

Abstract
Background
Cardiovascular disease is the leading cause of death in patients with chronic kidney disease. A body of evidence suggests that p-cresyl sulfate (PCS), a uremic toxin, is associated with the cardiovascular mortality rate of patients with chronic kidney disease; however, the molecular mechanisms underlying this feature have not yet been fully elucidated.
Methods and Results
We aimed to determine whether PCS accumulation could adversely affect cardiac dysfunction via direct cytotoxicity to cardiomyocytes. In mice that underwent 5/6 nephrectomy, PCS promoted cardiac apoptosis and affected the ratio of left ventricular transmitral early peak flow velocity to left ventricular transmitral late peak flow velocity (the E/A ratio) observed by echocardiography (n=8 in each group). Apocynin, an inhibitor of NADPH oxidase activity, attenuates this alteration of the E/A ratio (n=6 in each group). PCS also exhibited proapoptotic properties in H9c2 cells by upregulating the expression of p22phox and p47phox, NADPH oxidase subunits, and the production of reactive oxygen species. Apocynin and N-acetylcysteine were both able to suppress the effect of PCS, underscoring the importance of NADPH oxidase activation for the mechanism of action.
Conclusions
This study demonstrated that the cardiac toxicity of PCS is at least partially attributed to induced NADPH oxidase activity and reactive oxygen species production facilitating cardiac apoptosis and resulting in diastolic dysfunction.
Keywords: apoptosis, cardiac dysfunction, chronic kidney disease, nicotinamide adenine dinucleotide phosphate oxidase, p-cresyl sulfate
Cardiorenal syndrome refers to a broad spectrum of diseases involving both the heart and the kidneys.1 These 2 organs are closely involved in maintaining hemodynamic stability through an intricate network. Clinical studies have proven that chronic kidney disease (CKD) is a major risk factor for the development of cardiovascular complications, including atherosclerosis, vascular calcification, heart failure, and sudden cardiac death.2,3 Earlier stages of CKD are associated with an increased cardiovascular risk.4 Despite the clear clinical evidence addressing this correlation, the pathophysiology underlying the development and progression of cardiovascular impairment following renal dysfunction remains to be elucidated.
CKD is characterized by progressive loss of glomerular filtration rate over a period of months or years. Impaired urinary excretion of metabolites leads to accumulation of various uremic toxins, including middle-molecular-weight compounds (0.5 to 60 kD), free water-soluble low-molecular-weight compounds (<0.5 kD), and protein-bound solutes.5,6 Although chronic hemodialysis and peritoneal dialysis treatments are potent and efficient approaches to removing most free water-soluble compounds, the prognosis is still poor for CKD patients, and cardiovascular disease is a leading cause of death.7–9 A possible culprit involved in CKD is protein-bound solutes, a large family of compounds that cause various diseases.
p-Cresyl sulfate (PCS) is a protein-bound uremic toxin molecule. Plasma PCS concentrations are determined mainly by intestinal uptake of p-cresol, metabolism of p-cresol to PCS, and renal excretion of PCS.10,11 For CKD patients, albumin is the main binding protein of PCS in circulation.12 Previous studies have demonstrated that the accumulated PCS plays a role in activating leukocyte radical production, disturbing the renin–angiotensin–aldosterone system, promoting renal tubular cell damage, and interfering with insulin signaling pathways.13–17 The results of our previous study revealed that p-cresol disrupted proliferation, migration, tube formation, and cell-cycle kinetics in endothelial progenitor cells, but its metabolite PCS did not.18 An increasing body of clinical evidence suggests that serum PCS is a predictor of cardiovascular disease and mortality associated with CKD.19–21
Despite previous clinical implications, no study has focused on the role of PCS in cardiac injury; therefore, we investigated the effect of PCS in cardiotoxicity and cardiac dysfunction. We hope that our findings provide new insights into cardiorenal syndrome and facilitate the development of potential treatments.
Methods
Animals and Induction of Renal Insufficiency
Male C57BL/6J mice aged 5 weeks were obtained from the Model Animal Research Center of Nanjing University (Nanjing, People’s Republic of China) and housed in a specific pathogen–free facility under a 12-hour light/dark cycle with free access to food and water. After 1 week of accommodation, the mice underwent 5/6 nephrectomy (Nx) with a 2-step surgical procedure or a sham operation. Surgical procedures were performed under pentobarbital anesthesia, as described previously.22 Briefly, anesthetized mice underwent removal of approximately two-thirds of the left kidney and right subcapsular Nx after 1 week. Sham-operated mice underwent laparotomy in parallel by decapsulating the kidney before wound closure. The animal experimental protocol complied with the Animal Management Rules of the Chinese Ministry of Health (document no. 55, 2001) and was approved by the animal care committee of Shanghai Jiao Tong University.
Experimental Protocol
For animal study 1, experimental mice were randomly divided into 4 groups: sham control (n=8), sham treated with PCS (PCS; n=8), untreated 5/6 nephrectomized (Nx; n=8), and PCS-treated 5/6 nephrectomized mice (Nx/PCS; n=8). For animal study 2, Nx mice were randomly divided into 2 groups: 5/6 nephrectomized treated with PCS (Nx/PCS; n=6) and treated with both PCS and apocynin (Nx/PCS/apocynin; n=6). PCS and apocynin were administered by oral gavage for 8 weeks. The concentrations of PCS and apocynin were adjusted for a daily intake of 100 and 200 mg/kg, respectively. PCS was purchased from Shanghai Chempartner Co, Ltd. The product was >99% pure, and its structure was confirmed by nuclear magnetic resonance spectroscopy.
Echocardiography and Doppler Analysis
Transthoracic echocardiography was performed noninvasively with a high-resolution ultrasound imaging system (Vevo 2100; VisualSonics, Inc) equipped with a 30-MHz mechanical transducer. The mice were lightly anesthetized with 2% isoflurane/100% oxygen and placed on a warming platform (set to 37°C) for the duration of the recordings. The heart rate was monitored simultaneously by electrocardiography.
