

Abstract
Background
Multifunctional calcium/calmodulin-dependent kinase II (CaMKII) is activated by angiotensin II (Ang II) in cultured vascular smooth muscle cells (VSMCs), but its function in experimental hypertension has not been explored. The aim of this study was to determine the impact of CaMKII inhibition selectively in VSMCs on Ang II hypertension.
Methods and Results
Transgenic expression of a CaMKII peptide inhibitor in VSMCs (TG SM-CaMKIIN model) reduced the blood pressure response to chronic Ang II infusion. The aortic depressor nerve activity was reset in hypertensive versus normotensive wild-type animals but not in TG SM-CaMKIIN mice, suggesting that changes in baroreceptor activity account for the blood pressure difference between genotypes. Accordingly, aortic pulse wave velocity, a measure of arterial wall stiffness and a determinant of baroreceptor activity, increased in hypertensive versus normotensive wild-type animals but did not change in TG SM-CaMKIIN mice. Moreover, examination of blood pressure and heart rate under ganglionic blockade revealed that VSMC CaMKII inhibition abolished the augmented efferent sympathetic outflow and renal and splanchnic nerve activity in Ang II hypertension. Consequently, we hypothesized that VSMC CaMKII controls baroreceptor activity by modifying arterial wall remodeling in Ang II hypertension. Gene expression analysis in aortas from normotensive and Ang II–infused mice revealed that TG SM-CaMKIIN aortas were protected from Ang II–induced upregulation of genes that control extracellular matrix production, including collagen. VSMC CaMKII inhibition also strongly altered the expression of muscle contractile genes under Ang II.
Conclusions
CaMKII in VSMCs regulates blood pressure under Ang II hypertension by controlling structural gene expression, wall stiffness, and baroreceptor activity.
Keywords: angiotensin II, calcium/calmodulin-dependent kinase II, hypertension, sympathetic nerve activity, vascular remodeling
Multifunctional calcium ion Ca2+/calmodulin-dependent kinase II (CaMKII) is abundantly expressed in arterial smooth muscle cells in aorta and resistance blood vessels.1 CaMKII controls a variety of cellular processes, including intracellular Ca2+ handling and Ca2+-dependent gene transcription.2 Although many of these pathways have been elucidated in contractile cells, analysis of CaMKII function in vascular smooth muscle cells (VSMCs) has been limited to studies of dedifferentiated proliferative cells in vitro.3–5 Consequently, our understanding of CaMKII function in vivo is limited6 in part due to a paucity of genetic models that allow for unequivocal dissection of CaMKII function in the vascular wall.
Because several CaMKII downstream substrates identified in other excitable cells have also been implicated in blood pressure regulation,7,8 it is likely that CaMKII plays an integrative but unrecognized role in hypertension. Angiotensin II (Ang II) inhibition is an effective strategy to control blood pressure in humans, thus Ang II–induced hypertension is a clinically relevant model of experimental hypertension. CaMKII is acutely activated in isolated cultured VSMCs by vasoconstrictors such as Ang II or vasopressin9; however, it is unclear whether CaMKII mediates the effects of chronic Ang II administration in smooth muscle in vivo. In some studies, coadministration of the pharmacological CaMKII inhibitor KN-93 decreased arterial pressure in Ang II–dependent hypertension in rodents,1,10,11 but KN-93 has been recognized to have CaMKII-independent effects on ion channels that control vascular tone.12–14 Consequently, whether CaMKII regulates blood pressure in Ang II–induced hypertension remains unknown.
We recently reported on a model of specific and potent CaMKII inhibition with expression of the inhibitor peptide CaMKIIN limited to smooth muscle (TG SM-CaMKIIN model).15 In this model, we established that CaMKII does not affect vasoconstriction in aorta and mesenteric arteries despite controlling intracellular Ca2+ levels. In control experiments, we also ascertained that KN-93 strongly reduced vasoconstriction,15 as reported previously,16,17 suggesting that the earlier reports on KN-93 in Ang II–induced hypertension provide limited information on CaMKII function.
In this study, we investigated whether VSMC CaMKII modulates the blood pressure response in Ang II hypertension using our established model of TG SM-CaMKIIN mice and dissected the underlying process. Our data identify CaMKII as a key regulator of Ang II–induced hypertension through augmented expression of genes controlling extracellular matrix composition at the expense of smooth muscle contractile protein expression, leading to changes in functional and structural wall properties and baroreceptor resetting.
Materials and Methods
Mice
All experimental procedures were approved by the University of Iowa and the Iowa City VA Health Care System institutional animal care and use committee. All procedures were in compliance with the standards for the care and use of laboratory animals of the Institute of Laboratory Animal Research, National Academy of Sciences. To study specific CaMKII inhibition in VSMCs, cDNA for the hemagglutinin-tagged CaMKII inhibitor peptide CaMKIIN (CaMKIIN) was cloned into a construct containing the CX-1 promoter and a floxed enhanced green fluorescent protein sequence, as described previously.15,18,19 Transgenic hemagglutinin-tagged CaMKIIN mice were back-crossed with C57Bl/6 mice (No. 000664; Jackson Laboratory, Bar Harbor, ME) for 6 generations and mated with mice carrying a Cre recombinase gene controlled by the SM22α promoter (identifier 004746, Tg[Tagln-cre]1 Her/J; Jackson Laboratory) to generate TG SM-CaMKIIN mice.20 Littermates that did not carry the hemagglutinin-tagged CaMKIIN transgene served as wild-type (WT) controls for all experiments. Experiments were performed on mice aged between 10 and 12 weeks. Mice were housed in a room with controlled temperature (23°C) and a dark/light cycle of 12 hours. All mice had free access to water and standard rodent chow. Male and female mice were used for the experiments in equal proportions; no differences in blood pressure or echocardiographic parameters at baseline or after Ang II infusion were detected between sexes.
Osmotic Mini-Pumps
Chronic infusion of Ang II (AnaSpec) was performed using Alzet osmotic minipumps (model 1002; Durect Corporation). Pumps were filled following the manufacturer’s specifications with sterile PBS or Ang II (1.25 μg/kg per minute). Briefly, mice were anesthetized with ketamine and xylazine, and pumps were implanted subcutaneously through a subscapular incision that was closed using silk suture (Ethicon). The contents of the pumps were delivered at a rate of 0.25 μL/h for 14 days.
Telemetric Blood Pressure Measurement
Blood pressure was monitored by radiotelemetry (PA-C10; Data Science International) in conscious, unrestrained mice. Under ketamine–xylazine anesthesia, radiotelemetric catheters were implanted into the left common carotid artery through an anterior neck incision. The radiotelemeter transmitter was implanted subcutaneously into the left flank. After surgery, pain control was provided with buprenorphine or meloxicam, depending on the surgical protocol. After 12 days of recovery, arterial blood pressure and heart rate were recorded for a 10-second interval at 500 Hz every 10 minutes for 72 hours. To analyze the acute effects of smooth muscle–dependent vasodilation or sympathetic activity, sodium nitroprusside (SNP; 1 mg/kg; Sigma-Aldrich) or chlorisondamine (CND; 12 mg/kg; Sigma-Aldrich) was administered by tail vein, and data were collected continuously for 45 minutes. The standard deviation of arterial blood pressure recordings was calculated analogously to a previous publication.21
Immunoblotting
Aliquots of 25 µg whole-tissue lysate from aorta were resolved by SDS-PAGE and transferred to polyvinylidene difluoride membrane. After blocking with 5% BSA, the membranes were incubated with primary antibodies for collagen (Millipore) or GAPDH (Cell Signaling) followed by horseradish peroxidase–conjugated secondary antibody. The proteins were visualized with the ECL chemiluminescence system (Amersham; GE Healthcare). Densitometry was performed using National Institutes of Health ImageJ software.
Histology and Immunohistochemistry
Aortas were fixed in 4% paraformaldehyde and embedded in paraffin. Sections of 10 µg were collected on Superfrost Plus slides, and Masson’s trichrome and hematoxylin and eosin staining was performed. The medial area and the number of medial nuclei were determined in 10 consecutive hematoxylin and eosin–stained sections in the thoracic descending aorta in 5 to 7 mice. Other sections were preincubated in 5% goat serum for 30 minutes followed by incubation with primary antibodies against anti-α-smooth muscle actin (1:100; Santa Cruz Biotechnology), CaMKII (1:100; LSBio), or anti-hemagglutinin (1:100; Covance Laboratories) overnight at 4°C. Sections were then incubated with Alexa 568– or Alexa 488–conjugated secondary antibodies (Invitrogen; Thermo Fisher Scientific). Sections were counterstained with To-Pro-3 (Invitrogen; Thermo Fisher Scientific) or mounted in Vectashield containing DAPI (Vector Labs) to visualize nuclei. Images were captured with a Zeiss LSM 710 laser scanning microscope.
CaMKII Kinase Activity Assay
Autonomous and total CaMKII activity assays of fresh tissue lysates from TG SM-CaMKIIN and WT aortas were performed using 10 μg protein, as described previously.22 Autonomous CaMKII activity was measured under conditions that reflect the in vivo activation state of CaMKII (addition of ATP but no additional Ca2+/calmodulin). Total CaMKII activity was determined by adding an excess of Ca2+/calmodulin and is indicative of total levels of CaMKII in a sample.
Plasma Norepinephrine Levels
Mice were euthanized with pentobarbital. Immediately after death was confirmed, blood was collected by cardiac puncture. Plasma was isolated at the same time of day (10 am) from mice aged 12 weeks. Samples from 6 saline-treated and 9 Ang II–treated mice per genotype were collected. Norepinephrine levels were determined by ELISA, as recommended by the manufacturer (RE 59261; IBL International).
