

Abstract
Pathogenesis of thrombotic thrombocytopenic purpura (TTP) was a mystery for over half a century until the discovery of ADAMTS13. ADAMTS13 is primarily synthesized in the liver, and its main function is to cleave von Willebrand factor (VWF) anchored on the endothelial surface, in circulation, and at the sites of vascular injury. Deficiency of plasma ADAMTS13 activity (<10%) resulting from mutations of the ADAMTS13 gene or autoantibodies against ADAMTS13 causes hereditary or acquired (idiopathic) TTP. ADAMTS13 activity is usually normal or modestly reduced (>20%) in other forms of thrombotic microangiopathy secondary to hematopoietic progenitor cell transplantation, infection, and disseminated malignancy or in hemolytic uremic syndrome. Plasma infusion or exchange remains the initial treatment of choice to date, but novel therapeutics such as recombinant ADAMTS13 and gene therapy are under development. Moreover, ADAMTS13 deficiency has been shown to be a risk factor for the development of myocardial infarction, stroke, cerebral malaria, and preeclampsia.
Keywords: metalloprotease, arterial thrombosis, rare hematological disease, mutations, autoantibodies, autoimmune disorder
HISTORIC ASPECTS
In 1924, Moschcowitz described the case of a 16-year-old girl admitted to the Beth Israel Hospital in New York City who died within two weeks after an abrupt onset of petechiae, anemia, and pallor followed by paralysis and coma (1). Autopsy revealed occlusive hyaline thrombi disseminated in terminal arterioles and capillaries, later shown to be composed primarily of aggregated platelets. Moschcowitz suspected that the cause of this disorder was a powerful “poison” with agglutinative and hemolytic properties. In 1960, Schulman and colleagues reported the clinical course of an eight-year-old girl who had chronic episodes of anemia and thrombocytopenia; she responded transiently to plasma infusion (2). In 1978, Upshaw reported a 29-year-old woman with similar signs and symptoms, but her first episode had occurred at six months of age (3). These investigators proposed that lack of a plasma factor promoting platelet or red blood cell survival was the cause of the disease (2, 3). In 1979, Rennard & Abe suggested the use of the name Upshaw-Schulman syndrome to describe patients with these clinical features who responded to plasma infusion (4). The same condition was later referred to as congenital or hereditary thrombotic thrombocytopenic purpura (TTP) (5).
In 1966, a “pentad” of clinical and laboratory features was proposed as criteria for the diagnosis of TTP. This included microangiopathic hemolytic anemia with fragmentation of red blood cells (or schistocytes) in the peripheral blood smear, thrombocytopenia, neurological signs and symptoms, renal failure, and fever (6). However, subsequent studies demonstrated that neurological signs and symptoms, renal failure, and fever were not present in all patients with TTP (7, 8). Therefore, the presence of microangiopathic hemolytic anemia and thrombocytopenia in the absence of other likely etiologies is now considered sufficient to make a presumptive diagnosis of TTP. Plasma infusion and/or exchange therapy should be implemented urgently, and this treatment has reduced the mortality rate from 85–95% to 10–20% (7, 9).
How plasma therapy works to treat TTP is still not fully understood. The first clue to our current understanding of TTP pathogenesis came from a study by Moake and colleagues in 1982 (10). They reported the presence of circulating ultralarge VWF (ULVWF) multimers in four patients with chronic TTP that disappeared during acute episodes or relapses. They postulated that these patients lacked a plasma VWF “depolymerase” that led to the accumulation of ULVWF multimers in plasma. The ULVWF were hyperactive and capable of causing spontaneous platelet agglutination and thrombosis in small arterioles and capillaries (10). In 1997, Furlan and colleagues described four patients (including two brothers) with chronic relapsed TTP who had partial or complete deficiency of the plasma VWF depolymerase, also referred to as VWF-cleaving protease (VWF-cp). An inhibitor to the protease was not detected in a mixing study, so the deficiency of plasma VWF-cp was believed to be constitutional (11). One year later, Tsai & Lian (8) and Furlan et al. (12) independently reported an acquired deficiency of plasma VWF-cp in large cohorts of TTP patients, resulting from inhibitory immunoglobulin G (IgG) against the VWF-cp. Therefore, it became evident that the deficiency of plasma VWF-cp activity, either constitutional or acquired, was the common mechanism underlying the pathogenesis of congenital or acquired idiopathic TTP.
DISCOVERY OF ADAMTS13
Isolation of VWF-cp from plasma has been a daunting task. In 1996, Furlan (13) and Tsai (14) used chromatographic techniques to isolate VWF-cp from normal human plasma with only partial success. However, they were able to determine the cleavage site in VWF (the Tyr1605 and Met1606 bond) with partially purified materials once the protein had been denatured with 1.5 M urea (13) or guanidine-HCl (14). In addition, they demonstrated that plasma VWF-cp activity increased dramatically when a divalent metal ion was added to the reaction but was inhibited or abolished with the addition of ethylene diaminetetraacetic acid, which chelates divalent metal ions. Therefore, it was concluded that plasma VWF-cp is a metalloprotease.
In 2001, several groups independently isolated VWF-cp from plasma and determined its partial amino acid sequence. This led to the identification of VWF-cp as a novel member of the ADAMTS (a disintegrin and metalloprotease with thrombospondin type motifs) family of metalloproteases (15–17). The gene encoding VWF-cp was designated ADAMTS13 according to the HUGO Gene Nomenclature Committee (http://www.gene.ucl.ac.uk/nomenclature). The same gene was shown to be responsible for congenital TTP based on positional cloning and familial TTP pedigrees with subsequent linkage analysis (18). ADAMTS13, localized to chromosome 9q34, contains 29 exons spanning 37 kb in the genomic sequence. The primary translation product consists of 1,427 amino acid residues, comprising a signal peptide and a short propeptide, followed by a reprolysin-like metalloprotease domain, disintegrin domain, and first thrombospondin-1 (TSP1) repeat, and Cys-rich and spacer domains. The more distal C terminus contains seven additional TSP1 repeats and two CUB (for complement C1r/C1s, Uegf, Bmp1) domains (Figure 1). Human recombinant ADAMTS13 was quickly expressed in transfected cell lines as a 195-kDa protein, capable of cleaving soluble multimeric VWF (19, 20) and endothelium-released and anchored ULVWF strings (21). Success in gene identification, cloning, and expression of human ADAMTS13 has stimulated tremendous interest in and further investigation of its biology and the pathogenesis of TTP, which are reflected in the rapid increase in the number of publications per year over the past decade. This research has made it possible to develop novel diagnostic tools and therapeutics for what used to be a fatal disease.
Figure 1.
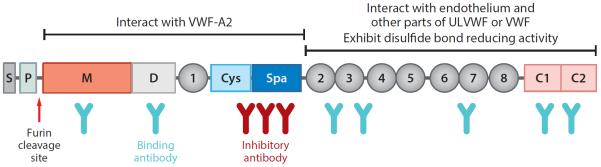
Schematic representation of ADAMTS13 domain organization, potential role in substrate recognition, and binding sites of autoantibodies in acquired TTP. S, signal peptide; P, propeptide; M, metalloprotease domain; D, disintegrin domain; 1, the first thrombospondin type 1 (TSP1) repeat; Cys, the cysteine-rich domain; Spa, the spacer domain; 2–8, TSP1 2–8 repeats; C1 and C2, the first and second CUB domain. Other abbreviations: ULVWF, ultralarge von Willebrand factor; VWF, von Willebrand factor; A2, the A2 domain of VWF.