Two-dimensional guided M-mode echocardiography was performed in the parasternal long-axis view. Left ventricular (LV) end-diastolic diameter and end-systolic diameter were measured. The LV end-diastolic volume and end-systolic volume were calculated, as described previously.23 Fractional shortening and ejection fraction were calculated using the following formulas: FS (%)=[(LVEDD−LVESD)/LVEDD]×100% and EF (%)=[(LVEDV−LVESV)/LVEDV]×100%. The formulas used the following abbreviations: FS, fractional shortening; LVEDD, LV end-diastolic diameter; LVESD, LV end-systolic diameter; EF, ejection fraction; LVEDV, LV end-diastolic volume; LVESV, LV end-systolic volume. Mitral valve flow Doppler was acquired in an apical 4-chamber view. LV diastolic function was assessed by measuring the wave ratio of the LV transmitral early peak flow velocity to LV transmitral late peak flow velocity (the E/A ratio). M-mode and Doppler measurement data represent 3 to 6 averaged cardiac cycles from at least 2 scans per mouse.
Blood Pressure Measurements
For animal study 1, the tail systolic blood pressure was measured in conscious mice using a Softron tail-cuff BP-98A noninvasive sphygmomanometer (Softron Inc).
Histological Analysis
Formalin-fixed myocardial sections were deparaffinized and rehydrated, as described in detail previously.24 Picrosirius red staining was used to assess the collagen content. The sections were photographed using a microscope (Olympus), and the percentage of picrosirius red stain in the sections was calculated.
Next, 5-μm sections of the left ventricle were prepared to detect apoptosis by in situ terminal deoxynucleotidyl-transferase-mediated 2′-deoxyuridine-5′-triphosphate nick-end labeling, or TUNEL. The procedures were performed according to the manufacturer’s instructions (TUNEL apoptosis assay kit; F. Hoffmann-La Roche Ltd). The percentage of apoptotic cells was determined as an apoptotic index, which was calculated by dividing the number of positively stained cardiomyocyte nuclei by the total number of cardiomyocyte nuclei and multiplying that value by 100.
Cell Culture and Treatment
H9c2 cardiac myoblasts were maintained in DMEM/high-glucose culture medium containing 10% FBS in a water-saturated atmosphere of 5% CO2 and 95% air at 37°C.The cells were starved in DMEM/high-glucose culture medium containing 0.5% FBS for 6 hours before exposure to PCS (0, 250, 500, and 1000 μmol/L) for the designated times. The cells were treated with 100 μmol/L apocynin or 10 mmol/L N-acetylcysteine (NAC) for 4 hours before they were incubated with 500 μmol/L PCS. H9c2 cells were obtained from Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, People’s Republic of China). DMEM/high-glucose culture medium and FBS were purchased from Gibco (Thermo Fisher Scientific). Apocynin and NAC were purchased from Sigma-Aldrich.
Annexin V–FITC and Propidium Iodide Staining for Detecting Phosphatidylserine Translocation
Cell apoptosis was measured with fluorescein isothiocyanate–labeled human recombinant annexin V (annexin V–FITC) and with propidium iodide (PI) using a detection kit (BD Pharmingen), according to the manufacturer’s instructions. Briefly, the cells were harvested, washed once with cold PBS, and resuspended in 1× binding buffer at a concentration of 1×106 cells/mL. Next, 100 μL of the solution (1×105 cells) was incubated with 5 μL annexin V–FITC and 5 μL PI solution for 15 minutes in the dark at room temperature, and 400 μL of 1× binding buffer was added. Samples were analyzed using flow cytometry (BD FACSCalibur; BD Biosciences) within 1 hour. Apoptotic cells, which stained positive for annexin V–FITC and negative or positive for PI, were counted and represented as a percentage of the total cell count.
Western Blot Analysis
H9c2 cells were cultured and treated in 6-well plates, washed twice with PBS, and lysed with the ProteoJET Mammalian Cell Lysis Reagent (Fermentas; Thermo Fisher Scientific) to extract cytoplasmic proteins. After the protein concentration was determined, the supernatants were stored at −80°C. After denaturation, equal amounts of protein extracts were subjected to SDS-PAGE and blotted onto polyvinylidene fluoride membranes. The membranes were blocked with Tris-buffered saline with Tween 20 containing 5% nonfat milk powder and subsequently probed overnight at 4°C with antibodies against B-cell lymphoma 2 (Bcl-2, 1:1000), Bcl-2 Associated X Protein (Bax, 1:1000), β-actin (1:2000), p22phox (1:500), and p47phox (1:500), followed by incubation with respective horseradish peroxidase–conjugated secondary antibodies (1:5000) for 1 hour at room temperature. Bands were detected using an enhanced chemiluminescense system (Millipore) and quantified by scanning densitometry. Each image was captured, and the intensity of each band was analyzed with Quantity One software (Bio-Rad Laboratories Inc).
Caspase-3 Activity Assay
This assay is based on the ability of caspase-3 to change acetyl-Asp-Glu-Val-Asp p-nitroanilide into p-nitroaniline, a yellow formazan product. Caspase-3 activity was determined using a caspase-3 activity kit (Beyotime), according to the manufacturer’s protocol. Briefly, treated cells were lysed with lysis buffer on ice after washing with cold Hanks’ balanced salt solution with calcium and magnesium. After the reaction buffer and caspase-3 substrate were incubated in 96-well plates at 37°C for 4 hours, the caspase-3 activity was quantified with a microplate spectrophotometer at 405 nm. Caspase-3 activity was expressed as the fold of enzyme activity compared with that of synchronized cells.
Reactive Oxygen Species Assay
Intracellular reactive oxygen species (ROS) levels were assayed on the basis of 5-(and-6)-carboxy-2′, 7′- dichlorodihydrofluorescein diacetate, or carboxy-H2DCFDA, a reliable fluorogenic marker for ROS in live cells.25 The Image-iT LIVE Green Reactive Oxygen Species Detection Kit (Molecular Probes; Thermo Fisher Scientific) was used, according to the manufacturer’s instructions. Briefly, cells were washed once with warm Hanks’ balanced salt solution with calcium and magnesium and incubated in a sufficient amount of the 25 μmol/L carboxy-H2DCFDA working solution for 30 minutes in the dark at 37°C. During the last 5 minutes of the incubation, 1.0 μmol/L Hoechst 33342 was added to the carboxy-H2DCFDA staining solution. The samples were washed 3 times with warm Hanks’ balanced salt solution with calcium and magnesium. Images were captured immediately with a microscope (Olympus).
Statistical Analysis
Data are presented as mean±SEM analyzed by paired or unpaired Student t test, unless otherwise stated. Statistical significance was determined with 1-way ANOVA followed by Student-Newman-Keuls post hoc analysis. A P value of <0.05 was considered statistically significant. Data were analyzed using GraphPad Prism 5 software (GraphPad Software Inc) and SPSS software (version 13.0; IBM Corp).
