradbiomed.2012.07.010. [DOI] [PubMed] [Google Scholar]
- Huynh K, Bernardo BC, McMullen JR, Ritchie RH. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther. 2014;142:372–415. doi: 10.1016/j.pharmthera.2014.01.003. [DOI] [PubMed] [Google Scholar]
- Rosenbaugh EG, Savalia KK, Manickam DS, Zimmerman MC. Antioxidant-based therapies for angiotensin II-associated cardiovascular diseases. Am J Physiol Regul Integr Comp Physiol. 2013;304:R917–R928. doi: 10.1152/ajpregu.00395.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Bolli R, Qiu Y, Tang XL, Guo Y, French BA. Gene therapy with extracellular superoxide dismutase protects conscious rabbits against myocardial infarction. Circulation. 2001;103:1893–1898. doi: 10.1161/01.cir.103.14.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Li J, Yuan Z. Effect of antioxidant vitamin supplementation on cardiovascular outcomes: a meta-analysis of randomized controlled trials. PLoS One. 2013;8:e56803. doi: 10.1371/journal.pone.0056803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007;93:903–907. doi: 10.1136/hrt.2005.068270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler AC, Sag CM, Maier LS. Reactive oxygen species and excitation-contraction coupling in the context of cardiac pathology. J Mol Cell Cardiol. 2014;73:92–102. doi: 10.1016/j.yjmcc.2014.03.001. [DOI] [PubMed] [Google Scholar]
- Simon JN, Duglan D, Casadei B, Carnicer R. Nitric oxide synthase regulation of cardiac excitation-contraction coupling in health and disease. J Mol Cell Cardiol. 2014;73:80–91. doi: 10.1016/j.yjmcc.2014.03.004. [DOI] [PubMed] [Google Scholar]
- Canton M, Menazza S, Sheeran FL, Polverino de Laureto P, Di LF, Pepe S. Oxidation of myofibrillar proteins in human heart failure. J Am Coll Cardiol. 2011;57:300–309. doi: 10.1016/j.jacc.2010.06.058. [DOI] [PubMed] [Google Scholar]
- Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, Chen K, Gabrielson KL, Blakely RD, Shih JC, Pacak K, Kass DA, Di Lisa F, Paolocci N. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res. 2010;106:193–202. doi: 10.1161/CIRCRESAHA.109.198366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Shah AM. Ros signalling between endothelial cells and cardiac cells. Cardiovasc Res. 2014;102:249–257. doi: 10.1093/cvr/cvu050. [DOI] [PubMed] [Google Scholar]
- Schulman IH, Hare JM. Regulation of cardiovascular cellular processes by S-nitrosylation. Biochim Biophys Acta. 2012;1820:752–762. doi: 10.1016/j.bbagen.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantalis V, Schulman IH, Hare JM. Nitroso-redox imbalance affects cardiac structure and function. J Am Coll Cardiol. 2013;61:933–935. doi: 10.1016/j.jacc.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Wang H, Ziolo MT. Targeting nos as a therapeutic approach for heart failure. Pharmacol Ther. 2014;142:306–315. doi: 10.1016/j.pharmthera.2013.12.013. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- Kohr MJ, Evangelista AM, Ferlito M, Steenbergen C, Murphy E. S-nitrosylation of TRIM72 at cysteine 144 is critical for protection against oxidation-induced protein degradation and cell death. J Mol Cell Cardiol. 2014;69:67–74. doi: 10.1016/j.yjmcc.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara MR, Thomas B, Cascio MB, Bae BI, Hester LD, Dawson VL, Dawson TM, Sawa A, Snyder SH. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc Natl Acad Sci U S A. 2006;103:3887–3889. doi: 10.1073/pnas.0511321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, Steenbergen C. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res. 2011;108:418–426. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Murphy E. Protein S-nitrosylation and cardioprotection. Circ Res. 2010;106:285–296. doi: 10.1161/CIRCRESAHA.109.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res. 2006;98:403–411. doi: 10.1161/01.RES.0000202707.79018.0a. [DOI] [PubMed] [Google Scholar]