BIOSYNTHESIS AND SECRETION OF ADAMTS13
The concentrations of ADAMTS13 in human plasma are 0.7–1.4 μg/ml (3.5–7.0 nM) (22). ADAMTS13 is synthesized primarily in the liver, demonstrated by in situ hybridization and immunohistochemistry. Human ADAMTS13 mRNA and protein are localized exclusively to hepatic stellate cells (HSCs), which reside in the interstitial space between hepatocytes (23). Also, isolated HSCs from mice (24) and rats (25) secrete an ~195-kDa ADAMTS13 protein, which is proteolytically active in the cleavage of multimeric VWF and its peptidyl derivatives. Expression of ADAMTS13 in rat HSCs increases as a function of culturing time during which cells are activated, as demonstrated by coexpression of α-smooth muscle actin. The rate of ADAMTS13 synthesis by HSCs is also increased after intravenous administration of carbon tetrachloride (25) and after bile duct ligation (26), which activates HSCs in vivo. Conversely, plasma ADAMTS13 antigen and activity are markedly reduced in rats treated with dimethylnitrosamine, which induces HSC apoptosis, or after partial hepatectomy, which reduces the number of functional HSCs (27). Together, these data support the hypothesis that HSCs are the major source of plasma ADAMTS13 in mammals.
ADAMTS13 is also produced in limited quantities by vascular endothelial cells (28), megakaryocytes and platelets (29), glomerular podocytes (30), and glial cells (31), although the physiological relevance of these sources remains to be determined. Considering their massive surface coverage, endothelial cells may contribute significantly to plasma levels of ADAMTS13. Platelets are specifically targeted to sites of vascular injury where they are activated and degranulated, releasing their granular contents (including VWF), which are prothrombotic and proinflammatory. Therefore, concurrent local release of even small amounts of active ADAMTS13 protease may have profound inhibitory effects on thrombosis and inflammation. Transgenic mice lacking plasma ADAMTS13 but expressing human ADAMTS13 in their platelets are protected from arterial thrombosis induced by ferric chloride and from TTP induced by shigatoxin or recombinant murine VWF (B. Pickens, X.L. Zheng, unpublished results).
How plasma levels of ADAMTS13 are regulated under physiological conditions remains poorly understood. In humans, VWF appears to be the major regulator of plasma ADAMTS13 concentration. For instance, patients with type 3 von Willebrand disease (lacking circulating VWF) have 30% higher plasma levels of ADAMTS13, whereas healthy volunteers who receive an intravenous infusion of 1-deamino-8-D-arginine vasopressin (DDAVP) that triggers release of endothelial VWF show a 20% reduction in plasma ADAMTS13 antigen (32). The mechanism of how VWF regulates plasma ADAMTS13 concentrations is not known, but it likely involves consumption.
In culture, ADAMTS13 synthesis by human umbilical vein endothelial cells and rat HSCs (33) is dramatically inhibited by inflammatory cytokines, including interferon-γ (INFγ), tumor necrosis factor-α (TNFα), and interleukin-4 (IL-4) or -6 (IL-6), which are variably released during systemic inflammation and acute episodes of TTP (34). In podocytes, IL-4 and IL-6 differentially regulate ADAMTS13 mRNA and protein, which is reversed by simvastatin, a widely used antiatherosclerotic agent (35). In glial cells (astrocytes and microglia), ADAMTS13 expression is significantly upregulated after spinal cord injury (31), suggesting a potential role for ADAMTS13 in the central nervous system. Together, the data available to date indicate that plasma concentrations of ADAMTS13 are regulated at the transcriptional and posttranslational levels under diverse (patho)physiological conditions through mechanisms that have not been thoroughly elucidated.
BIOSYNTHESIS AND SECRETION OF VON WILLEBRAND FACTOR
The mean concentration of VWF in human plasma is estimated to be ~10 μg/ml (μ50 nM) (36). VWF is produced primarily in vascular endothelial cells and megakaryocytes. In the endoplasmic reticulum, pro-VWF acquires high-mannose N-linked oligosaccharides and forms “tail-to-tail” dimers through the formation of C-terminal disulfide bonds. After reaching the Golgi apparatus, the high-mannose glycans on pro-VWF dimers are processed to the complex form and undergo sulfation. At acidic pH, pro-VWF dimers form “head-to-head” large multimers via N-terminal disulfide bonds facilitated by the propeptide. Prior to secretion and storage in the Weibel-Palade bodies of endothelial cells or in the α-granules of megakaryocytes and platelets, the propeptide is cleaved by furin, an enzyme that belongs to the subtilisin-like proprotein convertase family. The propeptide remains associated with the mature VWF subunits. In plasma, VWF is present as a series of repeating subunits, ranging in size from ~500 kDa to ~20,000 kDa. Each VWF subunit contains 2,050 amino acid residues with a molecular mass of ~250 kDa under reducing conditions. The VWF subunit is organized in the sequence D′–D3-A1-A2-A3-D4-C1–6-CK and capable of interacting with a variety of other plasma and matrix proteins, including coagulation factor VIII (D′-D3), platelet glycoprotein 1b (A1 domain), and collagen matrix (A3 domain) (Figure 2). The central A2 domain of VWF contains the binding sites and cleavage bond for ADAMTS13, which is buried in native VWF.
Figure 2.

Schematic representation of pro-VWF domain organization, proteins that interact with VWF (von Willebrand factor), and the ADAMTS13 cleavage site. The large propeptide (D1–D2) in pro-VWF is cleaved by furin to generate the mature VWF subunit, which includes FVIII-binding domains (D′-D3), GP1b- (A1) and collagen-binding (A3) domains, and a central A2 domain containing the ADAMTS13 cleavage site, as well as several C-terminal domains (D4-CK) that bind αIIβIII integrin.
The secretion of VWF from the Weibel-Palade bodies of endothelial cells can be induced by calcium ionophere A23187 and phorbol myristate acetate. VWF secretion may also be induced by agents that are of physiological relevance, including thrombin, histamine, fibrin, complement protein C5b-9 complexes (37), and bacterial shigatoxin (38). In vivo, VWF release can be triggered at the sites of vascular injury (39) or by DDAVP (40), used to treat mild hemophilia A and von Willebrand disease. VWF secreted from endothelial cells in culture and in vivo (39) rapidly forms “strings or bundle-like” structures through a mechanism referred to as lateral association, a process that requires the formation of disulfide bonds between preexisting or newly exposed free thiols on the surface of VWF under flow (41). This lateral association increases the length and thickness of VWF strings, which enhance the adhesive function of VWF in normal hemostasis.
The formation and elongation of VWF strings on endothelial surfaces are tightly regulated by ADAMTS13 through proteolytic cleavage of VWF strings (21) or inhibition of the formation of elongated VWF multimers by blocking lateral disulfide bond formation (39, 42). The length and thickness of VWF multimers strongly correlate with physiological hemostatic potential. Therefore, the cleavage or reduction of VWF multimers by ADAMTS13 is critical for maintaining normal hemostasis. In the absence of ADAMTS13 activity, as seen in patients with ADAMTS13 mutations or acquired autoantibodies that block plasma ADAMTS13 activity, endothelium-anchored ULVWF strings are capable of recruiting flowing platelets and causing uncontrolled thrombosis in terminal arterioles and capillaries.
Megakaryocytes and platelets also synthesize and secrete VWF, which contributes 10–20% of total VWF in platelet-rich plasma. Platelet VWF is found in a high-molecular-weight form that lacks N-linked sialylation and is relatively resistant to proteolysis by ADAMTS13 (43). As platelets are targeted directly to the sites of vascular injury, where they undergo degranulation and release of their contents, local delivery of high concentrations of ADAMTS13-resistant platelet VWF may play a significant physiological role in hemostasis. Consistent with this notion is that platelet storage of VWF is reduced in bleeding disorders, such as von Willebrand disease and essential thrombocythemia. Further investigation of platelet VWF and ADAMTS13 may shed new light on the mechanism of normal hemostasis and thrombosis.