Results
LV Dysfunction Assessed by Echocardiography and Doppler
As shown in Figure 1A through 1C, although ejection fraction and fractional shortening were decreased in 5/6 nephrectomized mice compared with untreated mice, the differences were not statistically significant. Echocardiography revealed no change in ejection fraction or fractional shortening regardless of whether or not PCS treatment was given. Other relevant data are summarized in Table1.
Figure 1.
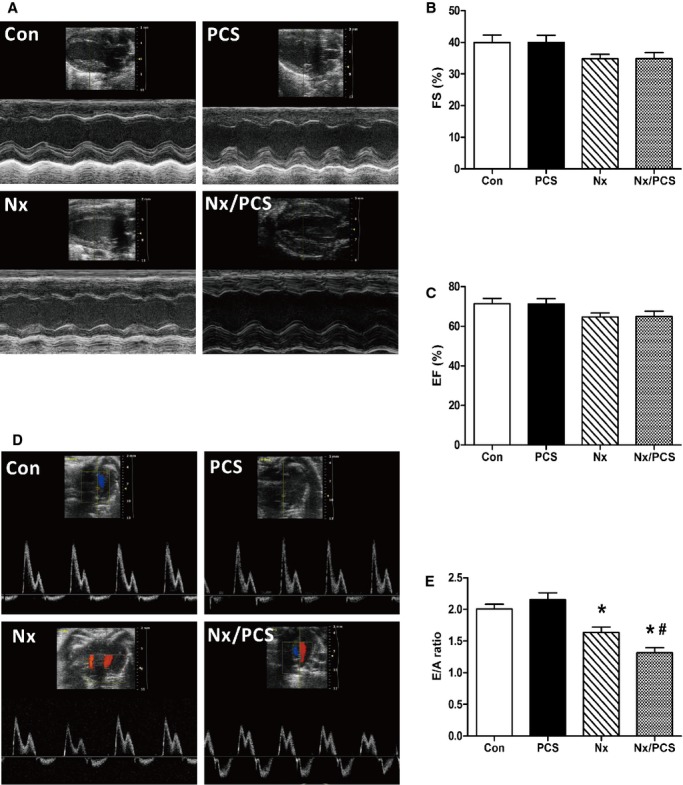
Functional analysis of the left ventricle with echocardiography. A, Representative M-mode echocardiograms from control, PCS-treated, Nx, and Nx/PCS-treated mice. B, The mean left ventricular FS in control, PCS, Nx, and Nx/PCS mice. Data are presented as mean±SEM (n=6 to 8). C, The mean left ventricular EF in control, PCS, Nx, and Nx/PCS mice. Data are presented as mean±SEM (n=6 to 8). D, Representative Doppler echocardiograms from control, PCS, Nx, and Nx/PCS mice. E, The mean E/A ratio in control, PCS, Nx, and Nx/PCS mice. Data are presented as mean±SEM (n=6 to 8). *P<0.05 vs control group. #P<0.05 vs Nx group. A indicates left ventricular transmitral late peak flow velocity; Con, control; E, left ventricular transmitral early peak flow velocity; EF, ejection fraction; FS, fractional shortening; Nx, 5/6 nephrectomy; PCS, p-cresyl sulfate.
Table 1.
Characteristics of Animal Models in Animal Study 1
| Parameters | Con Mice(n=8) | PCS Mice(n=8) | Nx Mice(n=8) | Nx/PCS Mice(n=8) |
|---|---|---|---|---|
| IVSd, mm | 0.856±0.032 | 0.864±0.050 | 0.979±0.046* | 0.983±0.050 |
| LVEDD, mm | 3.26±0.26 | 3.45±0.19 | 3.61±0.19 | 3.41±0.24 |
| LVPWd, mm | 0.847±0.043 | 0.853±0.042 | 0.897±0.031 | 0.925±0.048 |
| IVSs, mm | 1.481±0.086 | 1.497±0.069 | 1.624±0.080 | 1.691±0.070 |
| LVESD, mm | 1.99±0.19 | 2.07±0.14 | 2.37±0.16 | 2.22±0.18 |
| LVPWs, mm | 1.255±0.069 | 1.301±0.065 | 1.531±0.084* | 1.592±0.078* |
| LVW/BW, mg/g | 3.32±0.04 | 3.31±0.05 | 3.82±0.17* | 3.69±0.13* |
| SBP, mm Hg | 94.8±2.3 | 93.1±2.3 | 132.5±4.5* | 133.9±4.8* |
Values are mean±SEM. BW indicates body weight; Con, control; IVSd, interventricular septum diastole; IVSs, interventricular septum systole; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; LVPWd, left ventricular posterior wall diastole; LVPWs, left ventricular posterior wall systole; LVW, left ventricle weight; Nx, 5/6 nephrectomy; PCS, p-cresyl sulfate; SBP, systolic blood pressure.
P<0.05 vs Con mice.
Analysis of diastolic function revealed no change in the E/A ratio between the control and PCS-treated mice (control: 2.01±0.07; PCS: 2.15±0.11; P>0.05). Compared with the controls, 5/6 nephrectomized mice showed a significant reduction in the E/A ratio (control: 2.01±0.07; Nx: 1.64±0.08; P<0.05). The E/A ratio was further reduced in 5/6 nephrectomized mice treated with PCS (Nx: 1.64±0.08; Nx/PCS: 1.32±0.08; P<0.05) (Figure 1D and 1E).
Effect of PCS on Histological Changes
Specific staining for apoptosis using the TUNEL assay in the LV myocardium demonstrated significantly increased apoptosis following PCS administration (control: 3.75±0.95%; PCS: 7.75±0.85%; P<0.05). A similar phenomenon was observed in the PCS-treated 5/6 nephrectomized mice; the effect of the subtotal Nx was amplified (Nx: 12.5±1.94%; Nx/PCS: 19.0±1.22%; P<0.05) (Figure 2A).
Figure 2.