Vascular Reactivity Studies
Vascular responses in the descending thoracic aorta were measured, as described previously.23 The endothelium was removed, and vessel viability was determined by measurement of responses to KCl (50 mmol/L). Concentration response curves to Ang II (10−9 to 10−5 mol/L), phenylephrine (10−8 to 10−6 mol/L), serotonin (10−8 to 10−5 mol/L), and KCl (25 to 100 mmol/L) were performed. The passive vascular properties were assessed in the proximal common carotid artery, as reported previously.24 The arteries were dissected and placed in Krebs buffer with 118.3 mmol/L NaCl, 4.7 mmol/L KCl, 1.2 mmol/L MgSO4, 1.2 mmol/L KH2PO4, 25 mmol/L NaHCO3, 2.5 mmol/L CaCl2, and 11 mmol/L glucose (pH 7.4). Blood vessel viability was tested using 100 mmol/L KCl followed by 30-minute equilibration under no-flow conditions at an intraluminal pressure of 100 mm Hg in calcium-free Krebs buffer containing SNP (10−5 mol/L) and EGTA (2 mmol/L). Carotid arteries were then subjected to a series of pressures from 50 to 175 mm Hg. Internal and external diameters were recorded. Luminal cross-sectional area and cross-sectional compliance were calculated, as described previously.25 The calculations for stress, strain, and distensibility curves were performed according to published standard methods.25
Echocardiography
Transthoracic echocardiograms were acquired from conscious minimally sedated (midazolam 0.15 mg SC) mice, using a Vevo 2100 mainframe coupled to a 30-MHz linear array transducer (VisualSonics), as described previously.26 All images were acquired and analyzed by an operator blinded to mouse genotype. Endocardial and epicardial borders were traced in short-axis and long-axis planes at end diastole and end systole. Left ventricular mass, end-diastolic and end-systolic left ventricular volumes, and ejection fraction were calculated by the biplane area–length method.
Hydroxyproline Assay
Aortas were homogenized and acidified in 6 N HCl and hydrolyzed by heating to 120°C for 24 hours. Hydroxyproline measurements were performed, as reported previously,27 and normalized to aortic weight.
Renal and Splanchnic Sympathetic Nerve Recordings
Anesthesia was induced using ketamine and xylazine and maintained with α-chloralose (starting dose of 25 mg/kg IV, then a sustaining dose of 6 mg/kg IV per hour). Arterial blood pressure and heart rate were monitored in anesthetized mice with a catheter inserted into the left carotid artery. Through a left retroperitoneal flank incision, a renal nerve serving the left kidney and the nearby splanchnic nerve were identified and carefully isolated from the adjoining connective tissue. The renal sympathetic nerve was placed on a bipolar 36-gauge platinum–iridium electrode that was attached to a high-impedance probe (HIP-511; Grass Instruments) and then sealed with silicone gel (Kwik-Sil; WPI Instruments). The signal from the renal nerve was amplified (105) and filtered at 100- and 1000-Hz cutoffs with a Grass P5 preamplifier. After obtaining a stable basal recording of afferent and efferent renal sympathetic nerve activity for up to 30 minutes, the electrode was thoroughly cleaned and reused to record splanchnic nerve activity within the same mouse. Basal splanchnic sympathetic nerve activity (afferent and efferent) was recorded for up to 30 minutes with the identical amplification and filtering as used for renal nerves. The operator was unaware of the genotype or treatment group. Data acquisition was performed using the same settings for amplification, high- and low-filter cutoffs, and sampling rate for all mice.
Aortic Depressor Nerve Recordings
Aortic depressor nerve (ADN) activity was recorded, as described previously.28–30 Briefly, the left ADN was isolated, placed on a 40-gauge bipolar platinum–iridium electrode, and then encased with silicone gel. Nerve activity was amplified (105) and filtered at 100- and 1000-Hz cutoffs with a Grass P5 preamplifier, and its unique activity was identified as synchronized bursts with the beginning of the systolic cycle. Baseline ADN activity and hemodynamic parameters were recorded for 10 to 15 minutes, followed by slow infusion (20 μL/min) of SNP (1 to 5 μg/mL) immediately followed by phenylephrine (4 to 20 μg/mL). The nerve was then transected at a point caudal to the electrode. The nerve was stimulated with rectangular pulses of 10 V that were delivered at various frequencies for a period of 20 seconds while recording hemodynamic parameters.
Pulse Wave Velocity
Aortic pulse wave velocity (PWV) was measured, as described previously.31,32 Mice were anesthetized with 2% isoflurane and positioned supine on a heating board with limbs secured to ECG electrodes. Pulse waves were detected at the transverse aortic arch and the abdominal aorta using Doppler probes (MouseDoppler data acquisition system; Indus Instruments). The first 5 measurements were used to optimize the Doppler signal and were not included in the analysis. The following 20 measurements were recorded and analyzed. For this purpose, the time elapsed between the ECG R-wave and the foot of the Doppler signal was determined for each site, and PWV was calculated using the following equation: PWV=(distance between probes)/(Δtime abdominal − Δtime transverse). Two sets of measurements were recorded on consecutive days and averaged for each mouse.
Gene Array
Ascending aortas were harvested from WT and TG SM-CaMKIIN mice aged 14 weeks on day 14 of Ang II infusion or from saline-infused controls. RNA sample preparation for hybridization and subsequent hybridization to the Illumina MouseWG-6 v2.0 Expression BeadChips were performed at the University of Iowa, Iowa Institute of Human Genetics, Genomics Division, using the manufacturer’s recommended protocol. Briefly, 75 ng total RNA was converted to amplified biotin-aRNA using the Epicentre TargetAmp-Nano Labeling Kit for Illumina Expression BeadChip (catalog no. TAN07924; Illumina), according to the manufacturer’s recommended protocol. The amplified biotin-aRNA product was purified through a Qiagen RNeasy MinElute Cleanup column (catalog number 74204), according to modifications from Epicentre. Next, 1.5 μg of this product was mixed with Illumina hybridization buffer, placed onto Illumina MouseWG-6 v2.0 Expression BeadChips (part no. BD-201-0202) and incubated at 58°C for 17 hours, with rocking, in an Illumina Hybridization Oven. Following hybridization, the arrays were washed, blocked, and stained with streptavidin-Cy3 (Amersham/GE Healthcare) according to the Illumina Whole-Genome Gene Expression Direct Hybridization Assay protocol. Bead chips were scanned with the Illumina iScan System (identifier N0534), and data were collected using GenomeStudio software version 2011.1 (Illumina). All data are compliant with Minimum Information About a Microarray Experiment (MIAME), and the raw data were deposited in a MIAME-compliant database (Gene Expression Omnibus [GEO], accession number GSE64613).
Gene Array Analysis
Array data were analyzed using RStudio version 0.98.1083 running R version 3.1.2. Data exported from GenomeStudio were imported and normalized using neqc from the limma package,33 and normalized data were tested for differential expression, also using the limma package (version 3.22.1). Differential expression was considered if false discovery rate–adjusted P values were <0.01 and fold change >2-fold existed for specific contrasts. Heat maps were plotted using the gplots package34 (version 2.14.2).
Statistical Analysis
All values in the text and figures are presented as mean±SEM. The methods for the analysis of the gene array are described earlier. For all other experiments, we performed D’Agostino and Pearson omnibus normality tests. If the samples met criteria for normal distribution, statistical significance was determined using GraphPad Prism software version 6 by Student t test or ANOVA followed by Tukey’s or Bonferroni multiple comparison test, if appropriate. If the samples sizes were too small or the samples were not normally distributed, Mann–Whitney tests were performed instead of Student t tests and Kruskal–Wallis tests were used instead of 1-way ANOVA. The baroreflex sigmoidal curves, vasoconstriction, passive properties, and 24-hour blood pressure recordings were compared using 2-way ANOVA with repeated measures. Specifically, linear mixed-model analysis for repeated measures was used to compare dose response among treatment groups. The fixed effects in the model included group, dose, and group–dose interaction effect, with a significant group–dose interaction effect indicating differences in dose-response profile among the groups. In addition, to test for specific comparisons of interest (eg, pairwise comparison between group means at each dose), a test of mean contrast based on the fitted mixed model was performed with P values adjusted using Bonferroni’s method to account for the number of tests performed. P values <0.05 were considered significant.
Results
CaMKII Inhibition in VSMC Blunts Ang II Hypertension
We subjected WT and TG SM-CaMKIIN mice15 (Figure 1A) to 14 days of continuous infusion of Ang II or vehicle (normal saline). Chronic Ang II infusion enhanced total and autonomous CaMKII activity in the aorta from WT mice that was lowered by transgenic expression of CaMKIIN in VSMCs (Figure 1B and 1C).
Figure 1.
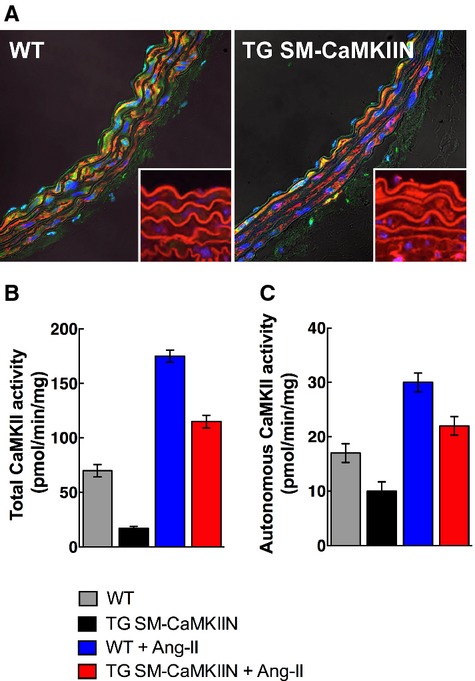
CaMKII activity and expression in normotensive and Ang II–hypertensive mice. A, Immunofluorescence for CaMKII and HA-tagged CaMKIIN in aortas of WT and TG SM-CaMKIIN mice (CaMKII, green; smooth muscle actin, red; nuclei, blue [insets: HA, red; nuclei, blue]). B and C, Assay for CaMKII activity with the synthetic substrate syntide in aortic lysates. B, Total activity, a measure of total available enzyme. C, Autonomous activity, a correlate of the active enzyme in the sample (4 aortas were pooled per sample, 3 independent experiments were conducted). Ang II indicates angiotensin II; CaMKII, calcium/calmodulin-dependent kinase II; HA, hemagglutinin; WT, wild type.