ADAMTS13 INTERACTION WITH VON WILLEBRAND FACTOR
ADAMTS13 binds soluble VWF adsorbed on surface with a dissociation constant (KD) of ~20 nM (44). ADAMTS13 also interacts with endothelium-anchored ULVWF, resulting in efficient cleavage of ULVWF strings or bundles (21). This cleavage can occur in the absence of flow (45) but is modestly enhanced by fluid shear stress (21), suggesting that cell-bound ULVWF is in its “open” conformation. Released ULVWF in solution exhibits a “closed” conformation which is not sensitive to ADAMTS13 cleavage until arterial shear (>20 dynes/cm2) is applied.
Binding platelet glycoprotein 1b (GP1b) and/or coagulation factor VIII (FVIII) to soluble VWF (46, 47) dramatically increases its rate of proteolysis by ADAMTS13 under shear. This effect is largely eliminated when VWF is predenatured with 1.5 M urea. These results suggest that the rate-enhancing effect of GP1b and FVIII on VWF proteolysis is likely mediated by their alteration of domain–domain interactions and destabilization of the VWF-A2 domain where the cleavage site resides. In vitro, destabilization or unfolding of the A2 domain is achieved by incubation with 1.5 M urea (48) or guanidine (8, 14) in a low ionic buffer or by mechanical force (49), which accelerates the cleavage of VWF by ADAMTS13. In contrast, binding of calcium to the A2 domain protects against unfolding by denaturants (50) or promotes refolding under a tensile force (51), thereby reducing the susceptibility of VWF to cleavage by ADAMTS13.
Structure-function analysis has demonstrated that the ADAMTS13 fragment comprising the metalloprotease (M), disintegrin (D), first TSP1 repeat (T), Cys-rich (C), and spacer (S) domains (i.e., MDTCS) (Figure 1) appears to be sufficient for cleavage of cell-bound ULVWF (45, 52) or soluble VWF (20, 53). An extensive exosite interaction between the DTCS domains and VWF-A2 domain governs substrate specificity and cleavage efficiency. Mutations in the highly variable regions of the DTCS domains are shown to significantly impair ADAMTS13 function (20, 53–55). However, the role of more distal C-terminal domains, including TSP1 2–8 and CUB domains, remains to be further elucidated. A C-terminal fragment comprising the TSP1 2–8 or 5–8 repeats and CUB domains can bind directly to multimeric VWF under flow (56). Peptides derived from the first CUB domain block the cleavage of endothelial ULVWF by ADAMTS13 under flow (57), suggesting the role of TSP1 repeats and CUB domains in ULVWF docking. Consistent with this hypothesis, mice lacking the seventh TSP1 and CUB domains because of a naturally occurring mutation exhibit a prothrombotic phenotype in ferric chloride–induced mesenteric arterial injury (58). However, human ADAMTS13 fragment lacking the TSP1 2–8 and CUB domains cleaves endothelial ULVWF normally in vitro (45, 52) and efficiently inhibits arterial thrombosis in vivo (59), suggesting that the distal C-terminal domains may be dispensable under certain assay conditions. The difference may be attributed to the sensitivity and specificity of various assays. More recently, Yeh et al. (42) and Bao et al. (39) have independently shown that C-terminal TSP1 2–8 and CUB domains of ADAMTS13 may have disulfide bond reductase activity. This activity is independent of ADAMTS13 proteolytic activity. It has been proposed that both proteolytic and disulfide bond–reducing activity may be required for optimal antithrombotic activity in vivo.
The findings of ADAMTS13 structure-function analyses are crucial for understanding the molecular mechanism of TTP, including autoantibody-mediated TTP. Most adult idiopathic TTP patients harbor polyclonal anti-ADAMTS13 IgGs that bind to multiple domains of ADAMTS13 (60). Importantly, the inhibitory activity of anti-ADAMTS13 antibodies in patients with acquired TTP appears to be primarily mediated by their binding to the spacer domain. This domain, highly conserved in ADAMTS13 from various species (human, mouse, and zebrafish) but different from other members of the ADAMTS family (61), appears to be critical for substrate recognition. For instance, deletion or alanine substitution of even a few surface-exposed residues in the spacer domain dramatically reduces its proteolytic activity (54) and binding to anti-ADAMTS13 antibodies (62). Conversely, conservative mutations in the same region retain or enhance ADAMTS13 activity and increase resistance to inhibition by autoantibodies (63). These gain-of-function ADAMTS13 variants might prove useful in the treatment of acquired TTP caused by inhibitors.
ADAMTS13 IN THE DIAGNOSIS OF THROMBOTIC THROMBOCYTOPENIC PURPURA
TTP must be distinguished from a similarly presenting clinical entity called hemolytic uremic syndrome (HUS). The most common cause (90%) of HUS, particularly in children, is shigatoxin-producing bacteria, typically Escherichia coli O157:H7 (64). This type of HUS is referred to as a diarrhea-positive HUS (D+HUS) or “typical” HUS. Clinical outcome is usually excellent with supportive therapy. Approximately 10% of cases are not caused by bacterial infections; these have been designated diarrhea-negative HUS (D–HUS) or “atypical” HUS (aHUS), which, until recently, has had a poor prognosis. Several precipitating factors have been postulated as secondary causes of aHUS, including infection, drugs, autoimmune diseases, transplantation, pregnancy, and malignancy. Approximately 60% of patients with aHUS have been shown to have mutations in complement regulatory proteins (65), including complement factor H (CFH, ~25%), factor I (CFI, 5–10%), and membrane cofactor protein (MCP, ~10%), or in complement proteins C3 and factor B (2–10%). Autoantibodies against CFH (66) and CFI (67) have also been identified in some patients with aHUS, as well as mutations in thrombomodulin (TM) (68) and diacylglycerol kinase varepsilon (DGKE) (69). TM is primarily expressed in the endothelium, whereas DGKE is found in endothelium, platelets, and podocytes. In addition to its well-established anticoagulant function, TM induces the generation of thrombin-activatable fibrinolysis inhibitor, a plasma carboxypeptidase B that cleaves C3a and C5a. TM also binds C3b to accelerate C3b inactivation by the CFI/CFH complex. The major function of DGKE is to downregulate arachidonic acid–containing diacylglycerols (DAG) signaling on platelets, thereby dampening thrombotic potential (69). Therefore, anticomplement therapy may have a role in aHUS patients with complement hyeractivation, but may not have a role in aHUS patients with DGKE mutations.
Assays for plasma ADAMTS13 activity and inhibitors are crucial to confirm the diagnosis of hereditary and autoimmune TTP. These tests also help differentiate TTP from aHUS and secondary thrombotic microangiopathy (TMA) occurring after hematopoietic progenitor transplantation, disseminated malignancies, certain drugs or chemotherapies, infections, etc. Nearly all patients with secondary TMA have normal or mildly reduced levels of plasma ADAMTS13 activity with no inhibitor present (70). Assays of plasma ADAMTS13 activity and inhibitors help not only in differential diagnosis, but also in stratifying TTP patients for more targeted therapy and in predicting the risk of relapse and long-term outcome (71). When provided in real time, the measurement of plasma ADAMTS13 activity may help to avoid unnecessary plasma-exchange therapy in patients who have other forms of TMA, thereby dramatically reducing overall healthcare costs (72).