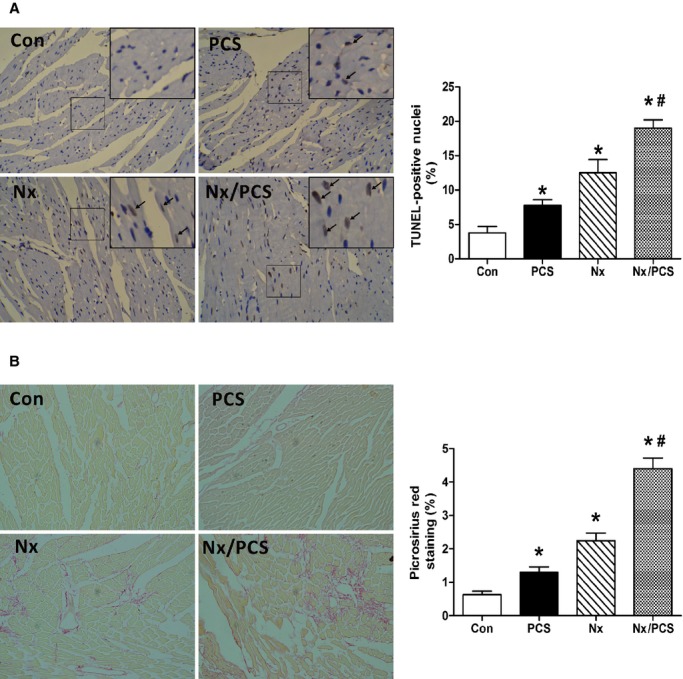
Histological analysis of cardiac sections from control, PCS-treated, Nx, and Nx/PCS mice. A, Representative images (×200) and analysis of TUNEL staining in all 4 groups. *P<0.05 vs control group. #P<0.05 vs Nx group. Data are presented as mean±SEM (n=4). B, Representative images (×200) and analysis of picrosirius red staining in the 4 groups. *P<0.05 vs control group. #P<0.05 vs Nx group. Data are presented as mean±SEM (n=4). Con indicates control; Nx, 5/6 nephrectomy; PCS, p-cresyl sulfate; TUNEL, terminal deoxynucleotidyl-transferase-mediated 2′-deoxyuridine-5′-triphosphate nick-end labeling.
In addition, we observed a significant increase in the interstitial and perivascular collagen in 5/6 nephrectomized mice compared with controls (control: 0.63±0.10%; Nx: 2.24±0.23%; P<0.05). Treatment with PCS further augmented the positive area of picrosirius red stain in the LV of 5/6 nephrectomized mice (Nx/PCS: 4.40±0.32%). Similarly, administration of PCS in sham-operated mice resulted in increased cardiac collagen content (PCS: 1.30±0.16%) (Figure 2B).
Effect of PCS on Apoptosis in H9c2 Cells
We then studied the effect of PCS on phosphatidylserine exposure in H9c2 cells using flow cytometry. Annexin V–FITC and PI were used to determine whether the cells were viable (negative for both annexin V–FITC and PI), damaged (positive for annexin V–FITC only), or dead (positive for both annexin V–FITC and PI or for PI only). Quantitative analysis showed that PCS treatment for 24 hours enhanced apoptosis of H9c2 cells in a dose-dependent manner. Specifically, compared with the control, PCS significantly increased the percentage of apoptosis at the concentrations of 500 and 1000 μmol/L (control: 15.37±1.43%; 250 μmol/L PCS: 20.90±2.08%; 500 μmol/L PCS: 28.03±1.21%; 1000 μmol/L PCS: 30.43±1.59%) (Figure 3A).
Figure 3.
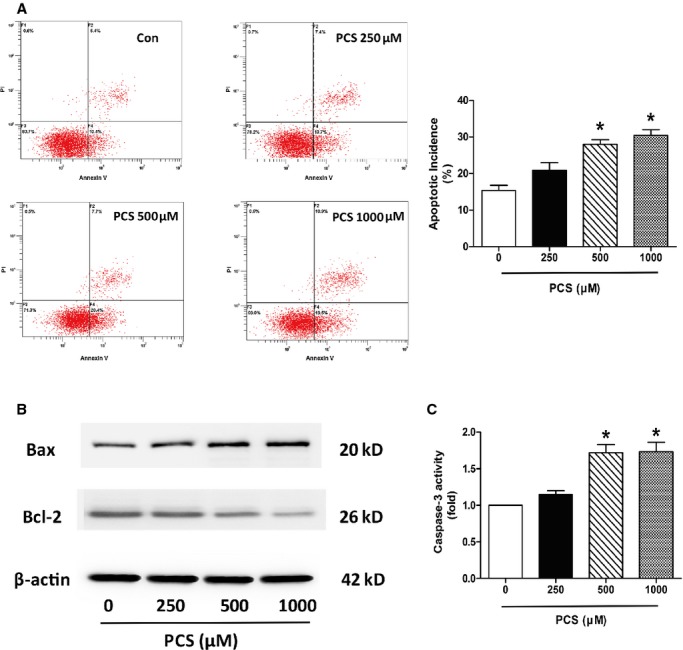
Apoptotic analysis in H9c2 cells after incubation with various concentrations of PCS (0, 250, 500, and 1000 μmol/L). A, Evaluation of apoptotic incidence by flow cytometry analysis via double staining with annexin V and fluorescein isothiocyanate and with propidium iodide. *P<0.05 vs control group. Data are presented as mean±SEM (n=3). B, Representative Western blots showing the expression of Bax and Bcl-2. C, Changes in caspase-3 activity. *P<0.05 vs control group. Data are presented as mean±SEM (n=3). Con indicates control; PCS, p-cresyl sulfate.
During the apoptotic process, the Bcl-2 protein family has emerged as a key regulator, consisting of death agonists (Bax and Bak) and antagonists (Bcl-2 and Bcl-xL). As shown in Figure 3B, Western blots from the present study revealed significant upregulation of Bax and downregulation of Bcl-2 expression following exposure to PCS.
Caspase-3 executes apoptosis and is involved in many important events leading to the completion of apoptosis. Activation of caspase-3 serves as an early marker of apoptosis in H9c2 cells. Our data showed that treatment with 500 and 1000 μmol/L PCS induced upregulation of caspase-3 activity compared with controls (250 μmol/L PCS: 1.15-fold; 500 μmol/L PCS: 1.72-fold; 1000 μmol/L PCS: 1.73-fold) (Figure 3C).
Effect of PCS on the NADPH Oxidase Subunit Expression and Intracellular ROS Production in H9c2 Cells
We examined whether nicotinamide adenine dinucleotide phosphate (NADPH) oxidase plays an important role in PCS-induced ROS production in H9c2 cells. The protein expression levels of p22phox and p47phox were evaluated by Western blotting. The results of the Western blot analysis revealed that PCS significantly increased the expression of p22phox in H9c2 cells at the concentrations of 250, 500, and 1000 μmol/L (1.59-, 2.21-, and 2.25-fold, respectively, over controls). Similarly, p47phox expression was significantly upregulated by PCS treatment, although to a lesser extent than p22phox (1.48-, 1.88-, and 1.97-fold, respectively, over controls) (Figure 4A). The fluorescent dye carboxy-H2DCFDA was used to monitor intracellular ROS production. The results revealed significant increase in fluorescence intensity in the PCS-treated cells (500 and 1000 μmol/L) relative to the controls, indicating that PCS can potentially increase the production of ROS (Figure 4B).