The hemodynamic parameters were recorded in the last 72 hours of Ang II infusion. As expected, Ang II produced a significant increase in mean arterial pressure in WT mice (97.8 mm Hg in normotensive versus 135.6 mm Hg in Ang II–infused WT mice) (Figure 2A). In contrast, CaMKII inhibition in VSMCs moderately but significantly blunted the blood pressure response (97.0 mm Hg in normotensive versus 123.5 mm Hg in Ang II–infused TG SM-CaMKIIN mice), suggesting that CaMKII is activated in Ang II hypertension. Analysis of blood pressure fluctuations over a 12-hour day/night cycle revealed the greatest differences between genotypes during the diurnal-activity phase at night (Figure 2B). The pulse pressure was not different between genotypes (Figure 2C). As reported before, Ang II infusion had little impact on heart rate (Figure 2D).35 VSMC CaMKII did not significantly alter heart rate or physical activity (Figure 2D and 2E). Consistent with the blood pressure differences, Ang II–induced hypertension increased heart weight/body weight ratios in WT but not in TG SM-CaMKIIN mice (Figure 2F). As expected, the left ventricular mass as determined by transthoracic echocardiography was increased in Ang II–infused WT but not TG SM-CaMKIIN mice, whereas the ejection fraction was not different between genotypes (Figure 2G and 2H). These data demonstrate that CaMKII in VSMCs contributes to the rise in blood pressure in Ang II hypertension. Others have demonstrated that chronic high-dose Ang II hypertension is neurogenic36 through decreases in baroreflex sensitivity.35 Based on the blunted diurnal blood pressure fluctuation in TG SM-CaMKIIN mice, we hypothesized that CaMKII in VSMCs modulated neurovascular coupling.
Figure 2.
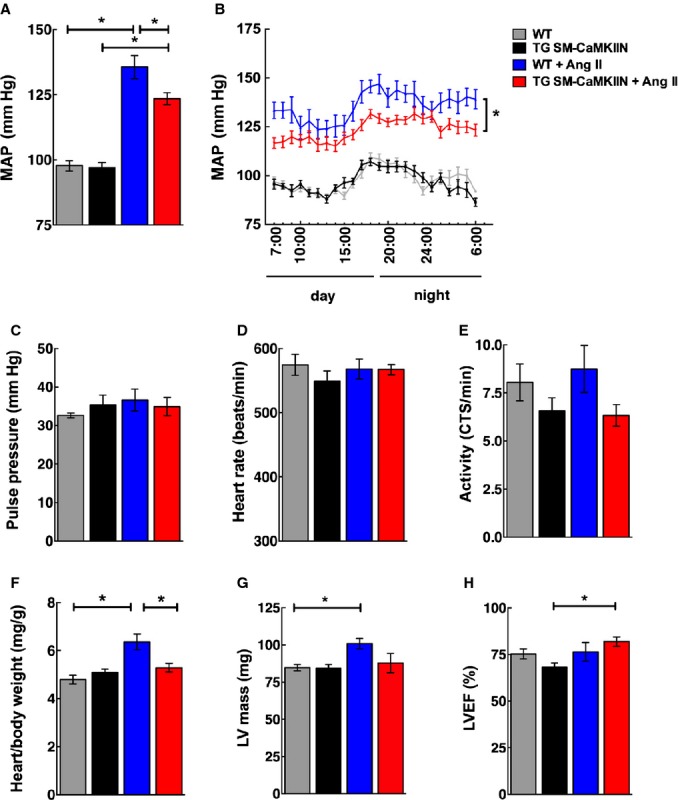
VSMC CaMKII inhibition alleviates Ang II hypertension. A, MAP recording by radiotelemetry after 14 days of infusion with Ang II or normal saline by minipump. MAP was sampled at 500 Hz for 10 seconds once every 10 minutes on days 12 to 14. B, Hourly MAP calculated as the average of measurements taken every 10 minutes for 60 minutes over a 24-hour time period on day 14. C, Pulse pressure derived from the data plotted in (A). D, Heart rate and (E) locomotor activity recorded by radiotelemetry after 12 to 14 days of Ang II or saline infusion. F, Cardiac hypertrophy determined by ex vivo heart/body weight ratio. G and H, Echocardiographic assessment of the effect of VSMC CaMKII inhibition on cardiac function. G, LV mass. H, LVEF was assessed in WT littermate and TG SM-CaMKIIN mice. The mice were matched for age and gender (16 WT, 16 TG SM-CaMKIIN, 21 WT plus Ang II, 21 TG SM-CaMKIIN plus Ang II mice per group for A through E; 13, 9, 16, and 18 mice per group for F, 14 mice per group for G and H). *P<0.05. Ang II indicates angiotensin II; CaMKII, calcium/calmodulin-dependent kinase II; CTS, counts; LV, left ventricular; LVEF, LV ejection fraction; MAP, mean arterial pressure; VSMC, vascular smooth muscle cells; WT, wild type.
Reduced Sympathetic Vasomotor Activity in Ang II–Infused TG SM-CaMKIIN Mice
To address whether VSMC CaMKII inhibition affects the increases in sympathetic efferent activity in Ang II hypertension, we examined changes in mean blood pressure after administration of the ganglionic blocker CND. As expected, CND decreased mean arterial pressure in hypertensive WT and TG SM-CaMKIIN mice (Figure 3A and 3B); however, VSMC CaMKII inhibition significantly blunted the blood pressure response to CND (Figure 3B). Similarly, the CND-induced decrease in heart rate was significantly lower under CaMKII inhibition (Figure 3C). Administration of the direct vasodilator SNP induced a similar effect on mean arterial pressure and heart rate in all groups (Figure 3D through 3F), indicating that the in vivo effect of VSMC CaMKII inhibition on arterial pressure is not explained by alterations in vascular tone.
Figure 3.
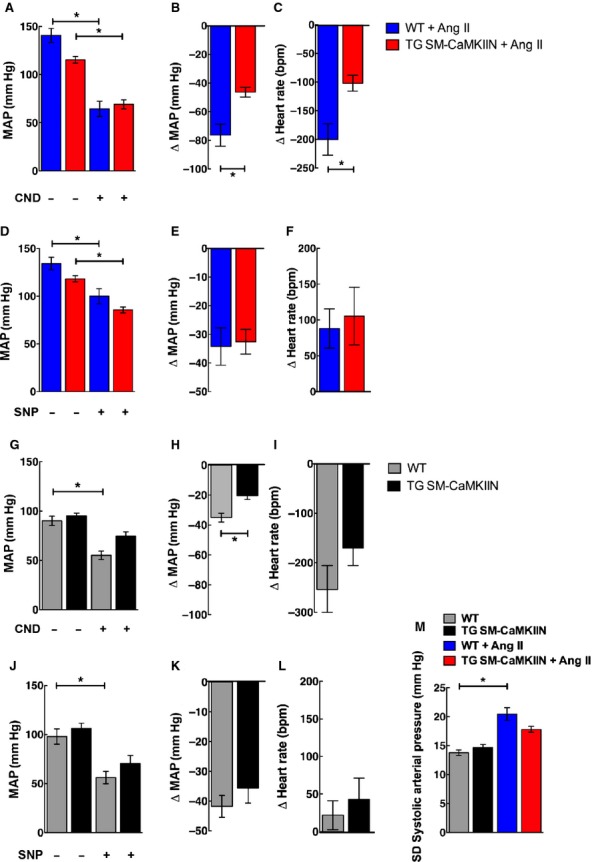
Ganglionic blockade reveals differences in sympathetic nerve activity in Ang II–infused control and TG SM-CaMKIIN mice. A, MAP after 2 weeks of Ang II infusion before and after administration of the ganglionic blocker CND. B, Decreases in MAP in (A) plotted as the difference before and after CND injection. C, Change in heart rate before and after CND injection. D, MAP after 2 weeks of Ang II infusion before and after administration of the vasodilator SNP. E, Decreases in MAP in (D) plotted as difference before and after SNP injection. F, Change in heart rate before and after SNP injection. G, MAP in saline-treated normotensive mice before and after administration of the ganglionic blocker CND. H, Decreases in MAP in (G) plotted as the difference before and after CND injection. I, Change in heart rate before and after CND injection. J, MAP after 2 weeks of Ang II infusion before and after administration of SNP. K, Decreases in MAP in (J) plotted as difference before and after SNP injection. L, Change in heart rate before and after SNP injection. M, Standard deviation of the systolic blood pressure over a 24-hour period on days 12 to 14 of saline or Ang II infusion (6 WT, 8 TG SM-CaMKIIN, 6 WT plus Ang II, 8 TG SM-CaMKIIN plus Ang II mice per group for A through K; 11 mice per group for M). *P<0.05. Ang II indicates angiotensin II; bpm, beats per minute; CND, chlorisondamine; MAP, mean arterial pressure; SNP, sodium nitroprusside; WT, wild type.
To evaluate whether VSMC CaMKIIN has any effect on sympathetic efferent activity at baseline, we measured blood pressure and heart response to CND and SNP in normotensive mice. At baseline, the response to CND was significantly greater in WT compared with TG SM-CaMKIIN mice (Figure 3G and 3H). Again, there were no differences in the response to SNP in normotensive mice (Figure 3J through 3L). As an additional measure of sympathetic activity, we examined the 24-hour standard deviation of blood pressure recordings and found that, as expected, Ang II infusion increased the 24-hour standard deviation in WT mice only (Figure 3M). These results confirm that Ang II hypertension increases efferent sympathetic activity and suggest that the reduced mean arterial pressure in Ang II–infused TG SM-CaMKIIN mice is driven by lower efferent signals of the sympathetic nervous system. In contrast, we did not find any evidence of a difference in vasodilator response. The results strongly suggest lower sympathetic nerve activity in hypertensive TG SM-CaMKIIN mice.
We next examined the effect of VSMC CaMKII inhibition on the afferent and efferent limbs of the sympathetic nervous system related to blood pressure regulation. In particular, we concentrated on the 2 locations at which the sympathetic nervous system and the vascular wall functionally interact. Because mechanical distension is sensed in the aortic arch by baroreceptors and converted into an afferent signal, we recorded ADN activity under anesthesia. ADN activity occurred in bursts and was synchronous with the arterial pressure (Figure 4A and 4B). The baroreceptor afferent function was interrogated by recording the ADN activity at different mean arterial pressures. Ang II hypertension shifted ADN activity to higher blood pressure in WT mice (Figure 4C), a typical Ang II effect related to baroreceptor resetting. In contrast, the baroreceptor activity was not changed after 14 days of Ang II infusion compared with baseline in TG SM-CaMKIIN mice (Figure 4D). To examine the central integration of the afferent signal, the ADN was transected, and the proximal nerve ending was stimulated at frequencies of 2.5 to 40 Hz with simultaneous measurements of blood pressure and heart rate. In all 4 groups, no differences in heart rate or blood pressure were recorded (Figure 4E and 4F), suggesting that the differences in ADN activity are not caused by central nervous system effects but rather by proximal sensing of aortic distension.
Figure 4.