ADAMTS13 testing has evolved over the past decade. The initial methods utilized a full-length VWF as a substrate, assayed in the presence of 1.5 M urea (73) or guanidine (8). These assays require agarose or SDS-polyacrylamide gel electrophoresis and Western blotting and are at best semiquantitative. The use of collagen-binding assay (CBA) (74) or an immunoturbidimetric method (75) for residual VWF activity has significantly improved the reliability of quantitation and turnaround time. More recently, an assay based on fluorescence energy resonance transfer (FRETS) and a peptide containing 73 amino acid residues from the central A2 domain of VWF (FRETS-VWF73) (76) has gained popularity owing to its simplicity and rapid turnaround time. However, the moderate correlation between the FRETS-based assay and the multimeric VWF-based assay (77) suggests the complexity of assessing ADAMTS13 activity in biological samples. Clinicians should be aware of which test method is used to obtain ADAMTS13 activity and inhibitor results before an appropriate interpretation can be made. Bilirubin (78) and free hemoglobin (79) in the blood sample may interfere with assays by masking the intensity of fluorescence generation, such as in a FRETS-based assay, and/or by directly affecting ADAMTS13-substrate interaction and enzymatic activity (80; R.N. Lu, X.L. Zheng, unpublished observation). Denaturants added to the reaction, such as urea or guanidine, may dissociate ADAMTS13-autoantibody complexes, which might falsely elevate plasma ADAMTS13 activity (81). In general, most samples with severe deficiency of ADAMTS13 activity (<5%) show a good agreement among CBA, FRETS-VWF73, and chromogenic VWF73 assays, but only 83% of these samples with <11% activity and 52% of samples with 11–55% activity show agreement between the CBA and FRETS-VWF73 or chromogenic VWF73 results (82). Such discrepant results indicate the presence of urea, bilirubin, and free hemoglobin, which may affect one assay but not another. A recent report suggests that measurement of plasma ADAMTS13 activity by the FRETS-VWF73 assay, but not a multimeric VWF-based assay in the presence of urea, more closely reflects plasma ADAMTS13 status in TTP patients (81).
The cut-off of plasma ADAMTS13 activity that should be used for diagnosis of TTP remains a question. The answer depends on the assay sensitivity and specificity. Most early studies demonstrated that a plasma ADAMTS13 activity of <5–10% appeared to be specific for TTP (83). Furthermore, if an autoantibody inhibitor is identified, the specificity for acquired TTP increases (8). However, a few patients with severe sepsis-induced disseminated intravascular coagulation have plasma ADAMTS13 activity of <5% (84). A recent retrospective study suggests that if an ADAMTS13 activity of <20% is used as the cut-off, the diagnostic sensitivity and specificity are 100% and 99%, respectively. The positive predictive value is 91%, and the negative predictive value is 100% (72).
Despite these advances, a commercial FRETS-VWF73 assay has certain limitations as it is performed under nonphysiological conditions, and there is potential for interference from free hemoglobin and bilirubin, both of which occur frequently in patients with TTP or in other critically ill patients. These issues may be partially addressed by the development of a new FRETS-rVWF71 substrate (79) that is similar to FRETS-VWF73 but with a brighter fluorescent dye. It emits at a wavelength where plasma is almost transparent. This substrate is compatible with physiological conditions and appears to be more sensitive to ADAMTS13 activity and inhibitors. However, whether this new assay is superior to other similar assays remains to be further investigated. Clearly, an easy and more physiological (under shear) assay for determining plasma ADAMTS13 activity is needed.
CURRENT AND POTENTIAL NOVEL THERAPEUTICS
Plasma infusion and exchange remain the mainstay of treatment for hereditary and acquired TTP, respectively, until recombinant ADAMTS13 and other novel therapeutics become available. Prior to plasma therapy, the TTP mortality rate was as high as ~85–95%, and this has been reduced to 10–20% (7, 9).
For patients with hereditary TTP, intermittent infusion of fresh frozen plasma (10–15 ml/kg) every 2–3 weeks is sufficient to raise trough plasma ADAMTS13 activity to 5–10% activity and prevent disease recurrence (85). However, lifelong treatment with fresh frozen plasma is inconvenient, and the risk associated with the use of plasma products remains a concern, particularly in the pediatric population. Gene therapy has been investigated in mouse models. Gene therapy approaches include in utero administration of a self-inactivated lentiviral vector encoding a full-length ADAMTS13 or MDTCS fragment (86), transplantation of hematopoietic progenitor cells reconstituted with a lentiviral vector encoding a full-length murine ADAMTS13 (87), and intravenous administration of an adeno-associated viral vector-8 (AAV8) encoding human MDTCS (88) for the correction of hereditary TTP. Of these three strategies, AAV8-mediated expression of human MDTCS under a liver-specific promoter, human alpha 1-antitrypsin (hAAT) promoter, shows the greatest promise (88), with sustained ADAMTS13 activity of 0.5–0.7 μg/ml (50–70% activity) without evidence of an immune response. These levels eliminate the circulating ULVWF, and recipients are protected from developing a TTP-like syndrome triggered by bacterial toxin.
Essentially all cases of acquired TTP with severe ADAMTS13 deficiency are caused by autoantibodies against ADAMTS13, which bind and inhibit plasma ADAMTS13. Plasma exchange with a replacement of 1.0–1.5× plasma volumes of fresh frozen plasma or cryosupernatant should be initiated once a presumptive diagnosis of TTP is made. A blood sample should be obtained prior to initiating plasma therapy to measure plasma ADAMTS13 activity and inhibitors. If for some reason this is not done, a blood sample should be obtained even if daily plasma exchange has been performed for several days. Low levels of plasma ADAMTS13 activity and inhibitory autoantibodies persist in patients with active disease (70). This is caused in part by redistribution of anti-ADAMTS13 IgGs from the extravascular space (~55%), which rapidly neutralizes the infused plasma ADAMTS13. In contrast, finding normal to moderately reduced levels of plasma ADAMTS13 activity after treatment has been initiated is not helpful for diagnosis.
How plasma works to treat TTP remains a bit of mystery. Current thought is that plasma exchange removes the “evil humors” that directly or indirectly cause acute episodes of TTP. These include autoantibodies against ADAMTS13 (primarily IgG, followed by IgA and rarely IgM), immune complexes, circulating ULVWF multimers, inflammatory cytokines, bacterial toxins, and activated complement components such as C3b and C5b, etc. Simultaneously, plasma exchange replenishes the deficient or inhibited ADAMTS13 enzyme essential to cleave and remove ULVWF from the endothelial surface, circulation, and the sites of thrombus formation. Plasma exchange may be discontinued after achieving complete resolution of neurological symptoms, normalization of platelet counts, and serum lactate dehydrogenase for three days. Tapering plasma exchange does not appear to reduce the relapse rate.
Approximately 30–40% of TTP patients may experience a relapse within 10 years after attaining a complete remission (77). In such cases, plasma exchange therapy should resume as early as possible. Patients with hereditary TTP and plasma ADAMTS13 activity of <3% are more likely to relapse than those with plasma ADAMTS13 of >9%. Similar findings have been reported in patients with acquired TTP in clinical remission, who are more likely to relapse if their plasma ADAMTS13 activity is <10%. If plasma therapy fails to induce remission or if renal function deteriorates despite an adequate volume of plasma exchanged even in the face of rising platelet count, the diagnosis of aHUS and the therapy that targets terminal complement C5b–9 complexes with eculizumab (a humanized monoclonal antibody against complement C5) should be considered (89). Plasma exchange alone does not prevent or reduce the risk of renal failure in patients with aHUS. A recent study has shown that patients with a clinical diagnosis of acquired idiopathic TTP also have elevated levels of serum or plasma C3a and soluble C5b–9 complexes, which correlate with poor outcome (90). Whether eculizumab should be used in patients with refractory or relapsing TTP merits study.