Figure 4.
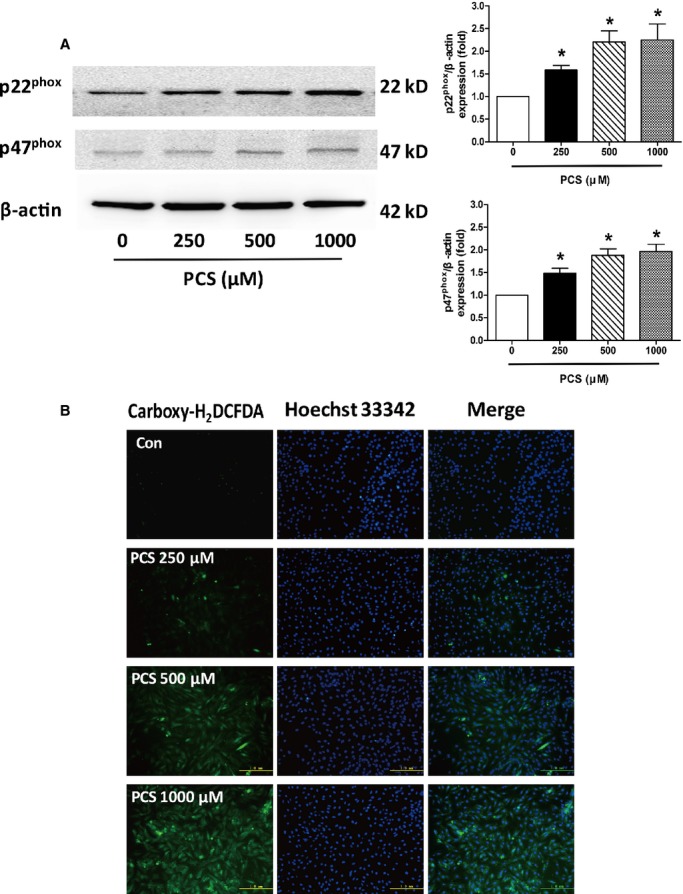
Evaluation of PCS on the expression of NADPH oxidase subunits and the production of ROS in H9c2 cells. A, Representative Western blots and analysis of p22phoxand p47phox expression. *P<0.05 vs control group. Data are presented as mean±SEM (n=3). B, Representative images of carboxy-H2DCFDA staining for detection of intracellular ROS. Carboxy-H2DCFDA indicates 5-(and-6)-carboxy-2′, 7′- dichlorodihydrofluorescein diacetate; Con, control; NADPH, nicotinamide adenine dinucleotide phosphate; PCS, p-cresyl sulfate; ROS, reactive oxygen species.
Effect of Apocynin and NAC on PCS-Induced Apoptosis in H9c2 Cells
Because oxidative stress is a key factor contributing to initiation of apoptosis in cardiomyocytes, we examined whether apocynin or NAC reduced PCS-induced cell damage. H9c2 cells were pretreated with 100 μmol/L apocynin or 10 mmol/L NAC for 3 hours and then exposed to 500 μmol/L PCS for 21 hours. Consistent with previous results, 500 μmol/L PCS elevated the expression of Bax and decreased that of Bcl-2, indicating increased apoptosis by PCS. The blots showed that pretreatment with apocynin or NAC partially restored the imbalance of Bax and Bcl-2 (Figure 5A).
Figure 5.
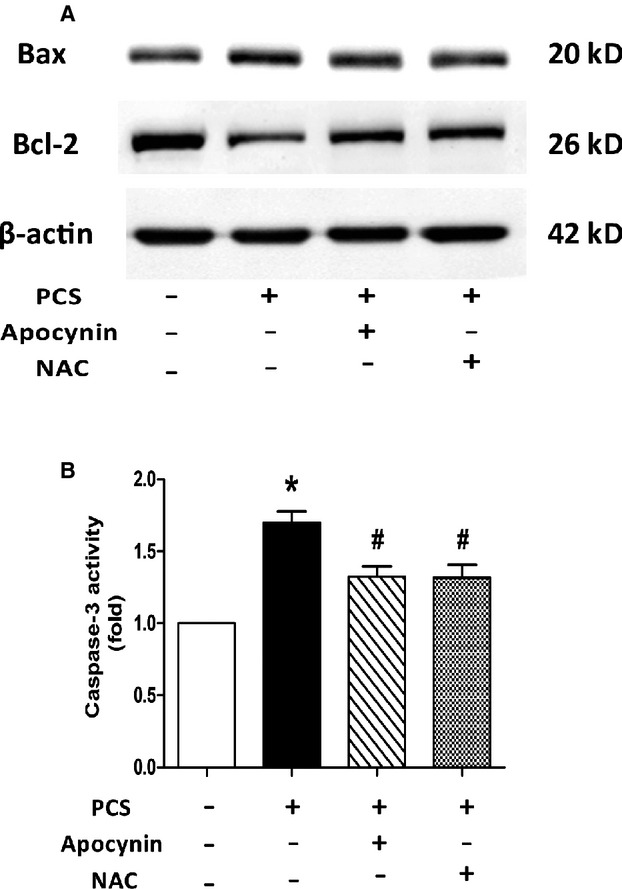
Effects of apocynin and NAC on PCS-induced apoptotic process in H9c2 cells. A, Representative Western blots of showing the expression of Bax and Bcl-2. B, Changes in caspase-3 activity. *P<0.05 vs control group. #P<0.05 vs PCS group. Data are presented as mean±SEM (n=3). NAC indicates N-acetylcysteine; PCS, p-cresyl sulfate.
The assay of caspase-3 activity proved again that apocynin or NAC could protect H9c2 cells from the detrimental effect of PCS. Compared with the controls, cells treated with 500 μmol/L PCS significantly elevated caspase-3 activity, whereas pretreatment with apocynin or NAC could restrict this activation (500 μmol/L PCS: 1.70-fold; PCS and apocynin: 1.32-fold; PCS and NAC: 1.32-fold) (Figure 5B).