- Wang G, Wu WW, Zeng W, Chou CL, Shen RF. Label-free protein quantification using LC-coupled ion trap or FT mass spectrometry: reproducibility, linearity, and application with complex proteomes. J Proteome Res. 2006;5:1214–1223. doi: 10.1021/pr050406g. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- Tong G, Aponte AM, Kohr MJ, Steenbergen C, Murphy E, Sun J. Postconditioning leads to an increase in protein S-nitrosylation. Am J Physiol Heart Circ Physiol. 2014;306:H825–H832. doi: 10.1152/ajpheart.00660.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huigsloot M, Nijtmans LG, Szklarczyk R, Baars MJ, van den Brand MA, Hendriksfranssen MG, van den Heuvel LP, Smeitink JA, Huynen MA, Rodenburg RJ. A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:488–493. doi: 10.1016/j.ajhg.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Steenbergen C, Murphy E, Sun J. Estrogen receptor-beta activation results in S-nitrosylation of proteins involved in cardioprotection. Circulation. 2009;120:245–254. doi: 10.1161/CIRCULATIONAHA.109.868729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haywood GA, Tsao PS, von der Leyen HE, Mann MJ, Keeling PJ, Trindade PT, Lewis NP, Byrne CD, Rickenbacher PR, Bishopric NH, Cooke JP, McKenna WJ, Fowler MB. Expression of inducible nitric oxide synthase in human heart failure. Circulation. 1996;93:1087–1094. doi: 10.1161/01.cir.93.6.1087. [DOI] [PubMed] [Google Scholar]
- Ago T, Matsushima S, Kuroda J, Zablocki D, Kitazono T, Sadoshima J. The NADPH oxidase NOX4 and aging in the heart. Aging. 2010;2:1012–1016. doi: 10.18632/aging.100261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarretta S, Yee D, Ammann P, Nagarajan N, Volpe M, Frati G, Sadoshima J. Role of NADPH oxidase in the regulation of autophagy in cardiomyocytes. Clin Sci. 2015;128:387–403. doi: 10.1042/CS20140336. [DOI] [PubMed] [Google Scholar]
- Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G, Ketner EA, Majmudar M, Gabrielson K, Halushka MK, Mitchell JB, Biswal S, Channon KM, Wolin MS, Alp NJ, Paolocci N, Champion HC, Kass DA. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation. 2008;117:2626–2636. doi: 10.1161/CIRCULATIONAHA.107.737031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest. 2005;115:509–517. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnicer R, Crabtree MJ, Sivakumaran V, Casadei B, Kass DA. Nitric oxide synthases in heart failure. Antioxid Redox Signal. 2013;18:1078–1099. doi: 10.1089/ars.2012.4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass DA, Shah AM. Redox and nitrosative regulation of cardiac remodeling. Antioxid Redox Signal. 2013;18:1021–1023. doi: 10.1089/ars.2012.4942. [DOI] [PubMed] [Google Scholar]
- Kohr MJ, Murphy E, Steenbergen C. Glyceraldehyde-3-phosphate dehydrogenase acts as a mitochondrial trans-S-nitrosylase in the heart. PLoS One. 2014;9:e111448. doi: 10.1371/journal.pone.0111448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Kohr MJ, Nguyen T, Aponte AM, Connelly PS, Esfahani SG, Gucek M, Daniels MP, Steenbergen C, Murphy E. Disruption of caveolae blocks ischemic preconditioning-mediated S-nitrosylation of mitochondrial proteins. Antioxid Redox Signal. 2012;16:45–56. doi: 10.1089/ars.2010.3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cocheme HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RA, Krieg T, Brookes PS, Murphy MP. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gucek M, Murphy E. What can we learn about carioprotection from the cardiac mitochondrial proteome. Cardiovasc Res. 2010;88:211–218. doi: 10.1093/cvr/cvq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C, Wu C, Ago T, Zhai P, Sadoshima J, Li H. Quantitiative analysis of redox-sensitive proteome with DIGE and ICAT. Mol. Cell Proteomics. 2009;8:1674–1687. [Google Scholar]
- Derakhshan B, Wille PC, Gross SS. Unbiased identification of cysteine S-nitrosylation sites on proteins. Nat Protoc. 2007;2:1685–1691. doi: 10.1038/nprot.2007.210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Peptides Showing Cysteine Oxidation as Identified by Oxidation Resin-Assisted Capture Proteomic Analysis
Table S2. Peptides Showing Cysteine S-Nitrosylation (SNO) as Identified by SNO Resin-Assisted Capture Proteomic Analysis
Table S3. SNO peptides identified by SNO-RAC proteomic analysis in nonfailing female, nonfailing male, DCM female and DCM male.