Refractory acquired TTP patients may benefit from other adjunctive therapies including corticosteroids, vincristine, cyclosporine, cyclophosphamide, and a humanized anti-CD20 monoclonal antibody (rituximab). Rituximab has gained popularity because of its high efficacy, low toxicity, and likely physician familiarity. In one review, each of 118 patients (64% refractory, 36% relapsed) achieved clinical remission after treatment with rituximab (91). Typically, rituximab is given in courses of four weekly doses of 375 mg/m2, which raises plasma ADAMTS13 activity and eliminates anti-ADAMTS13 autoantibodies in these patients.
Several novel therapeutic strategies to treat TTP have been evaluated in preclinical models or clinical trials with some success. These include the use of an antiplatelet glycoprotein 1b (GP1b) nanobody (92) and an anti-VWF A1 aptamer (93), both of which disrupt the interaction between the VWF-A1 domain and platelet GP1b, thereby preventing the formation of VWF-platelet aggregates, the pathological hallmark of TTP.
PERSPECTIVE
Deficiency of plasma ADAMTS13 activity and increased plasma VWF concentrations are also associated with increased risk for myocardial infarction (94), ischemic cerebral infarction (95), preeclampsia (96), and severe cerebral malaria (97). Studies in various animal models have confirmed the role of VWF and ADAMTS13 in the pathogenesis of these arterial thrombotic diseases. Mice lacking Adamts13 exhibit a greater propensity to develop postischemic/reperfusion injury to the heart and the brain than wild type mice or Adamts13−/− mice given recombinant human ADAMTS13 prior to injury (98). Moreover, Adamts13−/− mice develop more numerous and larger atherosclerotic plaques in the aorta and aortic arches than wild type mice after being fed normal chow or a high-fat diet (99, 100). A marked increase in leukocyte adhesion to the injured vessel walls in Adamts13−/− mice and macrophage infiltration into aortic tissues (99, 100) suggests a role for ADAMTS13 in attenuating acute and chronic inflammation in addition to its primary role in inhibiting thrombus formation.
Thus, the discovery of ADAMTS13 from the study of patients with a rare hematologic disorder, TTP, has provided us with a valuable tool to better understand the mechanism of hemostasis and thrombosis, as well as other related disease processes. Success in developing novel therapies for TTP may prove applicable to therapeutic intervention in more prevalent diseases, such as myocardial infarction, stroke, and malaria, associated with perturbations in the ADAMTS13/VWF axis.
SUMMARY POINTS.
ADAMTS13 is a key enzyme regulating VWF function.
Deficiency of plasma ADAMTS13 resulting from hereditary mutations or acquired autoantibodies causes TTP.
Biochemical studies of ADAMTS13 have provided valuable information for better understanding of the molecular mechanisms of TTP and other thrombotic diseases.
ADAMTS13 testing is critical for diagnosis and differential diagnosis of TTP. It helps stratify patients for a more targeted therapy and for prediction of relapse and outcome.
Plasma therapy remains the initial treatment of choice, but other adjunctive therapeutics should be considered for refractory or relapsed cases. Novel therapeutics for TTP are actively under development.
FUTURE ISSUES.
TTP is a rare disease, requiring large-scale collaboration for testing the efficacy of novel therapeutics.
Severe deficiency of ADAMTS13 activity defines a subset of hereditary or acquired TTP, but the mechanisms of secondary TTP or TMA remain to be elucidated.
Whether complement activation is an inciting factor or a prognostic marker for TTP and whether eculizumab has a role in treating refractory or relapsed TTP patients deserve further investigation.
ACKNOWLEDGMENTS
The author thanks Dr. Douglas Cines for his critical comments on the manuscript. Original work from the author's laboratory was supported in part by grants from the National Institutes of Health (R01HL-079027, P01HL-074124, and R01HL-115187) and by an Established Investigator Award (AHA-0940100N) from the American Heart Association.
Glossary
- ULVWF
ultralarge von Willebrand factor
- aHUS
atypical hemolytic uremic syndrome
- TMA
thrombotic microangiopathy
Footnotes
DISCLOSURE STATEMENT The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Moschcowitz E. Hyaline thrombosis of the terminal arterioles and capillaries: a hitherto undescribed disease. Proc. N.Y. Pathol. Soc. 1924;24:21–24. [Google Scholar]
- 2.Schulman I, Pierce M, Lukens A, et al. Studies on thrombopoiesis. I. A factor in normal human plasma required for platelet production; chronic thrombocytopenia due to its deficiency. Blood. 1960;16:943–57. [PubMed] [Google Scholar]
- 3.Upshaw JD., Jr. Congenital deficiency of a factor in normal plasma that reverses microangiopathic hemolysis and thrombocytopenia. N. Engl. J. Med. 1978;298:1350–25. doi: 10.1056/NEJM197806152982407. [DOI] [PubMed] [Google Scholar]
- 4.Rennard S, Abe S. Decreased cold-insoluble globulin in congenital thrombocytopenia (Upshaw-Schulman syndrome) N. Engl. J. Med. 1979;300:368. doi: 10.1056/NEJM197902153000718. [DOI] [PubMed] [Google Scholar]
- 5.Kinoshita S, Yoshioka A, Park YD, et al. Upshaw-Schulman syndrome revisited: a concept of congenital thrombotic thrombocytopenic purpura. Int. J. Hematol. 2001;74:101–8. doi: 10.1007/BF02982558. [DOI] [PubMed] [Google Scholar]
- 6.Amorosi E, Ultmann J. Thrombotic thrombocytopenic purpura: report of 16 cases and review of the literature. Medicine. 1966;45:139–59. [Google Scholar]
- 7.Bell WR, Braine HG, Ness PM, et al. Improved survival in thrombotic thrombocytopenic purpura–hemolytic uremic syndrome. Clinical experience in 108 patients. N. Engl. J. Med. 1991;325:398–403. doi: 10.1056/NEJM199108083250605. [DOI] [PubMed] [Google Scholar]
- 8.Tsai HM, Lian EC. Antibodies to von Willebrand factor–cleaving protease in acute thrombotic thrombocytopenic purpura. N. Engl. J. Med. 1998;339:1585–94. doi: 10.1056/NEJM199811263392203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N. Engl. J. Med. 1991;325:393–97. doi: 10.1056/NEJM199108083250604. [DOI] [PubMed] [Google Scholar]
- 10.Moake JL, Rudy CK, Troll JH, et al. Unusually large plasma factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N. Engl. J. Med. 1982;307:1432–35. doi: 10.1056/NEJM198212023072306. [DOI] [PubMed] [Google Scholar]
- 11.Furlan M, Robles R, Solenthaler M, et al. Deficient activity of von Willebrand factor–cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood. 1997;89:3097–103. [PubMed] [Google Scholar]
- 12.Furlan M, Robles R, Solenthaler M, et al. Acquired deficiency of von Willebrand factor–cleaving protease in a patient with thrombotic thrombocytopenic purpura. Blood. 1998;91:2839–46. [PubMed] [Google Scholar]
- 13.Furlan M, Robles R, Lammle B. Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood. 1996;87:4223–34. [PubMed] [Google Scholar]
- 14.Tsai HM. Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood. 1996;87:4235–44. [PubMed] [Google Scholar]