Effect of Apocynin on PCS-Induced LV Dysfunction
Finally, we analyzed whether apocynin treatment could ameliorate PCS-induced alteration of diastolic function in 5/6 nephrectomized mice. As shown in Figure 6, PCS treatment resulted in an E/A ratio of 1.30, whereas PCS and apocynin together elevated the ratio to 1.55 (Nx/PCS: 1.30±0.07; Nx/PCS/apocynin: 1.55±0.07). Statistical analysis revealed significant differences in the E/A ratios between the 2 groups. Other relevant data are summarized in Table2.
Figure 6.
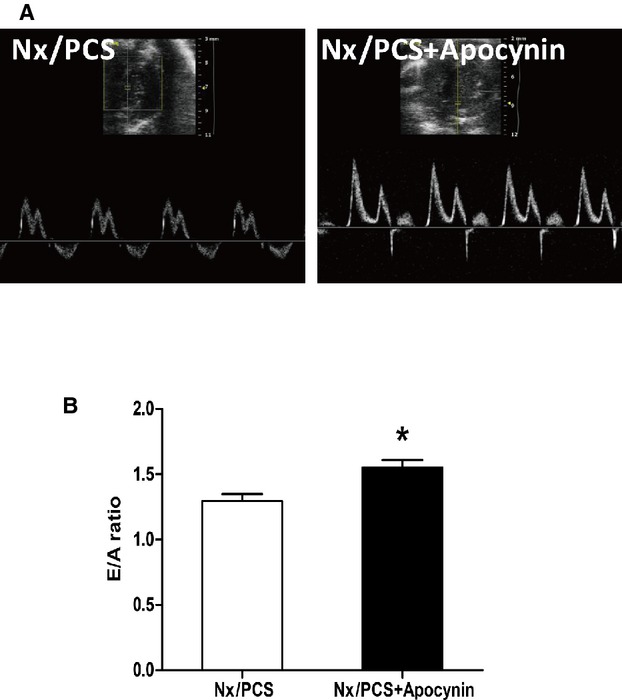
Left ventricular transmitral flow velocity analysis with echocardiography. A, Representative Doppler echocardiograms from Nx/PCS and with both PCS and apocynin (Nx/PCS/apocynin) mice. B, The mean E/A ratio from Nx/PCS and Nx/PCS/apocynin mice. Data are presented as mean±SEM (n=6). *P<0.05 vs Nx/PCS group. A indicates left ventricular transmitral late peak flow velocity; E, left ventricular transmitral early peak flow velocity; Nx indicates 5/6 nephrectomy; PCS, p-cresyl sulfate.
Table 2.
Characteristics of Animal Models in Animal Study 2
| Parameters | Nx/PCS Mice (n=6) | Nx/PCS/Apocynin Mice (n=6) |
|---|---|---|
| EF, % | 64.14±1.86 | 64.64±2.18 |
| FS, % | 34.22±1.36 | 34.89±1.68 |
| IVSd, mm | 0.985±0.055 | 0.981±0.046 |
| LVEDD, mm | 3.50±0.16 | 3.91±0.17 |
| LVPWd, mm | 0.928±0.048 | 0.911±0.038 |
| IVSs, mm | 1.708±0.068 | 1.650±0.090 |
| LVESD, mm | 2.26±0.10 | 2.54±0.11 |
| LVPWs, mm | 1.606±0.081 | 1.574±0.097 |
| LVW/BW, mg/g | 3.68±0.11 | 3.72±0.12 |
| SBP, mm Hg | 139.8±4.5 | 137.5±4.7 |
Values are means±SEM. BW indicates body weight; EF, ejection fraction; FS, fractional shortening; IVSd, interventricular septum diastole; IVSs, interventricular septum systole; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; LVPWd, left ventricular posterior wall diastole; LVPWs, left ventricular posterior wall systole; LVW, left ventricle weight; Nx, 5/6 nephrectomy; PCS, p-cresyl sulfate; SBP, systolic blood pressure.
Discussion
In the present study, PCS was found to promote NADPH oxidase–derived oxidative stress, resulting in an elevated percentage of apoptosis in H9c2 cells. In vivo, PCS was found to be involved in impaired LV diastolic function and increased cardiac apoptosis. We also observed an increase in cardiac synthesis of collagen, indicating possible compensation in the heart. Apocynin, an inhibitor of NADPH oxidase activity,26,27 was found to partially restore the PCS-induced decreased cardiac function.
PCS is of particular interest among the various uremic toxins because of its highly protein-bound property. PCS is about 94% bound to plasma protein, causing a predictable decrease in the measured dialytic clearance.28 An increasing body of evidence suggests that PCS is a predictor of cardiovascular and/or all-cause mortality in CKD patients; however, whether this biomarker plays a key role in the pathological process of cardiorenal syndrome is still under investigation.
In our study, Doppler-derived mitral flow velocities revealed an altered E/A ratio in 5/6 nephrectomized mice treated with PCS, and such alteration is always accompanied by diastolic relaxation abnormalities.29 Despite this finding, we did not observe any significant changes in systolic function but cannot exclude the possibility that systolic changes might have been detected had the animals been studied at later time points.
Previous experimental investigations have shown that renal impairment would mediate early cardiac apoptosis. Interestingly, Martin et al originally sought to assess LV structure and function in mild renal insufficiency; however, microarray gene analysis identified widespread changes in apoptotic pathway genes.30 In the present study, LV apoptosis was confirmed by TUNEL staining, suggesting that PCS facilitates the process of apoptosis when renal function is impaired. In vitro H9c2 cells cultured in medium supplemented with 0.5% FBS underwent apoptosis after PCS stimulation in a dose-dependent manner (250, 500, and 1000 μmol/L for 24 hours). An imbalance between the Bcl-2 and Bax expression levels was found to be closely correlated with apoptosis,31 and caspase-3 is a key molecule in the apoptosis pathway.32 The data from this study demonstrate that PCS significantly amplifies the effect of serum limitation on H9c2 cell apoptosis, as shown by the increased proportion of cells positively stained with annexin V–FITC, decreased Bcl-2 expression, and the increased Bax and caspase-3 fragment expression levels.