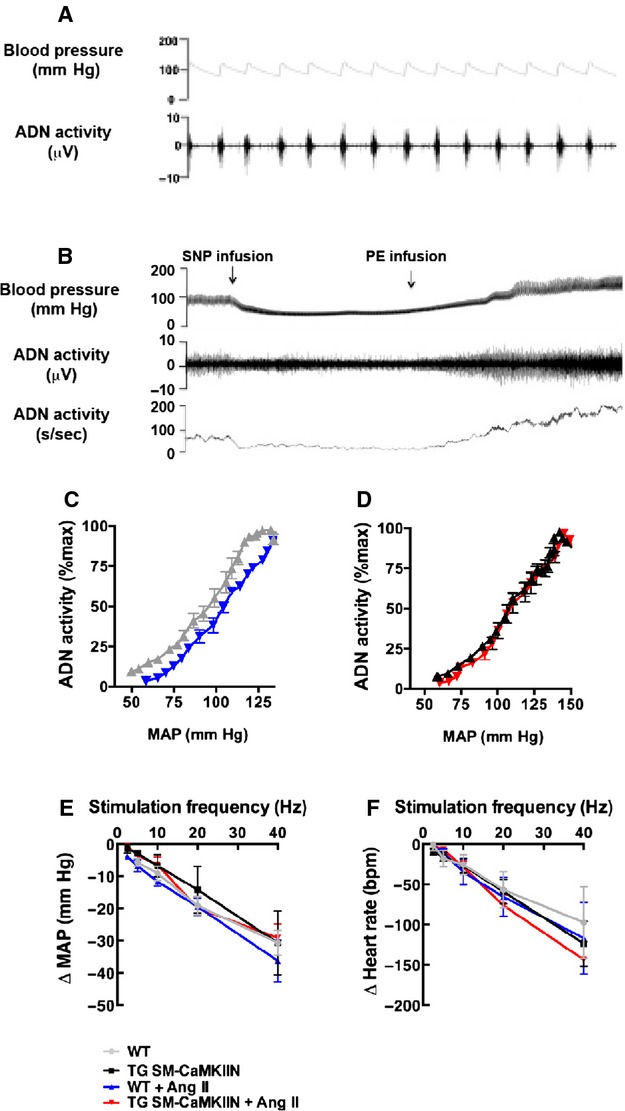
ADN recordings reveal differences in baroreceptor sensing in Ang II–infused TG SM-CaMKIIN and WT littermate aortas. A and B, Representative tracings of blood pressure and ADN activity in anesthetized WT mice demonstrate coupling between blood pressure and ADN firing. C and D, Baroreflex curves. Analysis of baroreceptor afferent function curves derived from ADN activity in response to intravenous infusion of SNP and PE in (C) WT and (D) TG SM-CaMKIIN mice. Studies were performed at 14 days after infusion of Ang II or saline. E and F, responses of (E) MAP and (F) heart rate to electric ADN stimulation (6 WT, 6 TG SM-CaMKIIN, 6 WT plus Ang II, 7 TG SM-CaMKIIN plus Ang II mice per group). ADN indicates aortic depressor nerve; Ang II, angiotensin II; MAP, mean arterial pressure; PE, phenylephrine; SNP, sodium nitroprusside; WT, wild type.
We next ascertained the effect of VSMC CaMKII inhibition on splanchnic nerve activity, a key mediator of blood pressure increases in Ang II hypertension.37 Representative tracings for all 4 groups are shown in Figure 5A. Whereas nerve activity was similar between genotypes at baseline, Ang II infusion greatly augmented splanchnic nerve activity in WT mice but had no effect in TG SM-CaMKIIN mice (Figure 5A through 5C). Similarly, the renal nerve activity was increased in Ang II–treated WT mice compared with saline treatment but was not increased in TG SM-CaMKIIN mice (Figure 5D through 5F). As an accessory measurement of sympathetic nerve activity, we also examined plasma norepinephrine concentrations and detected a significant increase in hypertensive versus saline-infused WT mice only (Figure 5G). No significant differences were seen between Ang II– and saline-infused TG SM-CaMKIIN mice. These data position VSMC CaMKII as a regulator of baroreceptor activity in Ang II hypertension, leading to changes in the activity of the sympathetic nervous system, specifically, in splanchnic and renal sympathetic nerve activity.
Figure 5.
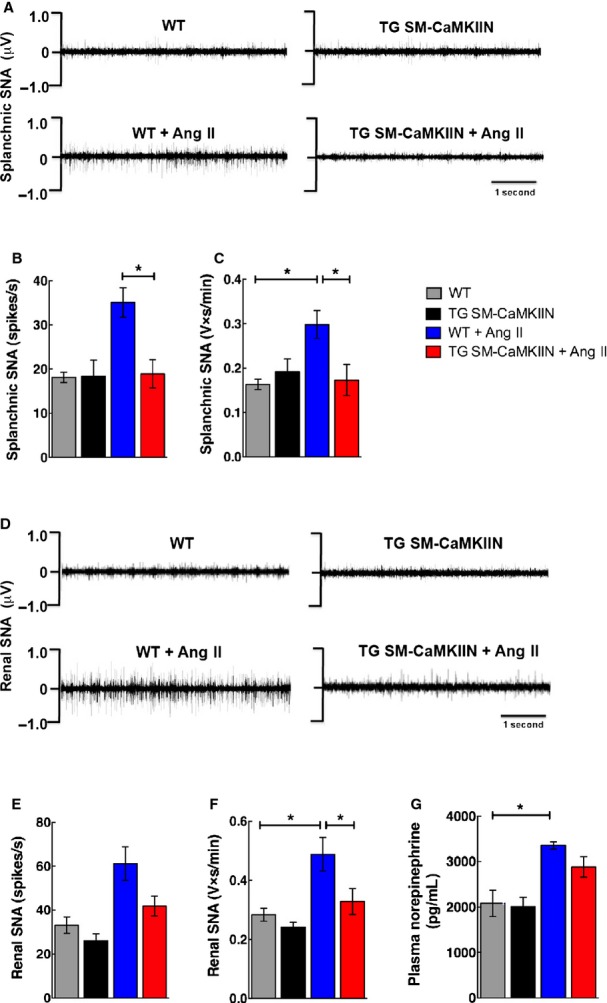
Reduced mean arterial pressure in Ang II–infused TG SM-CaMKIIN mice is related to lower sympathetic nervous system activity. A, Representative tracings of splanchnic SNA in WT and TG SM-CaMKIIN mice on day 14 of Ang II or saline infusion. B and C, Splanchnic SNA expressed as (B) spikes/s and (C) integrated SNA as V×s/min. D, Representative tracings of renal SNA in WT and TG SM-CaMKIIN mice on day 14 of Ang II or saline infusion. E and F, Renal SNA expressed as (E) spikes/s and (F) integrated SNA as V×s/min. G, Circulating plasma norepinephrine concentrations on day 14 of Ang II or saline infusion. (A through F: 6 per group, G: 6 WT, 6 TG SM-CaMKIIN, 9 WT plus Ang II, 9 TG SM-CaMKIIN plus Ang II). *P<0.05. Ang II indicates angiotensin II; SNA, sympathetic nerve activity; WT, wild type.
VSMC CaMKII Inhibition Prevents Ang II–Induced Aortic Wall Stiffening
To assess whether the changes in sympathetic activity are due to alterations in vessel function, we analyzed the mechanical properties of the aortic wall in vivo by recording the PWV, a correlate of vascular stiffness. At baseline, no differences between the genotypes were detected. As expected, under Ang II–induced hypertension, the PWV significantly increased in WT mice (Figure 6A). In striking contrast, the increase in PWV was abolished in hypertensive compared with normotensive TG SM-CaMKIIN mice. These data indicate, surprisingly, that CaMKII controls vascular wall properties in Ang II hypertension that determine increased wall stiffness in vivo.
Figure 6.
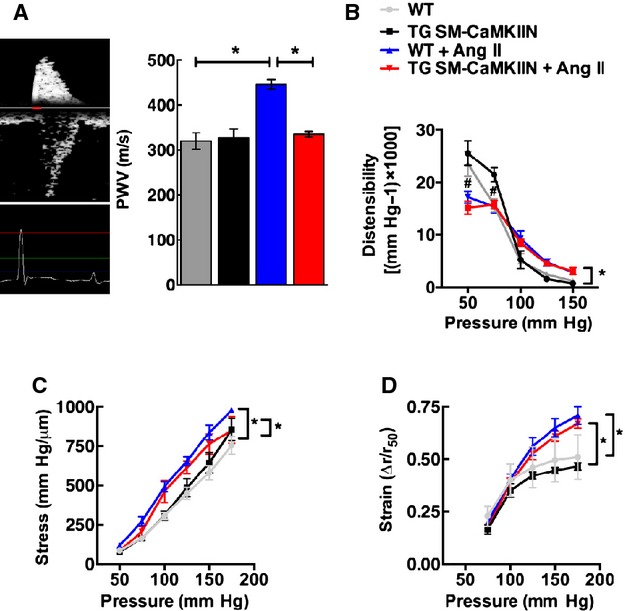
VSMC CaMKII inhibition protects against aortic stiffening in Ang II hypertension. A, Representative example of PWV recordings in the transverse aortic arch and the abdominal aorta. Analysis of PWV expressed as time elapsed between the ECG R-wave and the foot of the Doppler signal was determined for each site (5 to 6 per group). B through D, Passive elastic properties were measured in the proximal common carotid artery ex vivo under Ca2+-free conditions and expressed as (B) distensibility, (C) wall stress, and (D) strain, (A: 10 WT, 10 TG SM-CaMKIIN, 9 WT plus Ang II, 10 TG SM-CaMKIIN plus Ang II mice per group; B to D: 5 mice per group). *P<0.05. Ang II indicates angiotensin II; CaMKII, calcium/calmodulin-dependent kinase II; PWV, pulse wave velocity; VSMC, vascular smooth muscle cells; WT, wild-type.
Numerous studies in the past have reported on the mechanical vascular wall properties ex vivo. To correlate our in vivo findings by PWV with these techniques, we also analyzed the passive mechanical properties of proximal carotid arteries ex vivo in the absence of vascular tone. These studies revealed no differences between genotypes with regard to strain, stress, or distensibility (Figure 6B through 6D); however, as expected, chronic Ang II infusion altered stress and strain.