- 15.Fujikawa K, Suzuki H, McMullen B, et al. Purification of human von Willebrand factor–cleaving protease and its identification as a new member of the metalloproteinase family. Blood. 2001;98:1662–66. doi: 10.1182/blood.v98.6.1662. [DOI] [PubMed] [Google Scholar]
- 16.Gerritsen HE, Robles R, Lammle B, et al. Partial amino acid sequence of purified von Willebrand factor–cleaving protease. Blood. 2001;98:1654–61. doi: 10.1182/blood.v98.6.1654. [DOI] [PubMed] [Google Scholar]
- 17.Zheng X, Chung D, Takayama TK, et al. Structure of von Willebrand factor–cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J. Biol. Chem. 2001;276:41059–63. doi: 10.1074/jbc.C100515200. [DOI] [PubMed] [Google Scholar]
- 18.Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–94. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 19.Plaimauer B, Zimmermann K, Volkel D, et al. Cloning, expression, and functional characterization of the von Willebrand factor–cleaving protease (ADAMTS13) Blood. 2002;100:3626–32. doi: 10.1182/blood-2002-05-1397. [DOI] [PubMed] [Google Scholar]
- 20.Zheng X, Nishio K, Majerus EM, et al. Cleavage of von Willebrand factor requires the spacer domain of the metalloprotease ADAMTS13. J. Biol. Chem. 2003;278:30136–41. doi: 10.1074/jbc.M305331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–39. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 22.Rieger M, Ferrari S, Kremer Hovinga JA, et al. Relation between ADAMTS13 activity and ADAMTS13 antigen levels in healthy donors and patients with thrombotic microangiopathies (TMA) Thromb. Haemost. 2006;95:212–20. doi: 10.1160/TH05-08-0550. [DOI] [PubMed] [Google Scholar]
- 23.Uemura M, Tatsumi K, Matsumoto M, et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood. 2005;106:922–24. doi: 10.1182/blood-2005-01-0152. [DOI] [PubMed] [Google Scholar]
- 24.Zhou W, Inada M, Lee TP, et al. ADAMTS13 is expressed in hepatic stellate cells. Lab. Invest. 2005;85:780–88. doi: 10.1038/labinvest.3700275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niiya M, Uemura M, Zheng XW, et al. Increased ADAMTS-13 proteolytic activity in rat hepatic stellate cells upon activation in vitro and in vivo. J. Thromb. Haemost. 2006;4:1063–70. doi: 10.1111/j.1538-7836.2006.01893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe N, Ikeda H, Kume Y, et al. Increased production of ADAMTS13 in hepatic stellate cells contributes to enhanced plasma ADAMTS13 activity in rat models of cholestasis and steatohepatitis. Thromb. Haemost. 2009;102:389–96. doi: 10.1160/TH08-11-0732. [DOI] [PubMed] [Google Scholar]
- 27.Kume Y, Ikeda H, Inoue M, et al. Hepatic stellate cell damage may lead to decreased plasma ADAMTS13 activity in rats. FEBS Lett. 2007;581:1631–34. doi: 10.1016/j.febslet.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 28.Shang D, Zheng XW, Niiya M, et al. Apical sorting of ADAMTS13 in vascular endothelial cells and Madin-Darby canine kidney cells depends on the CUB domains and their association with lipid rafts. Blood. 2006;108:2207–15. doi: 10.1182/blood-2006-02-002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki M, Murata M, Matsubara Y, et al. Detection of von Willebrand factor–cleaving protease (ADAMTS-13) in human platelets. Biochem. Biophys. Res. Commun. 2004;313:212–16. doi: 10.1016/j.bbrc.2003.11.111. [DOI] [PubMed] [Google Scholar]
- 30.Manea M, Kristoffersson A, Schneppenheim R, et al. Podocytes express ADAMTS13 in normal renal cortex and in patients with thrombotic thrombocytopenic purpura. Br. J. Haematol. 2007;138:651–62. doi: 10.1111/j.1365-2141.2007.06694.x. [DOI] [PubMed] [Google Scholar]
- 31.Tauchi R, Imagama S, Ohgomori T, et al. ADAMTS-13 is produced by glial cells and upregulated after spinal cord injury. Neurosci. Lett. 2012;517:1–6. doi: 10.1016/j.neulet.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 32.Mannucci PM, Capoferri C, Canciani MT. Plasma levels of von Willebrand factor regulate ADAMTS-13, its major cleaving protease. Br. J. Haematol. 2004;126:213–18. doi: 10.1111/j.1365-2141.2004.05009.x. [DOI] [PubMed] [Google Scholar]
- 33.Cao WJ, Niiya M, Zheng XW, et al. Inflammatory cytokines inhibit ADAMTS13 synthesis in hepatic stellate cells and endothelial cells. J. Thromb. Haemost. 2008;6:1233–35. doi: 10.1111/j.1538-7836.2008.02989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Westwood JP, Langley K, Heelas E, et al. Complement and cytokine response in acute thrombotic thrombocytopenic purpura. Br. J. Haematol. 2014;164:858–66. doi: 10.1111/bjh.12707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen L, Lu G, Dong N, et al. Simvastatin increases ADAMTS13 expression in podocytes. Thromb. Res. 2013;132:94–99. doi: 10.1016/j.thromres.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 36.Borchiellini A, Fijnvandraat K, ten Cate JW, et al. Quantitative analysis of von Willebrand factor propeptide release in vivo: effect of experimental endotoxemia and administration of 1-deamino-8-D-arginine vasopressin in humans. Blood. 1996;88:2951–58. [PubMed] [Google Scholar]
- 37.Hattori R, Hamilton KK, McEver RP, et al. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J. Biol. Chem. 1989;264:9053–60. [PubMed] [Google Scholar]
- 38.Huang J, Motto DG, Bundle DR, et al. Shiga toxin B subunits induce VWF secretion by human endothelial cells and thrombotic microangiopathy in ADAMTS13-deficient mice. Blood. 2010;116:3653–59. doi: 10.1182/blood-2010-02-271957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bao J, Xiao J, Mao Y, et al. Carboxyl terminus of ADAMTS13 directly inhibits platelet aggregation and ultra large von Willebrand factor string formation under flow in a free-thiol-dependent manner. Arterioscler. Thromb. Vasc. Biol. 2014;34:397–407. doi: 10.1161/ATVBAHA.113.302547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mannucci PM. Desmopressin (DDAVP) in the treatment of bleeding disorders: the first twenty years. Haemophilia. 2000;6(Suppl. 1):60–67. doi: 10.1046/j.1365-2516.2000.00059.x. [DOI] [PubMed] [Google Scholar]
- 41.Choi H, Aboulfatova K, Pownall HJ, et al. Shear-induced disulfide bond formation regulates adhesion activity of von Willebrand factor. J. Biol. Chem. 2007;282:35604–11. doi: 10.1074/jbc.M704047200. [DOI] [PubMed] [Google Scholar]
- 42.Yeh HC, Zhou Z, Choi H, et al. Disulfide bond reduction of von Willebrand factor by ADAMTS-13. J. Thromb. Haemost. 2010;8:2778–88. doi: 10.1111/j.1538-7836.2010.04094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGrath RT, van den Biggelaar M, Byrne B, et al. Altered glycosylation of platelet-derived von Willebrand factor confers resistance to ADAMTS13 proteolysis. Blood. 2013;122:4107–10. doi: 10.1182/blood-2013-04-496851. [DOI] [PubMed] [Google Scholar]
- 44.Majerus EM, Anderson PJ, Sadler JE. Binding of ADAMTS13 to von Willebrand factor. J. Biol. Chem. 2005;280:21773–78. doi: 10.1074/jbc.M502529200. [DOI] [PubMed] [Google Scholar]