ROS-generating NADPH oxidase is a family of enzymes consisting mainly of 7 members: Nox1, Nox2, Nox3, Nox4, Nox5, Duox1, and Duox2.33,34 Of these, Nox2 and Nox4 are the predominant isoforms expressed in cardiomyocytes. Activation of these enzymes depends on the assembly of various subunits, including p22phox, p67phox, p40phox, p47phox, and Rac2.35 In our study, PCS treatment was found to upregulate the expression of the NADPH oxidase subunits p22phox and p47phox. An elevated intracellular ROS level was also observed. ROS and the related redox signaling potentially affect apoptosis at several levels, including at upstream signaling pathways that are proapoptotic and at the mitochondrial level.36,37 Indeed, apoptosis is markedly curbed in H9c2 cells pretreated with apocynin or NAC because either agent is capable of reversing the imbalance of Bcl-2 and Bax and restricting the activation of caspase-3. Based on these findings, we speculate that NADPH oxidase is at least partially involved in the observed PCS-related apoptosis and subsequent diastolic dysfunction.
Apocynin is a naturally occurring methoxy-substituted catechol with activity that relies on its oxidation and subsequent formation of the symmetrical dimer.27,38 This formatted diapocynin is metabolically active and inhibits the intracellular assembly of NADPH oxidases.27 Of note, apocynin also protects the heart from the E/A ratio alterations that are exacerbated by PCS, indicating that cardiac diastolic function is reserved by an inhibition of NADPH oxidase activity.
The present study has several limitations. First, we did not test our conclusion on gene knockout mouse models. If the toxicity of PCS is reduced in Nox2 or Nox4 knockout mice, the mechanisms should be more convincing. Second, our clinical data were limited. We are still working to establish a database of patients with cardiorenal syndrome. Such long-term and detailed studies, however, are difficult to execute, given the need for substantial follow-up durations.
In summary, the uremic toxin PCS was found to disturb the cellular redox balance and to contribute to detrimental cardiac effects in cell culture. Animal experiments confirm the adverse effects of PCS in cardiomyocytes. Such factors are attributable to the presence of diastolic dysfunction observed by echocardiography. These findings might provide new knowledge about cardiorenal syndrome and thus represent an important novel therapeutic target for the amelioration of the disease.
Acknowledgments
We thank Dr Yuehua Fang (Rui Jin Hospital, Shanghai, China) and Dr Jinquan Hu (Changzheng Hospital, Shanghai, China) for their excellent technical assistance and valuable comments on our study.
Sources of Funding
This study was supported by grants from the National Natural Science Foundation of China (81300178 and 81370401).
Disclosures
None.
References
- Cruz DN. Cardiorenal syndrome in critical care: the acute cardiorenal and renocardiac syndromes. Adv Chronic Kidney Dis. 2013;20:56–66. doi: 10.1053/j.ackd.2012.10.005. [DOI] [PubMed] [Google Scholar]
- Herzog CA, Asinger RW, Berger AK, Charytan DM, Diez J, Hart RG, Eckardt KU, Kasiske BL, McCullough PA, Passman RS, DeLoach SS, Pun PH, Ritz E. Cardiovascular disease in chronic kidney disease. A clinical update from kidney disease: improving global outcomes (KDIGO) Kidney Int. 2011;80:572–586. doi: 10.1038/ki.2011.223. [DOI] [PubMed] [Google Scholar]
- Matsushita K, Sang Y, Ballew SH, Shlipak M, Katz R, Rosas SE, Peralta CA, Woodward M, Kramer HJ, Jacobs DR, Sarnak MJ, Coresh J. Subclinical atherosclerosis measures for cardiovascular prediction in CKD. J Am Soc Nephrol. 2015;26:439–447. doi: 10.1681/ASN.2014020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb SS, Abraham W, Butler J, Forman DE, Loh E, Massie BM, O’Connor CM, Rich MW, Stevenson LW, Young J, Krumholz HM. The prognostic importance of different definitions of worsening renal function in congestive heart failure. J Cardiac Fail. 2002;8:136–141. doi: 10.1054/jcaf.2002.125289. [DOI] [PubMed] [Google Scholar]
- Kurella M, Lo JC, Chertow GM. Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. J Am Soc Nephrol. 2005;16:2134–2140. doi: 10.1681/ASN.2005010106. [DOI] [PubMed] [Google Scholar]
- Duranton F, Cohen G, De Smet R, Rodriguez M, Jankowski J, Vanholder R, Argiles A. Normal and pathologic concentrations of uremic toxins. J Am Soc Nephrol. 2012;23:1258–1270. doi: 10.1681/ASN.2011121175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998;32:S112–S119. doi: 10.1053/ajkd.1998.v32.pm9820470. [DOI] [PubMed] [Google Scholar]
- Anavekar NS, Pfeffer MA. Cardiovascular risk in chronic kidney disease. Kidney Int Suppl. 2004;66:S11–S15. doi: 10.1111/j.1523-1755.2004.09203.x. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, III, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer TW, Hostetter TH. Uremic solutes from colon microbes. Kidney Int. 2012;81:949–954. doi: 10.1038/ki.2011.504. [DOI] [PubMed] [Google Scholar]
- Poesen R, Viaene L, Verbeke K, Augustijns P, Bammens B, Claes K, Kuypers D, Evenepoel P, Meijers B. Cardiovascular disease relates to intestinal uptake of p-cresol in patients with chronic kidney disease. BMC Nephrol. 2014;15:87. doi: 10.1186/1471-2369-15-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaene L, Annaert P, de Loor H, Poesen R, Evenepoel P, Meijers B. Albumin is the main plasma binding protein for indoxyl sulfate and p-cresyl sulfate. Biopharm Drug Dispos. 2013;34:165–175. doi: 10.1002/bdd.1834. [DOI] [PubMed] [Google Scholar]
- Schepers E, Meert N, Glorieux G, Goeman J, Van der Eycken J, Vanholder R. p-Cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol Dial Transplant. 2007;22:592–596. doi: 10.1093/ndt/gfl584. [DOI] [PubMed] [Google Scholar]