Next, we examined ex vivo constriction of endothelium-denuded aortas from normotensive or hypertensive WT or TG SM-CaMKIIN mice. Compared with normotensive conditions, chronic Ang II infusion significantly altered the dose-response profile to phenylephrine and serotonin in WT and TG SM-CaMKIIN mice (Figure 7A through 7D); however, after chronic Ang II infusion, there was significantly greater Ang II–induced vasoconstriction in TG SM-CaMKIIN mice only compared with all other groups (Figure 7D).
Figure 7.
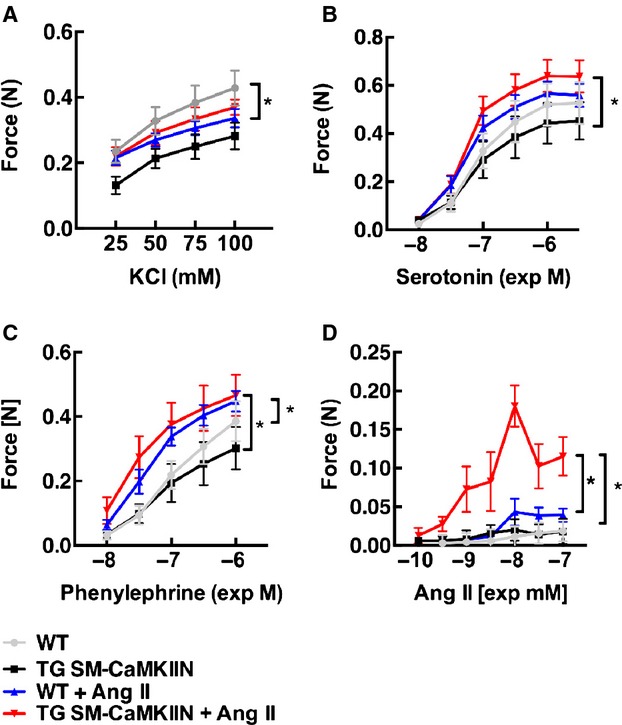
CaMKII inhibition increases agonist-stimulated vasoconstriction following Ang II hypertension. Vasoconstriction of aortas to (A) KCl, (B) serotonin, and (C) phenylephrine, and (D) Ang II after infusion of Ang II or saline for 14 days (2 rings were analyzed per mouse and data were averaged; 7 WT, 5 TG SM-CaMKIIN, 5 WT plus Ang II, 5 TG SM-CaMKIIN plus Ang II mice per group). *P<0.05. Ang II indicates angiotensin II; CaMKII, calcium/calmodulin-dependent kinase II; WT, wild type.
CaMKII Controls Vascular Wall Remodeling in Chronic Ang II Hypertension
Because we detected differences in vasoconstriction and wall stiffness in hypertensive TG SM-CaMKIIN versus control mice, we next asked how VSMC CaMKIIN affected aortic structure and gene and protein expression in Ang II hypertension. The number of nuclei in the media was lower in TG SM-CaMKIIN mice compared with WT littermates (Figure 8). These findings are in line with the concept that CaMKII controls cell proliferation.38 Under Ang II infusion, hypertrophic remodeling occurred in both genotypes but was greatly attenuated with CaMKII inhibition (Figure 8C). Consequently, we performed gene arrays in aortic arches of TG SM-CaMKIIN mice and littermates at baseline and after 14 days of Ang II hypertension. Data were analyzed under highly stringent conditions, adapted to low sample numbers. Minor differences were detected between genotypes at baseline (Figure 9A), consistent with low CaMKII activity (Figure 1A and 1B) and low transcriptional activity in normotensive aortic walls. As expected, Ang II profoundly affected the mRNA expression profiles. The transcripts of 91 genes were regulated by Ang II infusion in WT and 41 genes in TG SM-CaMKIIN mice compared with baseline, with an overlap of only 11 genes. Among the genes that were regulated in both genotypes, collagen type XII and troponin C were present. A breakdown of gene networks by Gene ontology analysis demonstrated that Ang II hypertension produced the anticipated upregulation of genes related to extracellular matrix and collagen remodeling in WT mice (Figure 9B and 9C). These findings are in line with the increased stiffness in hypertensive WT mice by PWV (Figure 6A). Consistent with gene array data, Ang II hypertension resulted in a significant increase in collagen synthesis by hydroxyproline assay in WT mice (Figure 9D and9E). These increases were significantly attenuated in hypertensive TG SM-CaMKIIN mice.
Figure 8.
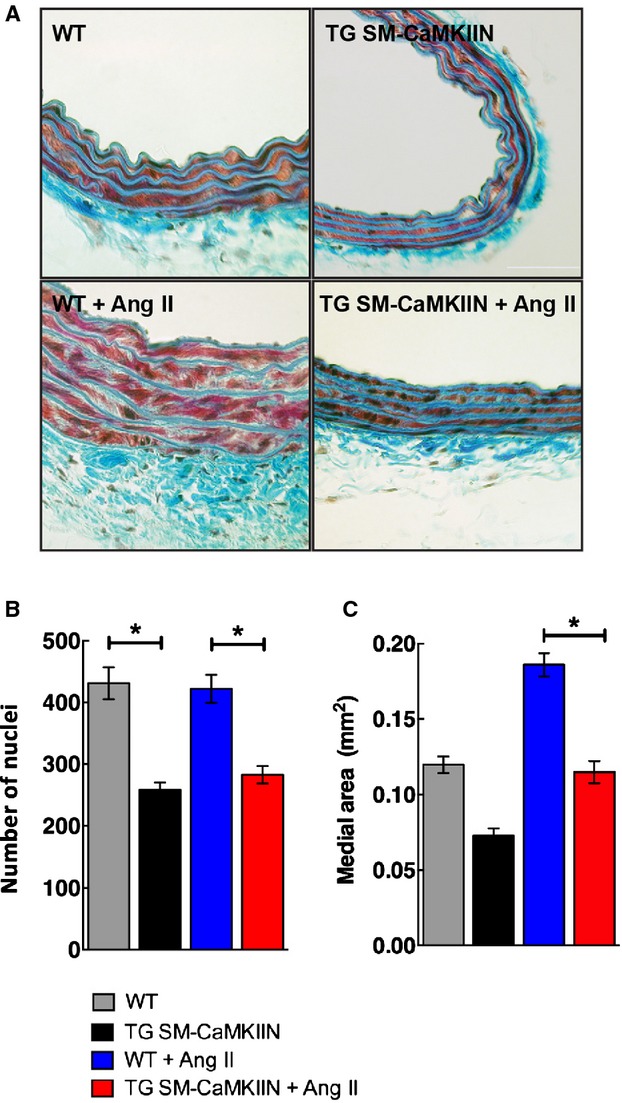
Aortic wall morphology. A, Representative images of Masson’s trichrome staining of aortas from normotensive and hypertensive WT and TG SM-CaMKIIN mice. B, Number of nuclei in the aortic media (15 mice per group). C, Medial area of the descending thoracic aorta in the sections analyzed in (B) (5 WT, 6 TG SM-CaMKIIN, 6 WT plus Ang II, 7 TG SM-CaMKIIN plus Ang II mice per group). *P<0.05. Ang II indicates angiotensin II; WT, wild type.
Figure 9.
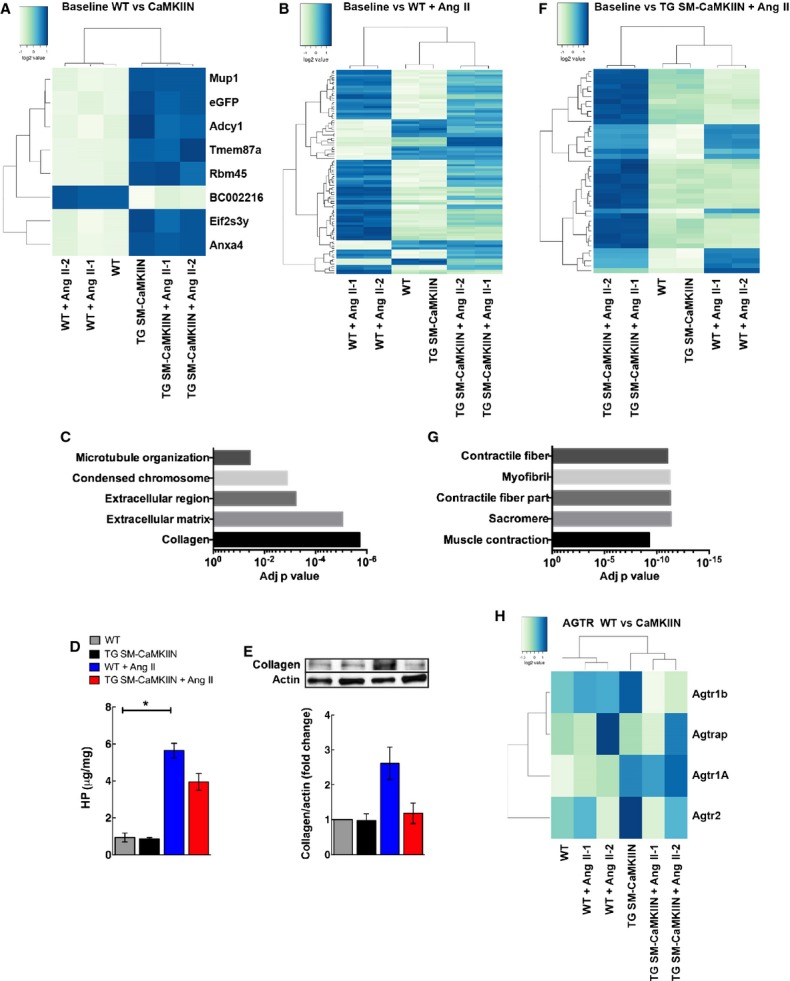
Gene expression analysis in Ang II–hypertensive WT littermate and TG SM-CaMKIIN mice reveals differences in expression of extracellular matrix and muscle structural proteins. A, Heat maps for differentially regulated genes between normotensive WT vs normotensive TG SM-CaMKIIN mice. B, Heat map for hypertensive WT mice compared with baseline, (C) GO analysis for gene array in (B). The 5 GO categories with the most significant adjusted P value are listed. D, HP assay in aortic arches from WT littermate and TG SM-CaMKIIN mice that were infused with saline or Ang II for 14 days (6 per group). E, Representative Western blot for collagen with quantification (5 aortic arches were pooled per group, 3 independent experiments were conducted). F, Heat map for hypertensive TG SM-CaMKIIN mice compared with baseline. G, GO analysis for gene array in (F). The 5 enriched GO categories with the most significant adjusted P value are listed. H, Gene expression heat map for AGTR–related genes. *P<0.05. AGTR indicates angiotensin II receptor; Ang II, angiotensin II; eGFP, enhanced green fluorescent protein; GO, gene ontology; HP, hydroxyproline; WT, wild type.