- 45.Jin SY, Skipwith CG, Shang D, et al. von Willebrand factor cleaved from endothelial cells by ADAMTS13 remains ultralarge in size. J. Thromb. Haemost. 2009;7:1749–52. doi: 10.1111/j.1538-7836.2009.03570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skipwith CG, Cao W, Zheng XL. Factor VIII and platelets synergistically accelerate cleavage of von Willebrand factor by ADAMTS13 under fluid shear stress. J. Biol. Chem. 2010;285:28596–603. doi: 10.1074/jbc.M110.131227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao W, Krishnaswamy S, Camire RM, et al. Factor VIII accelerates proteolytic cleavage of von Willebrand factor by ADAMTS13. Proc. Natl. Acad. Sci. USA. 2008;105:7416–21. doi: 10.1073/pnas.0801735105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Furlan M, Robles R, Galbusera M, et al. von Willebrand factor–cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N. Engl. J. Med. 1998;339:1578–84. doi: 10.1056/NEJM199811263392202. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Halvorsen K, Zhang CZ, et al. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. 2009;324:1330–34. doi: 10.1126/science.1170905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lynch CJ, Lane DA, Luken BM. Control of VWF A2 domain stability and ADAMTS13 access to the scissile bond of full-length VWF. Blood. 2014;123:2585–92. doi: 10.1182/blood-2013-11-538173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu AJ, Springer TA. Calcium stabilizes the von Willebrand factor A2 domain by promoting refolding. Proc. Natl. Acad. Sci. USA. 2012;109:3742–47. doi: 10.1073/pnas.1121261109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tao Z, Wang Y, Choi H, et al. Cleavage of ultralarge multimers of von Willebrand factor by C-terminal-truncated mutants of ADAMTS-13 under flow. Blood. 2005;106:141–43. doi: 10.1182/blood-2004-11-4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ai J, Smith P, Wang S, et al. The proximal carboxyl-terminal domains of ADAMTS13 determine substrate specificity and are all required for cleavage of von Willebrand factor. J. Biol. Chem. 2005;280:29428–34. doi: 10.1074/jbc.M505513200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin SY, Skipwith CG, Zheng XL. Amino acid residues Arg(659), Arg(660), and Tyr(661) in the spacer domain of ADAMTS13 are critical for cleavage of von Willebrand factor. Blood. 2010;115:2300–10. doi: 10.1182/blood-2009-07-235101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao W, Anderson PJ, Sadler JE. Extensive contacts between ADAMTS13 exosites and von Willebrand factor domain A2 contribute to substrate specificity. Blood. 2008;112:1713–19. doi: 10.1182/blood-2008-04-148759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang P, Pan W, Rux AH, et al. The cooperative activity between the carboxyl-terminal TSP1 repeats and the CUB domains of ADAMTS13 is crucial for recognition of von Willebrand factor under flow. Blood. 2007;110:1887–94. doi: 10.1182/blood-2007-04-083329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tao Z, Peng Y, Nolasco L, et al. Recombinant CUB-1 domain polypeptide inhibits the cleavage of ULVWF strings by ADAMTS13 under flow conditions. Blood. 2005;106:4139–45. doi: 10.1182/blood-2005-05-2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Banno F, Chauhan AK, Kokame K, et al. The distal carboxyl-terminal domains of ADAMTS13 are required for regulation of in vivo thrombus formation. Blood. 2009;113:5323–29. doi: 10.1182/blood-2008-07-169359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiao J, Jin SY, Xue J, et al. Essential domains of a disintegrin and metalloprotease with thrombospondin type 1 repeats-13 metalloprotease required for modulation of arterial thrombosis. Arterioscler. Thromb. Vasc. Biol. 2011;31:2261–69. doi: 10.1161/ATVBAHA.111.229609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng XL, Wu HM, Shang D, et al. Multiple domains of ADAMTS13 are targeted by autoanti-bodies against ADAMTS13 in patients with acquired idiopathic thrombotic thrombocytopenic purpura. Haematologica. 2010;95:1555–62. doi: 10.3324/haematol.2009.019299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng XL. Structure-function and regulation of ADAMTS-13 protease. J. Thromb. Haemost. 2013;11(Suppl. 1):11–23. doi: 10.1111/jth.12221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pos W, Sorvillo N, Fijnheer R, et al. Residues Arg568 and Phe592 contribute to an antigenic surface for anti-ADAMTS13 antibodies in the spacer domain. Haematologica. 2011;96:1670–77. doi: 10.3324/haematol.2010.036327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jian C, Xiao J, Gong L, et al. Gain-of-function ADAMTS13 variants that are resistant to autoantibodies against ADAMTS13 in patients with acquired thrombotic thrombocytopenic purpura. Blood. 2012;119:3836–43. doi: 10.1182/blood-2011-12-399501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arbus GS. Association of verotoxin-producing E. coli and verotoxin with hemolytic uremic syndrome. Kidney Int. Suppl. 1997;58:S91–96. [PubMed] [Google Scholar]
- 65.Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin. Nephrol. 2013;33:508–30. doi: 10.1016/j.semnephrol.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dragon-Durey MA, Blanc C, Garnier A, et al. Anti-factor H autoantibody-associated hemolytic uremic syndrome: review of literature of the autoimmune form of HUS. Semin. Thromb. Hemost. 2010;36:633–40. doi: 10.1055/s-0030-1262885. [DOI] [PubMed] [Google Scholar]
- 67.Kavanagh D, Pappworth IY, Anderson H, et al. Factor I autoantibodies in patients with atypical hemolytic uremic syndrome: disease-associated or an epiphenomenon? Clin. J. Am. Soc. Nephrol. 2012;7:417–26. doi: 10.2215/CJN.05750611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Delvaeye M, Noris M, De Vriese A, et al. Thrombomodulin mutations in atypical hemolyticuremic syndrome. N. Engl. J. Med. 2009;361:345–57. doi: 10.1056/NEJMoa0810739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lemaire M, Fremeaux-Bacchi V, Schaefer F, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat. Genet. 2013;45:531–36. doi: 10.1038/ng.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zheng XL, Kaufman RM, Goodnough LT, et al. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood. 2004;103:4043–49. doi: 10.1182/blood-2003-11-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Coppo P, Wolf M, Veyradier A, et al. Prognostic value of inhibitory anti-ADAMTS13 antibodies in adult-acquired thrombotic thrombocytopenic purpura. Br. J. Haematol. 2006;132:66–74. doi: 10.1111/j.1365-2141.2005.05837.x. [DOI] [PubMed] [Google Scholar]
- 72.Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch. Pathol. Lab. Med. 2014;138:546–49. doi: 10.5858/arpa.2013-0170-OA. [DOI] [PubMed] [Google Scholar]
- 73.Furlan M, Lammle B. Deficiency of von Willebrand factor–cleaving protease in familial and acquired thrombotic thrombocytopenic purpura. Baillieres Clin. Haemat. 1998;11:509–14. doi: 10.1016/s0950-3536(98)80064-4. [DOI] [PubMed] [Google Scholar]