- Sun CY, Chang SC, Wu MS. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS One. 2012;7:e34026. doi: 10.1371/journal.pone.0034026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Miyamoto Y, Honda D, Tanaka H, Wu Q, Endo M, Noguchi T, Kadowaki D, Ishima Y, Kotani S, Nakajima M, Kataoka K, Kim-Mitsuyama S, Tanaka M, Fukagawa M, Otagiri M, Maruyama T. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013;83:582–592. doi: 10.1038/ki.2012.448. [DOI] [PubMed] [Google Scholar]
- Sun CY, Hsu HH, Wu MS. p-Cresol sulfate and indoxyl sulfate induce similar cellular inflammatory gene expressions in cultured proximal renal tubular cells. Nephrol Dial Transplant. 2013;28:70–78. doi: 10.1093/ndt/gfs133. [DOI] [PubMed] [Google Scholar]
- Koppe L, Pillon NJ, Vella RE, Croze ML, Pelletier CC, Chambert S, Massy Z, Glorieux G, Vanholder R, Dugenet Y, Soula HA, Fouque D, Soulage CO. p-Cresyl sulfate promotes insulin resistance associated with ckd. J Am Soc Nephrol. 2013;24:88–99. doi: 10.1681/ASN.2012050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JZ, Zhang J, Yang K, Du R, Jing YJ, Lu L, Zhang RY. p-Cresol, but not p-cresylsulphate, disrupts endothelial progenitor cell function in vitro. Nephrol Dial Transplant. 2012;27:4323–4330. doi: 10.1093/ndt/gfs382. [DOI] [PubMed] [Google Scholar]
- Meijers BK, Bammens B, De Moor B, Verbeke K, Vanrenterghem Y, Evenepoel P. Free p-cresol is associated with cardiovascular disease in hemodialysis patients. Kidney Int. 2008;73:1174–1180. doi: 10.1038/ki.2008.31. [DOI] [PubMed] [Google Scholar]
- Liabeuf S, Barreto DV, Barreto FC, Meert N, Glorieux G, Schepers E, Temmar M, Choukroun G, Vanholder R, Massy ZA. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol Dial Transplant. 2010;25:1183–1191. doi: 10.1093/ndt/gfp592. [DOI] [PubMed] [Google Scholar]
- Lin CJ, Pan CF, Chuang CK, Sun FJ, Wang DJ, Chen HH, Liu HL, Wu CJ. p-Cresyl sulfate is a valuable predictor of clinical outcomes in pre-ESRD patients. Biomed Res Int. 2014;2014:526932. doi: 10.1155/2014/526932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J, Zhang W, Zhu Z, Zhu J, Du R, Jing Y, Lu L, Zhang R. In vivo kinetics of the uremic toxin p-cresyl sulfate in mice with variable renal function. Ther Apher Dial. 2014;18:637–642. doi: 10.1111/1744-9987.12185. [DOI] [PubMed] [Google Scholar]
- Wilson KD, Li Z, Wagner R, Yue P, Tsao P, Nestorova G, Huang M, Hirschberg DL, Yock PG, Quertermous T, Wu JC. Transcriptome alteration in the diabetic heart by rosiglitazone: implications for cardiovascular mortality. PLoS One. 2008;3:e2609. doi: 10.1371/journal.pone.0002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finckenberg P, Inkinen K, Ahonen J, Merasto S, Louhelainen M, Vapaatalo H, Muller D, Ganten D, Luft F, Mervaala E. Angiotensin II induces connective tissue growth factor gene expression via calcineurin-dependent pathways. Am J Pathol. 2003;163:355–366. doi: 10.1016/S0002-9440(10)63659-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konorev EA, Zhang H, Joseph J, Kennedy MC, Kalyanaraman B. Bicarbonate exacerbates oxidative injury induced by antitumor antibiotic doxorubicin in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2000;279:H2424–H2430. doi: 10.1152/ajpheart.2000.279.5.H2424. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li F, Zhi G, Zhang B, Chen YD. NADPH oxidase contributes to the left ventricular dysfunction induced by sinoaortic denervation in rats. Free Radical Res. 2015;49:57–66. doi: 10.3109/10715762.2014.978768. [DOI] [PubMed] [Google Scholar]
- Johnson DK, Schillinger KJ, Kwait DM, Hughes CV, McNamara EJ, Ishmael F, O’Donnell RW, Chang MM, Hogg MG, Dordick JS, Santhanam L, Ziegler LM, Holland JA. Inhibition of NADPH oxidase activation in endothelial cells by ortho-methoxy-substituted catechols. Endothelium. 2002;9:191–203. doi: 10.1080/10623320213638. [DOI] [PubMed] [Google Scholar]
- Martinez AW, Recht NS, Hostetter TH, Meyer TW. Removal of p-cresol sulfate by hemodialysis. J Am Soc Nephrol. 2005;16:3430–3436. doi: 10.1681/ASN.2005030310. [DOI] [PubMed] [Google Scholar]
- Oh JK, Appleton CP, Hatle LK, Nishimura RA, Seward JB, Tajik AJ. The noninvasive assessment of left ventricular diastolic function with two-dimensional and Doppler echocardiography. J Am Soc Echocardiogr. 1997;10:246–270. doi: 10.1016/s0894-7317(97)70062-2. [DOI] [PubMed] [Google Scholar]
- Martin FL, McKie PM, Cataliotti A, Sangaralingham SJ, Korinek J, Huntley BK, Oehler EA, Harders GE, Ichiki T, Mangiafico S, Nath KA, Redfield MM, Chen HH, Burnett JC., Jr Experimental mild renal insufficiency mediates early cardiac apoptosis, fibrosis, and diastolic dysfunction: a kidney-heart connection. Am J Physiol Regul Integr Comp Physiol. 2012;302:R292–R299. doi: 10.1152/ajpregu.00194.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Li X, Du W, Feng Y, Kong X, Li Y, Xiao L, Zhang P. Antitumor effects of ginkgolic acid in human cancer cell occur via cell cycle arrest and decrease the Bcl-2/Bax ratio to induce apoptosis. Chemotherapy. 2010;56:393–402. doi: 10.1159/000317750. [DOI] [PubMed] [Google Scholar]
- Yang C, Wang Y, Liu H, Li N, Sun Y, Liu Z, Yang P. Ghrelin protects H9c2 cardiomyocytes from angiotensin II-induced apoptosis through the endoplasmic reticulum stress pathway. J Cardiovasc Pharmacol. 2012;59:465–471. doi: 10.1097/FJC.0b013e31824a7b60. [DOI] [PubMed] [Google Scholar]
- Geiszt M. NADPH oxidases: new kids on the block. Cardiovasc Res. 2006;71:289–299. doi: 10.1016/j.cardiores.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Weissmann N, Schroder K. NADPH oxidases in cardiovascular disease. Free Radic Biol Med. 2010;49:687–706. doi: 10.1016/j.freeradbiomed.2010.04.030. [DOI] [PubMed] [Google Scholar]
- Filomeni G, Ciriolo MR. Redox control of apoptosis: an update. Antioxid Redox Signal. 2006;8:2187–2192. doi: 10.1089/ars.2006.8.2187. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Ichijo H. Stress-responsive protein kinases in redox-regulated apoptosis signaling. Antioxid Redox Signal. 2005;7:472–481. doi: 10.1089/ars.2005.7.472. [DOI] [PubMed] [Google Scholar]
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]