Among the 41 differentially regulated genes in hypertensive TG SM-CaMKIIN mice, numerous genes involved in muscle contractility were significantly altered (Figure 9F). Accordingly, gene ontology analysis revealed that VSMC CaMKII inhibition under Ang II hypertension significantly altered expression of genes related to contractile fiber structure and function (Figure 9G). Collectively, these data reveal CaMKII as a regulator of Ang II–induced VSMC gene reprogramming.
Discussion
In this study, we demonstrate that CaMKII inhibition in VSMCs in vivo blunts hypertension in the chronic Ang II infusion model. Whereas Ang II-induced hypertension caused the expected resetting of baroreceptors in WT mice, the baroreflex resetting was abolished in hypertensive TG SM-CaMKIIN mice. Consistent with these findings, WT mice experienced a significant increase in splanchnic and renal nerve activity, a key determinant of blood pressure in Ang II–induced hypertension37 that was absent under VSMC CaMKII inhibition. When the mean arterial pressure increases, baroreflex resetting facilitates an even greater increase in arterial pressure. Consequently, we believe that the absence of resetting is a reasonable explanation for the lower renal and splanchnic nerve activity and arterial pressure in the Ang II–infused TG SM-CaMKIIN versus WT mice. Viscoelastic coupling of the arterial wall to the baroreceptor endings has been considered as a mechanism contributing to baroreceptor resetting.39 The viscoelastic properties in vivo were assessed by PWV. The stiffness of aortas from hypertensive TG SM-CaMKIIN mice was significantly lower compared with their WT counterparts. Consistently, the gene array profile of aortas from hypertensive TG SM-CaMKIIN mice was altered toward expression of structural smooth muscle proteins, whereas in WT mice, greater changes were seen in extracellular matrix proteins and collagen that affect wall stiffness. Consequently, the differences in wall stiffness are likely explained by CaMKIIN-induced alterations in gene transcription of extracellular matrix and smooth muscle structural genes. In summary, our interpretation of the integrated findings in this study is that VSMC CaMKII inhibition affects the vascular wall structure in Ang II hypertension and alters viscoelastic coupling to the baroreceptor that ultimately determines blood pressure.
Endothelial, VSMC, and adventitia-based mechanisms are implicated in modulating baroreceptor activity.22,39–41 Endothelial dysfunction, for example, via inhibition of endogenous formation of prostacyclin in the isolated carotid sinus reduces baroreceptor activity.39 In addition, aortic medial hypertrophy has long been correlated with baroreceptor resetting in the spontaneously hypertensive rat model.42,43 A recent study revealed that expression of a dominant-negative PPARγ in VSMCs increases vascular contractility and hypertrophy and drives baroreceptor dysfunction, demonstrating that altered expression and function of a single arterial smooth muscle gene is sufficient to impair neurovascular coupling.40
Numerous studies in humans have linked arterial stiffness and hypertension in a bidirectional fashion.44,45 Similarly, in mice, Ang II–induced hypertension promotes adventitial collagen deposition in the aorta and stiffness and decreases arterial elasticity.44,46 Increased stiffness causes systolic hypertension, in part through changes in baroreceptor sensitivity.47 In view of the differences in baroreceptor activity in hypertensive WT versus TG SM-CaMKIIN mice, we investigated whether VSMC CaMKII inhibition affects aortic stiffness. The measurement of PWV by ultrasound is generally accepted as the most robust and reproducible method to determine arterial stiffness in humans.48 In fact, accelerated PWV constitutes a robust predictor of cardiovascular risk in hypertensive patients.49 In our experiments, we used an adaptation of this technique for the study of mice.31,32,50 As expected, the aortic PWV significantly increased in hypertensive versus normotensive WT mice46; however, no change in PWV was detected in hypertensive TG SM-CaMKIIN mice.
Ex vivo vasoreactivity and passive distensile properties in the murine aorta are frequently used as surrogate markers for elastic vascular wall properties in vivo.51,52 Similar to published data,53 the Ang II–induced constriction in hypertensive WT and TG SM-CaMKIIN aortas inversely correlated with PWV (Figures6A and 7). Interestingly, the vasoconstriction in aortas from hypertensive TG SM-CaMKIIN mice was greater than in littermates despite lower arterial pressure, demonstrating that ex vivo constriction of aortic rings does not consistently predict arterial pressure in vivo. In our experiments, we also did not find evidence for significant differences in wall stress or strain in the carotid artery between genotypes (Figure 6B through 6D). Regardless of genotype, the increase in stress and strain under Ang II hypertension is consistent with a rightward shift of the stress–strain curve, and that may be interpreted as decreased stiffness. Although unexpected, a previous study reported similar findings in a mouse model of angiotensinogen and renin overexpression.54 These results illustrated the diversity in mechanical properties of different vascular beds. Similar to our data, a recent study demonstrated baroreceptor resetting with expression of a PPARγ mutant limited to smooth muscle cells in the absence of differences in the stress–strain relationship in the carotid artery.40
Consistent with the concept of increased arterial wall stiffness in Ang II–induced hypertension,55 hydroxyproline levels were augmented in hypertensive WT mice. In contrast, decreased stiffness by PWV correlated with lower collagen depositions under smooth muscle CaMKII inhibition (Figure 9D and 9E). Medial VSMCs contribute to overall collagen synthesis in the aortic wall in Ang II–induced hypertension, likely by TGF-β–dependent mechanisms.56,57 In other tissues, CaMKII regulates collagen deposition.58,59 These findings strengthen our conclusion that VSMC properties determine baroreceptor sensitivity, in part, under Ang II–induced hypertension. Collectively, these data support the recent concept that, in addition to adventitial collagen deposition, vascular cell stiffness or adhesion properties are important determinants of elastic wall properties.60,61
As an underlying mechanism for the decreased blood pressure in hypertensive TG SM-CaMKIIN mice, we propose that Ang II–induced vascular remodeling is modulated by CaMKII inhibition, leading to changes in expression of structural proteins with the end result of preserved vascular wall mechanics. This interpretation is supported by our gene array data demonstrating significant CaMKIIN-mediated regulation of genes involved in extracellular matrix deposition and muscle structure and contractility. CaMKII modulates the activity of transcription factors, such as MEF-2, Creb, and myocardin/SRF,1,62–64 that are activated by Ang II and that control the expression of structural proteins. Consequently, the gene array results are likely due to direct effects of CaMKII on Ang II–induced gene transcription.
The augmented increase in vasoconstriction to Ang II in Ang II–hypertensive TG SM-CaMKIIN mice led us to investigate the expression levels of the angiotensin receptors in the aortic wall. Our gene array (Figure 9H) and confirmatory quantitative reverse transcription polymerase chain reaction data (data not shown) demonstrate that CaMKII inhibition has no consistent effect on angiotensin receptor transcript levels, including type 1B, which has been implicated to mediate constriction of the aorta to Ang-II.65–67
Because CaMKIIN is expressed in VSMCs in our model, we interpreted our data as evidence that CaMKII in VSMCs controls structural and functional responses to Ang II that lead to blood pressure differences in Ang II hypertension. CaMKII is expressed in neurons including vasomotor rostral ventrolateral medulla neurons and within the nucleus tractus solitaries and has been implicated in Ang II–induced increases in neuronal firing rate.68 Similarly, endogenous CaMKIIN expression occurs in the brain.69 An alternate interpretation of our data is that CaMKII controls neuronal activity; however, although some expression of CaMKIIN occurs in this Cre model in the brain, no evidence of recombination in structures that are critical for blood pressure regulation (ie, the medulla or the brainstem) was reported.70 In addition, our data showing that blood pressure and heart rate responses to direct stimulation of the transected ADN were similar support the hypothesis that the blood pressure differences between genotypes are not mediated by the central nervous system. Moreover, similar conclusions were reached in a recent report in another model of VSMC-specific gene modification that reported differences in baroreceptor sensitivity in the absence of differences in neuronal density or conclusive evidence for functional neuronal abnormalities.40
In summary, our data identify CaMKII as a multifaceted signaling integrator in Ang II hypertension in VSMCs. Interestingly, a recent study reported on a mathematical model of arterial pressure regulation that integrated numerous physiological variables and concluded that long-term control of arterial blood pressure is primarily through the baroreflex arc and the renin–angiotensin system.71 Moreover, the authors established that arterial stiffening provides a sufficient explanation for the etiology of primary hypertension associated with aging. Our data showing that aortic stiffness and its effect on the baroreflex reflex regulate arterial pressure validate these findings in our murine model and emphasize the relevance of this mechanism to understanding hypertension in humans. Although we investigated remodeling and sympathetic activity in depth, other important mechanisms for blood pressure regulation may be altered by VSMC CaMKII inhibition. We demonstrated that CaMKII inhibition abolishes the baroreceptor resetting. Nevertheless, Ang II–treated transgenic mice still experience a blood pressure increase, albeit to a lesser degree than WT mice (Figure 2A). These data suggest the presence of additional unidentified regulatory mechanisms, such as parasympathetic nerve activity and intravascular volume, that may affect blood pressure regulation in our model. These mechanisms may blunt the effect of CaMKII inhibition on blood pressure.
Sources of Funding
This work was supported by funding from the VA Office of Research and Development (1BX000163-01) and NIH (RO1 HL 108932) (Grumbach). Further funding was provided through VA 1BX000543-03 (Lamping), NIH T32 HL007121 (to Prasad), NIH OD019941 (to Weiss), NIH PPG HL1 4388 and VA Merit 1I01BX001414 (Chapleau) and NIH P01 HL084207 (Sigmund and Rahmouni). The content of this manuscript are solely the responsibility of the authors and does not necessarily represent the views of the granting agencies.
Acknowledgments
We thank Dr Kristina W. Thiel for assistance in the preparation of the manuscript, Dr Bridget Zimmermann for statistical advice, and Litao Xie for expert technical assistance. Transgenic mice were generated at the University of Iowa Transgenic Facility under the direction of Sigmund, PhD, and supported in part by grants from the NIH and from the Roy J. and Lucille A. Carver College of Medicine.