- 74.Gerritsen HE, Turecek PL, Schwarz HP, et al. Assay of von Willebrand factor (vWF)-cleaving protease based on decreased collagen binding affinity of degraded vWF: a tool for the diagnosis of thrombotic thrombocytopenic purpura (TTP) Thromb. Haemost. 1999;82:1386–89. [PubMed] [Google Scholar]
- 75.Knovich MA, Lawson HL, Burke MH, et al. Rapid quantitative assay of ADAMTS13 activity on an automated coagulation analyzer: clinical applications and comparison with immunoblot method. Am. J. Hematol. 2008;83:654–56. doi: 10.1002/ajh.21220. [DOI] [PubMed] [Google Scholar]
- 76.Kokame K, Nobe Y, Kokubo Y, et al. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br. J. Haematol. 2005;129:93–100. doi: 10.1111/j.1365-2141.2005.05420.x. [DOI] [PubMed] [Google Scholar]
- 77.Kremer Hovinga JA, Vesely SK, Terrell DR, et al. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood. 2010;115:1500–11. doi: 10.1182/blood-2009-09-243790. [DOI] [PubMed] [Google Scholar]
- 78.Meyer SC, Sulzer I, Lammle B, et al. Hyperbilirubinemia interferes with ADAMTS-13 activity measurement by FRETS-VWF73 assay: diagnostic relevance in patients suffering from acute thrombotic microangiopathies. J. Thromb. Haemost. 2007;5:866–67. doi: 10.1111/j.1538-7836.2007.02438.x. [DOI] [PubMed] [Google Scholar]
- 79.Muia J, Gao W, Haberichter SL, et al. An optimized fluorogenic ADAMTS13 assay with increased sensitivity for the investigation of patients with thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2013;11:1511–18. doi: 10.1111/jth.12319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Studt JD, Kremer Hovinga JA, Antoine G, et al. Fatal congenital thrombotic thrombocytopenic purpura with apparent ADAMTS13 inhibitor: in vitro inhibition of ADAMTS13 activity by hemoglobin. Blood. 2005;105:542–44. doi: 10.1182/blood-2004-06-2096. [DOI] [PubMed] [Google Scholar]
- 81.Mancini I, Valsecchi C, Lotta LA, et al. FRETS-VWF73 rather than CBA assay reflects ADAMTS13 proteolytic activity in acquired thrombotic thrombocytopenic purpura patients. Thromb. Haemost. 2014;112:297–303. doi: 10.1160/TH13-08-0688. [DOI] [PubMed] [Google Scholar]
- 82.Mackie I, Langley K, Chitolie A, et al. Discrepancies between ADAMTS13 activity assays in patients with thrombotic microangiopathies. Thromb. Haemost. 2013;109:488–96. doi: 10.1160/TH12-08-0565. [DOI] [PubMed] [Google Scholar]
- 83.Bianchi V, Robles R, Alberio L, et al. Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: a severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood. 2002;100:710–13. doi: 10.1182/blood-2002-02-0344. [DOI] [PubMed] [Google Scholar]
- 84.Ono T, Mimuro J, Madoiwa S, et al. Severe secondary deficiency of von Willebrand factor–cleaving protease (ADAMTS13) in patients with sepsis-induced disseminated intravascular coagulation: its correlation with development of renal failure. Blood. 2006;107:528–34. doi: 10.1182/blood-2005-03-1087. [DOI] [PubMed] [Google Scholar]
- 85.Furlan M, Lammle B. Aetiology and pathogenesis of thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome: the role of von Willebrand factor-cleaving protease. Best Prac. Res. Clin. Haemat. 2001;14:437–54. doi: 10.1053/beha.2001.0142. [DOI] [PubMed] [Google Scholar]
- 86.Niiya M, Endo M, Shang D, et al. Correction of ADAMTS13 deficiency by in utero gene transfer of lentiviral vector encoding ADAMTS13 genes. Mol. Ther. 2009;17:34–41. doi: 10.1038/mt.2008.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Laje P, Shang D, Cao W, et al. Correction of murine ADAMTS13 deficiency by hematopoietic progenitor cell-mediated gene therapy. Blood. 2009;113:2172–80. doi: 10.1182/blood-2008-08-173021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jin SY, Xiao J, Bao J, et al. AAV-mediated expression of an ADAMTS13 variant prevents shigatoxin-induced thrombotic thrombocytopenic purpura. Blood. 2013;121:3825–29. S1–3. doi: 10.1182/blood-2013-02-486779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cataland SR, Wu HM. How I treat: the clinical differentiation and initial treatment of adult patients with atypical hemolytic uremic syndrome. Blood. 2014;123:2478–84. doi: 10.1182/blood-2013-11-516237. [DOI] [PubMed] [Google Scholar]
- 90.Wu TC, Yang S, Haven S, et al. Complement activation and mortality during an acute episode of thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2013;11:1925–27. doi: 10.1111/jth.12369. [DOI] [PubMed] [Google Scholar]
- 91.Caramazza D, Quintini G, Abbene I, et al. Relapsing or refractory idiopathic thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: the role of rituximab. Transfusion. 2010;50:2753–60. doi: 10.1111/j.1537-2995.2010.02763.x. [DOI] [PubMed] [Google Scholar]
- 92.Callewaert F, Roodt J, Ulrichts H, et al. Evaluation of efficacy and safety of the anti-VWF nanobody ALX-0681 in a preclinical baboon model of acquired thrombotic thrombocytopenic purpura. Blood. 2012;120:3603–10. doi: 10.1182/blood-2012-04-420943. [DOI] [PubMed] [Google Scholar]
- 93.Knobl P, Jilma B, Gilbert JC, et al. Anti-von Willebrand factor aptamer ARC1779 for refractory thrombotic thrombocytopenic purpura. Transfusion. 2009;49:2181–85. doi: 10.1111/j.1537-2995.2009.02232.x. [DOI] [PubMed] [Google Scholar]
- 94.Matsukawa M, Kaikita K, Soejima K, et al. Serial changes in von Willebrand factor-cleaving protease (ADAMTS13) and prognosis after acute myocardial infarction. Am. J. Cardiol. 2007;100:758–63. doi: 10.1016/j.amjcard.2007.03.095. [DOI] [PubMed] [Google Scholar]
- 95.Hanson E, Jood K, Nilsson S, et al. Association between genetic variation at the ADAMTS13 locus and ischemic stroke. J. Thromb. Haemost. 2009;7:2147–48. doi: 10.1111/j.1538-7836.2009.03617.x. [DOI] [PubMed] [Google Scholar]
- 96.Stepanian A, Cohen-Moatti M, Sanglier T, et al. Von Willebrand factor and ADAMTS13: a candidate couple for preeclampsia pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2011;31:1703–9. doi: 10.1161/ATVBAHA.111.223610. [DOI] [PubMed] [Google Scholar]
- 97.Larkin D, de Laat B, Jenkins PV, et al. Severe Plasmodium falciparum malaria is associated with circulating ultra-large von Willebrand multimers and ADAMTS13 inhibition. PLOS Pathog. 2009;5:e1000349. doi: 10.1371/journal.ppat.1000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fujioka M, Hayakawa K, Mishima K, et al. ADAMTS13 gene deletion aggravates ischemic brain damage: a possible neuroprotective role of ADAMTS13 by ameliorating postischemic hypoperfusion. Blood. 2010;115:1650–53. doi: 10.1182/blood-2009-06-230110. [DOI] [PubMed] [Google Scholar]
- 99.Gandhi C, Khan MM, Lentz SR, et al. ADAMTS13 reduces vascular inflammation and the development of early atherosclerosis in mice. Blood. 2012;119:2385–91. doi: 10.1182/blood-2011-09-376202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jin SY, Tohyama J, Bauer RC, et al. Genetic ablation of Adamts13 gene dramatically accelerates the formation of early atherosclerosis in a murine model. Arterioscler. Thromb. Vasc. Biol. 2012;32:1817–23. doi: 10.1161/ATVBAHA.112.247262. [DOI] [PMC free article] [PubMed] [Google Scholar]