Disclosures
Grumbach receives grant funding from the VA Merit Program for this work. Lamping and Chapleau serve as consultant on this research grant.
References
- Li H, Li W, Gupta AK, Mohler PJ, Anderson ME, Grumbach IM. Calmodulin kinase II is required for angiotensin II-mediated vascular smooth muscle hypertrophy. Am J Physiol Heart Circ Physiol. 2010;298:H688–H698. doi: 10.1152/ajpheart.01014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- House SJ, Ginnan RG, Armstrong SE, Singer HA. Calcium/calmodulin-dependent protein kinase II-delta isoform regulation of vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2007;292:C2276–C2287. doi: 10.1152/ajpcell.00606.2006. [DOI] [PubMed] [Google Scholar]
- Mercure MZ, Ginnan R, Singer HA. CaM kinase II delta2-dependent regulation of vascular smooth muscle cell polarization and migration. Am J Physiol Cell Physiol. 2008;294:C1465–C1475. doi: 10.1152/ajpcell.90638.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Kahn AM. Insulin-inhibited and stimulated cultured vascular smooth muscle cell migration are related to divergent effects on protein phosphatase-2A and autonomous calcium/calmodulin-dependent protein kinase II. Atherosclerosis. 2008;196:227–233. doi: 10.1016/j.atherosclerosis.2007.04.050. [DOI] [PubMed] [Google Scholar]
- House SJ, Singer HA. CaMKII-delta isoform regulation of neointima formation after vascular injury. Arterioscler Thromb Vasc Biol. 2008;28:441–447. doi: 10.1161/ATVBAHA.107.156810. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Schulla V, Welling A, Feil R, Feil S, Wegener JW, Hofmann F, Klugbauer N. Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. EMBO J. 2003;22:6027–6034. doi: 10.1093/emboj/cdg583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieves-Cintron M, Amberg GC, Nichols CB, Molkentin JD, Santana LF. Activation of NFATc3 down-regulates the beta1 subunit of large conductance, calcium-activated K+ channels in arterial smooth muscle and contributes to hypertension. J Biol Chem. 2007;282:3231–3240. doi: 10.1074/jbc.M608822200. [DOI] [PubMed] [Google Scholar]
- Abraham ST, Benscoter H, Schworer CM, Singer HA. In situ Ca2+ dependence for activation of Ca2+/calmodulin-dependent protein kinase II in vascular smooth muscle cells. J Biol Chem. 1996;271:2506–2513. doi: 10.1074/jbc.271.5.2506. [DOI] [PubMed] [Google Scholar]
- Muthalif MM, Karzoun NA, Benter IF, Gaber L, Ljuca F, Uddin MR, Khandekar Z, Estes A, Malik KU. Functional significance of activation of calcium/calmodulin-dependent protein kinase II in angiotensin II–induced vascular hyperplasia and hypertension. Hypertension. 2002;39:704–709. doi: 10.1161/hy0202.103823. [DOI] [PubMed] [Google Scholar]
- Saura M, Marquez S, Reventun P, Olea-Herrero N, Arenas MI, Moreno-Gomez-Toledano R, Gomez-Parrizas M, Munoz-Moreno C, Gonzalez-Santander M, Zaragoza C, Bosch RJ. Oral administration of bisphenol A induces high blood pressure through angiotensin II/CaMKII-dependent uncoupling of eNOS. FASEB J. 2014;28:4719–4728. doi: 10.1096/fj.14-252460. [DOI] [PubMed] [Google Scholar]
- Rezazadeh S, Claydon TW, Fedida D. KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinn amyl)-N-methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J Pharmacol Exp Ther. 2006;317:292–299. doi: 10.1124/jpet.105.097618. [DOI] [PubMed] [Google Scholar]
- Gao L, Blair LA, Marshall J. CaMKII-independent effects of KN93 and its inactive analog KN92: reversible inhibition of L-type calcium channels. Biochem Biophys Res Commun. 2006;345:1606–1610. doi: 10.1016/j.bbrc.2006.05.066. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Braun AP, Schulman H, Premack BA. Multifunctional Ca2+/calmodulin-dependent protein kinase mediates Ca(2+)-induced enhancement of the L-type Ca2+ current in rabbit ventricular myocytes. Circ Res. 1994;75:854–861. doi: 10.1161/01.res.75.5.854. [DOI] [PubMed] [Google Scholar]
- Prasad AM, Nuno DW, Koval OM, Ketsawatsomkron P, Li W, Li H, Shen FY, Joiner ML, Kutschke W, Weiss RM, Sigmund CD, Anderson ME, Lamping KG, Grumbach IM. Differential control of calcium homeostasis and vascular reactivity by Ca2+/calmodulin-dependent kinase II. Hypertension. 2013;62:434–441. doi: 10.1161/HYPERTENSIONAHA.113.01508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Je HD, Gallant C, Zhan Q, Riper DV, Badwey JA, Singer HA, Morgan KG. Ca2+-calmodulin-dependent protein kinase II-dependent activation of contractility in ferret aorta. J Physiol. 2000;526(Pt 2):367–374. doi: 10.1111/j.1469-7793.2000.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokolya A, Singer HA. Inhibition of CaM kinase II activation and force maintenance by KN-93 in arterial smooth muscle. Am J Physiol Cell Physiol. 2000;278:C537–C545. doi: 10.1152/ajpcell.2000.278.3.C537. [DOI] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- Regan CP, Manabe I, Owens GK. Development of a smooth muscle-targeted cre recombinase mouse reveals novel insights regarding smooth muscle myosin heavy chain promoter regulation. Circ Res. 2000;87:363–369. doi: 10.1161/01.res.87.5.363. [DOI] [PubMed] [Google Scholar]
- Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–332. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- Kudo H, Kai H, Kajimoto H, Koga M, Takayama N, Mori T, Ikeda A, Yasuoka S, Anegawa T, Mifune H, Kato S, Hirooka Y, Imaizumi T. Exaggerated blood pressure variability superimposed on hypertension aggravates cardiac remodeling in rats via angiotensin II system-mediated chronic inflammation. Hypertension. 2009;54:832–838. doi: 10.1161/HYPERTENSIONAHA.109.135905. [DOI] [PubMed] [Google Scholar]
- Wu Y, MacMillan LB, McNeill RB, Colbran RJ, Anderson ME. CaM kinase augments cardiac L-type Ca2+ current: a cellular mechanism for long Q-T arrhythmias. Am J Physiol. 1999;276:H2168–H2178. doi: 10.1152/ajpheart.1999.276.6.H2168. [DOI] [PubMed] [Google Scholar]
- Lamping KG, Faraci FM. Role of sex differences and effects of endothelial NO synthase deficiency in responses of carotid arteries to serotonin. Arterioscler Thromb Vasc Biol. 2001;21:523–528. doi: 10.1161/01.atv.21.4.523. [DOI] [PubMed] [Google Scholar]
- Ketsawatsomkron P, Lorca RA, Keen HL, Weatherford ET, Liu X, Pelham CJ, Grobe JL, Faraci FM, England SK, Sigmund CD. PPARγ regulates resistance vessel tone through a mechanism involving RGS5-mediated control of protein kinase C and BKCa channel activity. Circ Res. 2012;111:1446–1458. doi: 10.1161/CIRCRESAHA.112.271577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen MC, Krauhs JM, Brown AM. Relationship of aortic wall and baroreceptor properties during development in normotensive and spontaneously hypertensive rats. Circ Res. 1978;43:728–738. doi: 10.1161/01.res.43.5.728. [DOI] [PubMed] [Google Scholar]
- Berry CJ, Thedens DR, Light-McGroary K, Miller JD, Kutschke W, Zimmerman KA, Weiss RM. Effects of deep sedation or general anesthesia on cardiac function in mice undergoing cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2009;11:16. doi: 10.1186/1532-429X-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffer OA, Carter AB, Sanders PN, Dibbern ME, Winters CJ, Murthy S, Ryan AJ, Rokita AG, Prasad AM, Zabner J, Kline JN, Grumbach IM, Anderson ME. Mitochondrial-targeted antioxidant therapy decreases TGFβ mediated collagen production in a murine asthma model. Am J Respir Cell Mol Biol. 2015;52:106–115. doi: 10.1165/rcmb.2013-0519OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney MJ, Morgan DA, Mark AL. Prolonged renal sympathoinhibition following sustained elevation in arterial pressure. Am J Physiol. 1990;258:H1476–H1481. doi: 10.1152/ajpheart.1990.258.5.H1476. [DOI] [PubMed] [Google Scholar]
- Kenney MJ, Morgan DA. Sustained increases in aortic depressor nerve activity after acute elevation in arterial pressure. J Hypertens. 1994;12:1171–1176. [PubMed] [Google Scholar]
- Ma X, Abboud FM, Chapleau MW. Analysis of afferent, central, and efferent components of the baroreceptor reflex in mice. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1033–R1040. doi: 10.1152/ajpregu.00768.2001. [DOI] [PubMed] [Google Scholar]
- Fleenor BS, Sindler AL, Eng JS, Nair DP, Dodson RB, Seals DR. Sodium nitrite de-stiffening of large elastic arteries with aging: role of normalization of advanced glycation end-products. Exp Gerontol. 2012;47:588–594. doi: 10.1016/j.exger.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindler AL, Fleenor BS, Calvert JW, Marshall KD, Zigler ML, Lefer DJ, Seals DR. Nitrite supplementation reverses vascular endothelial dysfunction and large elastic artery stiffness with aging. Aging Cell. 2011;10:429–437. doi: 10.1111/j.1474-9726.2011.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- Warnes GR, Bolker B, Bonebakker L, Gentleman R, Liaw WHA, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M. Gplots: Various R programming tools for plotting data. Vienna, Austria: R Foundation for Statistical Computing; 2013. R package version 2.11.3. [Google Scholar]
- Sabharwal R, Zhang Z, Lu Y, Abboud FM, Russo AF, Chapleau MW. Receptor activity-modifying protein 1 increases baroreflex sensitivity and attenuates angiotensin-induced hypertension. Hypertension. 2010;55:627–635. doi: 10.1161/HYPERTENSIONAHA.109.148171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Dale WE, Hasser EM, Blaine EH. Acute and chronic angiotensin hypertension: neural and nonneural components, time course, and dose dependency. Am J Physiol. 1996;271:R200–R207. doi: 10.1152/ajpregu.1996.271.1.R200. [DOI] [PubMed] [Google Scholar]
- King AJ, Osborn JW, Fink GD. Splanchnic circulation is a critical neural target in angiotensin II salt hypertension in rats. Hypertension. 2007;50:547–556. doi: 10.1161/HYPERTENSIONAHA.107.090696. [DOI] [PubMed] [Google Scholar]
- Skelding KA, Rostas JA, Verrills NM. Controlling the cell cycle: the role of calcium/calmodulin-stimulated protein kinases I and II. Cell Cycle. 2011;10:631–639. doi: 10.4161/cc.10.4.14798. [DOI] [PubMed] [Google Scholar]
- Chapleau MW, Cunningham JT, Sullivan MJ, Wachtel RE, Abboud FM. Structural versus functional modulation of the arterial baroreflex. Hypertension. 1995;26:341–347. doi: 10.1161/01.hyp.26.2.341. [DOI] [PubMed] [Google Scholar]
- Borges GR, Morgan DA, Ketsawatsomkron P, Mickle AD, Thompson AP, Cassell MD, Mohapatra DP, Rahmouni K, Sigmund CD. Interference with peroxisome proliferator-activated receptor-gamma in vascular smooth muscle causes baroreflex impairment and autonomic dysfunction. Hypertension. 2014;64:590–596. doi: 10.1161/HYPERTENSIONAHA.114.03553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelat M, Dessy C, Massion P, Desager JP, Feron O, Balligand JL. Rosuvastatin decreases caveolin-1 and improves nitric oxide-dependent heart rate and blood pressure variability in apolipoprotein E-/- mice in vivo. Circulation. 2003;107:2480–2486. doi: 10.1161/01.CIR.0000065601.83526.3E. [DOI] [PubMed] [Google Scholar]
- Wellens D, Borgers M, Verheyen A. Structural basis for resetting of baroreceptor regulation in spontaneously hypertensive rats (SHR) Experientia. 1973;29:1268–1271. doi: 10.1007/BF01935111. [DOI] [PubMed] [Google Scholar]
- Sapru HN, Wang SC. Modification of aortic barorecptor resetting in the spontaneously hypertensive rat. Am J Physiol. 1976;230:664–674. doi: 10.1152/ajplegacy.1976.230.3.664. [DOI] [PubMed] [Google Scholar]
- Payne RA, Wilkinson IB, Webb DJ. Arterial stiffness and hypertension: emerging concepts. Hypertension. 2010;55:9–14. doi: 10.1161/HYPERTENSIONAHA.107.090464. [DOI] [PubMed] [Google Scholar]
- Mitchell GF. Arterial stiffness and hypertension: chicken or egg? Hypertension. 2014;64:210–214. doi: 10.1161/HYPERTENSIONAHA.114.03449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham DM, Martin-McNulty B, Wang YX, Da Cunha V, Wilson DW, Athanassious CN, Powers AF, Sullivan ME, Rutledge JC. Angiotensin II injures the arterial wall causing increased aortic stiffening in apolipoprotein E-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1442–R1449. doi: 10.1152/ajpregu.00295.2002. [DOI] [PubMed] [Google Scholar]
- Chesterton LJ, Sigrist MK, Bennett T, Taal MW, McIntyre CW. Reduced baroreflex sensitivity is associated with increased vascular calcification and arterial stiffness. Nephrol Dial Transplant. 2005;20:1140–1147. doi: 10.1093/ndt/gfh808. [DOI] [PubMed] [Google Scholar]
- Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, Pannier B, Vlachopoulos C, Wilkinson I, Struijker-Boudier H European Network for Non-invasive Investigation of Large A. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. 2006;27:2588–2605. doi: 10.1093/eurheartj/ehl254. [DOI] [PubMed] [Google Scholar]
- Blacher J, Asmar R, Djane S, London GM, Safar ME. Aortic pulse wave velocity as a marker of cardiovascular risk in hypertensive patients. Hypertension. 1999;33:1111–1117. doi: 10.1161/01.hyp.33.5.1111. [DOI] [PubMed] [Google Scholar]
- Reddy AK, Taffet GE, Li YH, Lim SW, Pham TT, Pocius JS, Entman ML, Michael LH, Hartley CJ. Pulsed Doppler signal processing for use in mice: applications. IEEE Trans Biomed Eng. 2005;52:1771–1783. doi: 10.1109/TBME.2005.855709. [DOI] [PubMed] [Google Scholar]
- Cox RH. Basis for the altered arterial wall mechanics in the spontaneously hypertensive rat. Hypertension. 1981;3:485–495. doi: 10.1161/01.hyp.3.4.485. [DOI] [PubMed] [Google Scholar]
- Cox RH. Alterations in active and passive mechanics of rat carotid artery with experimental hypertension. Am J Physiol. 1979;237:H597–H605. doi: 10.1152/ajpheart.1979.237.5.H597. [DOI] [PubMed] [Google Scholar]
- Fitch RM, Rutledge JC, Wang YX, Powers AF, Tseng JL, Clary T, Rubanyi GM. Synergistic effect of angiotensin II and nitric oxide synthase inhibitor in increasing aortic stiffness in mice. Am J Physiol Heart Circ Physiol. 2006;290:H1190–H1198. doi: 10.1152/ajpheart.00327.2005. [DOI] [PubMed] [Google Scholar]
- Baumbach GL, Sigmund CD, Faraci FM. Cerebral arteriolar structure in mice overexpressing human renin and angiotensinogen. Hypertension. 2003;41:50–55. doi: 10.1161/01.hyp.0000042427.05390.5c. [DOI] [PubMed] [Google Scholar]
- Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W, Harrison DG. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res. 2014;114:616–625. doi: 10.1161/CIRCRESAHA.114.302157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocnik EF, Chan BM, Pickering JG. Evidence for a role of collagen synthesis in arterial smooth muscle cell migration. J Clin Invest. 1998;101:1889–1898. doi: 10.1172/JCI1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CM, Li S, Pickering JG. Angiotensin II stimulates collagen synthesis in human vascular smooth muscle cells. Involvement of the AT(1) receptor, transforming growth factor-beta, and tyrosine phosphorylation. Arterioscler Thromb Vasc Biol. 1999;19:1843–1851. doi: 10.1161/01.atv.19.8.1843. [DOI] [PubMed] [Google Scholar]
- An P, Tian Y, Chen M, Luo H. Ca(2+)/calmodulin- dependent protein kinase II mediates transforming growth factor-beta-induced hepatic stellate cells proliferation but not in collagen alpha1(I) production. Hepatol Res. 2012;42:806–818. doi: 10.1111/j.1872-034X.2012.00983.x. [DOI] [PubMed] [Google Scholar]
- Zhang W, Chen DQ, Qi F, Wang J, Xiao WY, Zhu WZ. Inhibition of calcium-calmodulin-dependent kinase II suppresses cardiac fibroblast proliferation and extracellular matrix secretion. J Cardiovasc Pharmacol. 2010;55:96–105. doi: 10.1097/FJC.0b013e3181c9548b. [DOI] [PubMed] [Google Scholar]
- Sehgel NL, Sun Z, Hong Z, Hunter WC, Hill MA, Vatner DE, Vatner SF, Meininger GA. Augmented vascular smooth muscle cell stiffness and adhesion when hypertension is superimposed on aging. Hypertension. 2015;65:370–377. doi: 10.1161/HYPERTENSIONAHA.114.04456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgel NL, Zhu Y, Sun Z, Trzeciakowski JP, Hong Z, Hunter WC, Vatner DE, Meininger GA, Vatner SF. Increased vascular smooth muscle cell stiffness: a novel mechanism for aortic stiffness in hypertension. Am J Physiol Heart Circ Physiol. 2013;305:H1281–H1287. doi: 10.1152/ajpheart.00232.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sun LY, Singer DV, Ginnan R, Singer HA. CaMKIIdelta-dependent inhibition of cAMP-response element-binding protein activity in vascular smooth muscle. J Biol Chem. 2013;288:33519–33529. doi: 10.1074/jbc.M113.490870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun P, Enslen H, Myung PS, Maurer RA. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8:2527–2539. doi: 10.1101/gad.8.21.2527. [DOI] [PubMed] [Google Scholar]
- Fluck M, Booth FW, Waxham MN. Skeletal muscle CaMKII enriches in nuclei and phosphorylates myogenic factor SRF at multiple sites. Biochem Biophys Res Commun. 2000;270:488–494. doi: 10.1006/bbrc.2000.2457. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Chen Y, Dirksen WP, Morris M, Periasamy M. AT1b receptor predominantly mediates contractions in major mouse blood vessels. Circ Res. 2003;93:1089–1094. doi: 10.1161/01.RES.0000101912.01071.FF. [DOI] [PubMed] [Google Scholar]
- Ryan MJ, Didion SP, Mathur S, Faraci FM, Sigmund CD. Angiotensin II-induced vascular dysfunction is mediated by the AT1A receptor in mice. Hypertension. 2004;43:1074–1079. doi: 10.1161/01.HYP.0000123074.89717.3d. [DOI] [PubMed] [Google Scholar]
- Cavalli A, Lattion AL, Hummler E, Nenniger M, Pedrazzini T, Aubert JF, Michel MC, Yang M, Lembo G, Vecchione C, Mostardini M, Schmidt A, Beermann F, Cotecchia S. Decreased blood pressure response in mice deficient of the alpha1b-adrenergic receptor. Proc Natl Acad Sci USA. 1997;94:11589–11594. doi: 10.1073/pnas.94.21.11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Sumners C, Raizada MK. Chronotropic action of angiotensin II in neurons via protein kinase C and CaMKII. Hypertension. 2002;39:562–566. doi: 10.1161/hy0202.103057. [DOI] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase II inhibitor protein in brain. Proc Natl Acad Sci USA. 1998;95:10890–10895. doi: 10.1073/pnas.95.18.10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox EA, Biddinger JE, Jones KR, McAdams J, Worman A. Mechanism of hyperphagia contributing to obesity in brain-derived neurotrophic factor knockout mice. Neuroscience. 2013;229:176–199. doi: 10.1016/j.neuroscience.2012.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard DA, Pettersen KH, Carlson BE, Omholt SW, Bugenhagen SM. A computational analysis of the long-term regulation of arterial pressure. F1000Res. 2013;2:208. doi: 10.12688/f1000research.2-208.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]